

Abstract
Astrocytes play numerous vital roles in the central nervous system. Accordingly, it is of merit to identify structural and functional properties of astrocytes in both health and disease. The majority of studies examining the morphology of astrocytes have employed immunoassays for markers such as GFAP, which are insufficient to encapsulate the considerable structural complexity of these cells. Herein, we describe a method utilizing a commercially available and validated, genetically-encoded membrane-associated fluorescent marker of astrocytes, AAV-GfaABC1D-Lck-GFP. This tool and approach allows for visualization of a single isolated astrocyte in its entirety, including fine peripheral processes. Astrocytes are imaged using confocal microscopy, and reconstructed in three dimensions to obtain detailed morphometric data. We further provide an immunohistochemistry procedure to assess the colocalization of isolated astrocytes with synaptic markers throughout the z-plane. This technique, which can be utilized on a standard laboratory confocal microscope and Imaris software, allows for detailed analysis of the morphology and synaptic colocalization of astrocytes in fixed tissue.
Keywords: astrocyte, peripheral astrocyte processes, confocal microscopy, morphology, synaptic colocalization, Lck-GFP
Introduction:
Astrocytes perform diverse functions within the central nervous system including regulation of transmitter uptake and release, ion buffering, and fortification of the blood brain barrier, among others (Allen & Barres 2009). Together with pre and post-synaptic membranes, astrocytes form the tripartite synapse, allowing for bidirectional communication between astrocytes and neuronal synapses (Araque et al 1999, Durkee & Araque 2019, Perea et al 2009). By virtue of the tripartite synapse, astrocytes regulate the development and maturation of neuronal synapses, as well as synaptic transmission and plasticity (Allen & Eroglu 2017, De Pitta et al 2016, Durkee & Araque 2019, Perez-Alvarez & Araque 2013). Moreover, structural and physiological adaptations of astrocytes have been implicated in a wide range of both neurodegenerative and neuropsychiatric diseases in which abnormal synaptic function is observed (Blanco-Suarez et al 2017, Kim et al 2018).
The majority of studies that have investigated the structural changes in astrocytes have typically employed immunoassays for astrocyte markers. One of the more popular markers of astrocytes is glial fibrillary acidic protein (GFAP) (Hol & Pekny 2015, Middeldorp & Hol 2011), an intermediate filament protein and cytoskeletal marker for astrocytes. However, not all astrocytes express GFAP, and variance in the levels of GFAP expression is observed across brain regions (Kettenmann & Verkhratsky 2011, Sofroniew & Vinters 2010). Moreover, GFAP does not encapsulate the structural complexity of astrocytes, including any changes that occur along the peripheral processes and end feet of astrocytes.
In order to more completely observe structural properties of astrocytes, our lab and others have utilized a lymphocyte specific protein tyrosine kinase (Lck) green fluorescent protein (Scofield et al 2016, Shigetomi et al 2010b, Testen et al 2019, Testen et al 2018). Post-translational modification on the N-terminus of Lck renders association to the plasma membrane. When under the control of the astrocyte-specific GfaABC1D promoter, GFP is expressed along the plasma membrane of astrocytes, allowing for GFP labeling of peripheral processes not observed with a cytosolic eGFP (Benediktsson et al 2005, Shigetomi et al 2013, Shigetomi et al 2010b). The area of Lck-GFP-labeled astrocytes is approximately 10-fold greater than astrocytes that only express GFAP (Shigetomi et al 2013), demonstrating the efficacy of Lck-GFP in studying the structural plasticity of astrocytes.
Using high-resolution confocal microscopy, a single, isolated Lck-GFP-expressing astrocyte can be imaged in its entirety in three dimensions. Bitplane IMARIS software is then used to obtain measurements of the morphometric properties of the astrocyte including surface area and volume. In addition, immunohistochemistry for synaptic markers such as synapsin-1 and post-synaptic density 95 can be employed to assess synaptic colocalization of Lck-GFP. Using these methods, our lab and others have shown changes in nucleus accumbens astrocytes associated with self-administration of cocaine (Scofield et al 2016, Testen et al 2018), heroin, and methamphetamine (Kruyer et al 2019, Siemsen et al 2019), as well as in other structures across development (Testen et al 2019) and in response to severe stress (Jones et al 2018). The general methods employed in these above-mentioned studies are described in the protocols below. Basic protocol 1 describes surgical methods to microinject AAV-Lck-GFP. Basic protocol 2 illustrates tissue processing methods including brain extraction, sectioning, and immunohistochemistry procedures. Basic protocol 3 describes image acquisition of Lck-GFP expressing astrocytes using confocal microscopy. Basic protocol 4 details image analysis using IMARIS software.
BASIC PROTOCOL 1
MICROINJECTION OF AAV5-GFAABC1D-LCK GFP INTO THE NUCLEUS ACCUMBENS (NAC) OF RATS
Within this protocol, we detail how infusion pumps are primed to microinject AAV-Lck-GFP into brain tissue. We have generally found that a minimum of 3 weeks is needed for sufficient viral expression and visualization of Lck-GFP expressing astrocytes. Furthermore, we have found suitable viral expression at longer timepoints (~30 days (Scofield et al 2016, Testen et al 2018), ~60 days (Kim et al, in preparation) and 70 days (Testen et al 2019)), without evidence of reactive astrocytes.
NOTE: All animal procedures were performed with approval from the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill, and in accordance with the Guide for the Care and Use of Laboratory Animals.
Materials
Adeno-associated virus (University of North Carolina Vector Core): AAV5-GfaABC1D-Lck GFP 6.2 × 1012 virus molecules/ml, 1 μl microinjected into the NAc core (the plasmid (#105598) and AAV in serotype 5 (105598-AAV5) are now commercially available through Addgene)
Polyethylene tubing- 0.015” inner diameter, 0.043” outer diameter, PE #20 (Becton Dickinson 427406)
Internal cannula microinjectors- 33 gauge, cut 17 mm below pedestal (Plastics One C315IA-5/SPC)
Hamilton syringes- 10 μl (Hamilton Company 80366)
Sterile saline (Med-Vet International RXSAL-POD1LT)
1 ml syringes (BD 309659)
26-gauge needles (BD 305110)
Infusion pump (Harvard Apparatus 70-4501)
Dual arm stereotaxic instrument- Small animal stereotaxic instrument with digital display (Kopf model 942)
Heating pad (ALA Scientific Instruments ALA HEATINGPAD-2)
Sterile drapes (Dynarex 4410)
Sterile surgical gloves (Kimberly Clark 55092)
Sterile surgical tools (scalpel (Fine Science Tools 10004-13), forceps (Fine Science Tools 11373-12, hemostats (Fine Science Tools 13020-12), scissors (Fine Science Tools 14060-09))
Scalpel blade (Fine Science Tools 10021-00)
Sterile cotton swabs (Fisher Scientific 2340019)
Rat (Envigo, male Sprague Dawley, 6–8 weeks old weighing 250–275 g; RGD Cat# 70508, RRID: RGD_70508)
Ketamine (Med-Vet International RXKETAMINE)
Xylazine (Med-Vet International RXANASED-20)
Meloxicam (Med-Vet International RXMELOX-INJ20)
95–100% ethanol (Fisher Scientific 04355222)
Betadine Surgical Scrub (Purdue Products 6761815101)
Shaver (Oster 78004-011)
Ophthalmic ointment (Med-Vet International PH-PARALUBE-VET)
Laboratory tape (Fisher Scientific 15-901-5R)
Sutures (Ethicon PERMA-HAND SILK 683G)
Vetbond tissue adhesive (3M Vetbond 14695B)
Topical antibiotic ointment (Med Vet International CUR001231)
Absorbent underpads (VWR 56617)
Paper towel (VWR 470308-268)
Micro drill with drill bit (Dremel 200 series, 26150106AC)
Prepare microinjectors
-
1Cut polyethylene tubing to appropriate length.The length of the tubing should be long enough to reach from the Hamilton syringe on the infusion pump to the stereotaxic arm.
-
2
Attach microinjectors (unilateral infusion cannula) to polyethylene tubing. Attach the other end to a Hamilton syringe.
-
3Remove the plunger on the Hamilton syringe. Replace the plunger with a 26-gauge needle attached to 1 ml syringe filled with saline. Flush approximately 1 ml of saline through the Hamilton syringe/tubing/microinjector.The flow of saline should not be obstructed. If there is a barrier to the flow of saline, it may be necessary to replace the microinjectors. Furthermore, there should be no air bubbles within the Hamilton syringe/tubing/microinjector. If air bubbles are present, flush an additional 1 ml of saline through the setup.For bilateral microinjections, repeat steps 1–3.
-
4
Place the Hamilton syringe(s) securely on the infusion pump. Push the pump forward to ensure that saline flows through the microinjector(s).
-
5Reverse the flow on the infusion pump to create a small (~ 1 μl) air bubble within the setup.The air bubble will separate the saline from the AAV, and will also be used to visually track movement of the virus into tissue.
-
6Load the AAV into the setup by drawing virus up through the microinjector(s).Before loading, the virus should be first left to thaw on ice for 30 min. After that, mix the virus gently using a pipette. Draw the virus directly from the tube, making sure it is enough for all microinjections. For example, for bilateral microinjections in two rats, draw ~4–5 μl. Any remaining thawed aliquots of the AAV should be kept on ice when not in the tubing.
-
7
Secure microinjector(s) onto stereotaxic arm by using laboratory tape and cannula holders (Fig. 1).
Figure 1.

Stereotaxic microinjection setup. A) Stereotaxic instrument (Kopf) with 33G microinjectors secured bilaterally to the stereotaxic arms and connected to a microinjection PE20 tubing. B) A close up of microinjectors being secured to a stereotaxic arm using laboratory tape.
Prepare rat
-
8Anesthetize rat with 100 mg/kg ketamine and 7 mg/kg xylazine.The rat is fully anesthetized if there is no physiological response following a toe pinch. The absence of a toe pinch response should be confirmed approximately every 20 min during the surgery, in all four quadrants. If a toe pinch response is observed, a booster dose of ketamine (20–40 mg/kg) can be administered. The plane of anesthesia can be monitored by assessment of heart rate and breathing.
-
9Inject rat with 4 mg/kg meloxicam (i.p.).Other analgesics (buprenorphine, ketorolac, carprofen, acetaminophen, ibuprofen, etc.) can be used as a substitute for meloxicam, as per your IACUC protocol.
-
10
Remove the fur on the top of the head using a shaver. This includes the fur from behind the ears to the tip of the nose.
-
11
Place rat in the stereotaxic frame. Maintain a body temperature of approximately 37°C by placing the rat on top of a heating pad (a sterile surgical drape can be placed on top of the heating pad).
-
12
Apply ophthalmic ointment to both eyes. To keep the eyes moist during surgery, ophthalmic ointment can be re-applied as needed.
-
13
Disinfect the scalp using three applications of alternating 95–100% ethanol and betadine (povidone-iodine).
-
14
Using a scalpel, expose the skull by making an incision along the midline of the scalp. Using hemostats, pull the skin edges laterally to expose bregma and lambda. Remove the fascia using forceps and sterile cotton swabs.
-
15
Confirm that the head is level within the stereotaxic frame. This can be accomplished by recording dorsal-ventral coordinates of bregma and lambda.
AAV5-GfaABC1D-Lck-GFP microinjections into the NAc
All surgical techniques are done using aseptic and sterile techniques. This includes the use of sterile drapes, sterile surgical gloves and autoclaved surgical tools.
-
16
Locate bregma using the tip of the microinjectors. Mark bregma and record the coordinates.
-
17
Move microinjectors to desired anterior-posterior (AP) and medial-lateral (ML) coordinates (for NAc: 6° angle, AP: + 1.5 mm, ML: +/− 2.6 mm). Mark new positions of the microinjectors on the surface of the skull.
-
18Drill through the surface of the skull using a micro drill.If excessive bone dust is present, use a saline-dipped cotton swab to remove it.
-
19Lower microinjectors to the desired dorsal-ventral (DV) coordinates (for NAc: 6° angle, DV: −7.2 mm).To avoid clogs within the microinjectors, slowly lower the microinjectors (over 1–2 min) through the skull hole.
-
20Set up the infusion pump to microinject virus (1 μl total volume per hemisphere for NAc) at an infusion rate of 0.05 – 0.1 μl / min. Leave microinjectors in place upon completion of virus infusion for approximately 15 min, to allow diffusion of the virus. Slowly (over 1–2 min) remove the microinjectors from the brain.If performing more than one infusion, gently wipe the microinjectors and mark new positions of the air bubbles. Confirm that the flow through the microinjectors is not obstructed, and that there is enough virus for the next infusion.
-
21
Suture the skin closed and apply a small amount of vetbond tissue adhesive. Apply antibiotic ointment above the skin incision line.
-
22
Place the rat on a paper towel on top of a heating pad. Once ambulatory, return the rat to its home cage.
-
23
Administer analgesics, and/or antibiotics and post-operative care for a minimum of 2–3 days, based on your approved IACUC protocol.
Basic Protocol 2
TISSUE PROCESSING AND IMMUNOHISTOCHEMISTRY (IHC) FOR PSD-95
This protocol describes tissue processing methods for analysis of Lck-GFP expressing astrocytes using confocal microscopy. Deeply anesthetized rats are first perfused with paraformaldehyde and brains are extracted. Brains are then cut into 100 μm sections from the region of interest. Thick sections ensure that astrocytes are imaged in their entirety. Below, we describe our IHC procedure for maximum penetration of the synaptic marker post-synaptic density-95 (PSD-95) on 100 μm coronal slices containing the NAc.
Materials:
Perfusion
Perfusion pump (Cole-Parmer 77410)
Tubing for perfusion pump (Fisher Scientific 14-169-7F)
18-gauge blunted perfusion needle (BD 309659)
1x phosphate buffer (PB)
4% paraformaldehyde (PFA) in 1x PB (Paraformaldehyde from Sigma 158127)
Forceps (Fine Science Tools 11373-12)
Hemostats (Fine Science Tools 13020-12)
Scissors (Fine Science Tools 14060-09)
Rongeurs (Fine Science Tools 16000-14)
Spatula (Fine Science Tools 10094-13)
Vials (Fisher Scientific 03-337-7)
Sodium pentobarbital
1 ml syringes (BD 309659)
26-gauge needles (BD 305110)
Guillotine (World Precision Instruments DCAP-L)
Tools for brain extraction (scissor, forceps, rongeurs, spatulas)
Cryosectioning
30% sucrose in 1x PBS (Sucrose from Sigma S8501)
Cryostat (Leica CM3050S)
Specimen stage (Pathlabs 25M-LE)
Cryostat blade (C.L. Sturkey DT315R50)
O.C.T. compound (Fisher Scientific 23-730-571)
24 well plate (Fisher Scientific 07201590)
50% glycerol/1x PBS solution (Glycerol from Sigma G9012)
Red Sable Brush (Ted Pella 11806)
Razor blade (Thermo Fisher Scientific s17302)
Slides (Fisher Scientific 22-034979)
1x PBS (prepared fresh using CSH Protocol(2006))
Immunohistochemistry
PBST (1x PBS + Triton) (Triton from Fisher Scientific BP151-500)
Normal goat serum (NGS) (Sigma-Aldrich G9023)
Mouse anti PSD-95 antibody (Thermo Fisher Scientific Cat# MA1-045, RRID:AB_325399)
Rabbit anti-GFAP antibody (Agilent Cat# Z0334, RRID:AB_10013382)
Goat anti mouse IgG (H+L) alexa 594 secondary antibody (Thermo Fisher Scientific Cat# A-11032, RRID:AB_2534091)
Goat anti rabbit IgG (H+L) alexa 647 secondary antibody (Thermo Fisher Scientific Cat# A-21245, RRID:AB_2535813)
Laboratory shaker (Fisher Scientific 02-217-987)
Microcentrifuge (Cole-Parmer C1008-B)
Laboratory refrigerator (VWR GDM-23)
Perfusion
For a 300 g rat, the flow rate on the perfusion pump is set to approximately 20–25 ml/min. It generally takes about 200–250 ml of 1x PB and 200–250 ml of 4% paraformaldehyde to perfuse a 300 g rat. The flow rate and volume of each solution can be adjusted based on the body weight of the rat.
-
1
Inject the rat with 100 mg/kg sodium pentobarbital, i.p. Confirm full anesthesia via the absence of a toe pinch response before proceeding.
-
2
Make a horizontal incision just below the diaphragm. Cut rostrally along the lateral edges of the body until the heart is fully exposed.
-
3
Insert perfusion needle into the left ventricle through the apex of the heart. Clip the right atrium to allow flow and turn on perfusion pump.
-
4
Transcardially perfuse the rat with ~200–250 ml of 1x PB.
-
5
Transcardially perfuse the rat with ~200 – 250 ml of 4% PFA made in 1x PB.
-
6Decapitate the rat using a guillotine. Cut through the skin above the skull and remove the skull using rongeurs. Extract the brain and store it in a vial containing ~15 ml of 4% PFA for a maximum of 4 hours.We have found that post-fixation of brains in 4% PFA for more than 4 hours makes it difficult for antibodies to penetrate thick slices during IHC. We recommend post-fixation between 2–4 hours in 4% PFA for optimal IHC staining.
-
7Cryoprotect the brain by transferring it to a new vial containing a 30% sucrose solution. Wait for the brain to sink to the bottom of the vial before beginning cryosectioning (~2 days).As an alternative to cryosectioning (and hence, cryoprotecting the brain in 30% sucrose), brains can be sectioned using a vibratome. In this case, following post-fixation of brains in 4% PFA, brains can be stored in 1x PBS until sectioning.
Cryosectioning
This protocol describes cryosectioning of the brain into coronal slices. Alternatively, the brain can be sectioned into sagittal and horizonal slices.
-
8
Remove the brain from the 30% sucrose solution and place on a flat surface. Using a razor blade, cut off the cerebellum.
-
9Place a small amount of O.C.T. compound on the specimen disk. Using forceps, pick up the brain and place the posterior part of the brain within the O.C.T. compound on the specimen disk. Return to cryostat and let the O.C.T. compound solidify at −20°C for approximately 5 min.At −20°C, the O.C.T. compound will solidify and turn white.
-
10Cover the brain with O.C.T. compound and let it solidify at −20°C for approximately 20 min.It is important to avoid air bubbles when covering the brain with the O.C.T. compound. Any space within the solidified O.C.T. compound will result in uneven sectioning of the brain.
-
11Place the specimen disk on the cryostat stage.At this point, the posterior part of the brain is on the specimen disk, which is secured on the cryostat stage. The anterior part of the brain should be facing the cryostat blade. The dorsal part of the brain should face the top of the cryostat, and the ventral part should face the bottom of the cryostat. The cryostat stage can be adjusted to ensure even sectioning of the brain.
-
12Cut 100 μm thick coronal slices containing the region of interest. Place slices in a 24-well plate containing 50% 1x PBS/glycerol.To confirm that slices are stored properly, each well should contain at least 1 ml of the 50% 1x PBS/glycerol solution. Approximately 3–4 100 μm coronal slices can be stored in each well. Once sectioning is completed, the well plate can be stored at −20°C until IHC.
IHC protocol for 100 μm coronal slices
To assess synaptic colocalization of Lck-GFP expressing astrocytes, IHC for the synaptic marker PSD-95 is performed on coronal slices. Furthermore, to aid in the analysis of Lck-GFP expressing astrocytes, IHC for the astrocyte marker GFAP is also conducted.
Prior to starting the IHC protocol, it is recommended to check coronal slices for GFP expression. Only slices with suitable GFP fluorescence within the region of interest should be used for IHC staining. To check GFP fluorescence, slices near the region of interest are mounted on slides (~2–3 slices per slide) and coverslipped with 1x PBS. Using the 10x objective on the confocal microscope, ensure that slices have an adequate number of astrocytes for image analysis. Once GFP fluorescence is confirmed, the coverslip can be removed, and slices can be transferred to new well plates for IHC. A detailed explanation on use of the confocal microscope is described in the next section. We have typically found that 2 slices per animal (resulting in ~ 8–10 astrocytes per animal) is sufficient for analysis of the morphometric properties and synaptic colocalization of astrocytes, and to power statistical assessment of differences between groups.
To maximize penetration of primary antibodies, each well contains only 1 slice. Furthermore, it is imperative that there is enough solution in the well to completely submerge the slice. We have found that 500 – 750 μl of solution per well is sufficient. Furthermore, the well plate should be covered with aluminum foil at each step to minimize loss of GFP fluorescence.
-
13
Wash slices 3 × 5 min in 1x PBST (0.2% triton x-100) at room temperature on a laboratory shaker at ~ 100 rpm.
-
14
Transfer slices to new wells containing 5% normal goat serum (NGS) in 1x PBST (2% triton x-100). Block slices for 1 hour at room temperature on a laboratory shaker.
-
15Transfer slices to new wells containing blocking solution plus primary antibodies (mouse anti-PSD-95 @ 1:500; rabbit anti-GFAP @ 1:500). Incubate slices in solution for 72 hours at 4°C on a laboratory shaker.For optimal penetration of primary antibodies, flip the slices halfway through the incubation period.
-
16
Transfer slices to new wells for washing. Wash 3 × 5 min in 1x PBST (0.2 % triton x-100) at room temperature on a laboratory shaker.
-
17Transfer slices to new wells containing blocking solution plus secondary antibodies (goat anti-mouse Alexa Fluor 594 @ 1:1000; goat anti-rabbit Alexa Fluor 647 @ 1:1000). Incubate slices in solution for 72 hours at 4°C on a laboratory shaker.For optimal penetration of primary antibodies, flip the slices halfway through the incubation period.
-
18
Transfer slices to new wells for washing. Wash 3 × 10 min in 1x PBST (0.2% triton x-100) at room temperature on a laboratory shaker.
-
19Transfer slices to new wells. Wash 1 × 10 min in 1x PBS.Slices can be stored in 1x PBS at 4°C until imaging.
BASIC PROTOCOL 3
SINGLE-CELL IMAGE ACQUISITION
Single-cell acquisition is performed using fluorescent confocal microscopy. Confocal microscopy enables high-resolution imaging throughout the thickness of fixed sections, facilitating focused images of individual optical planes with minimal amount of light scattering from planes above and below. Care should be taken to select only cells that are within brain region of interest and can be imaged in their entirety. Imaging parameters should be adjusted according to individual requirement and resources. Imaging time can vary greatly based on the size of the cell, selected resolution, number of selected channels (fluorophores used), laser scanning speed and z-axis step size. For this protocol, in addition to astrocyte-specific Lck-GFP reporter, astrocytes were also visualized with anti-GFAP antibody, nuclear stain (DAPI) and neuronal anti-PSD-95 marker. This requires a confocal microscope with 4 distinct fluorescent channels as well as a motorized stage for automatic z-axis adjustment. Our setup included a Zeiss LSM 800 system with accompanied ZEN software. This said, the protocol is compatible with any modern confocal system. The goal is to acquire a complete, isolated z-stack of an astrocyte to be later used for 3-dimensional reconstruction.
Materials:
Microscope slides – Superfrost Plus (Catalog No. 22-034-979, Fisherbrand)
Microscope cover glass (Catalog No. 12545M, Fisherbrand)
Mounting media
DAPI-Fluoromount-G (Catalog No. 0100-20, SouthernBiotech)
OR Fluoromount-G (Catalog No. 0100-01, SouthernBiotech)
Confocal microscope ZEISS LSM 800:
2 gallium arsenide phosphide (GaAsP) detectors
4 diode lasers with 405, 488, 561 and 640 nm excitation wavelength, respectively
470, 545, 620 nm short pass (SP) and 655 nm long pass (LP) filters
10x (“Plan-Neofluar” 10x/0.30, Catalog No. 420340-9901-000), 20x (“Plan-Apochromat” 20x/0.8, Catalog No. 420650-9901-000), 40x (“C-Apochromat” 40x/1.20 water immersion, Catalog no. 421767-9971-711) and 63x (“Plan-Apochromat” 63x/1.4 Oil immersion, Catalog No. 420782-9900-000) objectives (all Zeiss)
Motorized stage (Catalog No. 90-24-550-0000, Märzhäuser Wetzlar)
Fluorescence lamp (X-CITE Series 120PC Q, formerly LUMEN DYNAMICS, now Excelitas)
Immersion oil (Immersol, Zeiss)
Image acquisition workstation running Windows operating system and ZEN blue software.
- Coverslip pre-selected stained sectionsEach slide should contain 1–4 pre-selected, stained sections from Basic Protocol 2. Number of sections depends on quality and spread of Lck-GFP expression. Glycerol-based mounting media containing DAPI is normally used with this protocol. Conversely, if the blue (405 nm) channel is already occupied by any of the antibodies, DAPI-free mounting media should be used instead. Care should be taken to coverslip wet sections without drying them, introducing air bubbles into the specimen and to apply enough mounting media to not squeeze or damage the tissue. The latter could directly interfere with morphometric measurement.
- Locate the region of interestUse 10x magnification and fluorescence lamp to locate the region of interest. Lck-GFP signal observed through the ocular at 10x magnification looks diffuse, without sharp or readily discernable structural features (Fig. 2).
- Locate an individual cell to imageOnce a cell has been identified, switch to 63x magnification for single cell acquisition. Use immersion oil. At 63x magnification, Lck-GFP expressing astrocytes are easily identified by their distinct cloud-like morphology. It is paramount to select a discrete astrocyte whose domain does not border with another Lck-GFP expressing astrocyte. Even though astrocyte domains are generally non-overlapping (tiled) (Freeman 2010), two adjacent astrocytes can be extremely hard to separate and to measure properly, and should thus be avoided. Lck-GFP expression is relatively sparse, making this step of the protocol effortless. Another important criteria for choosing a suitable astrocyte is their uncompromised morphology; in other words, only astrocytes that can be collected in their entirety should be acquired. Sliced (in z-axis) or compressed cells should not be acquired.
- Set general acquisition parametersAll the acquisition parameters should be carefully considered so to enable best image quality in a reasonable time frame. For this protocol, images were acquired with 1080 × 1080 pixel resolution, 16 bits per pixel bit depth, 1.52 μsec pixel dwell time and 4x averaging.
- Set z-axis parametersFirst and last optical sections of the z-stack should be determined in a manner to assure the cell is acquired in its entirety, which can be simply achieved by encompassing a few micrometers of space above and below the cell into the z-stack. The most import part of acquiring a z-stack is setting a step size. Step sizes ranging from 0.3 to 2 μm have been used previously (Scofield et al 2016, Siemsen et al 2019) for similar procedures. Whole cell 3-dimensional imaging can be time prohibitive; thus the selection of a step size for a z-stack should reflect the end objective of the analysis. If only morphometric measurements are to be taken, more generous step size is allowed while for more detailed analysis (e.g. branching complexity analysis), a smaller step size is in order. For the purpose of this protocol, where the measurements of morphometric features are the end objective, a 1 μm step size is sufficient. This said, the most objective option of selecting the step size is the employment of the Nyquist-Shannon sampling theorem (Pawley 2006). The theorem establishes the minimal sampling density, ensuring the data is not over- or under sampled. It is based on the inherent properties of the system’s optics and sampling capabilities and as such should be individually determined for each confocal system.
- Adjust signal intensity for each individual fluorescent channel usedSignal intensity adjustment should be performed in the middle of the selected z-stack. The channel order is arbitrary. The objective here is to get strong fluorescent signal with minimal background. For most genetically expressed fluorophores (including Lck-GFP) and Alexa-branded secondary antibodies, laser strength and pinhole size are locked at 1% and 1 AU, respectively, for the duration of the imaging. Sensitivity of the detectors is adjusted using “master gain” and “digital offset” with “range indicator” mode on. When satisfactory intensity is achieved on each channel, the settings should be kept constant throughout the experiment. Changes to detector gain are allowed only for green Lck-GFP channel since expression levels of the Lck-GFP are known to vary from one astrocyte to another.
Acquire a z-stack
Save a z-stack on a local drive in a raw file format (ZEN (.czi) file in case of this protocol).
Figure 2.
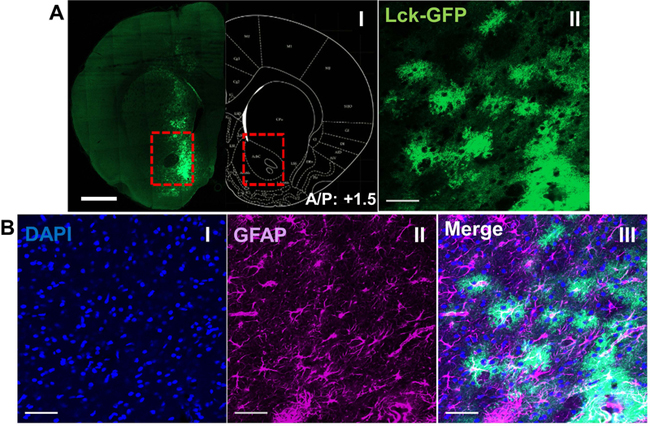
Confirmation of viral spread and intensity. A) A single coronal section of rat brain showing the expression and the spread of viral GfaABC1D-Lck-GFP in the NAc core (I) and a 10x multiple intensity projection (MIP) from the same section, showing sparsely distributed Lck-GFP positive astrocytes and their signature “cloudy” morphology (II). Scale bar: 1 mm (I) and 50 μm (II), respectively. B) IHC confirmation of astrocyte-specific expression of Lck-GFP showing MIP of DAPI (I) and GFAP (II) merged Lck-GFP (III). Scale bar: 50 μm.
BASIC PROTOCOL 4
3-DIMENSIONAL RECONSTRUCTION OF SINGLE CELLS
3-dimensional reconstruction uses the previously acquired astrocyte z-stack as a scaffold to re-build a cell in 3-dimensions. The surface function within Imaris software is used afterwards, employing membrane Lck-GFP signal intensity to build a functional surface, in order to extract surface area and volume information for each cell.
Materials:
Sufficiently powerful image processing workstation running the latest version of Imaris and deconvolution software (such as AutoQuant X3 etc.).
Our setup consisted of:
CPU: Intel(R) Core(TM) i7-8700 @ 3.20 GHz
GPU: Radeon RX 580 8 GB
RAM: 64 GB DDR4 - 2666 MHz
Storage: 1 TB Samsung SSD 960 EVO (OS, Imaris, AutoQuant X3), 3× 8 TB Seagate BarraCuda HDD (file storage)
- Import raw ZEN file in the deconvolution softwareDeconvolution resolves convolutions arising from imperfect optics, such as out of focus light. These imperfections result in a fluorescence signal not being spatially limited to a point but being spread in a point spread function (PSF), limiting resolution of the image. Deconvolution applies a restorative algorithm that reassigns out-of-focus fluorescence back to the source, improving image contrast and resolution. For the purpose of this protocol, 64-bit edition of AutoQuant X3 v.3.0.5 was used. Other deconvolution software was not tested but it can be reasonably expected to be compatible with this protocol.
- Set deconvolution parameters and apply the algorithm for the entire z-stackExcellent results are usually obtained employing 10 iterations of deconvolution using the adaptive point of spread function (PSF) (aka blind deconvolution) and medium level of noise setting.
- Save final deconvolution data as Imaris (.ims) fileIf deconvolution is for some reason skipped, Imaris File Converter should be used to convert ZEN files to IMARIS file. Since version 9.3, Imaris has a built in native deconvolution software, which can be used instead of a dedicated third party software. However, Imaris deconvolution is not part of the normal package and must be purchased separately.
Import post-deconvolution data into Imaris.
- Click “Auto Adjust all Channels” within “Display Adjustment” setting to improve default visualization of the z-stackRaw intensity values are not altered with this step
Select “Add new Surfaces” feature from the tab menu.
Check the “Segment only a Region of Interest” box under “Algorithm Settings” (step 1/5). Proceed to the next step using blue arrow button at the bottom of selection (Fig. 3A).
- Select region of interest (step 2/5). Proceed to the next step using blue arrow button at the bottom of selection (Fig. 3B).Region of interest should include the astrocyte in its entirety with minimum amounts of background. This will decrease computational intensity of surface building as well as decrease time to completion. Use “pull arrows” to isolate the cell. To quickly switch between “Navigate” and “Select” mode, use Esc key shortcut.
Select the source channel and unselect “Smooth” option (step 3/5). Proceed to the next step using blue arrow button at the bottom of selection (Fig. 3C).
- Build a surface by adjusting signal threshold corresponding to it (step 4/5). Proceed to the next step using the blue arrow button at the bottom of selection (Fig. 3D).Using the slide bar, manually select the intensity threshold of the Lck-GFP channel corresponding to the new surface. Sufficient fluorescence signal intensity and isolation of the cell are paramount for the successful completion of this step. Final threshold level should be visually confirmed, with the newly built surface completely blotting out the Lck-GFP signal of the previously selected cell. The new surface should also not be in direct contact with any “background surfaces” potentially neighboring the primary target surface.
- Select the primary surface (5/5) (Fig. 3E).Background Lck-GFP signal can sometimes exceed threshold level and generate small neighboring surfaces that are of no use to morphometric analysis. To streamline analysis, all but the primary surface are filtered out at this last step by using the slide bar and moving it to the right.
Finish the surface building process by clicking green “finish” button.
- Select “Statistics” tab within a newly built surface menu and export morphometric measurementsMorphometric data can be exported individually, by selecting the measurement value in question from the drop-down menu under Statistics/Detailed/ and clicking “Export Statistics on Tab Display to File” variable from the drop-down menu, or wholesale, by exporting all of the variables with “Export All Statistics to File”. Data is saved locally in a form of .csv files. After collecting morphometric data for all the cells, the master excel file is usually created to hold all the extracted data. This can then be fed into statistical tool of choice.
Select “Edit” (pencil icon) under a newly built surface and click “Mask all” (Fig. 3G).
- Select the Lck-GFP channel from the drop-down menu and click “OK”.Using a newly built surface as a mask for the Lck-GFP signal will create a new channel (with a prefix “Masked”) which will be completely devoid of any background signal, consisting only of the signal corresponding to the analyzed cell. This will greatly streamline the following colocalization analysis.
Figure 3.
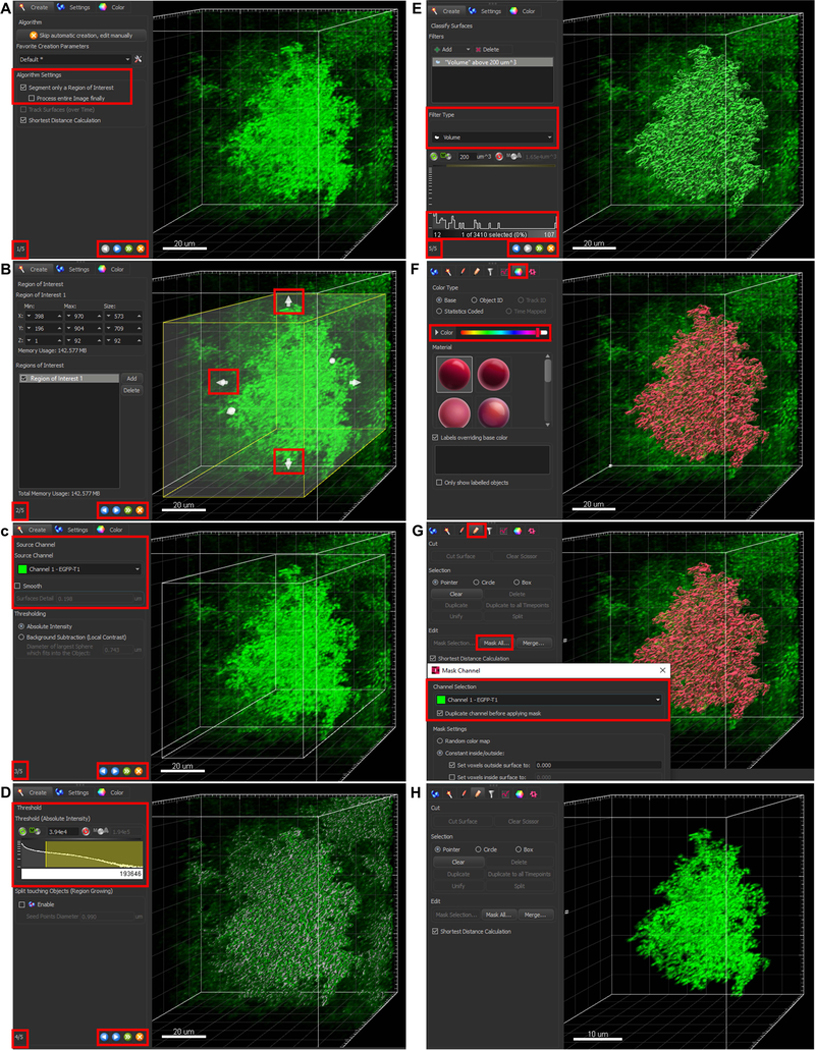
A visual guide through surface building using Imaris 9.5. A) In step 1/5, select “Segment only a Region of Interest”. This will help with the surface building (especially if the background signal is moderate) and decrease computational intensity of the task. Blue arrow button advances to the next step. B) In step 2/5, define the region of interest by manipulating white arrows on the side of the opaque rectangular. Coordinates of the limits can also be inputted manually. C) In step 3/5, select the source channel for the surface (in this case green channel). The “Smooth” option should be unselected to avoid averaging. D) In step 4/5, select the threshold intensity for selected channel. Automatic option is available although it proved unreliable in our hands. Manual threshold is set by manipulating the highlighted section of the intensity curve. If the cell is of the uniform intensity and isolated, the default threshold value presented should be very close to the correct final value. Every cell should be visually closely inspected, and threshold adjusted if under/over-sampling is observed. If threshold value is predetermined, it can be manually inputted directly. E) In final step 5/5, a single astrocyte surface is selected. Correct thresholding sometimes produces small nearby surface, where background intensity is sufficiently high. These unwanted surfaces should be filtered out by volume so only the main surface remains. Green arrow button will finish the building process. F) By default, the color of the surface matches the color of the source channel. For better contrast, the color can be changed under the “Color” tab. G) Besides taking measurements, the surface also serves as a mask to isolate astrocyte-only signal for presentation purposes or later use for colocalization analysis. To use surface as a mask, click “Mask All” under the “Edit” tab and select the source channel (same one as used for the surface) in pop-up window. H) Final isolated signal of a single astrocyte.
Basic Protocol 5
3-DIMENSIONAL COLOCALIZATION ANALYSIS
Colocalization is a widely used method in fluorescence microscopy and can be traditionally broken down into two components: a) co-occurrence of two signals, which constitute the basic spatial overlap and b) correlation, where two probes not only spatially overlap but also co-distribute proportionally between each other’s structures (Dunn et al 2011). It is important not to overestimate the power of colocalization analysis. It is beyond the scope of classical light microscopy colocalization analysis to evaluate direct contacts between two structures when used in non-super resolution microscopy. It can, however, provide a great proxy regarding the association of two fluorescent structures. Here we introduce colocalization analysis of the previously acquired and isolated astrocyte-specific Lck-GFP channel and selected neuronal marker. Presented is analysis involving post-synaptic marker, PSD-95 although others may be employed. The result of the analysis is percentage of region of interest (ROI) colocalized with selected neuronal marker, where ROI is isolated Lck-GFP signal of the individual astrocyte.
Select the “Coloc” tab within Imaris software
Select PSD-95 channel as channel A
- Set the intensity threshold value for channel A (Fig. 4A).This is the most critical step of the analysis. The selected threshold value for channel A will directly affect final colocalization data. This protocol employs manual threshold selection. It should be noted there is an option to select threshold automatically (Automatic thresholding/Calculate threshold), but in our experience this produces inconsistent and unreproducible results. Final threshold is thus selected by averaging intensity values of unambiguous positive puncta, randomly selected throughout the z-stack. This can be achieved by selecting positive puncta with the assistance of the outlining tool, integrated within Imaris colocalization analysis (left click and drag). The entirety of the signal should be encapsulated within the outlining border (Fig. 4A, white arrow). Values of individual puncta should be recorded and their average calculated. The average value is then manually keyed in the “Threshold” box of channel A.
Set the masked Lck-GFP channel as channel B
- Set the intensity threshold value for channel BSince all the background Lck-GFP signal was already removed by threshold masking of the original Lck-GFP channel, the threshold for channel B should be set at 1. This will make all of the masked Lck-GFP channel available for colocalization analysis. After setting both threshold levels, the 2D histogram on the right should encompass whole of the channel B and most of the channel A signal, bar from background that would appear red on the heat map.
- Define region of interest (ROI) (Fig. 4B)Check the “Mask Dataset” checkbox. ROI is the whole of masked Lck-GFP signal, so the said channel should be selected and threshold set as 1.
- Click “Build Coloc Channel”Colocalization results for a newly built colocalization channel can be extracted by going to the “Display adjustment” tab, clicking on the channel name and clicking “Export” after the pop-up channel window opens. Colocalization data for this single cell is then saved in the local directory as a .csv file. From the selection of extracted data (Pearson’s and Mander’s coefficient etc.) within the .csv save file we pick “% of ROI colocalized”. This tells us what portion of the region of interest (ROI, which in our case is a single isolated Lck-GFP-positive astrocyte) is colocalized with above-threshold PSD-95 signal. After collecting colocalization data for all the cells, similarly as with morphometric data, the master excel file can be made to store data or feed them to statistical software. Results are later plotted (see Figure 3 of (Testen et al 2018) and Figure 4&5 of (Testen et al 2019) for more information and examples of the types of data one can obtain.
Figure 4.

A visual guide through colocalization analysis using Imaris 9.5. A) Threshold for channel A (PSD-95 in case of this protocol) is selected by acquiring fluorescence intensity of multiple positive puncta and averaging their value. Measurements are acquired by left clicking and capturing the positive puncta within region of interest (see white arrow). The average is manually keyed into the correct “Threshold” box. B) Using previously created masked channel containing astrocyte-only signal as a region of interest assures that percentage of reported colocalization will apply only to the volume of the astrocyte and not the entire z-stack (see the crossed-out portion of the z-stack not containing the masked astrocyte-only channel). C) 2D Histogram is showing the total amount of the signal captured employing the current threshold. The histogram can be used to manually adjust the threshold level but this is done more precisely by the process described in (A). Thus, the histogram should be observed at the very end only to confirm that the selected threshold value for channel A indeed captures the correct signal (all of the signal should be collected excluding the red portion of the heat map representing the PSD-95 background signal).
Figure 5.

Representative reconstruction of a single astrocyte. Image of a 3D reconstruction of a representative z-stack showing Lck-GFP expressing astrocyte (green), neuronal and astrocyte nuclei stained with DAPI (blue) as well as IHC for GFAP (magenta) (I). The white signal represents colocalization of GFAP with Lck-GFP, clearly confirming the cell is indeed GFAP-positive astrocyte. Lck-GFP signal can be used to create a functional surface, capable of reporting volumetric features (II), which can be further used to completely isolate relevant signal and remove the background (III). Scale bar: 15 μm
COMMENTARY
Background Information
Detailed descriptions of astrocyte morphology have existed since Cajal first recorded his observations (Garcia-Marin et al 2007). As technology and computational power progressed, an astrocyte-specific morphological marker capable of recapitulating the complexity of astrocytes has remained elusive. The Golgi-Cox stain used by Cajal reveals detailed branching complexity, but fails to capture small peripheral astrocyte processes (PAP) that interact with synapses and vasculature (Ranjan & Mallick 2012). Information regarding astrocyte volume is also lost (or highly simplified) when using Golgi stain. The same caveat applies for GFAP, the most widely used structural astrocyte marker or the newest small molecule astrocyte markers, like cationic pyridinium family of markers (Preston et al 2019, Preston et al 2018). The majority of structural astrocyte analyses is performed using GFAP exclusively (SheikhBahaei et al 2018, Viola et al 2009) or injectable intracellular fluorescence dyes (Butt & Ransom 1989, Moye et al 2019). GFAP is found in primary, secondary and some tertiary astrocyte branches, but not in higher order processes, excluding significant portions of the astrocyte from analysis.
The main advantage of the Lck-GFP reporter used here is its association with the membrane, which enables uniform dissociation across the membrane thus ensuring faithful representation and reconstruction of the entire cell. This is made possible due to Lck fusion tag attached to the GFP protein. Lck (lymphocyte-specific protein tyrosine kinase) is a 56 kDa protein belonging to the Src family of tyrosine kinases. The N-terminal of Lck is myristoylated, as well as palmitoylated, which renders membrane association (Zlatkine et al 1997). Use of a fusion a short amino acid sequence from Lck to generate Lck-GFP has previously revealed more detailed morphology of astrocytes in hippocampal slice cultures (Benediktsson et al 2005) and cortical cultures (Heller et al 2017) compared to more commonly used cytosolic GFP while Lck-GCaMP constructs have revealed dynamics of astrocyte calcium signaling at the PAPs (Shigetomi et al 2013, Shigetomi et al 2010a). Recently 3D-reconstructions of Lck-GFP transduced astrocytes were used to explore morphometric changes after administration of various drugs of abuse (Kruyer et al 2019, Scofield et al 2016, Siemsen et al 2019, Testen et al 2018) as well as for measurements of developmental changes (Testen et al 2019).
Beyond use of Lck-GFP, other studies have utilized astrocyte reporter mouse lines or fluorescent dyes to label and identify astrocytes in greater detail than that afforded by GFAP immunohistochemistry (Morel et al 2017, Morel et al 2019, Moye et al 2019). Moreover, a companion protocol provided in this same issue (cite) details use of live cell FRET to quantify proximal interactions between neurons and astrocytes (Octeau et al 2018). As a complement to these approaches, the protocol provided here provides a readily available approach to investigation of structural adaptations and synaptic colocalization of astrocytes following injury or disease.
Critical Parameters
Each step in this protocol carries potential to influence the quality of final results. Because 100 μm sections are used, it is necessary for antibodies used to penetrate the complete thickness of the section. The staining protocol presented is optimized to accomplish this although a few other parameters were found to affect antibody penetration efficiency beside the time of the antibody incubation and the temperature at which it is done. These are, namely, the quality of the perfusion and the tissue post-fixation time. Inadequate perfusions will reproducibly produce poor IHC staining results and thwarted future imaging endeavors. Well-known indicators of sufficiently performed perfusion such as fixation tremors and post-fixation whole-body stiffness should be observed. We also found that minimal (2–4h) post-fixation of the collected tissue is optimal. Interestingly, the length of the post-fixation period appears to be inversely proportional with the efficiency of antibody tissue penetration, with longer post-fixation producing suboptimal results. During antibody incubation, the tissue should be flipped at least once during the each incubation period, to minimize the possibility of sections folding, in essence doubling the volume and making successful penetration highly unlikely.
Careful attention should be given to sample preparation. Tissue should be mounted and coverslipped directly out of wash, and immediately before the imaging session. Care should be taken to prevent any drying of tissue sections. Sufficient mounting media should be used to prevent tissue deformation or damage during coverslipping. Applying any pressure during this process will compress the tissue, producing variable volumetric measurements. After coverslipping, the imaging should follow immediately to avoid any risk of inconsistency within the data set arising due to sample handling and/or storage. Sections should be left in the wash buffer until immediately before imaging. At this point, samples should also be encrypted for the duration of the study for blind data inquisition and analysis.
During imaging, it is of upmost importance to collect the cell in its entirety. A good rule of thumb is to allow for a few micrometers of “buffer space” above and below the cell. Attention should also be paid to assuring the cell is indeed a single astrocyte and not multiple astrocyte domains connected together. This is sometimes hard during imaging and it becomes clear only later, during the Imaris analysis. Even if two neighboring cells are both transduced with Lck-GFP they can be separated if the signal intensity is sufficiently different. This requires significantly longer post-acquisition analysis but has a potential of producing two cells with a single z-stack acquisition. It is up to the experimenter to weigh potential pros and cons of such acquisition.
Imaging parameters, namely master gain, should be left constant throughout the imaging session for all immunohistochemistry. This will ensure the validity of intensity-based colocalization analysis. The gain for Lck-GFP can be altered during the imaging if necessary, since acquiring the complete structure of an astrocyte is paramount to later analysis while absolute value of signal intensity is not. Level of signal intensity of Lck-GFP transduced astrocytes can also vary significantly thus making an application of a constant gain value unpractical. During the colocalization analysis, however, there should be a consistency in deciding what constitutes a positive PSD-95 puncta. Even small differences in measuring and determining threshold value for PSD-95 can significantly affect colocalization analysis.
Troubleshooting
| Issue | Potential causes and solutions |
|---|---|
| Poor or nonexistent virus expression | Missing viral expression may indicate unsuccessful microinjection, most likely due to clogging of microinjector. Another possibility is decreased transduction efficiency of a virus due to poor storage conditions or too many thaw/freezing cycles. |
| Saturated virus expression | Some anatomically insulated brain structures (like hippocampus) sometimes show Lck-GFP signal saturation. The virus used in this protocol can be diluted with saline if needed to produce optimal expression |
| Poor antibody penetration | Poor perfusion and/or too long period of post-fixation (2–4h max). Folding of tissue during antibody incubation period. For colocalization antibodies other than those referenced here, the incubation time and/or level of detergent used during IHC can be varied. |
| Ambiguity regarding the number of collected astrocytes | Sometimes it can be hard to assess if the collected astrocyte is indeed singular using only visual observation. In these instances, colocalization analyses between Lck-GFP and DAPI as well as Lck-GFP and GFAP can be beneficial. A single astrocyte can be colocalized with multiple nuclei but only one of them should be encapsulated in GFAP signal. |
| Diminishing fluorescence intensity along z-axis | Sometimes acquired Lck-GFP signal shows clear decrease in fluorescence signal intensity along the z-axis due to diminishing excitation potential of a light source deeper in the tissue. Since this can affect surface building, the signal should be attenuated along the z-axis with a built-in Imaris XTension called “Attenuation Correction”. Imaris XTensions are scripts that compliment Imaris analysis, enabling features that are not yet part of a stand-alone Imaris experience. This is achieved by running a part of a code outside Imaris ecosystem (in MATLAB) and importing results back in. Attenuation correction itself is a part of a default Imaris installation package and does not require an additional installation but it does require a working MATLAB installation. See Figure 6 for comparison. The same correction can be applied to colocalization signal or any other IHC staining. |
Figure 6.

Advantage of attenuation correction. Representative reconstruction of a single Lck-GFP positive astrocyte (shown from the side; x- and z-axis) before (I) and after (II) employing “Attenuation Correction” option. Scale bar: 10 μm
Understanding Results
Successful image acquisition requires a good viral spread in the pre-selected areas of interest (Fig. 2A I), with high signal-to-noise ratio and sparsely transduced cells (Fig. 2A II). Verification of astrocyte specificity is done using GFAP immunohistochemistry (Fig. 2B II, III). Following acquisition and deconvolution, a newly imported z-stack should be seamlessly recreated with Imaris software. For better visualization, “Auto Adjust All Channels” should be selected (Fig. 5 I). Selecting appropriate threshold for Lck-GFP intensity, a functional surface should be built around the Lck-GFP signal, encapsulating the entirety of the cell, including small peripheral sections (Fig. 5 II). Sequential masking of the Lck-GFP signal on a newly built surface should reveal the single isolated astrocyte, without any background present (Fig. 5 III). This said, the most satisfying results, where the astrocyte is isolated with high precision when it comes to peripheral parts, can only be achieved if fluorescence intensity of Lck-GFP signal is uniform throughout the z-stack. This can be hard in cases where the imaged astrocyte is located near the middle of the brain section. Setting a correct threshold for these astrocytes is near impossible since an appropriate threshold for the upper portion completely misses bottom signal. In these cases the Imaris XTension (custom script running parallel in MATLAB) called “Attenuation Correction” should be used. The package corrects the artefact, making signal uniform in all dimensions (Fig. 6). The said isolated (and attenuated if necessary) astrocyte is a starting point for the following colocalization analysis (Fig. 7 I). As emphasized before, IHC for PSD-95 (or any other marker) should produce consistent fluorescence intensity throughout the thickness of the section (Fig. 7 II), allowing the colocalization analysis to capture astrocyte-neuronal interaction along the entirety of the z-axis. During the analysis itself, when PSD-95 (channel 1) and masked Lck-GFP (channel 2) are selected, the changing of the threshold produces real-time colocalization visualization in a form of white colocalization signal, overlaid on top of the two primary channels (Fig. 7 III). Finally, after completing the analysis, the colocalization channel can be accessed and visualized from the “Display Adjustment” window (Fig. 7 IV). A correctly constructed channel features individual puncta that can be easily analyzed by Imaris software. If the colocalization signal is fused, threshold was most likely set too low.
Figure 7.

Colocalization between astrocytes and post-synaptic marker PSD-95 in NAc core. Previously presented isolated astrocyte (I) now showing IHC for a neuronal marker PSD-95 (II). Merger of the channels (III) produces white colocalization signal which can be further isolated and analyzed (IV).
Time Considerations
Approximate time required for each protocol is as follows:
Microinjection surgeries: animals require a week of acclimatization before surgery as well as a week of recovery afterwards. Virus microinjections take 10 min (on 0.1 μl/min setting), followed by 15 min diffusion period. The entire surgery lasts approximately 45 min for a single bilateral microinjection (depending on the experience and skill level of the experimenter).
Tissue processing and IHC: rat perfusion and brain sectioning depends also on the experience and skill level of the experimenter. It takes generally approximately 10–20 min for a single rat perfusion. As mentioned before, post-fixation should be limited to 2–4 h. Sectioning time depends on a number of brain regions collected and is roughly similar per brain to perfusion time. If collected in 50% 1x PBS/glycerol, the sections can be stored at −20 °C for a prolonged period of time (up to a year). IHC takes 6 days (3 days primary antibody incubation, 3 days secondary antibody incubation). After the last wash, the sections can wait in PBS at 4 °C until mounting time.
Imaging and analysis: imaging time depends on multiple settings including number of channels selected, required resolution, z-stack size, scanning time selected etc. With the imaging system and settings described in this protocol, a single, average z-stack (~ 60 μm along z-axis) takes approximately 25 min to acquire (2 channels per track). In the best-case scenario, time should be allocated to complete the imaging of the currently mounted tissue in a single sitting to avoid drying and compression artifacts. Deconvolution time depends on a z-stack size as well as on computational power of the imaging workstation and takes on average 5 min. The most unpredictable is time needed for reconstruction and colocalization analysis. If images collected are of good quality, with high signal-to-noise ratio, the analysis is straightforward and takes 10–15 min per cell. Images of lower quality can still produce valid results yet require significantly more time for extraction of said results.
ACKNOWLEDGEMENTS:
This work was supported by NIH R01 DA041455 (KJR) and NIH NRSA F31 DA044695 (RK).
References
- 2006. Phosphate-buffered saline (PBS). Cold Spring Harbor Protocols 2006: pdb.rec8247 [Google Scholar]
- Allen NJ, Barres BA. 2009. Neuroscience: Glia - more than just brain glue. Nature 457: 675–7 [DOI] [PubMed] [Google Scholar]
- Allen NJ, Eroglu C. 2017. Cell Biology of Astrocyte-Synapse Interactions. Neuron 96: 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. 1999. Tripartite synapses: glia, the unacknowledged partner. Trends in neurosciences 22: 208–15 [DOI] [PubMed] [Google Scholar]
- Benediktsson AM, Schachtele SJ, Green SH, Dailey ME. 2005. Ballistic labeling and dynamic imaging of astrocytes in organotypic hippocampal slice cultures. J Neurosci Methods 141: 41–53 [DOI] [PubMed] [Google Scholar]
- Blanco-Suarez E, Caldwell AL, Allen NJ. 2017. Role of astrocyte-synapse interactions in CNS disorders. J Physiol 595: 1903–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM, Ransom BR. 1989. Visualization of oligodendrocytes and astrocytes in the intact rat optic nerve by intracellular injection of lucifer yellow and horseradish peroxidase. Glia 2: 470–5 [DOI] [PubMed] [Google Scholar]
- De Pitta M, Brunel N, Volterra A. 2016. Astrocytes: Orchestrating synaptic plasticity? Neuroscience 323: 43–61 [DOI] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. 2011. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300: C723–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkee CA, Araque A. 2019. Diversity and Specificity of Astrocyte-neuron Communication. Neuroscience 396: 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR. 2010. Specification and morphogenesis of astrocytes. Science 330: 774–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marin V, Garcia-Lopez P, Freire M. 2007. Cajal’s contributions to glia research. Trends in neurosciences 30: 479–87 [DOI] [PubMed] [Google Scholar]
- Heller JP, Michaluk P, Sugao K, Rusakov DA. 2017. Probing nano-organization of astroglia with multi-color super-resolution microscopy. J Neurosci Res 95: 2159–71 [DOI] [PubMed] [Google Scholar]
- Hol EM, Pekny M. 2015. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Current opinion in cell biology 32: 121–30 [DOI] [PubMed] [Google Scholar]
- Jones ME, Paniccia JE, Lebonville CL, Reissner KJ, Lysle DT. 2018. Chemogenetic Manipulation of Dorsal Hippocampal Astrocytes Protects Against the Development of Stress-enhanced Fear Learning. Neuroscience 388: 45–56 [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Verkhratsky A. 2011. Neuroglia, der lebende Nervenkitt. Fortschr Neurol Psychiatr 79: 588–97 [DOI] [PubMed] [Google Scholar]
- Kim R, Healey KL, Sepulveda-Orengo MT, Reissner KJ. 2018. Astroglial correlates of neuropsychiatric disease: From astrocytopathy to astrogliosis. Progress in neuro-psychopharmacology & biological psychiatry 87: 126–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruyer A, Scofield MD, Wood D, Reissner KJ, Kalivas PW. 2019. Heroin Cue-Evoked Astrocytic Structural Plasticity at Nucleus Accumbens Synapses Inhibits Heroin Seeking. Biological psychiatry 86: 811–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J, Hol EM. 2011. GFAP in health and disease. Progress in neurobiology 93: 421–43 [DOI] [PubMed] [Google Scholar]
- Morel L, Chiang MSR, Higashimori H, Shoneye T, Iyer LK, et al. 2017. Molecular and Functional Properties of Regional Astrocytes in the Adult Brain. J Neurosci 37: 8706–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel L, Men Y, Chiang MSR, Tian Y, Jin S, et al. 2019. Intracortical astrocyte subpopulations defined by astrocyte reporter Mice in the adult brain. Glia 67: 171–81 [DOI] [PubMed] [Google Scholar]
- Moye SL, Diaz-Castro B, Gangwani MR, Khakh BS. 2019. Visualizing Astrocyte Morphology Using Lucifer Yellow Iontophoresis. J Vis Exp [DOI] [PubMed] [Google Scholar]
- Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, Khakh BS. 2018. An Optical Neuron-Astrocyte Proximity Assay at Synaptic Distance Scales. Neuron 98: 49–66 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawley JB. 2006. Handbook of biological confocal microscopy New York, NY: Springer; xxviii, 985 p. pp. [Google Scholar]
- Perea G, Navarrete M, Araque A. 2009. Tripartite synapses: astrocytes process and control synaptic information. Trends in neurosciences 32: 421–31 [DOI] [PubMed] [Google Scholar]
- Perez-Alvarez A, Araque A. 2013. Astrocyte-neuron interaction at tripartite synapses. Current drug targets 14: 1220–4 [DOI] [PubMed] [Google Scholar]
- Preston AN, Cervasio DA, Laughlin ST. 2019. Visualizing the brain’s astrocytes. Methods Enzymol 622: 129–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston AN, Farr JD, O’Neill BK, Thompson KK, Tsirka SE, Laughlin ST. 2018. Visualizing the Brain’s Astrocytes with Diverse Chemical Scaffolds. ACS Chem Biol 13: 1493–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Mallick BN. 2012. Differential staining of glia and neurons by modified Golgi-Cox method. J Neurosci Methods 209: 269–79 [DOI] [PubMed] [Google Scholar]
- Scofield MD, Li H, Siemsen BM, Healey KL, Tran PK, et al. 2016. Cocaine Self-Administration and Extinction Leads to Reduced Glial Fibrillary Acidic Protein Expression and Morphometric Features of Astrocytes in the Nucleus Accumbens Core. Biological psychiatry 80: 207–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SheikhBahaei S, Morris B, Collina J, Anjum S, Znati S, et al. 2018. Morphometric analysis of astrocytes in brainstem respiratory regions. J Comp Neurol 526: 2032–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, et al. 2013. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol 141: 633–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Kracun S, Khakh BS. 2010a. Monitoring astrocyte calcium microdomains with improved membrane targeted GCaMP reporters. Neuron Glia Biol 6: 183–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Kracun S, Sofroniew MV, Khakh BS. 2010b. A genetically targeted optical sensor to monitor calcium signals in astrocyte processes. Nature neuroscience 13: 759–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemsen BM, Reichel CM, Leong KC, Garcia-Keller C, Gipson CD, et al. 2019. Effects of Methamphetamine Self-Administration and Extinction on Astrocyte Structure and Function in the Nucleus Accumbens Core. Neuroscience 406: 528–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. 2010. Astrocytes: biology and pathology. Acta Neuropathol 119: 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testen A, Ali M, Sexton HG, Hodges S, Dubester K, et al. 2019. Region-Specific Differences in Morphometric Features and Synaptic Colocalization of Astrocytes During Development. Neuroscience 400: 98–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testen A, Sepulveda-Orengo MT, Gaines CH, Reissner KJ. 2018. Region-Specific Reductions in Morphometric Properties and Synaptic Colocalization of Astrocytes Following Cocaine Self-Administration and Extinction. Front Cell Neurosci 12: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola GG, Rodrigues L, Americo JC, Hansel G, Vargas RS, et al. 2009. Morphological changes in hippocampal astrocytes induced by environmental enrichment in mice. Brain Res 1274: 47–54 [DOI] [PubMed] [Google Scholar]
- Zlatkine P, Mehul B, Magee AI. 1997. Retargeting of cytosolic proteins to the plasma membrane by the Lck protein tyrosine kinase dual acylation motif. J Cell Sci 110 ( Pt 5): 673–9 [DOI] [PubMed] [Google Scholar]