

Abstract
Background
Neuromuscular disorders (NMDs) comprise a group of heterogeneous genetic diseases with a broad spectrum of overlapping the clinical presentations that makes diagnosis challenging. Notably, the recent introduction of whole‐exome sequencing (WES) is introducing rapid changes on the genetic diagnosis of NMDs. We aimed to investigate the diagnostic value of WES for pediatric‐onset NMDs.
Methods
We applied integrated diagnostic approach and performed WES in 50 Chinese subjects (30 males, 20 females) with undiagnosed pediatric‐onset NMDs despite previous specific tests. The patients were categorized in four subgroups according to phenotyping and investigation findings. Variants on NMDs gene list and open exome analysis for those with initial negative findings were identified.
Results
WES identified causative variants in ACTA1 (n = 2), POMT1, COL6A1 (n = 2), MTMR2, LMNA, SELENON, DNM2, TGFB1, MPZ, IGHMBP2, and LAMA2 in 13 patients. Two subjects have variants of uncertain significance (VUSs) in TTN and SCN11A, unlikely to be pathogenic due to incompatible phenotypes. The mean interval time from symptom onset to genetic diagnosis was 10.4 years (range from 1 month to 33 years). The overall diagnostic yield of WES in our cohort was 26%. Open exome analysis was necessary to identify the pathogenic variant in TGFB1 that caused skeletal dysplasia with neuromuscular presentation.
Conclusion
Our study shows a clear role of WES in the pathway of integrated diagnostic approach to shorten the diagnostic odyssey in patients with rare NMDs.
Keywords: integrated approach, neuromuscular disorders, pediatric‐onset, whole‐exome sequencing
Our study achieved a diagnostic yield from WES of 26% (13/50). These cases were diagnostically challenging as prior investigations failed to give clues on specific genetic diagnosis. Our findings are compatible to previous studies observing the diagnostic yield tends to be lower in cohorts that have already undergone prior extensive evaluations.

1. INTRODUCTION
Neuromuscular disorders (NMDs) comprise a group of heterogeneous diseases of the peripheral nervous system. These diseases are rare, with prevalence rates of 1–10 per 100,000 people worldwide (Deenen, Horlings, Verschuuren, Verbeek, & van Engelen, 2015) and an estimated prevalence of 1 per 4,669 residents in Hong Kong (Chung, Wong, & Ip, 2003). The diagnosis of NMDs is challenging as the phenotypic spectrum is broad, frequently overlapping, and the symptom onset, the clinical features and disease progression are often variable. Nevertheless, establishing a diagnosis is important to enable early treatment, recruitment of patient registries and clinical trials, and genetic counseling.
However, a precise diagnosis remains challenging. Sequential targeted gene Sanger sequencing is the conventional approach in our current clinical setting. This method is tedious and ineffective due to the genotypic heterogeneity of NMDs and huge size of the disease‐associated genes. Moreover, many NMDs‐related genes have not been identified yet.
Most of the above challenges may be resolved by the next generation sequencing (NGS)‐based gene panel test or whole‐exome sequencing (WES). NGS‐based gene panel has the advantage of high coverage, and it is able to detect various types of pathogenic variants including single nucleotide variants (SNVs), small indels, deletions, and duplications, in both the coding region and noncoding region in the known disease‐associated genes. Ankala et al. performed a study in a cohort of NMDs patients using a comprehensive NMDs gene panel. Comparing with other clinical tests, for example, single gene testing and disease‐targeted panels, comprehensive NMDs gene panel has the highest diagnostic rate of 46%. Further, they proposed that 11%–18% of the pathogenic variants would be missed by WES due to the low coverage (Ankala et al., 2015).
Moreover, due to gene‐disease association discovery over the years, gene panel test has its limitation. The study by Park et al. illustrated that a gene panel which includes a more comprehensive list of genes has a higher diagnostic yield when compared to the commercially available gene panel (Park et al., 2019). Frequent comprehensive literature search is required for designing the gene panel up to date. Also, there may be non‐NMDs with phenotypes mimicking NMDs, which would probably be missed by NGS‐gene panel approach. It was believed that WES can overcome these limitations as all the coding regions in the entire human genome is sequenced at one time.
The diagnostic value of WES in NMDs has been demonstrated in previous studies. Haskell et al. performed WES in 93 NMDs pediatric and adult patients with overall diagnostic yield of 12.9%, and only 63% prior phenotyping testing, including invasive muscle biopsy, is informative to reach the diagnosis (Haskell et al., 2018). Waldrop et al. performed trio WES in 31 pediatric patients yielded a diagnostic rate of 39%. Two rare genetic cases, Vici syndrome associated with EGP5, infantile hypotonia with psychomotor retardation, and characteristic facies 3 caused by TBCK pathogenic variants, were identified. With positive genetic diagnosis and proper surveillance, treatment could be provided (Waldrop et al., 2019).
In this study, we report the diagnostic approach applied to a cohort of Chinese patients with undiagnosed pediatric‐onset NMDs and the diagnostic yield associated with WES.
2. METHODOLOGY
2.1. Ethical compliance
The study was approved by the Institutional Review Board of the University of Hong Kong (UW 15‐603). Written informed consent was obtained from all patients or parents of patients recruited in this study.
2.2. Patient cohort
This prospective study recruited 50 Chinese patients with undiagnosed pediatric‐onset hereditary NMDs referred to our hospital between September 2016 and August 2018. All recruited patients were examined by a pediatric neurologist and neuromuscular specialist at one or more clinic visit(s) to gather information on disease presentation, family history, cardiac and lung function assessment, and for a thorough physical examination and investigation analysis including blood assay, imaging, muscle and nerve electrophysiological study, genetic testing, and muscle biopsy.
After evaluation, the patients were categorized into four subgroups: hereditary congenital myopathy subgroup (n = 24), hereditary muscular dystrophy subgroup (n = 11), hereditary peripheral neuropathy subgroup (n = 11), and complex condition with neuromuscular involvement subgroup (n = 4).
2.3. Whole‐exome sequencing: Genomic analysis, NGS data processing, and variant analysis
WES was performed for all recruited subjects as described previously (Tsang et al., 2019). Genomic DNA was extracted from the peripheral blood samples using a Qiagen Blood Mini Kit (Qiagen). Subsequently, an exome library was prepared using the TruSeq Rapid Exome Library Prep Kit (Illumina Inc.). All DNA preparations and library quality control protocols were performed according to the manufacturers’ instructions. The DNA libraries were sequenced using the Illumina NextSeq500 sequencing platform. Our in‐house‐developed bioinformatics pipeline was used for variant calling and data analysis. The raw reads were aligned to the hg19 reference human genome (University of California Santa Cruz, UCSC) using BWA 0.7.10 software (Li & Durbin, 2009). Variant calling workflow was performed according to the GATK best practices, v3.4‐46 (DePristo et al., 2011). The output files were annotated using ANNOVAR software.
The 2018 version of the gene table of monogenic NMDs (nuclear genome) was used for the first‐tier analysis for all patients (Table 1; Bonne, Rivier, & Hamroun, 2017). Subsequently, cases with negative findings were subjected to open exome analysis. Here, we targeted variants located in the coding and canonical splice‐site regions with population frequencies of <1% in control population databases such as the Exome Aggregation Consortium (ExAC) and Genome Aggregation Database (gnomAD). The pathogenicity of the remaining rare variants was assessed using the American College of Medical Genetics and Genomics (ACMG) guideline (Richards et al., 2015). Finally, the potential disease‐causing variants were confirmed and segregated using Sanger sequencing.
Table 1.
Genes in the neuromuscular disorders gene panel for first‐tier analysis (Bonne et al., 2017)
| AARS | AARS2 | ABCC9 | ABHD5 | ACAD9 | ACADVL | ACTA1 | ACTC1 |
| ACTN2 | ACVR1 | ADCK3 | ADSSL1 | AFG3L2 | AGL | AGRN | AHNAK2 |
| AIFM1 | AKAP9 | ALDH18A1 | ALDH3A2 | ALG13 | ALG14 | ALG2 | ALPK3 |
| ALS2 | AMPD2 | ANG | ANK2 | ANKRD1 | ANO10 | ANO5 | ANXA11 |
| AP4B1 | AP4E1 | AP4M1 | AP4S1 | AP5Z1 | APTX | AR | ARHGEF10 |
| ARL6IP1 | ASAH1 | ASCC1 | ATG5 | ATL1 | ATL3 | ATM | ATP13A2 |
| ATP1A1 | ATP1A2 | ATP2A1 | ATP7A | ATXN1 | ATXN10 | ATXN2 | ATXN3 |
| ATXN7 | ATXN8OS | B3GALNT2 | B4GALNT1 | B4GAT1 | BAG3 | BEAN1 | BICD2 |
| BIN1 | BSCL2 | BVES | C12orf65 | C19orf12 | C1orf194 | C9orf72 | CACNA1A |
| CACNA1C | CACNA1G | CACNA1H | CACNA1S | CACNB2 | CACNB4 | CALM1 | CALM2 |
| CALR3 | CAPN1 | CAPN3 | CASQ1 | CASQ2 | CAV3 | CCDC78 | CCDC88C |
| CCT5 | CFL2 | CHAT | CHCHD10 | CHKB | CHMP2B | CHP1 | CHRNA1 |
| CHRNB1 | CHRND | CHRNE | CHRNG | CLCN1 | CLN3 | CLTCL1 | CNBP |
| CNTN1 | CNTNAP1 | COA7 | COL12A1 | COL13A1 | COL25A1 | COL6A1 | COL6A2 |
| COL6A3 | COLQ | COQ2 | COQ4 | COQ6 | COQ7 | COQ9 | COX15 |
| COX6A1 | COX6A2 | CPT1C | CPT2 | CRYAB | CSRP3 | CTDP1 | CTNNA3 |
| CWF19L1 | CYP2U1 | CYP7B1 | DAG1 | DCAF8 | DCTN1 | DDHD1 | DDHD2 |
| DES | DGAT2 | DGUOK | DHTKD1 | DMD | DMPK | DNA2 | DNAJB2 |
| DNAJB6 | DNM2 | DNMT1 | DOK7 | DOLK | DPAGT1 | DPM1 | DPM2 |
| DPM3 | DSC2 | DSG2 | DSP | DST | DTNA | DUX4 | DYNC1H1 |
| DYSF | ECEL1 | EEF2 | EGR2 | ELOVL4 | ELOVL5 | ELP1 | EMD |
| ENO3 | ENTPD1 | ERBB3 | ERBB4 | ERLIN1 | ERLIN2 | ETFA | ETFB |
| ETFDH | EXOSC3 | EXOSC8 | EYA4 | FA2H | FAM111B | FARS2 | FASTKD2 |
| FAT2 | FBLN5 | FBXL4 | FBXO38 | FDX2 | FGD4 | FGF14 | FHL1 |
| FIG4 | FKRP | FKTN | FLAD1 | FLNA | FLNC | FLVCR1 | FUS |
| FXN | FXR1 | GAA | GAN1 | GARS | GATAD1 | GBA2 | GBE1 |
| GDAP1 | GDAP2 | GFPT1 | GJA5 | GJB1 | GJB3 | GJC2 | GLE1 |
| GMPPB | GNB4 | GNE | GOLGA2 | GOSR2 | GPD1L | GRID2 | GRM1 |
| GYG1 | GYS1 | HACD1 | HACE1 | HARS | HCN4 | HEXB | HINT1 |
| HK1 | HNRNPA1 | HNRNPA2B1 | HNRNPDL | HOXD10 | HRAS | HSPB1 | HSPB3 |
| HSPB8 | HSPD1 | HSPG2 | IBA57 | IFRD1 | IGHMBP2 | ILK | INF2 |
| INPP5K | ISCU | ISPD | ITGA7 | ITPR1 | JPH2 | JUP | KARS |
| KBTBD13 | KCNA1 | KCNA5 | KCNC3 | KCND3 | KCNE1 | KCNE2 | KCNE3 |
| KCNH2 | KCNJ18 | KCNJ2 | KCNJ5 | KCNQ1 | KIAA0196 | KIDINS220 | KIF1A |
| KIF1B | KIF1C | KIF21A | KIF26B | KIF5A | KLC2 | KLHL40 | KLHL41 |
| KLHL9 | KY | L1CAM | LAMA2 | LAMA4 | LAMA5 | LAMB2 | LAMP2 |
| LARGE1 | LDB3 | LDHA | LIMS2 | LITAF | LMNA | LMOD3 | LPIN1 |
| LRP12 | LRP4 | LRSAM1 | MAG | MAP3K20 | MAPT | MARS | MARS2 |
| MATR3 | MB | MEGF10 | MET | MFN2 | MGME1 | MIB1 | MME |
| MORC2 | MPV17 | MPZ | MRE11A | MRPL3 | MRPL44 | MRPS25 | MSTN |
| MSTO1 | MTM1 | MTMR2 | MTO1 | MTPAP | MURC | MUSK | MYBPC1 |
| MYBPC3 | MYH14 | MYH2 | MYH3 | MYH6 | MYH7 | MYH8 | MYL1 |
| MYL2 | MYL3 | MYL4 | MYLK2 | MYMK | MYO18B | MYO9A | MYOT |
| MYOZ2 | MYPN | NAGLU | NDRG1 | NDUFAF1 | NEB | NEFH | NEFL |
| NEK1 | NEXN | NGF | NIPA1 | NKX6‐2 | NOP56 | NOTCH2NLC | NPPA |
| NT5C2 | NTRK1 | NUP155 | NUP88 | OPA1 | OPTN | ORAI1 | PABPN1 |
| PAX7 | PCNA | PDK3 | PDYN | PEX7 | PFKM | PFN1 | PGAM2 |
| PGK1 | PGM1 | PHKA1 | PHOX2A | PHYH | PIEZO2 | PIP5K1C | PKP2 |
| PLD3 | PLEC | PLEKHG5 | PLN | PLP1 | PMP2 | PMP22 | PNKP |
| PNPLA2 | PNPLA6 | PNPLA8 | POGLUT1 | POLG | POLG2 | POMGNT1 | POMGNT2 |
| POMK | POMT1 | POMT2 | PPP2R2B | PRDM12 | PRDM16 | PREPL | PRKAG2 |
| PRKCG | PRPH | PRPS1 | PRUNE1 | PRX | PSEN1 | PSEN2 | PTRF |
| PTRH2 | PUM1 | PUS1 | PYGM | PYROXD1 | RAB7A | RAF1 | RAPSN |
| RBCK1 | RBM20 | RBM7 | REEP1 | REEP2 | RETREG1 | RFC1 | RNASEH1 |
| RNF216 | RPH3A | RRM2B | RTN2 | RUBCN | RXYLT1 | RYR1 | RYR2 |
| RYR3 | SACS | SBF1 | SBF2 | SCN11A | SCN1B | SCN2B | SCN3B |
| SCN4A | SCN4B | SCN5A | SCN9A | SCO2 | SCYL1 | SDHA | SELENON |
| SEPT9 | SETX | SGCA | SGCB | SGCD | SGCE | SGCG | SGPL1 |
| SH3TC2 | SIGMAR1 | SIL1 | SLC12A6 | SLC16A1 | SLC18A3 | SLC1A3 | SLC22A5 |
| SLC25A1 | SLC25A20 | SLC25A4 | SLC25A42 | SLC25A46 | SLC33A1 | SLC52A2 | SLC52A3 |
| SLC5A7 | SLC9A1 | SMCHD1 | SMN1 | SNAP25 | SNTA1 | SNX14 | SOD1 |
| SPAST | SPEG | SPG11 | SPG20 | SPG21 | SPG7 | SPTAN1 | SPTBN2 |
| SPTBN4 | SPTLC1 | SPTLC2 | SQSTM1 | STAC3 | STIM1 | STUB1 | SUCLA2 |
| SUCLG1 | SURF1 | SYNE1 | SYNE2 | SYT14 | SYT2 | TARDBP | TAZ |
| TBK1 | TBP | TCAP | TDP1 | TDP2 | TECPR2 | TECRL | TFG |
| TGFB3 | TGM6 | TIA1 | TIMM22 | TK2 | TMEM240 | TMEM43 | TMEM65 |
| TMPO | TNNC1 | TNNI2 | TNNI3 | TNNT1 | TNNT2 | TNNT3 | TNPO3 |
| TOP3A | TOR1A | TOR1AIP1 | TPM1 | TPM2 | TPM3 | TPP1 | TRAPPC11 |
| TRDN | TRIM2 | TRIM32 | TRIM54 | TRIM63 | TRIP4 | TRPC3 | TRPV4 |
| TSFM | TTBK2 | TTN | TTPA | TTR | TUBA4A | TUBB3 | TWNK |
| TYMP | UBA1 | UBA5 | UBAP1 | UBQLN2 | UCHL1 | UNC13A | VAMP1 |
| VAPB | VCL | VCP | VMA21 | VPS13D | VPS37A | VRK1 | VWA3B |
| WARS | WDR73 | WNK1 | WWOX | XRCC1 | YARS | YARS2 | ZFHX2 |
| ZFYVE26 | ZFYVE27 |
3. RESULTS
3.1. Overall
Fifty Chinese patients (30 males and 20 females, male: female ratio: 1.5:1) of ages ranging from 1 month to 37 years old, were recruited. Thirty‐six patients (72%) were below 18 years old at the time of recruitment.
An average coverage of 89× was achieved by WES. Twelve patients (12/50, 24%) were found to harbor either pathogenic or likely pathogenic variants compatible with the clinical diagnosis using the NMDs gene panel. One additional diagnosis was made using open exome analysis (1/50, 2%). The overall WES diagnostic yield was 26% (13/50; Table 2). Two patients were found to harbor VUSs incompatible with the disease phenotype (Table S1).
Table 2.
Detail clinical information, prior investigation findings, WES result and diagnosis of the 13 patients with positive findings in this cohort
| Project No. | Sex/ Age | Onset | NMD sub group | Motor | Clinical presentation | Systemic involvement (respiratory, cardiac, cognition, musculoskeletal) | Investigations | WES findings |
ACMG CLN Post_P value by Bayesian calculation |
Reported/ Novel (diagnosis) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MRI brain | MRI muscles |
CK U/L |
Muscle biopsy | NCS & EMG | Prior normal gene studies | |||||||||||
|
NMD 006 |
F/18 | B | CM | Delayed walking since 24 months | Arthrogyposis multiplex congenita and delay walking, followed by stabilization of motor performance since then. Exam showed facial, neck flexor and limb girdle weakness, pectus excavatum, rigid spine, and club feet. |
Respiratory: Restrictive lung function Cardiac: Mitral valve prolapse and regurgitation with left ventricular dilatation on Lisinopril Cognition: Normal Musculoskeletal: rigid spine, pes cavus |
NAD | ABN | 54–200 | Nonspecific myopathic change |
NCV: Absent bilateral peroneal CMAP responses EMG: Myopathic |
— | ACTA1 (NM_001100.3) | c.874A>G, p.(Arg292Gly) de novo | Likely pathogenic (0.975) | Novel (Congenital myopathy) |
|
NMD 037 |
F/1 month | AN | CM |
Non‐ambulatory Minimal active movement |
Antenatal polyhydramnios and decrease fetal movement. Born prematurely at 32 weeks of gestation, required intubation, and resuscitation at birth. Exam showed generalized weakness with poor respiratory effort. Developed chylothorax required chest drain. Died at 46 days old. |
Respiratory: Intubated and ventilated since birth Cardiac: Normal Cognition: Uncertain Musculoskeletal: Bilateral shoulder, wrist, finger, knee, ankle contractures |
ND | ND | 298 | ND | ND | SMN1 | ACTA1 (NM_001100.3) | c.1001C>G, p.(Pro334Arg) de novo | Likely pathogenic (0.975) | Reported (Laing et al., 2009) Nemaline myopathy (Congenital myopathy) |
|
NMD 033 |
M/22 | B | CM |
Walking before 18 months Fell easily at early age Lost ambulation at 13 years |
Neonatal‐onset hypotonia and mild generalized weakness. Independent walking at a young age with waddling and easy falling. Lost ambulation at 13 years and required nocturnal NIV since aged 14 and continuous use since aged 19. Developed progressive scoliosis, and lost ability to sit at aged 22. Exam revealed facial weakness with ptosis, ophthalmoplegia, generalized weakness, muscle wasting, marked scoliosis. |
Respiratory: Nocturnal NIV since 14 years; Continuous NIV since 19 years Cardiac: Normal Cognition: Normal Musculoskeletal: Cavovarus feet with surgeries; Scoliosis with initial brace use; refused scoliosis surgery |
ND | ND | 21–72 | congenital fiber‐type dispropor‐tion | Refused | — | DNM2 (NM_001005360.2) | c.1852G>A, p.(Ala618Thr) de novo | Likely pathogenic (0.975) | Reported (Bitoun et al., 2007) Centronuclear myopathy (Congenital myopathy) |
|
NMD 016 |
M/16 | CH | CM | Delayed walking since 21 months | Delayed walking and persistent motor clumsiness with increased walking difficulty at 10 years. Developed type II respiratory failure required BIPAP at 13 years. Rigid spine with neck hyperextension observed at 14.5 years. Exam showed limb girdle weakness (lower limb > upper limb) and contractures. |
Respiratory: Nocturnal NIV since 13 years Cardiac: Normal Cognition: Normal Musculoskeletal: Progressive rigid spine, contractures at shoulders, elbows, ankles |
NAD | ABN |
↑ 116–360 |
Nonspecific myopathic change |
NCV: Small CMAPs EMG: ND |
SELENON (NM_206926.1) |
c.238delG, p.(Asp80Thrfs*20) unknown inheritance c.1304G>A, p.(Arg435Gln) maternal inherited |
Likely pathogenic and VUS (0.994 and 0.900) | Novel (Rigid spine syndrome) | |
|
NMD 046 |
F/19 | IN | MD | Delayed walking since 18 months | Noted hypotonia & dislocated hip in first year of life. Delayed walking at 18 months old. Required orthopaedic intervention to both hip dislocation at aged 2. Persistent waddling gait and failed to climb stairs unassisted at 6 years of age. Exam revealed facial rash, pilaris keratosis, distal hyperlaxity, marked limb girdle weakness, elbow, and finger flexor contracture, marked equinovarus. |
Respiratory: No need for ventilator Cardiac: Normal Cognition: Normal Musculoskeletal: Dislocated hip before 1 years; progressive long finger flexor, elbow flexor and knee flexion contractures, tight Achilles tendons; scoliosis without brace |
NAD | ND | 61–299 | Dystrophic change, severe deletion of collagen VI in sarcolemma |
NCV: Normal EMG: Myopathic |
— | COL6A1 (NM_001848.3) |
c.850G>A, p.(Gly284Arg) de novo |
Pathogenic (1.000) | Reported (Giusti et al., 2005) Ullrich CMD (Ullrich CMD) |
|
NMD 008 |
M/18 | CH | MD | Walking since 12 months | Normal walking at 1 years old, but persistent easy falling and motor clumsiness after 6 years. Examination revealed mild limb girdle weakness (lower limb > upper limb), distal hyperlaxity. Facial rash, follicular hyperkeratosis, contractures over finger flexors, elbows, and tendoachilles, pes cavus. |
Respiratory: Mild obstructive sleep apnea syndrome Cardiac: Normal Cognition: Normal Musculoskeletal: Progressive long finger flexor, elbow flexion and knee flexion contractures, tight Achilles tendons |
NAD | ABN |
↑ 299–1,311 |
Dystrophic changes ↓ alpha‐dystrogly‐can no collagen VI staining done |
NCV: Normal EMG: Myopathic |
DMD FKTN FKRP |
COL6A1 (NM_001848.3) |
c.1056 + 2dupT de novo |
Pathogenic (1.000) | Novel (Bethlam myopathy) |
|
NMD 013 |
M/8 | CH | MD | Walking since 12 months | History of persistent clumsiness since walking began. Noted lower limb weakness at 2 years. Exam showed thin body build, limb girdle weakness (lower limb > upper limb), elbow & finger contractures, and tight tendoachilles. |
Respiratory: Normal Cardiac: Normal Cognition: Normal Musculoskeletal: Contractures at elbows and ankles |
NAD | ABN |
↑ 699–1,259 |
Myopathic change Core‐like structures |
NCV: Normal EMG: Myopathic |
SElENON | LMNA (NM_170707.4) |
c.1357C>T, p.(Arg453Trp) de novo |
Likely pathogenic (0.999) |
Reported (Bonne et al., 1999) EDMD (EDMD) |
|
NMD 048 |
F/1 | NN | MD |
Sitting Walking with support |
Noted hypotonia, hyporeflexia, significant head lag and decreased active limb movement in the first month of life. Persistent gross motor delay. Sat unsupported at 9 months. |
Respiratory: Normal Cardiac: Normal Cognition: Normal Musculoskeletal: No contractures |
ABN | ND |
↑ 2,253–2,887 |
ND |
NCV: Small CMAPs EMG: ND |
— | LAMA2 (NM_000426.3) |
c.250C>T, p.(Arg84*) unknown inheritance c.4157A>T, p.(Tyr1386Phe) unknown inheritance |
Likely pathogenic and VUS 0.994 and 0.325 |
Novel (Merosin deficient CMD) |
|
NMD 007 |
F/12 | CH | MD | Walking since 16 months | History of developmental delays & mild joint laxity. Asymptomatic hyperCKaemia with no clinical weakness. Microcephaly. Mild autism spectrum disorder. |
Respiratory: Normal Cardiac: Normal Cognition: Mild intellectual disabilities, autism spectrum disorder Musculoskeletal: Normal |
NAD | AB | ↑1408–3,883 | ND | Both NCV and EMG normal | DMD | POMT1 (NM_007171.3) |
c.685C>T, p.(Gln229*) maternally inherited c.1024C>T, p.(His342Tyr) paternally inherited |
Likely pathogenic and VUS (0.997 and 0.812) | Novel (Asymptomatic HyperCKaemia) |
|
NMD 010 |
F/33 | CH | PN |
Walking since normal age Independent walking until 17 years |
History of easy falling and foot deformity requiring orthopedic surgeries during childhood. Progressive weakness with loss of ambulation at 17 years, nocturnal NIV since 29 years. Exam revealed ptosis, facial weakness, soft & hoarse voice, generalized limb weakness (distal > proximal), muscle wasting in hands, and calves. |
Respiratory: Nocturnal NIV since 29 years Cardiac: Normal Cognition: Normal Musculoskeletal: Contractures at fingers, knees and ankles, mild scoliosis |
ND | ND | 30–91 |
End stage changes suspected decrease collagen VI |
NCV: Refused initially, agreed after genetic result available – no SNAP, CMAP responses EMG: ND |
COL6A1, COL6A2, COL6A3 | MTMR2 (NM_016156.5) |
c.1797_1798insAGAA, p.(Leu600fs*5) maternal inherited c.1387−2A>G unknown inheritance |
Pathogenic (0.994 and 0.994) | Novel (CMT 4B1) |
|
NMD 060 |
F/7 | CH | PN |
Walking since 15 months Independent walking |
History of easy falling and unsteady gait since walking began. Exam showed mild proximal girdle weakness but significant distal weakness with muscle wasting and mild tightness in Achilles tendons. |
Respiratory: Normal Cardiac: Normal Cognition: Unknown Musculoskeletal: Normal |
ND | ND | 83 | ND |
NCV: SM axonal polyneuro‐pathy EMG: Neurogenic |
— | IGHMBP2 (NM_002180.2) |
c.1060G>A, p.(Gly354Ser) paternally inherited c.2356delG, p.(Arg786fs*45) unknown inheritance |
Likely pathogenic (0.900 and 0.994) |
Reported (Giannini et al., 2006; Liu et al., 2017) SMARD type I (CMT 2) |
|
NMD 042 |
M/14 | CH | PN |
Normal walking age Independent walking |
History of intermittent lower limb pain and motor clumsiness since 8 years. Stable motor performance with mild progressive distal muscle weakness, especially weak ankle dorsiflexors with varus deformity of the feet over time. |
Respiratory: Normal Cardiac: Normal Cognition: Normal Musculoskeletal: Contractures at ankles |
ND | ND | 48–127 | ND |
NCV: SM demyelina‐ting polneuro‐pathy EMG: ND |
PMP22 | MPZ (NM_000530.8) | c.737A>G, p.(Asp246Gly) de novo | Likely pathogenic (0.975) | Novel (CMT1B) |
|
NMD 034 |
F/8 | CH | CN | Walking since 12 months | Walked at a normal age but had persistent easy falling and motor clumsiness after 2 years. Very mild lower limb girdle weakness noted on examination. |
Respiratory: Normal Cardiac: Normal Cognition: Normal Musculoskeletal: Skeletal dysplasia |
NAD | NAD | 100 | ND |
NCV: Normal EMG: neurogenic |
SMN1 | TGFB1 (NM_000660.6) |
c.653G>A, p.(Arg218His) de novo |
Likely pathogenic (0.975) | Reported (Kinoshita et al., 2000) Camurati‐Engelmann disease (Camurati‐Engelmann disease) |
Abbreviations: ↑, increased; ABN, abnormal; ACMG CLN, American College of Medical Genetics and Genomics Classification; AD, adulthood; Adol, adolescence; AN, antenatal; B, birth; BIPAP, Bilevel positive airway pressure; BMD, Becker muscular dystrophy; CH, childhood; CK, creatine kinase; CM, hereditary congenital myopathy subgroup; CMAP, compound muscle action potentials; CMD, Congenital muscular dystrophy; CN, complex condition with neuromuscular involvement subgroup; EDMD, Emery Dreifuss muscular dystrophy; EMG, electromyography; F, female; IN, infantile; LGMD, limb‐girdle muscular dystrophy; M, male; MD, hereditary muscular dystrophy subgroup; MRI, magnetic resonance imaging; NAD, normal; NCS, nerve conduction study; NCV, nerve conduction study; ND, not done; NIV, noninvasive ventilation; NMDs, Neuromuscular disorders; NN, neonatal; PN, hereditary peripheral neuropathy subgroup; SM, sensorimotor; SMARD, spinal muscular atrophy with respiratory distress; SNAP, sensory nerve action potentials; VUS, variant of unknown sequencing; WES, whole‐exome sequencing.
3.2. Hereditary congenital myopathy subgroup
Four out of 24 patients in congenital myopathy subgroup were found to have disease‐causing variants in ACTA1 (OMIM 102610) (n = 2), SELENON (OMIM 606210), and DNM2 (OMIM 602378), giving a diagnostic yield of 17%.
One of the patients with ACTA1 variant had a mild motor phenotype (NMD006; Figure 1a,b), whereas the other subject presented with antenatal onset fetal akinesia with poor prognosis and was electively extubated after detailed discussion with parents (NMD037; Figure 1c,d). One patient has SELENON rigid spine syndrome with typical disease presentation (NMD016; Figure 1e). One patient has DMN2‐related congenital myopathy with progressive disease course (NMD033).
Figure 1.
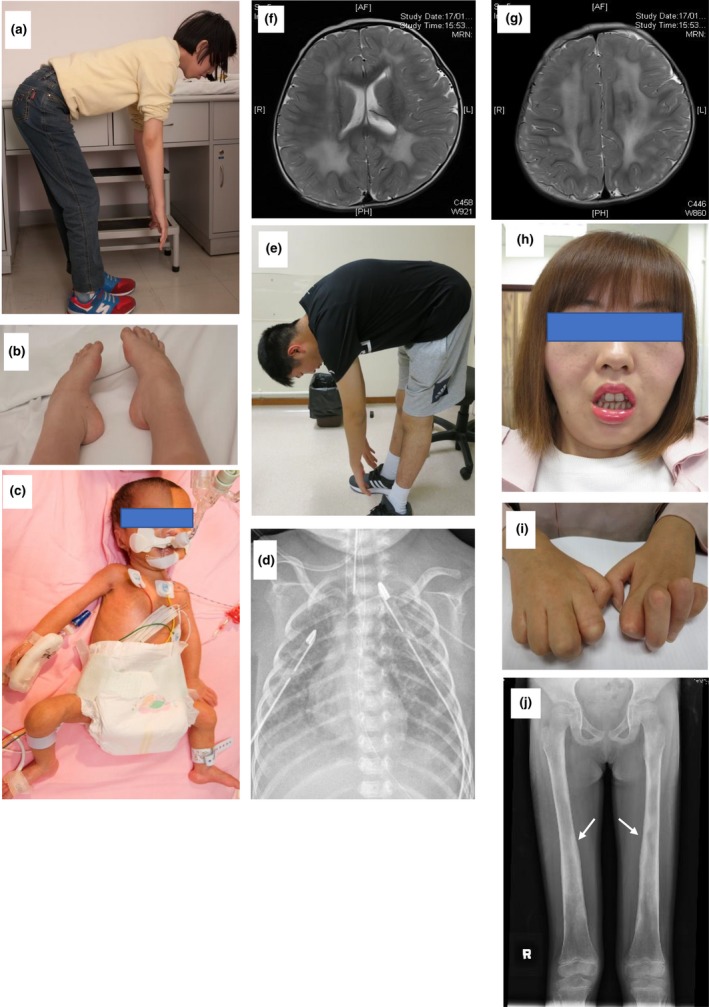
(a, b) Patient with heterozygous novel variant in ACTA1 has rigid spine and pes cavus. (c, d) Patient with heterozygous reported variant in ACTA1 had flaccid posture with minimal skin creases. Chest X‐ray demonstrated bilateral chest drains with residual chylothorax on the right side. (e) Patient with compound heterozygous variants in SELENON has rigid spine. (f, g) Patient with compound heterozygous variants in LAMA2 has the axial view of her T2 weighted MRI brain images shown diffuse cerebral white matter signal changes compatible to merosin‐deficient congenital muscular dystrophy. (h, i) Patient with compound heterozygous loss‐of‐function variants in the MTMR2, with facial weakness and marked finger contractures with distal hand wasting. (j) Patient with heterozygous reported pathogenic variant in TGFB1 with thickening of the diaphyseal bones (white arrows) on her X‐ray of bilateral femurs
3.3. Hereditary muscular dystrophy subgroup
Among the 11 patients in this subgroup, WES identified four disease‐causative genes, including COL6A1 (OMIM 120220; n = 2), LMNA (OMIM 150330), POMT1 (OMIM 607423), and LAMA2 (OMIM 156225), in five patients giving a diagnostic yield of 45% (5/11).
One girl has POMT1‐related asymptomatic hyperCKaemia with no major weakness but mild joint hyperlaxity (NMD007). She also has mild intellectual disability, autism spectrum disorder, and microcephaly, although her brain MRI findings were normal. WES found compound heterozygous variants in POMT1 (NM_007171.3) The nonsense variant c.685C>T, p.(Gln229Ter), which has a population frequency of 0.000004061, was classified as likely pathogenic. The missense variant c.1024C>T, p.(His342Tyr) was a VUS with multiple in silico prediction as damaging.
Another young girl with LAMA2‐related congenital muscular dystrophy (NMD048), she has infantile onset hypotonia with isolated gross motor delay. Brain MRI revealed diffuse cerebral white matter changes (Figure 1f,g) characteristic of merosin‐deficient congenital muscular dystrophy.(Kumar, Aroor, Mundkur, & Kumar, 2014) WES identified compound heterozygous variants in LAMA2 (NM_000426.3). The variant c.250C>T, p. (Arg84Ter) was classified as likely pathogenic, whereas c.4157A>T, p.(Tyr1386Phe) was considered as a VUS with a population frequency of 0.00009693 and a contradictory in silico prediction.
3.4. Hereditary peripheral neuropathy subgroup
WES identified disease‐causing variants in MTMR2 (OMIM 603557), MPZ (OMIM 159440), and IGHMBP2 (OMIM 600502) in three patients, giving a diagnostic yield of 27% (3/11). One patient had a VUS in SCN11A (OMIM 604385). Two related patients with negative WES had subsequent neuropathy gene panel revealed a reported disease causing variant in the noncoding region of GJB1 (OMIM 304040).
Here we reported the first Charcot Marie Tooth Disease (CMT) type 4B1 in Chinese (NMD010) (Figure 1h,i). WES found compound heterozygous loss‐of‐function variants over the MTMR2 gene (NM_016156.5), c.1794_1797dup, p.(Leu600Argfs*5), and c.1387‐2A>G. Both variants were novel and absent in the control population. Both variants are classified as pathogenic, supported by in vivo studies of mtmr2‐null mice revealed myelin alterations that produced a less severe version of the neuropathy observed in humans (Bolino et al., 2004; Bonneick et al., 2005).
3.5. Complex condition with associated neuromuscular involvement
Among the four patients in this subgroup, WES identified one disease‐causative variant in, TGFB1 (OMIM 190180) by open exome analysis, giving a diagnostic yield of 25% (1/4).
The female patient (NMD034) had persistent motor clumsiness and limb pain, mild girdle weakness. WES analysis using NMDs gene panel did not identify any pathogenic variant. We subsequently proceeded with open exome analysis, which revealed a previously reported pathogenic variant, c.653G>A, p.(Arg218His), in TGFB1 (NM_000660.6). Segregation study using Sanger sequencing confirms the variant is de novo. This variant was associated with Camurati–Engelmann disorder (Kinoshita et al., 2000). Patients with this condition exhibit progressive diaphyseal dysplasia with thickening of the diaphyseal bones, sclerosis of the skull base, and bone pain. Many cases are initially classified as neuromuscular disease because this condition often manifests with features of myopathy. Her subsequent skeletal survey confirmed skeletal dysplasia (Figure 1j).
4. DISCUSSION
4.1. Our study confirms the importance of WES in the diagnosis of patients with NMDs
4.1.1. Diagnostic yield
Our study achieved an overall diagnostic yield from WES of 26% (13/50), with 12 cases (24%) by NMDs gene panel analysis, and one (2%) by open exome analysis. These cases were diagnostically challenging as prior investigations failed to give clues on specific genetic diagnosis. Our findings are compatible to previous studies where the diagnostic yield tends to be lower in cohorts that have undergone prior comprehensive evaluations, with diagnostic yields as low as 12.9% (Haskell et al., 2018) comparing to 79% in a newly diagnostic cohorts without prior extensive investigation (Schofield et al., 2017). The integrated approach, with deep phenotyping complemented by investigation findings, support the categorization of four distinct subgroups, with the muscular dystrophy subgroup had the highest diagnostic yield (45%).
4.2. Finding of pathogenic variants previously unreported in Chinese populations
Few studies based on WES have been performed in Chinese NMDs patients. To our knowledge, this is the first cohort study to report pathogenic variants in a Chinese patient with MTMR2‐related CMT type 4B1. This finding highlights the usefulness of WES in such cases and expands the genetic spectrum relevant to the Chinese population.
4.3. Impact on clinical management
A timely diagnosis enables accurate prognostication and provides guidance regarding the clinical management and family counseling. For example, the confirmed genetic diagnosis of antenatal‐onset ACTA1 myopathy in our patient within the first month of life enabled an accurate prognostication to support parents to understand the infant's condition. This led to timely counseling on redirection of care, and provided the parents with relevant knowledge needed to prepare for future pregnancies. A genetic diagnosis also enables relevant surveillance and early detection of NMD‐related complications. As in our child with Emery–Dreifuss muscular dystrophy, though he has not yet developed any cardiac involvement, the genetic diagnosis highlighted the need for close monitoring of cardiac symptoms to enable the early detection of arrhythmia or cardiomyopathy. In another child with Camurati–Engelmann disease, the finding of bony dysplasia that mimicked a neuromuscular condition allowed a timely referral for orthopedic monitoring of skeletal complications. At the same time, routine echocardiogram was discontinued.
4.4. Expanding the phenotype of known pathogenic variant
POMT1 variant has not been reported to be associated with asymptomatic hyperCKaemia. The findings of our patient have expanded the clinical spectrum of this known pathogenic variant.
4.5. Role and limitations of WES
WES has shortened the diagnostic odyssey for our patients. The time from symptom onset to achieve a genetic diagnosis from WES of our 13 patients ranged from 1 month to 30 years with a mean interval time of 10.4 years. All our recruited patients are diagnostically challenging with no causative genetic diagnosis despite highly specialized tests including muscle biopsy, gene test, and imaging (Figure 2). WES also identifies new conditions that mimic neuromuscular disorders, as observed in our patient with skeletal dysplasia due to TGFB1 pathogenic variant.
Figure 2.
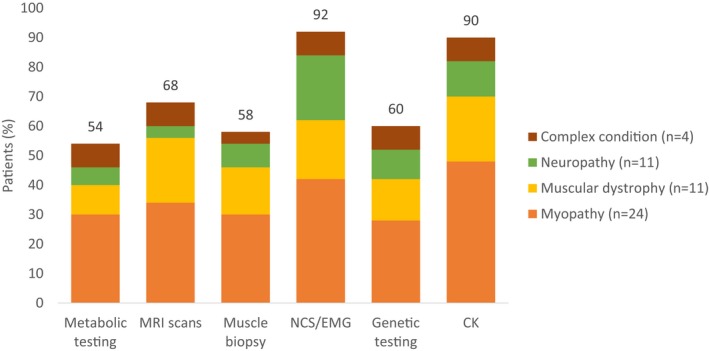
Summary of patients in each phenotypic category with prior neuromuscular investigations including metabolic testing, MRI scan, muscle biopsy, nerve conduction study/electromyography, genetic testing, and creatine kinase testing. CK, creatine kinase; EMG, electromyography; MRI, magnetic Resonance Imaging; NCS, nerve conduction study
However, WES also has its limitations. One major challenge is the interpretation of VUSs, which represents a potential bottleneck in the genetic diagnostic pipeline (Bertier, Hétu, & Joly, 2016). In our cohort, we identified 2 VUSs not compatible with the patients' phenotypes. The patient with heterozygous TTN variant might be either carrying a second undetected TTN disease‐causing variant and therefore has a recessive titnopathy, or is simply carrying this heterozygous variant that does not explain his condition (Table S1).
Short‐read WES is of limited usefulness for detecting variants other than SNVs and small indels, such as copy number variations (CNVs), expansions, or contractions in repetitive regions, chromosomal rearrangements and deep intronic variants. CNVs include exon deletion in SMN1 in spinal muscular atrophy, exon deletion, or duplication in dystrophinopathy, PMP22 duplication in Charcot‐Marie‐tooth diseases, could be evaluated by multiple ligation probe analysis (MLPA) or whole‐genome sequencing. Expansion or contraction in repetitive regions include CTG triplet repeat in myotonic dystrophy and contraction of the D4Z4 macrosatellite repeat in DUX4 in facioscapulohumeral muscular dystrophy could be evaluated by fragment analysis. Correct clinical diagnosis of these distinctive NMDs guiding the appropriate target gene study would avoid unnecessary WES that could not detect these variants.
WES may also miss the variant outside the exome that arise in the deep intronic or untranslated regions (UTR). This is illustrated by the two brothers, with heterozygous variant in the 5´ UTR of GJB1 with c.‐103C>T variant have been identified as causative for X‐linked CMT (Tomaselli et al., 2017) that could not be picked up by WES but revealed by neuropathy gene panel that specifically looked up this region. Furthermore, pseudogenes may lead to WES errors and WES coverage is nonoptimal at the beginnings or ends of exons, in GC‐rich regions, and homopolymeric regions.
5. CONCLUSION
Deep phenotyping is needed to direct the WES analysis. While there are challenges in using WES, its application has shortened the diagnostic odyssey for patients with monogenic neuromuscular disorders. Given the declining cost, we recommend considering WES in the diagnostic workflow in patients with NMDs (Figure 3).
Figure 3.
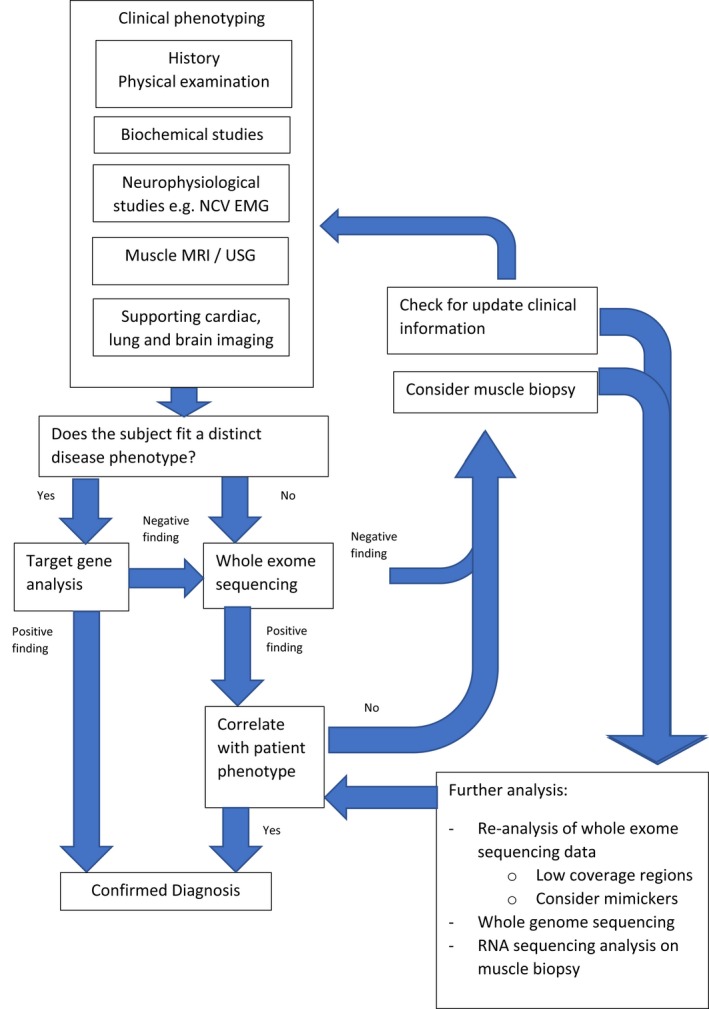
Proposed integrated diagnostic approach of neuromuscular disorders
CONSENT FOR PUBLICATION
All subjects in this study provided signed consent for the publication of their data.
CONFLICT OF INTEREST
All authors have no conflict of interest to declare.
AUTHOR CONTRIBUTION
Mandy Ho Yin Tsang and Annie Ting Gee Chiu should be considered joint first author. Brian Hon Yin Chung, Sophelia Hoi Shan Chan should be considered joint corresponding authors. Mandy Ho Yin Tsang designed and performed the WES analysis and prepared the manuscript. Annie Ting Gee Chiu prepared the manuscript. Bernard Ming Hong Kwong designed and performed the WES analysis. Rui Liang assisted patient recruitment, data entry, DNA extraction, and WES preparation. Wetor Hok Lai Ho helped with the DNA extraction and WES preparation. Mullin Ho Chung Yu, Kit San Yeung, Christopher Chun Yu Mak, Gordon Ka Chun Leung, Steven Lim Cho Pei, and Jasmine Lee Fong Fung assisted the WES analysis. Virginia Chun Nei Wong and Francesco Muntoni reviewed the manuscript. Brian Hon Yin Chung design and conducted the WES analysis. Sophelia Hoi Shan Chan recruited the patients, designed the study, and prepared the manuscript. All co‐authors approved the submitted manuscript.
Supporting information
Table S1
ACKNOWLEDGMENTS
We thank all the pediatric neurology colleagues who referred the patients to our neuromuscular diagnostic program, and all the participating patients and families of this study.
Tsang MHY, Chiu ATG, Kwong BMH, et al. Diagnostic value of whole‐exome sequencing in Chinese pediatric‐onset neuromuscular patients. Mol Genet Genomic Med. 2020;8:e1205 10.1002/mgg3.1205
Mandy H. Y. Tsang and Annie T. G. Chiu are co‐first authors.
Funding information
The WES study was funded by a grant from the Health and Medical Research Fund (HMRF).
Contributor Information
Brian H. Y. Chung, Email: bhychung@hku.hk.
Sophelia H. S. Chan, Email: sophehs@hku.hk.
DATA AVAILABILITY STATEMENT
The dataset analyzed during the current study is not publicly available for privacy reasons. However, the corresponding authors will provide access upon reasonable request.
REFERENCES
- Ankala, A. , da Silva, C. , Gualandi, F. , Ferlini, A. , Bean, L. J. H. , Collins, C. , … Hegde, M. R. (2015). A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Annals of Neurology, 77(2), 206–214. 10.1002/ana.24303 [DOI] [PubMed] [Google Scholar]
- Bertier, G. , Hétu, M. , & Joly, Y. (2016). Unsolved challenges of clinical whole‐exome sequencing: A systematic literature review of end‐users' views. BMC Medical Genomics, 9(1), 52–52. 10.1186/s12920-016-0213-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitoun, M. , Bevilacqua, J. A. , Prudhon, B. , Maugenre, S. , Taratuto, A. L. , Monges, S. , … Guicheney, P. (2007). Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Annals of Neurology, 62(6), 666–670. 10.1002/ana.21235 [DOI] [PubMed] [Google Scholar]
- Bolino, A. , Bolis, A. , Previtali, S. C. , Dina, G. , Bussini, S. , Dati, G. , … Wrabetz, L. (2004). Disruption of Mtmr2 produces CMT4B1‐like neuropathy with myelin outfolding and impaired spermatogenesis. The Journal of Cell Biology., 167(4), 711–721. 10.1083/jcb.200407010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonne, G. , Barletta, M. R. D. , Varnous, S. , Bécane, H.‐M. , Hammouda, E.‐H. , Merlini, L. , … Schwartz, K. (1999). Mutations in the gene encoding lamin A/C cause autosomal dominant Emery‐Dreifuss muscular dystrophy. Nature Genetics, 21(3), 285 10.1038/6799 [DOI] [PubMed] [Google Scholar]
- Bonne, G. , Rivier, F. , & Hamroun, D. (2017). The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscular Disorders, 27(12), 1152–1183. 10.1016/j.nmd.2017.10.005 [DOI] [PubMed] [Google Scholar]
- Bonneick, S. , Boentert, M. , Berger, P. , Atanasoski, S. , Mantei, N. , Wessig, C. , … Suter, U. (2005). An animal model for Charcot–Marie–Tooth disease type 4B1. Human Molecular Genetics, 14(23), 3685–3695. 10.1093/hmg/ddi400 [DOI] [PubMed] [Google Scholar]
- Chung, B. , Wong, V. , & Ip, P. (2003). Prevalence of neuromuscular diseases in Chinese children: A study in southern China. Journal of Child Neurology, 18(3), 217–219. 10.1177/08830738030180030201 [DOI] [PubMed] [Google Scholar]
- Deenen, J. C. , Horlings, C. G. , Verschuuren, J. J. , Verbeek, A. L. , & van Engelen, B. G. (2015). The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. Journal of Neuromuscular Diseases, 2(1), 73–85. 10.3233/JND-140045 [DOI] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , … Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43(5), 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini, A. , Pinto, A. M. , Rossetti, G. , Prandi, E. , Tiziano, D. , Brahe, C. , & Nardocci, N. (2006). Respiratory failure in infants due to spinal muscular atrophy with respiratory distress type 1. Intensive Care Medicine, 32(11), 1851–1855. 10.1007/s00134-006-0346-8 [DOI] [PubMed] [Google Scholar]
- Giusti, B. , Lucarini, L. , Pietroni, V. , Lucioli, S. , Bandinelli, B. , Sabatelli, P. , … Pepe, G. (2005). Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Annals of Neurology, 58(3), 400–410. 10.1002/ana.20586 [DOI] [PubMed] [Google Scholar]
- Haskell, G. T. , Adams, M. C. , Fan, Z. , Amin, K. , Guzman Badillo, R. J. , Zhou, L. , … Berg, J. S. (2018). Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurology Genetics, 4(1), e212 10.1212/nxg.0000000000000212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, A. , Saito, T. , Tomita, H.‐A. , Makita, Y. , Yoshida, K. , Ghadami, M. , … Yoshiura, K.‐I. (2000). Domain‐specific mutations in TGFB1 result in Camurati‐Engelmann disease. Nature Genetics, 26(1), 19–20. 10.1038/79128 [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Aroor, S. , Mundkur, S. , & Kumar, M. (2014). Merosin‐deficient congenital muscular dystrophy with cerebral white matter changes. Bmj Case Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing, N. G. , Dye, D. E. , Wallgren‐Pettersson, C. , Richard, G. , Monnier, N. , Lillis, S. , … Mitrani‐Rosenbaum, S. (2009). Mutations and polymorphisms of the skeletal muscle α‐actin gene (ACTA1). Human Mutation, 30(9), 1267–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Li, X. , Hu, Z. , Mao, X. , Zi, X. , Xia, K. , … Zhang, R. (2017). IGHMBP2‐related clinical and genetic features in a cohort of Chinese Charcot‐Marie‐Tooth disease type 2 patients. Neuromuscular Disorders, 27(2), 193–199. 10.1016/j.nmd.2016.11.008 [DOI] [PubMed] [Google Scholar]
- Park, J. , Oh, H. M. , Park, H. J. , Cho, A. R. , Lee, D. W. , Jang, J. H. , & Jang, D. H. (2019). Usefulness of comprehensive targeted multigene panel sequencing for neuromuscular disorders in Korean patients. Molecular Genetics & Genomic Medicine, 7(10), e00947 10.1002/mgg3.947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield, D. , Alam, K. , Douglas, L. , Shrestha, R. , MacArthur, D. G. , Davis, M. , … O’Grady, G. L. (2017). Cost‐effectiveness of massively parallel sequencing for diagnosis of paediatric muscle diseases. NPJ Genomic Medicine, 2, 10.1038/s41525-017-0006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli, P. J. , Rossor, A. M. , Horga, A. , Jaunmuktane, Z. , Carr, A. , Saveri, P. , … Reilly, M. M. (2017). Mutations in noncoding regions of GJB1 are a major cause of X‐linked CMT. Neurology, 88(15), 1445–1453. 10.1212/wnl.0000000000003819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang, M.‐Y. , Leung, G.‐C. , Ho, A.‐C. , Yeung, K.‐S. , Mak, C.‐Y. , Pei, S.‐C. , … Chung, B.‐Y. (2019). Exome sequencing identifies molecular diagnosis in children with drug‐resistant epilepsy. Epilepsia Open, 4(1), 63–72. 10.1002/epi4.12282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldrop, M. A. , Pastore, M. , Schrader, R. , Sites, E. , Bartholomew, D. , Tsao, C. Y. , & Flanigan, K. M. (2019). Diagnostic utility of whole exome sequencing in the neuromuscular clinic. Neuropediatrics, 50(2), 96–102. 10.1055/s-0039-1677734 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The dataset analyzed during the current study is not publicly available for privacy reasons. However, the corresponding authors will provide access upon reasonable request.