

Abstract
Background
Musculocontractural Ehlers–Danlos Syndrome (mcEDS) is a rare connective tissue disorder caused by biallelic loss‐of‐function variants in CHST14 (mcEDS‐CHST14) or DSE (mcEDS‐DSE), both of which result in defective dermatan sulfate biosynthesis. Forty‐one patients with mcEDS‐CHST14 and three patients with mcEDS‐DSE have been described in the literature.
Methods
Clinical, molecular, and glycobiological findings in three additional patients with mcEDS‐DSE were investigated.
Results
Three patients from two families shared craniofacial characteristics (hypertelorism, blue sclera, midfacial hypoplasia), skeletal features (pectus and spinal deformities, characteristic finger shapes, progressive talipes deformities), skin features (fine or acrogeria‐like palmar creases), and ocular refractive errors. Homozygous pathogenic variants in DSE were found: c.960T>A/p.Tyr320* in patient 1 and c.996dupT/p.Val333Cysfs*4 in patients 2 and 3. No dermatan sulfate was detected in the urine sample from patient 1, suggesting a complete depletion of DS.
Conclusion
McEDS‐DSE is a congenital multisystem disorder with progressive symptoms involving craniofacial, skeletal, cutaneous, and cardiovascular systems, similar to the symptoms of mcEDS‐CHST14. However, the burden of symptoms seems lower in patients with mcEDS‐DSE.
Keywords: clinical features, delineation, dermatan sulfate, musculocontractural EDS‐DSE
Clinical, molecular, and glycobiological findings in three additional patients from two families with musculocontractural Ehlers–Danlos syndrome (mcEDS‐DSE) are described. McEDS‐DSE is a congenital multisystem disorder with progressive symptoms involving craniofacial, skeletal, cutaneous, and cardiovascular systems, similar to the symptoms of mcEDS‐CHST14. However, the burden of symptoms seems lower in patients with mcEDS‐DSE.

1. INTRODUCTION
Musculocontractural Ehlers–Danlos Syndrome (mcEDS) is a rare connective tissue disorder, caused by biallelic loss‐of‐function variants in CHST14 (mcEDS‐CHST14) (MIM#601776) or in DSE (mcEDS‐DSE) (MIM#615539), both of which result in defective dermatan sulfate (DS) biosynthesis (Brady, Demirdas, & Fournel‐Gigleux, 2017; Malfait, Francomano, & Byers, 2017). Hallmarks of mcEDS include altered craniofacial features, multiple congenital contractures (e.g., adducted thumbs, talipes equinovarus), characteristic fine palmar creases, peculiar finger shapes, progressive spinal and foot deformities, large subcutaneous hematomas, and ophthalmological and urogenital involvement (Brady et al., 2017). Only five patients with mcEDS‐DSE have been described in the literature (Müller, Mizumoto, & Suresh, 2013; Schirwani et al., 2019; Syx, Van Damme, & Symoens, 2015) in contrast to 41 patients with mcEDS‐CHST14 (Brady et al., 2017; Kono, Hasegawa‐Murakami, & Sugiura, 2016; Sandal & Kaur, 2018). Here, we report detailed clinical, molecular, and glycobiological findings in three additional patients with mcEDS‐DSE.
2. CLINICAL REPORT
Patient 1, a 19‐year‐old man and average high school student, was born of consanguineous Turkish parents. Pregnancy was complicated by premature rupture of membranes and he was delivered by emergency cesarean section at gestational week 29. At birth, he exhibited cyanosis and bradycardia with Apgar scores of 7 at 1 min and 10 at 5 min. His birth weight was 1,215 g (−1 standard deviation [SD]), length was 35 cm (−3 SDs), and occipitofrontal circumference (OFC) was 26.7 cm (−0.5 SD).
In infancy to early childhood, he exhibited large anterior fontanel, brachycephaly, low hairline, wide neck, hypertelorism, blue sclera, short nose with hypoplastic columella, long philtrum, and thin upper lip vermilion. He had bilateral adducted thumbs, arachnodactyly, and rocker bottom feet, but did not exhibit talipes equinovarus. He had umbilical hernia, diastasis recti, bilateral hydronephrosis, and cryptorchidism. He experienced recurrent constipation, which was treated with laxatives. Echocardiography revealed an atrial septal defect and patent ductus arteriosus. He exhibited delayed motor development and mild hypotonia; he sat without support at age 8 months and walked unassisted at 18 months. He underwent surgeries for strabismus and cryptorchidism at 3 and 5 years of age, respectively.
At 8 years of age, brain magnetic resonance imaging showed cortical heterotopia. He was diagnosed with myopia and strabismus, and later diagnosed with unilateral 40 dB high‐frequency sensorineural hearing loss. During his school years, he experienced repeated shoulder luxation and large subcutaneous hematomas at various locations (each antebrachium and the gluteal region) after minor traumas and without the evidence of coagulation defects. He had mild scoliosis and joint hypermobility, and underwent surgery for pes cavus at 11 years of age. His skin was hyperextensible with delayed wound healing. At 15 years of age, he had a self‐limiting hemoptysis with no evidence of tuberculosis.
At 18 years of age, his height was 178 cm (−0.2 SD), weight was 77 kg (+0.5 SD), and OFC was 58 cm (+2 SDs). He had downslanting palpebral fissures, grey sclera, but no hypertelorism. His ears were not rotated or low‐set; however, his right ear showed underfolded helix, lower insertion of the lobule than on the left side, and pits on the posterior conchae. He had dental crowding, and his skin was hyperextensible, with fine palmar (Figure 1b) and solar creases, and broadened surgical scars on the right foot (Figure 1d). His fingers were long and slender with joint laxity and a swan‐neck deformity, as well as the radial deviation of the bilateral second to fourth fingers (Figure 1a). He had bilateral pes cavus (Figure 1e), mild pectus excavatum (Figure 1c), and lumbar scoliosis. He had no echocardiographic abnormalities, but was diagnosed with high blood pressure and treated with an angiotensin‐converting enzyme inhibitor.
Figure 1.
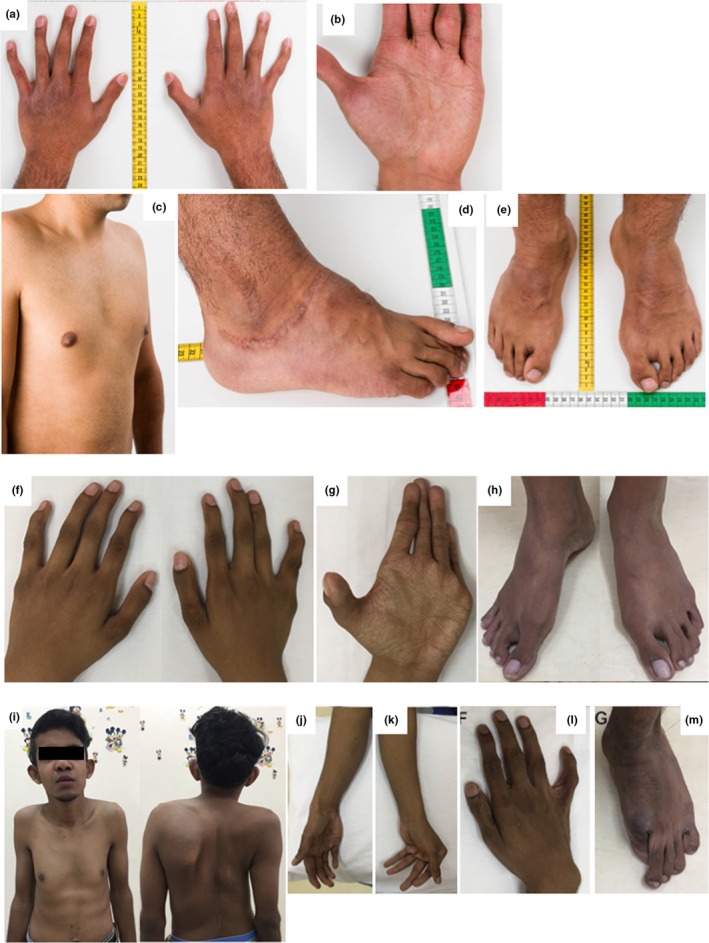
Clinical photographs of patient 1 at 18 years of age (a−e), patient 2 at 12.5 years of age (f−h), and patient 3 at 21 years of age (i−m)
Patient 2, a 14‐year‐old girl and average student, was born of consanguineous Indian parents. After an uncomplicated pregnancy, she had been delivered vaginally at term with a birth weight of 2.5 kg; she had normal developmental milestones. She experienced intermittent joint pain and swelling after exercise (left knee and right ankle) and exhibited scoliosis at 11 years of age. She had astigmatism, but normal hearing.
At 12.5 years of age, her height was 141 cm, weight was 27.9 kg, and OFC was 48 cm (all below the third centile). She had hypertelorism, blue sclera, and a broad tall nasal bridge; she had no skin hyperextensibility or apparent scars. Her fingers and toes were long with mild finger webbing (Figure 1f,h), and crisscrossing palmar and solar creases (Figure 1g). Joint laxity was limited to the dorsal subluxation of the metacarpophalangeal joints, especially the thumbs. Her chest was asymmetric with pectus carinatum and she had thoracic kyphoscoliosis. Her bone mineral density, measured by dual‐energy X‐ray absorptiometry, was reduced to 0.659 g/cm2 at the lumbar spine (normal range for 20‐year‐old women, 0.8−1.2 g/cm2), and 0.513 g/cm2 at the femoral neck (normal range for 20‐year‐old women, 0.6−1.0 g/cm2). She had no echocardiographic abnormalities or muscle weakness.
Patient 3, a 22‐year‐old man and the older brother of patient 2, was born at term after an uncomplicated pregnancy, with a birth weight of 2.9 kg. He had bilateral talipes equinovarus, which was treated with serial casting and surgical correction at 1 year of age. His early psychomotor development was normal. He experienced right arm fracture after a fall at 4 years of age and right fifth finger fracture at 7 years of age. He developed a large subcutaneous hematoma in his left calf twice, both requiring surgical evacuation. He also had intermittent bruises over his shins. He exhibited scoliosis at 14 years of age, which progressed and required surgical correction at 20 years of age. He had myopia, but normal hearing. He attended a normal school, but did not perform well.
At 21 years of age, his height was 151 cm and weight was 40 kg (both below the third centile). He had small simple ears, mild hypertelorism, blue sclera, a broad tall nasal bridge (Figure 1i), and retained primary teeth. Atrophic surgical scars, finger webbing, and abnormal palmar (Figure 1j,k) and solar creases were present; skin hyperextensibility was absent. He had an asymmetric chest (Figure 1i), long fingers and toes (Figure 1l,m), hallux valgus, and plantar subluxation of the metatarsophalangeal joint of the halluces, as well as prominent calcaneus (Figure 1m). He developed multiple joint contractures resulting in reduced ankle dorsiflexion, hip flexion, elbow extension, and wrist dorsiflexion.
3. MOLECULAR INVESTIGATION
Sanger sequencing of CHST14 and DSE was performed for patient 1 based on clinical suspicion of mcEDS. No pathogenic variants were detected in CHST14; a homozygous nonsense variant was identified in DSE (NM_013352.4, c.960T>A, p.(Tyr320*)) (Figure 2a). For patient 2, whole exome sequencing was performed (Data S1; Miyake, Tsurusaki, & Koshimizu, 2016) and a homozygous frameshift variant was identified in DSE (NM_013352.4, c.996dupT, p.(Val333Cysfs*4)); patient 3 exhibited homozygosity for this variant, whereas their parents exhibited heterozygosity (Figure 2a). None of the identified DSE variants were registered in ExAC, Exome Variant Server, or Human Genetic Variant Database.
Figure 2.
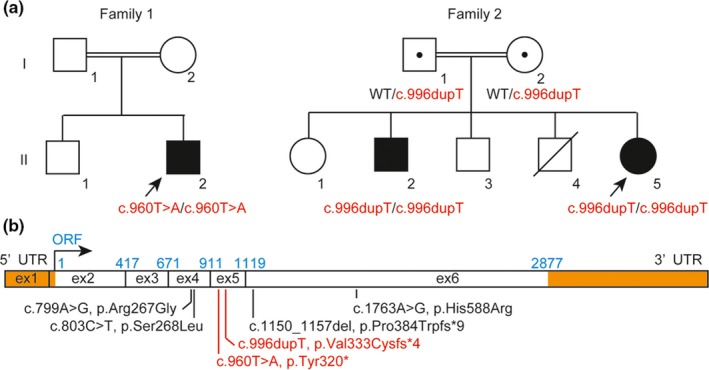
Molecular investigation. (a) Pedigree information and intrafamilial segregation of detected variants in DSE. (b) cDNA structure of DSE and pathogenic variants (mcEDS‐DSE). Untranslated regions (UTRs) are shown as orange bars. The first base and each position of exon‐exon junctions in open reading frames (ORFs) are shown in light blue. Pathogenic variants reported in this study are shown in red characters. ex: exon
4. GLYCOBIOLOGICAL ANALYSIS
Disaccharide compositions of DS and chondroitin sulfate (CS) chains in urine samples from patient 1 and an age‐matched healthy man were analyzed as described previously (Data S2; Mizumoto, Kosho, & Hatamochi, 2017). DS disaccharide was not detected in the urine of patient 1, whereas it was present in the urine of the age‐matched healthy man (Figure S1; Table S1A). In contrast, the amount of CS disaccharides in the urine was similar between patient 1 and the age‐matched healthy man (Figure S1; Table S1B).
5. DISCUSSION
We presented three patients with mcEDS‐DSE from two new families. Clinical and molecular features of the five previously reported patients (Müller et al., 2013; Schirwani et al., 2019; Syx et al., 2015), including additional information of the sisters reported by Syx et al. (2015) and the three patients in this series are reviewed in Table 1. Truncating variants in DSE were identified in the three current patients, whereas previously detected variants were missense in three families and a frameshift variant in one family. Significant reduction or loss of epimerase activity and a marked reduction in DS disaccharides were demonstrated in the patient with p.(Ser268Leu) (Müller et al., 2013). A minor fraction of DS was detected as the glycosaminoglycan component of decorin in the patient with p.(Arg267Gly) (Syx et al., 2015). The preservation of DS moieties in mcEDS‐DSE, in contrast to the complete loss of DS in mcEDS‐CHST14 (Dündar, Müller, & Zhang, 2009; Miyake, Kosho, & Mizumoto, 2010), suggested the residual activity of mutant DSE or partially compensating activity of dermatan sulfate epimerase‐like protein (encoded by DSEL) (Syx et al., 2015). The measurement of the disaccharide compositions of DS and CS chains in a urine sample is a recently established non‐invasive method to screen for mcEDS‐CHST14 through the assessment of DS biosynthesis (Mizumoto et al., 2017). The lack of DS in the urine sample from patient 1 in this study suggests that this test may also be useful for screening of mcEDS‐DSE.
Table 1.
Clinical and molecular features of patients with mcEDS‐DSE
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Total (n = 8) | mcEDS‐CHST14 (n = 41) |
|---|---|---|---|---|---|---|---|---|---|---|
| Family | 1 | 2 | 3 | 4 | 5 | 6 | ||||
| Publication | Müller et al. (2013) | Syx et al. (2015) | Schirwani et al. (2019) | This study | ||||||
| Patient 1 | Patient 2 | Patient 1 | Patient 2 | Patient 3 | ||||||
| Mutation (cDNA) | c.803C>T/homo | c.799A>G/homo | c.1150_1157del/homo | c.1763A>G/homo | c.960T>A/homo | c.996dupT/homo | ||||
| Amino acid change (amino acid) | p.Ser268Leu | p.Arg267Gly | p.Pro384Trpfs*9 | p.His588Arg | p.Tyr320* | p.Val333Cysfs*4 | ||||
| Age at the publication (years) | 2 | 48 | 39 | 33 | 2 | 19 | 14 | 22 | ||
| Sex | M | F | F | M | M | M | F | M | M 5, F 3 | M 22, F 19 |
| Origin | Indian | Spanish | Portuguese | Pakistani | Turkish | Indian | ||||
| Craniofacial | ||||||||||
| Large fontanel with delayed closure (early childhood) | Yes | NR | NR | NR | Yes | Yes | NR | NR | 3/3 (100%) | 23 |
| Small mouth/micro‐retrognathia (infancy) | NR | Yes | Yes | Yes | NR | NR | NR | NR | 3/3 (100%) | |
| Slender face/protruding jaw (from adolescence) | NR | NR | NR | Yes a | NR | No | No | Yes | 2/4 (50%) | 11 |
| Facial asymmetricity (from adolescence) | NR | NR | NR | No a | NR | Yes | No | No | 1/4 (25%) | 10 |
| Hypertelorism | Yes | Yes | Yes | NR | NR | Yes | Yes | Yes | 6/6 (100%) | 35 |
| Downslanting palpebral fissures | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | 7/8 (88%) | 34 |
| Short palpebral fissures | NR | Yes | Yes | No a | Yes | Yes | No | No | 4/7 (57%) | |
| Blue sclera | Yes | Yes | Yes | NR | NR | Yes | Yes | Yes | 6/6 (100%) | 25 |
| Midfacial hypoplasia | NR | Yes | Yes | No a | NR | Yes | Yes | Yes | 5/6 (83%) | |
| Short nose with hypoplastic columella | NR | Yes | Yes | No a | No a | No | No | No | 2/7 (29%) | 16 |
| Ear deformity (e.g. low‐set, posterior rotation, prominent) | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | 7/8 (88%) | 33 |
| Palatal abnormalities (e.g. high, cleft) | Yes | NR | NR | Yes | No | No | Yes | NR | 3/5 (60%) | 25 |
| Long philtrum and/or thin upper lip vermilion | NR | Yes (thin upper lip vermilion) | Yes (both) | No a | Yes (thin upper lip vermilion) a | No | No | No | 3/7 (43%) | 24 |
| Crowded teeth | Yes | NR | NR | Yes | NR | Yes | NR | NR | 3/3 (100%) | |
| Brachycephaly/flat occiput | Yes | NR | NR | No a | Yes | Yes | No | No | 3/6 (50%) | |
| Others | Hypotonic face with wrinkled and saggy eyelids, cheeks, and neck | Prominent forehead | ||||||||
| Skeletal | ||||||||||
| Congenital multiple contractures a | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | 7/8 (88%) | 41 |
| Adducted thumbs | Yes (bil) | No | No | Yes (bil) | Yes (bil) | Yes (bil) | No | No | 4/8 (50%) | 33 |
| Talipes equinovarus | Yes (bil) | Yes (bil) | Yes (bil) | Yes (bil) | Yes (bil) | No | No | Yes (bil) | 6/8 (75%) | 41 |
| Recurrent/chronic joint dislocations | NR | No | NR | No | No | Yes (shoulder) | No | No | 1/6 (17%) | 20 |
| Pectus deformilties | NR | NR | NR | NR | No | Yes (mild excavatum) | Yes (excavatum, asymmetric) | Yes (excavatum, asymmetric) | 3/4 (75%) | 18 |
| Spinal deformities | NR | No | Yes (mild to moderate scoliosis) | No | No | Yes (mild lumbar scoliosis) | Yes (scoliosis, thoracic kyphoscoliosis) | Yes (scoliosis) | 4/7 (57%) | 22 |
| Finger shape characteristics | Yes (long, tapering) | Yes (long, slender, tapering) | Yes (long, slender, tapering) | Yes (cylingdrical) | Yes (long, slender) | Yes (long, slender) | Yes (long, slender) | Yes (long, slender) | 8/8 (100%) | 35 |
| Progressive foot deformities | NR | Yes (short, broad feet with short toes) | NR | Yes (wide feet with clawed toes) | Yes | Yes (cavus) | Yes (uni. planus) | Yes (hallux valgus, planus, cavus) | 6/6 (100%) | 26 |
| Marfanoid habitus/slender build | NR | NR | NR | No | NR | No | Yes | Yes | 2/4 (50%) | 13 |
| Joint hypermobility | Yes | Yes b | Yes b | No | NR | Yes | No | No | 4/7 (57%) | |
| Osteoporosis | NR | NR | NR | NR | NR | NR | Yes | NR | 1/1 (100%) | |
| Others | Joint pain | Chronic pain, brachydactyly, Madelung deformity | Torticollis | Joint pain | Fractures | |||||
| Skin | ||||||||||
| Hyperextensibility | NR | Yes | Yes | No c | No | Yes | No | No | 3/7 (43%) | 24 |
| Bruisability | NR | Yes | Yes | NR | Yes | No | No | Yes | 4/6 (67%) | 21 |
| Fragility | NR | Yes | Yes | No | No | No | No | No | 2/7 (29%) | 21 |
| Atrophic scars | Yes | NR | NR | NR | No | No | No | Yes | 2/5 (40%) | |
| Hyperalgesia to pressure | NR | NR | NR | NR | NR | NR | NR | NR | 0/0 (0%) | 8 |
| Fine or acrogeria‐like palmar creases | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | 7/8 (88%) | 28 |
| Recurrent subcutaneous infections | NR | NR | NR | NR | NR | No | No | No | 0/3 (0%) | 8 |
| Fistula formation | NR | NR | NR | NR | NR | No | No | No | 0/3 (0%) | |
| Delayed wound healing | Yes | NR | NR | NR | NR | Yes | No | No | 2/4 (50%) | |
| Umbilical hernia | NR | NR | NR | NR | NR | Yes | No | No | 1/3 (33%) | |
| Others | Transparent, thin | Transparent, thin | Piezogenic pedal papules | |||||||
| Cardiovascular | ||||||||||
| Congenital heart defects | Yes (PFO) | NR | No | No | Yes (ASD) | Yes (ASD, PDA) | No | NR | 3/6 (50%) | 6 |
| Valve abnormalities | No | Yes (MVP, myxomatous valve with ruptureed chordae, severe MR) | No | No | NR | No | No | NR | 1/6 (17%) | 7 |
| Enlargement of ascending aorta | No | No | No | No | NR | No | No | NR | 0/6 (0%) | 2 |
| Large subcutaneous hematoma | NR | Yes | Yes | NR | No | Yes (elbow, arm, forehead, knee, gluteal region) | NR | Yes (calf) | 4/5 (80%) | 20 |
| Respiratory | ||||||||||
| Pneumothorax/Hemopneumothorax | NR | NR | NR | No | No | No | No | No | 0/5 (0%) | 3 |
| Gastrointestinal | ||||||||||
| Constipation | NR | NR | NR | No | No | Yes | No | No | 1/5 (20%) | 9 |
| Diverticula (e.g. perforation, infection) | NR | NR | NR | No | No | No | No | No | 0/5 (0%) | 4 |
| Others | Eventration after gallbladder surgery | |||||||||
| Urological | ||||||||||
| Nephrolithiasis or cystolithiasis | NR | NR | NR | NR | NR | No | NR | NR | 0/1 (0%) | 7 |
| Hydronephrosis | NR | NR | NR | No | NR | Yes (bil) | NR | NR | 1/2 (50%) | 10 |
| Bladder dysfunction | NR | Yes (prolapse after two deliveries) | NR | NR | NR | No | NR | NR | 1/2 (50%) | 2 |
| Recurrent urinary tract infection | NR | NR | NR | NR | NR | No | NR | NR | 0/1 (0%) | 3 |
| Inguinal hernia | Yes (lt) | NR | Yes | Yes (bil) | NR | Yes (lt) | No | No | 4/6 (67%) | 2 |
| Cryptorchidism in male | NR | Yes | No | Yes (bil) | No | 2/4 (50%) | 17 | |||
| Ophthalmological | ||||||||||
| Strabismus | NR | No | No | No | Yes | Yes (esotropia) | No | No | 2/7 (29%) | 14 |
| Glaucoma or elevated intraocular pressure | NR | No | No | No | No | No | No | No | 0/7 (0%) | 8 |
| Refractive error | NR | No | No | Yes (my) | No | Yes (my) | Yes (as) | Yes (my) | 4/7 (57%) | 16 (hy 4, my 12, as 5) |
| Retinal detachment | NR | No | No | NR | NR | No | No | No | 0/5 (0%) | 6 |
| Otological | ||||||||||
| Hearing impairment | NR | NR | NR | NR | NR | Yes (mild, uni, SNHL for high‐pitched sound) | No | No | 1/3 (33%) | 10 |
| Sexual development‐related | ||||||||||
| Hypogonadism | NR | NR | NR | NR | NR | No | NR | No | 0/2 (0%) | |
| Others | Uterine prolapse after two deliveries | |||||||||
| Central nervous system | ||||||||||
| Ventricular abnormalities (enlargement, asymmetry) | No | NR | NR | NR | No | No | NR | NR | 0/3 (0%) | 8 |
| Hypoplasia of septum pellucidum | No | NR | NR | NR | No | No | NR | NR | 0/3 (0%) | |
| Dandy‐Walker variant | No | NR | NR | NR | No | No | NR | NR | 0/3 (0%) | |
| Muscular system | ||||||||||
| Hypotonia | NR | Yes | Yes | NR | NR | Yes | NR | NR | 3/3 (100%) | 14 |
| Muscle weakness | Yes | Yes | NR | NR | NR | Yes | No | No | 3/5 (60%) | |
| Development | ||||||||||
| Motor developmental delay | Yes | NR | NR | NR | Yes | Yes | No | NR | 3/4 (75%) | 23 |
| Intellectual disabilities | No | NR | NR | No | NR | No | No | Yes | 1/5 (20%) | 4 |
Abbreviations: as, astigmatism; ASD, atrial septal defect; bil, bilateral; F, female; hy, hyperopia; lt, left; M, male; MR, mitral valve regurgitation; MVP, mitral valve prolapse; my, myopia; No, absent; NR, not recorded; PDA, patent ductus arteriosus; PFO, persistent foramen ovale; SNHL, sensorineural hearing loss; uni, unilateral; Yes, present.
Judged from images in the relevant report.
In younger ages but not in adulthood.
Only at the elbows.
Frequent (affecting at least three patients) craniofacial and skeletal features in mcEDS‐DSE are shown in Table 1. Two reported patients (Schirwani et al., 2019; Syx et al., 2015) and patient 2 in the present report had joint pain. Frequent skin, vascular, ocular, nervous, and muscle features are also shown in Table 1. Even though the number of patients with mcEDS‐DSE is small and an accurate frequency of each feature in patients with mcEDS‐CHST14 is unavailable, the general patterns of symptoms seem to be similar between the two subtypes. However, several patients with mcEDS‐CHST14 had life‐threatening complications (e.g., infectious endocarditis, Kono et al., 2016; Kosho, Miyake, & Hatamochi, 2010, fulminant gastric ulcer, Kosho et al., 2010; diverticular perforation, Kosho et al., 2010; Mochida, Amano, & Miyake, 2016), and five patients died (Dündar, Kurtoglu, & Elmas, 2001; Janecke, Li, & Boehm, 2016). No such serious complication has yet been observed in patients with mcEDS‐DSE. Syx et al. (2015) hypothesized that the residual availability of some DS structures, including iduronic acid‐containing disaccharide units, might contribute to an attenuated phenotype.
In conclusion, mcEDS‐DSE constitutes a multisystem disorder with congenital and progressive features, as well as the depletion of DS, similar to mcEDS‐CHST14. However, symptoms tend to be milder in patients with mcEDS‐DSE.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
FigS1
TableS1
TableS2
DataS1
DataS2
ACKNOWLEDGMENTS
We thank the patients and their families for participating in this study. We thank Ryan Chastain‐Gross, Ph.D., from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript. This study was supported by Grant‐in‐Aid for Scientific Research (C) from the Japan Society for the Promotion of Science, Japan (#16K08251 and #19K07054); Grant‐in‐Aid for Scientific Research (B) from the Japan Society for the Promotion of Science, Japan (#19H03616); Problem‐Solving Oriented Training Program for Advanced Medical Personnel: NGSD (Next Generation Super Doctor) Project; Grant‐in‐Aid for Research Center for Pathogenesis of Intractable Diseases from the Research Institute of Meijo University; and the Research Foundation, Flanders, Belgium.
Lautrup CK, Teik KW, Unzaki A, et al. Delineation of musculocontractural Ehlers–Danlos Syndrome caused by dermatan sulfate epimerase deficiency. Mol Genet Genomic Med. 2020;8:e1197 10.1002/mgg3.1197
Charlotte K. Lautrup, Keng W. Teik and Ai Unzaki contributed equally to this work.
Contributor Information
Noriko Miyake, Email: nmiyake@yokohama-cu.ac.jp.
Tomoki Kosho, Email: ktomoki@shinshu-u.ac.jp.
DATA AVAILABILITY STATEMENT
The data are not available for public access because of patient privacy concerns, but are available from the corresponding author on reasonable request.
REFERENCES
- Brady, A. F. , Demirdas, S. , Fournel‐Gigleux, S. , Ghali, N. , Giunta, C. , Kapferer‐Seebacher, I. , … Malfait, F. (2017). The Ehlers‐Danlos syndromes, rare types. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175(1), 70–115. 10.1002/ajmg.c.31550 [DOI] [PubMed] [Google Scholar]
- Dundar, M. , Kurtoglu, S. , Elmas, B. , Demiryilmaz, F. , Candemir, Z. , Ozkul, Y. , & Durak, A. C. (2001). A case with adducted thumb and club foot syndrome. Clinical Dysmorphology, 10(4), 291–293. 10.1097/00019605-200110000-00012 [DOI] [PubMed] [Google Scholar]
- Dündar, M. , Müller, T. , Zhang, Q. I. , Pan, J. , Steinmann, B. , Vodopiutz, J. , … Janecke, A. R. (2009). Loss of dermatan‐4‐sulfotransferase 1 function results in adducted thumb‐clubfoot syndrome. American Journal of Human Genetics, 85(6), 873–882. 10.1016/j.ajhg.2009.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecke, A. R. , Li, B. , Boehm, M. , Krabichler, B. , Rohrbach, M. , Müller, T. , … Steinmann, B. (2016). The phenotype of the musculocontractural type of Ehlers‐Danlos syndrome due to CHST14 mutations. American Journal of Medical Genetics. Part A, 170A(1), 103–115. 10.1002/ajmg.a.37383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono, M. , Hasegawa‐Murakami, Y. , Sugiura, K. , Ono, M. , Toriyama, K. , Miyake, N. , … Akiyama, M. (2016). A 45‐year‐old woman with Ehlers‐Danlos syndrome caused by dermatan 4‐O‐sulfotransferase‐1 deficiency: Implications for early ageing. Acta Dermato Venereologica, 96(6), 830–831. 10.2340/00015555-2390 [DOI] [PubMed] [Google Scholar]
- Kosho, T. , Miyake, N. , Hatamochi, A. , Takahashi, J. , Kato, H. , Miyahara, T. , … Matsumoto, N. (2010). A new Ehlers‐Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility‐related manifestations. American Journal of Medical Genetics, 152A(6), 1333–1346. [DOI] [PubMed] [Google Scholar]
- Malfait, F. , Francomano, C. , Byers, P. , Belmont, J. , Berglund, B. , Black, J. , … Tinkle, B. (2017). The 2017 international classification of the Ehlers‐Danlos syndromes. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175(1), 8–26. 10.1002/ajmg.c.31552 [DOI] [PubMed] [Google Scholar]
- Miyake, N. , Kosho, T. , Mizumoto, S. , Furuichi, T. , Hatamochi, A. , Nagashima, Y. , … Matsumoto, N. (2010). Loss‐of‐function mutations of CHST14 in a new type of Ehles‐Danlos syndrome. Human Mutation, 31(8), 966–974. 10.1002/humu.21300 [DOI] [PubMed] [Google Scholar]
- Miyake, N. , Tsurusaki, Y. , Koshimizu, E. , Okamoto, N. , Kosho, T. , Brown, N. J. , … Matsumoto, N. (2016). Delineation of clinical features in Wiedemann‐Steiner syndrome caused by KMT2A mutations. Clinical Genetics, 89(1), 115–119. 10.1111/cge.12586 [DOI] [PubMed] [Google Scholar]
- Mizumoto, S. , Kosho, T. , Hatamochi, A. , Honda, T. , Yamaguchi, T. , Okamoto, N. , … Sugahara, K. (2017). Defect in dermatan sulfate in urine of patients with Ehlers‐Danlos syndrome caused by a CHST14/D4ST1 deficiency. Clinical Biochemistry, 50(12), 670–677. 10.1016/j.clinbiochem.2017.02.018 [DOI] [PubMed] [Google Scholar]
- Mochida, K. , Amano, M. , Miyake, N. , Matsumoto, N. , Hatamochi, A. , & Kosho, T. (2016). Dermatan 4‐O‐sulfotransferase 1‐deficient Ehlers‐Danlos syndrome complicated by a large subcutaneous hematoma on the back. Journal of Dermatology, 43(7), 832–833. 10.1111/1346-8138.13273 [DOI] [PubMed] [Google Scholar]
- Müller, T. , Mizumoto, S. , Suresh, I. , Komatsu, Y. , Vodopiutz, J. , Dundar, M. , … Janecke, A. R. (2013). Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers‐Danlos syndrome. Human Molecular Genetics, 22(18), 3761–3772. 10.1093/hmg/ddt227 [DOI] [PubMed] [Google Scholar]
- Sandal, S. , & Kaur, A. (2018). Panigrahi I. Novel mutation in the CHST14 gene causing musculocontractural type of Ehlers‐Danlos syndrome. BMJ Case Reports, pii, bcr‐2018‐226165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirwani, S. , Metcalfe, K. , Wagner, B. , Berry, I. , Sobey, G. , & Jewell, R. (2019). DSE associated musculocontractural EDS, a milder phenotype or phenotypic variability. European Journal of Medical Genetics, 10.1016/j.ejmg.2019.103798 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Syx, D. , Van Damme, T. , Symoens, S. , Maiburg, M. C. , van de Laar, I. , Morton, J. , … Malfait, F. (2015). Genetic heterogeneity and clinical variability in musculocontractural Ehlers‐Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Human Mutation, 36(5), 535–547. 10.1002/humu.22774 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigS1
TableS1
TableS2
DataS1
DataS2
Data Availability Statement
The data are not available for public access because of patient privacy concerns, but are available from the corresponding author on reasonable request.