

Abstract
Background
Hemophilia B (HB) is a coagulation disorder with an X‐linked recessive inheritance pattern, caused by plasma FIX deficiency. In Colombia, HB is considered a rare and high‐cost disease, with 362 males reported in 2017.
Methods
Here, we characterized 20 HB apparently unrelated families by PCR amplification and Sanger sequencing.
Results
Fourteen unique variants were identified: seven missense, three nonsense, one variant in the 3′ UTR region, two large deletions >50 bp, and one intronic substitution that affects splicing c.520+13A>G that was present in 7/20 patients (35%). All these variants have been previously reported in the literature, except for exons 3 and 4, deletions, present in one patient. The genotype‐phenotype association correlates with the reported in the literature, with the exception of one patient.
Conclusion
This molecular analysis allowed us to establish the causal variant of HB in 100% of patients, to provide the appropriate genetic counseling to each of the families, and to propose a more cost‐effective carrier analysis. Here, we reported the first variants in Colombian population with Hemophilia B, finding a new variant and one intron recurrent variant present in 35% of patients.
Keywords: coagulation factor IX, Colombia, F9, genetics diagnosis, hemophilia B
This is the first molecular characterization of patients with Hemophilia B in Colombia. Using Sanger sequencing we found the patogenic variant in all patients. One large deletion of exon 3 and 4 hasn't been reported previously in international databases.
1. INTRODUCTION
Plasma Factor IX protein deficiency causes a coagulation disorder known as Hemophilia B (HB) (OMIM 306900), characterized by spontaneous or secondary bleedings in response to traumatic or surgical events. It is caused by alterations in the DNA sequence of the Factor IX gene (F9). This gene is 32 Kilobases in size, with 8 exons and 7 introns, and it is located on the X chromosome (Xq27.1). HB presents an X linked recessive inheritance pattern (Goodeve, 2015). HB is classified based on the percentage of residual activity of FIX in plasma in: severe (<1%), moderate (1%–5%) and mild (5%–40%), and it has a high phenotypic heterogeneity (Srivastava et al., 2013).
There is a high allelic heterogeneity in HB, and it is possible to find almost any type of variant. The point variants are observed more frequently (73% of the cases), followed by deletions (16%) and, in minor proportion, insertions, duplications, small indels and large rearrangements (Belvini et al., 2005).
Sixty‐five percent of patients with HB have missense variants, approximately 83% of the pathogenic variants are located in exonic regions, and 12% in intronic regions (Rallapalli, Kemball‐Cook, Tuddenham, Gomez, & Perkins, 2013).
The 2017 Epidemiological Report, shows a total of 362 men with HB in Colombia, out of which 38.1% had a severe phenotype, 36.4% moderate phenotype, and 23.4% mild phenotype (Fondo Colombiano de enfermedades de alto costo, 2018).
Different studies in countries worldwide, such as Spain, United States, Italy, Argentina, and Brazil, have identified the pathogenic variants of the F9 in their population, these countries have reference centers for diagnosis as well, (Belvini et al., 2005; Li, Miller, Payne, & Craig Hooper, 2013; Meireles et al., 2017; Radic, 2010). This study is the first molecular analysis of Hemophilia B in Colombian population.
2. MATERIALS, SUBJECTS, AND METHODS
2.1. Subjects
Twenty unrelated men samples (ages between 5 and 68 years) with a confirmed HB diagnosis and receiving treatment in a comprehensive management program for Hemophilia B, were analyzed. The study was previously approved by the Ethics Committees of the Faculty of Medicine of the National University of Colombia and the participating health care entities.
Table 1 shows patients' data such as: age, severity of HB, percentage of factor IX activity (coagulometric analysis), history of inhibitor development, and whether they have a family history of HB or not.
Table 1.
Characterization of Colombian patients with HB
| ID | Age | Severity | FIX:C (coagulometric analysis) | Prophylaxis | Family history | Inhibitors |
|---|---|---|---|---|---|---|
| HB_01 | 18 | Moderate | 4.8 | No | Yes | No |
| HB_02 | 14 | Moderate | 1.8 | Yes | Yes | No |
| HB_03 | 18 | Moderate | 2.9 | Yes | Yes | No |
| HB_04 | 7 | Severe | 0.2 | No | Yes | No |
| HB_05 | 14 | Severe | 0.1 | Yes | No | No |
| HB_06 | 5 | Moderate | 4.4 | No | Yes | No |
| HB_07 | 16 | Mild | 28.4 | No | No | No |
| HB_08 | 7 | Severe | 0.2 | Yes | Yes | No |
| HB_09 | 35 | Moderate | 1.5 | Yes | Yes | No |
| HB_10 | 20 | Moderate | 4.8 | Yes | Yes | No |
| HB_11 | 34 | Moderate | 4.4 | No | Yes | No |
| HB_12 | 42 | Mild | 8.4 | No | Yes | No |
| HB_13 | 18 | Severe | 0.7 | Yes | Yes | No |
| HB_14 | 38 | Moderate | 4.8 | No | Yes | No |
| HB_15 | 68 | Moderate | 1.9 | No | Yes | No |
| HB_16 | 24 | Mild | 11 | No | Yes | No |
| HB_17 | 45 | Severe | 0.3 | No | Yes | No |
| HB_18 | 53 | Mild | 8.5 | No | No | No |
| HB_19 | 55 | Mild | 7.9 | No | Yes | No |
| HB_20 | 57 | Mild | 5.9 | No | Yes | No |
2.2. Collection of blood sample and DNA isolation
Four milliliters of peripheral blood were collected from each patient in EDTA tubes. Genomic DNA extraction was carried out with 200 μl of the collected blood, using the “DNA Mini kit Quiagen” (Quiagen Corporation) following the manufacturer's instructions; the quantification of the isolated DNA was performed by spectrometry in a “Nanodrop 2000”. Subsequently, the DNA was stored at −4°C.
2.3. F9 sequencing
The most important F9 regions were amplified: promoter, eight exons, exon‐intron junction regions, and the 3′ UTR, in 12 PCR reactions, with primers previously reported (Radic, 2010). In each PCR reaction, we used the sample of a normal individual as a positive control.
PCR was carried out with 3 μl of DNA, 5 μl Buffer 5X GoTaq (Promega), 2 μl dNTPS (200 μM), 1.5/2.0 μl MgCL2 (Stock), 8.375 μl of H2O, 0.125 μl of GoTaq Polymerase, and 5 μl of each primer pair, with a final reaction volume of 25 μl. Thermocycling conditions for the 12 PCR reactions were: Hot start 94°C for three minutes, followed by 30 cycles of: denaturation at 94°C for 30 s, annealing at 55°C for 30 s, elongation at 72°C for 60 s, and a final extension at 72°C for five minutes (Radic, 2010). PCR amplification was confirmed through electrophoresis in Nusieve agarose gel 3:1 at 2% at 100 V for one hour.
Purification of the amplified regions was performed using "Purelink Quick PCR Purification Kit" (Invitrogen), following the manufacturer's instructions. Quantification was performed by spectrometry with “Nanodrop 2000”.
The sequence of each fragment was amplified with the “BigDye 3.1 Terminator Cycle Sequencing Kit” (Applied Biosystems), using 3.5 ng/μl of the PCR products purified with 1.2 μl of the primer forward or reverse (10 pmol/μl); capillary electrophoresis was performed in a “ABI3500” (Applied Biosystems).
2.4. Sequence analysis
For sequence quality analysis and variants identification, the “Chromas 2.6.5” (BioEdit) and “SeqScape V5.4” (Applied Biosystems) software packages were used applying the reference sequence. The Genebank accession numbers are: NG_007994.1 (Homo sapiens coagulation Factor IX (F9), RefSeqGen LRG 556 on chromosome X), NM_000133.3 and NP_000124.1.
The variants were reported following the guidelines of the Human Genome Variation Society (HGVS) (den Dunnen et al., 2016), the international F9 databases were consulted (http://://www.factorix.org/; http://://www.cdc.gov/ncbddd/hemophilia/champs; http://://www.hemobase.com/) to verify if they were previously reported.
2.5. Bioinformatics analysis
The pathogenicity and stability of the protein in the missense variants found were analyzed through the use of different software and online bioinformatics tools, such as: “POLYPHEN2,” “I‐Mutant2.0,” “PANTHER,” “Mutation Taster,” and “iStable.” “Project HOPE” (http://://www.cmbi.ru.nl/hope/input) allowed us to make a 3D modeling of the protein, and simultaneously showed us both mutated and wild residue was (Capriotti, Calabrese, & Casadio, 2006; Chen, Lin, & Chu, 2013; Ramensky, 2002; Schwarz, Cooper, Schuelke, & Seelow, 2014; Tang & Thomas., 2016).
The analysis for variants located in intron 5, and 3′ UTR, were performed with the “Human Splicing Finder HSF 3.0” tools which combines 12 different algorithms to predict the impact of variants in acceptor sites, splicing donors, branch points, possible breaking points, and auxiliary sequences of potentiators or silencers of both: exonic and intronic splicing (Tang, Prosser, & Love, 2016), another tool used to analyze these variants was NNSplicing (Reese, Eeckman, Kulp, & Haussler, 1997).
3. RESULTS
The pathogenic variant was identified in all samples tested. We identified 14 unique pathogenic variants, of which, 7 were missense, 3 nonsense, 2 large deletions, 1 substitution affecting splicing, and 1 substitution at the 3′ UTR (Table 2). Of these variants, only the large deletion of exon 3 and 4 have not been previously reported. Eighteen patients presented a single variant and two had a double variant (HB_07 and 14). Seven patients (35%) presented the same intronic pathogenic variant that affects splicing (HB_01, 06, 10, 12, 18, 19, and 20), and two patients have an identical missense pathogenic variant (HB_15 and 17).
Table 2.
Variants identified in Colombian patients with HB
| ID | Nucleotide change NG_007994.1 | Nucleotide change NM_000133.3 | Location | Aminoacid change | Mutation effect | Previously reported | Domain |
|---|---|---|---|---|---|---|---|
| HB_01 | g.22769A>G | c.520+13A>G | IVS 5 | N/A | Substitution | Yes | Splicing |
| HB_02 | g.37493A>G | c.*1157A>G | Exon 8 | 3′ UTR | Substitution | Yes | 3′ UTR |
| HB_03 | g.36085C>T | c.1135C>T | Exon 8 | p. R379* | Nonsense | Yes | Protease |
| HB_04 | g.11409C>T | c.223C>T | Exon 2 | p. R75* | Nonsense | Yes | Gla |
| HB_05 | g.22754C>T | c.518C>T | Exon 5 | p.A173V | Missense | Yes | EGF2 |
| HB_06 | g.22769A>G | c.520+13A>G | IVS 5 | N/A | Substitution | Yes | Splicing |
| HB_07 |
g.35917G>A g.36288G>C |
c.967G>A c.1338G>C |
Exon 8 |
p.E323K p.K446N |
Double missense | Yes | Protease |
| HB_08 | Del. exons 5 and 6 | N/A | Exon 5 and 6 | N/A | Deletion | Yes | N/A |
| HB_09 | Del. exons 3 and 4 | N/A | Exon 3 and 4 | N/A | Deletion | No | N/A |
| HB_10 | g.22769A>G | c.520+13A>G | IVS 5 | N/A | Substitution | Yes | Splicing |
| HB_11 | g.35831G>A | c.881G>A | Exon 8 | p.R294Q | Missense | Yes | Protease |
| HB_12 | g.22769A>G | c.520+13A>G | IVS 5 | N/A | Substitution | Yes | Splicing |
| HB_13 | g.35842C>T | c.892C>T | Exon 8 | p.R298* | Nonsense | Yes | Protease |
| HB_14 | g.25527C>A g.25386G>A |
c.721C>A c.580A>G |
Exon 6 | p.Q241K p.T194A* | Missense | Yes | Protease |
| HB_15 | g.25377C>T | c571C>T | Exon 6 | p.R191C | Missense | Yes | Activation peptide |
| HB_16 | g.11646A>G | c.272A>G | Exon 3 | p.Y91C | Missense | Yes | Gla |
| HB_17 | g.25377C>T | c.571C>T | Exon 6 | p.R191C | Missense | Yes | Activation peptide |
| HB_18 | g.22769A>G | c.520+13A>G | IVS 5 | N/A | Substitution | Yes | Splicing |
| HB_19 | g.22769A>G | c.520+13A>G | IVS 5 | N/A | Substitution | Yes | Splicing |
| HB_20 | g.22769 A>G | c.520+13A>G | IVS 5 | N/A | Substitution | Yes | Splicing |
Acording to HGVS nomenclature, it represent a stop codon.
3.1. Missense variants
In seven patients, seven missense variants were identified, two of these were found in exon 6, three in exon 8, one in exon 5, and one in exon 3 (See Figure 1). Only two of these variants, p.Arg294Gln and p.Arg191Cys, are located in CpG dinucleotides considered variant hot spots. The p.Arg191Cys variant was found in two unrelated patients.
Figure 1.
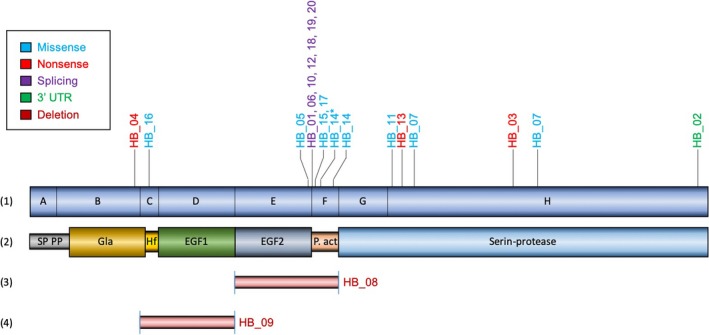
Distribution of variants in F9 in Colombian patients. (1) Schematic representation of the F9 gene showing its 8 exons (A‐H), with the location of the mutations found in the patients studied, type: missense (Blue color), nonsense (Red color), splicing (Purple color) and 3′ UTR variant (Green color). HB_14* Benign variant. (2) Factor IX protein, SP domain: signal peptide, PP: Pro peptide, Gla: gamma‐carboxy‐glutamic domain, EGF1 and EGF2: domains with homology to epidermal growth factor 1 and 2 respectively. (3) large deletion of exons 5 and 6, and (4) of exons 3 and 4
Two patients (HB_07 and 14) had a double variant. Patient HB_07 has both variants in exon 8, g.35917G>A and g.36288G>C, located in the catalytic domain of the protein (p.Glu323Lys; p.Lys446Asn). The two variants found in patient HB_14 are located in exon 6, g.25527C>A and g.25386G>A (p.Gln241Lys and p.Thr194Ala). One of them, p.Thr194Ala, is a benign variant (HB_14* in Figure 1).
All these variants, according to the in‐silico analysis and literature, are classified as deleterious or probably deleterious, and affecting the stability of the protein.
3.2. Nonsense variants
Nonsense variants were found in 15% of the patients, two variants located in exon 8: g.36085 C>T identified in patient HB_03, and g.35842 C>T identified in patient HB_13; one variant in exon 2: g.11409 C>T was observed in patient HB_04. All three are in CpG dinucleotides, for which there are numerous reports of patients with these same variants in the literature.
3.3. Substitution in the intron 5 and 3′ UTR
In seven apparently unrelated patients (35%), the same variant was observed in intron 5, 13 nucleotides from the donor splice site (g.22769A>G; c.520+13A>G). The bioinformatics analysis with “NNSplicing,” predicted a new splicing donor site with a score of 0.41 compared to the wild‐type sequence of 0.2; with “Human Splicing Finder, HSF”, we identified the formation of a new potential splicer donor site with a +50.25% variation, in favor of the mutant sequence versus the wild sequence; additionally, two new splice enhancers binding sites are also formed. This variant presumably generates a mRNA with the inclusion of 12 bp in exon 5 that will introduce 4 additional codons, altering protein translation by introducing 4 new amino acids (Gly, His, Asn, and Leu), also in the exon 6 the first amino acid Val would change to Met, resulting in the possible decrease of FIX activity.
A variant in exon 8 was detected in patient HB_02, g.37493 A>G, which corresponds to the 3′ UTR region of the F9. Bioinformatics analysis with “Human Splicing Finder, HSF” showed that this variant generates a new cryptic donor splicing site, and additionally, generates an exonic splicing enhancer (ESE).
3.4. Large deletions
Two patients (HB_08 and 09), 10% of the analyzed sample, had large deletions (>50 pb), evidenced by the absence of PCR amplicon bands in agarose gel electrophoresis from the amplified exons.
Patient HB_08 showed the absence of fragments E and F amplification, corresponding to exon 5 and exon 6, respectively.
A deletion of exons 3 and 4 (fragments C and D, respectively) was observed in patient HB_09, who presents moderate HB. This variant has not been reported in the literature to date. There are deletions reports that include these two exons with other additional exons. The exact location of the break in the F9 is not known since the Primers used for the amplification are found in adjacent intronic regions.
These results were confirmed in duplicate with a new DNA isolation for each patient, where the amplicon absence of the fragments was evidenced again. Each PCR was carried out simultaneously with the DNA sample from a normal individual (+ control), that showed successful amplification of those fragments, in addition, multiplex PCR was performed on both cases with primers involved in the deletion sequence (HB_08: exons 5 and 6; HB_09: exons 3 and 4) and the primers non involving (F9: exon 1 and beta globin gene NM 000518.4), the result confirmed the absence of amplification of the deleted exons (data not shown), therefore we ruled out the lack of amplifications due to defective primers.
3.5. Genotype‐phenotype correlation
Five patients (25%) had a severe phenotype. From these, two have a nonsense variant, two a missense variant and one a deletion of exons 5 and 6. Nine patients (45%) have moderate HB. Three of them presented a variant in intron 5 that affects splicing, one has a double missense variant, two have a missense variant, one has a nonsense variant, one has a variant in the 3′ UTR, and one presented the newly detected variant, a deletion of exons 3 and 4. Six patients (30%) were reported to have mild HB, four of them have the same variant that affects splicing in intron 5, one has a double missense, and one has a missense variant.
None of the 20 patients have developed inhibitors to date. Eighty‐five percent of the patients have a family history of HB. From the three patients without a family history of HB, one has severe, one moderate, and one mild phenotype. Seven patients (35%) received treatment in the prophylactic scheme and 13 (65%) received on demand treatment.
4. DISCUSSION
This is the first molecular HB study in Colombia. The pathogenic variant for HB was identified in 100% of the analyzed cohort, similar to other reports in the literature (Abla et al., 2018; Khan et al, 2018). Ninety percent of the studied patients presented single nucleotide variants, which is slightly higher than what is reported in other studies (Abla et al., 2018; Khan et al, 2018). Ten percent of patients had large structural variants, similar to previous studies (Li et al., 2014), and to the European database (EAHAD‐CFDB).
The pathogenic variant frequencies found in this series are different from those reported in the literature. For example, missense variants represent 35% of the variants in this study, compared to 65%–70% from other studies (Li et al., 2014; Mårtensson, Letelier, Halldén, & Ljung, 2016; Rallapalli et al., 2013). The frequency of nonsense variants (15%) are similar to other studies and databases (Li et al., 2014; Radic, 2010). The largest difference in mutational frequencies was found in variants that affect splicing, with a frequency of 35% of the patients analyzed. These results contrast strongly with other reports. Splice variants had been found in frequencies of 3 and 8% in the Argentinean and Brazilian cohort, respectively (Li et al., 2014; Mårtensson et al., 2016; Meireles et al., 2017; Radic et al., 2013; Rallapalli et al., 2013). This great difference is due to the presence of the same pathogenic variant (g.22769A>G) in seven patients, who are unrelated based on genetic interviews and genealogies. These unexpected findings suggest the possible existence of a founder effect. Further single nucleotide variant studies of X chromosome would allow us to determine if the variant is associated with the same haplotype to confirm this hypothesis.
The variant found in patient HB_02 in the 3′ UTR region was previously described by Vielhaber and collaborators in four individuals of different ethnicities. They pointed out the high possibility that this new splicing site generates a bypass in the polyadenylation signal which would produce an unstable mRNA that could easily be degraded in the cytoplasm (Vielhaber, Jacobson, Ketterling, Liu, & Sommer, 1993).
The only pathogenic variant not reported previously in the literature corresponds to a large deletion of exons 3 and 4. This deletion is responsible for the absence of the Gla and EGF1 protein domains involved in calcium‐dependent platelet surface adhesion and cellular interactions (Thompson, 1986).
Some variants identified in the present study have been reported previously in Argentinian (Radic, 2010), and Brazilian populations (Meireles et al., 2017). Variants p.Arg75* and p.Arg298* have been identified in Brazil and Argentina, while p.Arg191Cys variant, p.Arg294Gln, p.Arg379*, and exon 5–6 deletion have been reported only in Argentina.
The phenotype/genotype correlations are quite heterogeneous compared to other studies (Abla et al., 2016; Awidi et al., 2011; Radic et al., 2013).
In patient HB_16, with a mild phenotype, the genotype identified (a missense variant in exon 3) differs from that reported in the literature: three patients, two with moderate phenotype and one with unknown phenotype.
5. CONCLUSIONS
We have performed molecular analysis for the identification of pathogenic variants in HB patients for the first time in Colombia. The pathogenic variants were identified in 100% of the studied cohort. Remarkably, 35% (7/20) of apparently non related patients carried the same intronic variant affecting splicing (g.22769A>G; c.520+13A>G), which suggests a possible founder effect. In addition, a new variant involving a large exon deletion (exons 3 and 4), was found.
ACKNOWLEDGMENTS
We wish to thank each of the patients and their families for agreeing to participate in this study; to the Comprehensive Hemophilia Treatment Center of Colsubsidio Children's Clinic, to the Clinical Research Center of Colsubsidio; to the Institute of Human Genetics of the National University of Colombia where the processing and analysis of the samples was carried out; to the Sequencing Service of the Institute of Genetics of the National University of Colombia SSIGMOL; to the nurses Claudia Suarez and Yadira Valderrama for their contribution collecting the samples and their assistance in the evaluation of patient's pedigrees. This project was financed entirely by the National University of Colombia HERMES Code 34708 directed by Juan J. Yunis L.
Parrado Jara YA, Yunis Hazbun LK, Linares A, Yunis Londoño JJ. Molecular characterization of hemophilia B patients in Colombia. Mol Genet Genomic Med. 2020;8:e1210 10.1002/mgg3.1210
References
REFERENCES
- Abla, Z. , Mouloud, Y. , Hejer, E. L. M. , Emna, G. , Meriem, A. , Yamina, O. , & Naouel, S. (2018). Mutations causing hemophilia B in Algeria: Identification of two novel mutations of the factor 9 gene. Biodiversitas, 19(1), 52–58. 10.13057/biodiv/d190109 [DOI] [Google Scholar]
- Awidi, A. , Alhattab, D. , Bsoul, N. , Magablah, A. , Mefleh, R. , Dweiri, M. , & Fauori, A. S. (2011). FIX mutation spectrum in haemophilia B patients from Jordan: Identification of three novel mutations. Haemophilia, 17(1), 162–163. 10.1111/j.1365-2516.2010.02365.x [DOI] [PubMed] [Google Scholar]
- Belvini, D. , Salviato, R. , Radossi, P. , Pierobon, F. , Mori, P. , Castaldo, G. , & Tagariello, G. (2005). Molecular genotyping of the Italian cohort of patients with hemophilia B. Haematologica, 90(5), 635–642. [PubMed] [Google Scholar]
- Capriotti, E. , Calabrese, R. , & Casadio, R. (2006). Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics, 22(22), 2729–2734. 10.1093/bioinformatics/btl423 [DOI] [PubMed] [Google Scholar]
- Chen, C. W. , Lin, J. , & Chu, Y. W. (2013). iStable: Off‐the‐shelf predictor integration for predicting protein stability changes. BMC Bioinformatics, 14(S5). 10.1186/1471-2105-14-S2-S5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen, J. T. , Dalgleish, R. , Maglott, D. R. , Hart, R. K. , Greenblatt, M. S. , McGowan‐Jordan, J. , … Taschner, P. E. (2016). HGVS recommendations for the description of sequence variants: 2016 Update. Human Mutation, 37, 564–569. 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- Fondo colombiano de enfermedades de alto costo, C. de alto costo . (2018). Situación de la hemofilia en Colombia 2017. Bogotá Colombia: Retrieved from http://www.cuentadealtocosto.org [Google Scholar]
- Goodeve, A. C. (2015). Hemophilia B: Molecular pathogenesis and mutation analysis. Journal of Thrombosis and Haemostasis, 13(7), 1184–1195. 10.1111/jth.12958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. T. M. , Naz, A. , Ahmed, J. , Shamsi, T. , Ahmed, S. , … Taj, A. S. (2018). Mutation spectrum and genotype – Phenotype analyses in a Pakistani cohort with hemophilia B. Clinical and Applied Thrombosis/Hemostasis, 24, 741–748. 10.1177/1076029617721011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T. , Miller, C. H. , Driggers, J. , Payne, A. B. , Ellingsen, D. , & Hooper, W. C. (2014). Mutation analysis of a cohort of US patients with hemophilia B. American Journal of Hematology, 89(4), 375–379. 10.1002/ajh.23645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T. , Miller, C. H. , Payne, A. B. , & Craig Hooper, W. (2013). The CDC hemophilia B mutation project mutation list: A new online resource. Molecular Genetics & Genomic Medicine, 1(4), 238–245. 10.1002/mgg3.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårtensson, A. , Letelier, A. , Halldén, C. , & Ljung, R. (2016). Mutation analysis of Swedish haemophilia B families ‐ High frequency of unique mutations. Haemophilia, 22(3), 440–445. 10.1111/hae.12854 [DOI] [PubMed] [Google Scholar]
- Meireles, M. R. , Pantoja, A. G. , Ornaghi, A. P. M. , Vieira, G. F. , Salzano, F. M. , & Bandinelli, E. (2017). Molecular characterization of haemophilia B patients in southern Brazil. Haemophilia, 5, 457–461. 10.1111/hae.13277 [DOI] [PubMed] [Google Scholar]
- Radic, C. P. (2010). Genética molecular de hemofilia: caracterización de mutaciones en hemofilia B, expresión de hemofilia en mujeres y desarrollo de nuevos métodos de análisis de inversiones. Tesis Doctoral, Universidad de Buenos Aires. Facultad de Ciencias Exactas y Naturales. Retrieved from http://hdl.handle.net/20.500.12110/tesis_n4794_Radic [Google Scholar]
- Radic, C. P. , Rossetti, L. C. , Abelleyro, M. M. , Candela, M. , Bianco, R. P. , Pinto, M. D. T. , … Brasi, C. D. D. (2013). Assessment of the F9 genotype‐specific FIX inhibitor risks and characterisation of 10 novel severe F9 defects in the first molecular series of Argentinian patients with haemophilia B. Thrombosis and Haemostasis, 109(1), 24–33. 10.1160/TH12-05-0302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallapalli, P. M. , Kemball‐Cook, G. , Tuddenham, E. G. , Gomez, K. , & Perkins, S. J. (2013). An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. Journal of Thrombosis and Haemostasis, 11(7), 1329–1340. 10.1111/jth.12276 [DOI] [PubMed] [Google Scholar]
- Ramensky, V. (2002). Human non‐synonymous SNPs: Server and survey. Nucleic Acids Research, 30(17), 3894–3900. 10.1093/nar/gkf493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese, M. G. , Eeckman, F. H. , Kulp, D. , & Haussler, D. (1997). Improved splice site detection in genie. Journal of Computational Biology, 4(3), 311–323. 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). Mutationtaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Srivastava, A. , Brewer, A. K. , Mauser‐Bunschoten, E. P. , Key, N. S. , Kitchen, S. , Llinas, A. , & Street, A. (2013). Guidelines for the management of hemophilia. Haemophilia, 19(1), e1–e47. 10.2217/fmeb2013.13.148 [DOI] [PubMed] [Google Scholar]
- Tang, H. , & Thomas, P. D. (2016). PANTHER‐PSEP: Predicting disease‐causing genetic variants using position‐specific evolutionary preservation. Bioinformatics, 32(14), 2230–2232. 10.1093/bioinformatics/btw222 [DOI] [PubMed] [Google Scholar]
- Tang, R. , Prosser, D. O. , & Love, D. R. (2016). Evaluation of bioinformatic programmes for the analysis of variants within splice site consensus regions. Advances in Bioinformatics, 2016, 5614058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, A. (1986). Structure, function, and molecular defects of factor IX. Blood, 67(3), 565–572. 10.1182/blood.V67.3.565.565 [DOI] [PubMed] [Google Scholar]
- Vielhaber, E. , Jacobson, D. P. , Ketterling, R. P. , Liu, J. Z. , & Sommer, S. S. (1993). A mutation in the 3′ untranslated region of the factor IX gene in four families with hemophilia B. Human Molecular Genetics, 2(8), 1309–1310. 10.1093/hmg/2.8.1309 [DOI] [PubMed] [Google Scholar]