

Summary
Understanding the impact of elevated CO2 (eCO2) in global agriculture is important given climate change projections. Breeding climate‐resilient crops depends on genetic variation within naturally varying populations. The effect of genetic variation in response to eCO2 is poorly understood, especially in crop species. We describe the different ways in which Solanum lycopersicum and its wild relative S. pennellii respond to eCO2, from cell anatomy, to the transcriptome, and metabolome. We further validate the importance of translational regulation as a potential mechanism for plants to adaptively respond to rising levels of atmospheric CO2.
Keywords: elevated carbon dioxide, Solanum, genetic variation, root, transcriptome, translatome, metabolome, regulation
Significance Statement
In a domesticated and a wild Solanum species, translational and transcriptional regulation feedback onto each other and show interspecific differences in response to elevated CO2.
Introduction
Atmospheric CO2 has increased more than 40% since pre‐industrial times and a similar magnitude of increase is predicted by the end of this century (IPCC, 2013). Plants are autotrophic and their prime substrate for photosynthetic production of sugars is CO2. In C3 species, photosynthetic CO2 assimilation is limited by the diffusion of CO2 from the atmosphere into the chloroplast and the biochemical capacity for CO2 fixation in the chloroplast. Consequently, growth at elevated CO2 concentration (eCO2) generally stimulates photosynthetic CO2 assimilation, which in turn leads to greater leaf respiration and plant growth when sufficient nutrients and water are available (Drake et al., 1997; Ainsworth and Long, 2004; Leakey et al., 2009a,b).
Meta‐analyses of C3 species investigated at 12 Free Air CO2 Enrichment (FACE) facilities with long‐term application of eCO2 supports this general biomass stimulation with an average increase in above‐ground biomass of 20% in response to eCO2. Yet, different plant species respond in different ways to CO2 (Bishop et al., 2014). Presumably, this diversity of responses involves complex interactions among plant carbon, water, and nutrient relations, which are also constrained by plant developmental programmes and gene regulation (Leakey and Lau, 2012; Gray and Brady, 2016). As a consequence, improving plant performance under future growing conditions requires a global understanding of both the physiological, developmental and regulatory mechanisms that govern the response to eCO2 across organs and cell types in diverse plant species. Capturing genetic variation associated with enhanced yield under eCO2 would be highly valuable for breeding crop varieties for the future.
The output of genetic variation is phenotypic variation in the form of physiological, developmental, cellular, and molecular traits. The physiological traits most directly impacted by elevated CO2, with significant subsequent impacts on growth and yield, are photosynthetic carbon assimilation and stomatal conductance (Ainsworth and Long, 2004). Much of the previous research investigating the effects of eCO2 on plant development has focused on above‐ground tissues (Taylor, 2003; Dermody et al., 2006; Gray and Brady, 2016). However, because yield responses to eCO2 ultimately depend on nutrient and water status as well as sink‐source balance for carbon, root function is recognized to be very important for plant responses to eCO2 (Rogers et al., 1994; Zak et al., 2000; Gray et al., 2012). Nevertheless, knowledge of how root development is altered in response to eCO2 lags far behind our understanding of shoot responses.
Stimulatory effects of eCO2 on root biomass have been observed in several plant species, including Arabidopsis (Crookshanks et al., 1998; Jauregui et al., 2015), wheat (Vicente et al., 2016), and tomato (Cohen et al., 2018). Whether or not genes that determine root system architecture do so in a Mendelian manner in response to eCO2 across plant species is less clear. At the cellular level, changes in root and shoot function can occur through modification of xylem cell development or hydraulic conductivity. As the majority of water acquired by roots is transported from roots to shoots before diffusing into the atmosphere via stomata in exchange for CO2, understanding xylem vessel diameter is also critically important to understanding plant water relations. Root xylem vessel diameter was shown to increase in tomato (cv Ikram) in response to eCO2 in different concentrations of nitrate (Cohen et al., 2018). These studies demonstrate that root cellular anatomy should be an additional phenotypic trait used to quantify genetic variation in a plant's response to eCO2.
Molecular mechanisms underlie these morphological and physiological responses. Monitoring the transcriptome in leaves has revealed gene expression patterns that closely mirror changes in metabolism (Kaplan et al., 2012; Watanabe et al., 2014; Markelz et al., 2014a; Jauregui et al., 2015; Vicente et al., 2016; Zhang et al., 2018; Vicente et al., 2018). In soybean, eCO2 also induced significant changes in the expression of genes associated with carbohydrate metabolism and respiration (Leakey et al., 2009a,b). In Arabidopsis, eCO2 induced expression of genes encoding respiratory machinery, components of the glycolytic cycle, the tri‐carboxylic acid (TCA) cycle, and the mitochondrial electron transport chain, which is consistent with increased leaf respiration and foliar starch and sugar content (Leakey et al., 2009a,b). The root transcriptome of Arabidopsis in response to eCO2 has recently been characterized and indicates potential mechanisms for differences in organ‐level N assimilation (Jauregui et al., 2016).
In cereal crops, grain protein content is reduced in response to elevated carbon dioxide (Medek et al., 2017). Despite these organ‐specific changes in protein level, to the best of our knowledge, translational regulation in response to eCO2 has not been analyzed previously. The association of specific transcripts with ribosomes is dynamic, and changes spatiotemporally (Mustroph et al., 2009; Lin et al., 2014; Tian et al., 2019) as well as in response to many abiotic stresses including light (Juntawong and Bailey‐Serres, 2012; Liu et al., 2013), heat (Yángüez et al., 2013; Lukoszek et al., 2016), salt (Li et al., 2018), hypoxia (Mustroph et al., 2009), dehydration (Kawaguchi et al., 2004), and phosphate deficiency (Bazin et al., 2017). In these cases, transcripts associated with primary metabolism and translation often show reduced ribosomal association, while genes associated with the stress response show increased ribosomal association. These changes are thought to be adaptive and present an additional regulatory mechanism by which a plant responds to stress.
Given the importance of identifying genetic variation to breed plants with an improved response to eCO2, we characterized the eCO2 response of the domesticated tomato species, Solanum lycopersicum, and its wild relative, S. pennellii. S. pennellii is a desert‐adapted species, that displays salt and drought stress tolerance (Dehan and Tal, 1978; Koca et al., 2006; Easlon and Richards, 2009). Furthermore, these two species have striking differences in root anatomy, morphology, and gene expression; these are likely to contribute to their differences in response to stress (Ron et al., 2013; Toal et al., 2018) and provide a compelling opportunity to assess how closely related species with significant differences in root morphology and anatomy respond differentially to eCO2. We used S. lycopersicum and S. pennellii to address the following question: Which physiological, cell anatomical and molecular mechanisms are different between a crop species and a wild drought‐tolerant relative in response to eCO2? While extensive genetic variation in a range of stress responses has been observed between these two species (Gong et al., 2010; Frary et al., 2010; Frary et al., 2011), they respond in a similar physiological manner in response to eCO2. Specific differences in how these species respond include changes in xylem vessel anatomy, a small number of metabolites, and transcriptional regulation, particularly in genes associated with translation. We determined that these transcriptional differences lead to changes in translation, and present a newly described mode of regulation in response to eCO2, that is, differential polysome occupancy of transcripts. Furthermore, this translational regulation serves to dampen transcriptional regulation. These results have implications for improving our basic understanding of species‐specific responses to eCO2 and the genetic variation available for breeding climate change‐resilient crops.
Results and Discussion
Elevated CO2 increases photosynthesis and biomass in both S. lycopersicum and S. pennellii
We measured changes in root and shoot biomass, the photosynthetic rate of leaves, and stomatal conductance under ambient and eCO2 in S. lycopersicum cv M82 (written as S. lycopersicum throughout) and S. pennellii at 7, 10, and 13 days after planting (DAP) (Figure 1a). It should be noted that differences in plant morphology between these species do not represent a developmental delay (Ron et al., 2013; Koenig et al., 2013). Samples were collected at equivalent times of the day, although this does not preclude small differences in developmental timing between samples. We observed a significant net increase of dry biomass in shoots (F 1,92 = 45.8, P < 0.05) and roots (F 1,92 = 4.5, P < 0.05) in both species in response to eCO2 (Figure 1; Table S1). The photosynthetic carbon assimilation rate increased significantly in response to eCO2 (F 1,92 = 24.5, P < 0.05), consistent with other studies (Markelz et al., 2014b; Gray and Brady, 2016) (Figure 1c and Table S1). Stomatal conductance had a significant net decrease (F 1,92 = 23.9, P < 0.05) (Figure 1d and Table S1), also as previously reported in other species (Hussain et al., 2013; Gray and Brady, 2016). There was no difference in how these two species responded to eCO2 for dry biomass of shoots and roots, photosynthetic carbon assimilation rate and stomatal conductance, as determined by the lack of a significant interaction effect (P > 0.05) between species and CO2. Thus, these two species respond in a similar physiological manner to other C3 plant species: an increase in plant biomass in response to eCO2 due to an increase in photosynthesis and decrease in stomatal conductance (Crookshanks et al., 1998; Markelz et al., 2014a), but there is no variation in their response.
Figure 1.
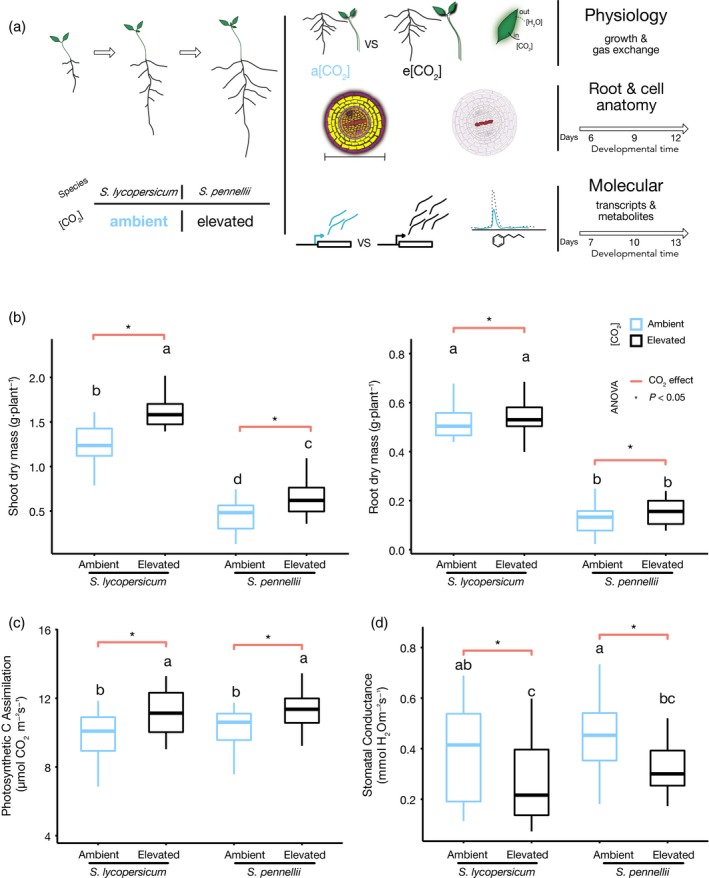
Experimental design and Solanum lycopersicum/S. pennellii physiological and growth responses to eCO2.
(a) Experimental design to characterize the interspecific response to elevated CO2 of S. lycopersicum and S. pennellii. Root system architecture, cell anatomy, and physiological measurements were recorded. Cell anatomy measurements were sampled at 6, 9 and 12 days after planting (DAP). Molecular data included metabolites and transcriptional profiles sampled at 7, 10 and 13 DAP (right side).
(b–d) Blue = ambient CO2; black = elevated CO2. (b) Elevated CO2 has a significant (P < 0.05) effect in shoots (left panel) and roots (right panel) for dry mass in both species (two‐way anova). (c) Photosynthetic rate is increased in response to elevated CO2 (two‐way anova). (d) Stomatal conductance decreased significantly due to elevated CO2 (two‐way anova). The letters above the boxplots are the results of a Tukey honest significant difference (HSD) test. Orange lines above boxplots indicate if there was a significant effect of CO2 (anova, P < 0.05; Table S1).
Elevated CO2 alters root cell anatomy in Solanum species
Although these species respond in a similar physiological manner, it is possible that they do so through the execution of different regulatory pathways. This hypothesis is further supported by the existing differences in development between S. lycopersicum and S. pennellii. Root biomass changes in a very minimal manner in response to eCO2. Thus, we chose to only characterize root anatomical changes. We compared the area and diameter of the root radial axis and metaxylem in response to eCO2 treatment (700 ppm versus 400 ppm for ambient conditions) for 7, 10, and 13 DAP (Figure 2a). There was no difference in these traits for either species (Figure 2b,c and Table S2) in the root radial axis. In contrast, we found a difference in two aspects of metaxylem development in response to eCO2; a net increase in the developing metaxylem area (F 1,35 = 11.35, P < 0.05) and diameter (F 1,35 = 8.3, P < 0.05) (Figure 2d,e and Table S2). The way in which metaxylem area changes differed between species. Metaxylem area was much larger and had more variation in S. lycopersicum at 10 DAP relative to S. pennellii (Figures 2e and S1; there was a significant interaction effect between species and developmental stage in response to eCO2; F 2,35 = 3.6, P < 0.05). Furthermore, this change in metaxylem area was not due to the change in root diameter or area (Figure S1 and Table S2). This contrasts with our photosynthesis and biomass measurements for which both Solanum species responded in a similar manner. The metaxylem differences between species provide support that a different mode of regulation in the eCO2 response might exist between both species.
Figure 2.
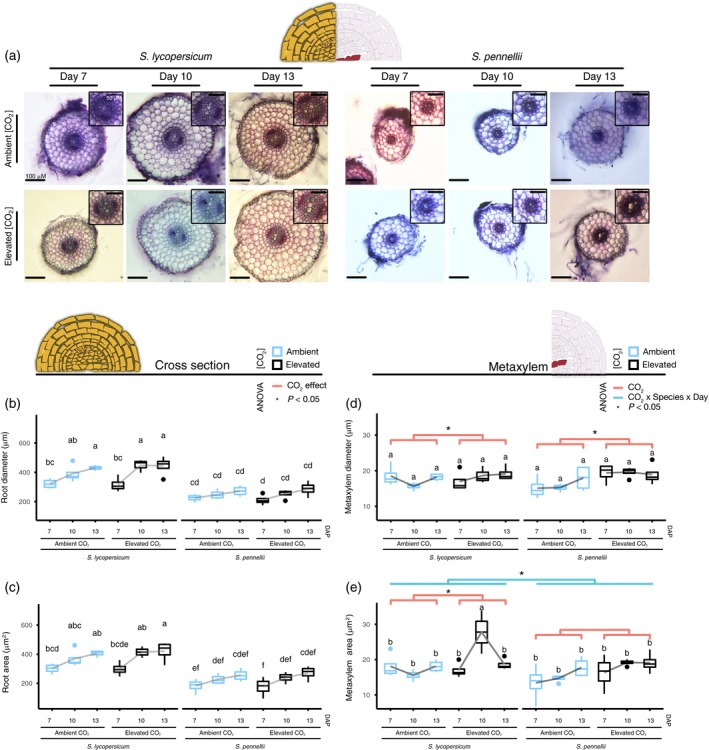
Metaxylem development changes in different ways between species in response to eCO2.
(a) Cross‐sections taken at 1 cm from the root tip of Solanum lycopersicum (left half) or S. pennellii (right half) at 7, 10, or 13 days after planting (DAP), under ambient (upper row) or elevated (bottom row) CO2. Inset = vascular area. Scale bars (main figure) = 100 μm (0.1 mm); scale bars (inset) = 50 μm (0.05 mm).
A 3‐way anova revealed no significant effect of [CO2] on root diameter (b) or area (c). Conversely, metaxylem diameter (d) and area (e) were affected by [CO2]. Letters above the boxplots are the results of a Tukey HSD test. Lines above boxplots indicate if there was a significant effect of CO2 (orange) or an interaction (CO2 × Species × Day in blue) effect (anova, P < 0.05; Table S2).
To further understand the effect of eCO2 in the xylem of mature plants, we measured xylem vessel area and vessel number in 12‐week‐old plants; however, these traits were no longer eCO2 responsive (Figure S2 and Table S2). Taken together, these results point to a targeted effect of eCO2 in the root that is specific to the early developing metaxylem. The eCO2‐dependent increase of the metaxylem area could increase hydraulic conductivity and enable the increase in shoot biomass.
Elevated CO2 perturbs the accumulation of a subset of metabolites
Elevated CO2 is known to target primary metabolism. To address potential changes between species in metabolite accumulation in response to eCO2 in both species, we profiled primary metabolites in the shoots and roots of S. lycopersicum and S. pennellii at three time points 6, 9, and 12 DAP; staggered from the anatomical measurements for ease of sampling (Figure 3a). Our analysis identified 180 metabolites in the roots and 140 metabolites in the shoots whose concentration changed significantly (q‐value <0.05; Table S3) in a species, day, or CO2‐dependent manner (either as a main effect or interaction). Only three out of the 140 metabolites that changed in the shoots, changed in response to eCO2: glutamic acid (CO2; logFC = 0.97, qVal < 0.05), ornithine (CO2; logFC = 1.44, qVal < 0.05), and aspartic acid (CO2; logFC = 1.56, qVal < 0.05) (Figure 3a,b and Table S3). Three of the 180 differentially accumulated root metabolites changed their abundance in response to eCO2, two of which changed in distinct ways between the two species, as well as in developmental time (Figure 3b and Table S3): fumaric acid (CO2 × Species × Day 12; logFC = −1.11, qVal < 0.05) and phenylalanine (CO2 × Species; logFC = 0.69, qVal < 0.05). Hexuronic acid (CO2 × Day 12; logFC = −2.44, qVal < 0.05) abundance changed in response to eCO2 in a similar way in both species. These results provided further evidence of interspecies variation in response to eCO2 at the molecular level. The observed changes in accumulation of glutamic acid and ornithine in response to eCO2 are similar to previous observations in Arabidopsis, in which the metabolites changed in response to eCO2 but not between ecotypes (Li et al., 2008a). No previous studies have reported changes of individual metabolites whose response to eCO2 varied between genotypes or species.
Figure 3.
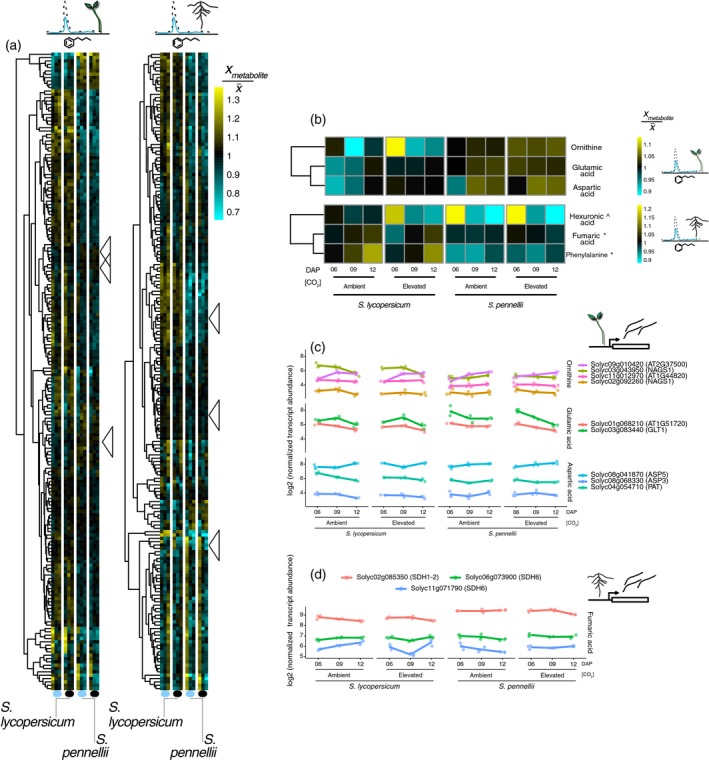
Elevated CO2‐responsive metabolites and associated pathways in shoots and roots.
(a) Heatmap of the 191 metabolites profiled. Colour scale represents the mean‐normalized ratio of the log2 metabolite concentration.
Significant metabolites with a CO2 (main or interaction) effect (q‐value <0.05) are marked with arrowheads: glutamic acid, ornithine and aspartic acid in shoots (b, upper panel); in the roots, a CO2‐related effect was significant for phenylalanine, fumaric acid, and hexuronic acid (b, lower panel. Superscript indicates the type of interaction term: ^CO2 × Day; *CO2 × Species).
Mean‐normalized transcript abundance from total RNA‐seq of selected genes in shoots (c) and roots (d) related to the metabolite synthetic pathways of differentially accumulated metabolites.
Distinct CO2‐dependent transcriptional reprogramming of S. lycopersicum and S. pennellii
To link the molecular mechanisms underlying our observed physiological, developmental, and metabolic plant responses to eCO2, we analyzed the transcriptomic landscape of shoots and roots in both species over three developmental time points (6, 9, and 12 DAP). We explored the transcript accumulation of genes involved in glutamate to ornithine biosynthesis, 2‐oxoglutarate to glutamic acid (l‐glutamate) biosynthesis, aspartic acid biosynthesis, and succinate to fumarate biosynthesis to determine if transcriptional regulation of metabolic enzymes is responsible for the differential accumulation of metabolites. The transcript accumulation patterns of these enzymes revealed no discernible pattern of transcript accumulation in relation to metabolite concentration for either species in response to eCO2 (Figure 3c,d). Therefore, differential accumulation of these metabolites is likely to not be transcriptionally regulated.
Our results suggested that, although S. lycopersicum and S. pennellii have similar physiological responses to eCO2 (increase of photosynthesis and biomass with a decrease in stomatal conductance), the root anatomic and metabolomic data provided evidence that distinct biological programmes can be employed to give rise to these similarities. We therefore hypothesized that the underlying transcriptional regulatory network coordinating the eCO2 response may be different between the Solanum species. This hypothesis was supported by analyzing whole transcriptome expression patterns. The two species primarily diverged in their eCO2‐responsive transcript accumulation at 12 DAP (Figure S3a,b). Clustering of these divergently expressed genes between species combined with Gene Ontology (GO) enrichment analysis revealed the putative functions of these genes (Figure 4a,b). In the root, four clusters (patterns 1, 2, 3, and 7) were associated with ribosome biogenesis and translation; other clusters were associated with the plasma or vacuolar membrane, GTP‐associated signaling, and the cell wall (Figure S4b–d and Table S4). As anticipated (Watanabe et al., 2014; Markelz et al., 2014a; Jauregui et al., 2015), shoot genes associated with glycolysis and the TCA cycle were enriched, in addition to photosynthesis (Figure 4b). Surprisingly, genes associated with translation or ribosome biogenesis (Figure 4b) were significantly differentially expressed in five co‐expressed gene groups (patterns 3, 5, 9, 10, and 11; Figure 4a). These processes differed slightly between the root and shoot; processes associated with ribosome protein subunit biogenesis were enriched in the root (Figure S4d), while processes associated with translational initiation and elongation were enriched in the shoot (Figure 4b). We further analyzed the promoters of genes assigned to these dominant patterns (3, 5, 9, 10, and 11) in shoots. We found 41 significantly enriched motifs (adjusted P‐value <0.01, Bonferroni correction; Table S4) that represent potential transcription factor binding sites common to all patterns (Figure S4e). The TFIIIA binding motif was highly enriched in patterns 3, 5, 10, and 11. TFIIIA is specifically required for the transcription of 5S ribosomal RNA, an important component of the ribosome (Layat et al., 2013). Overexpression of TFIIIA in rice enhanced its tolerance to drought, cold, and salt stresses (Huang et al., 2012).
Figure 4.
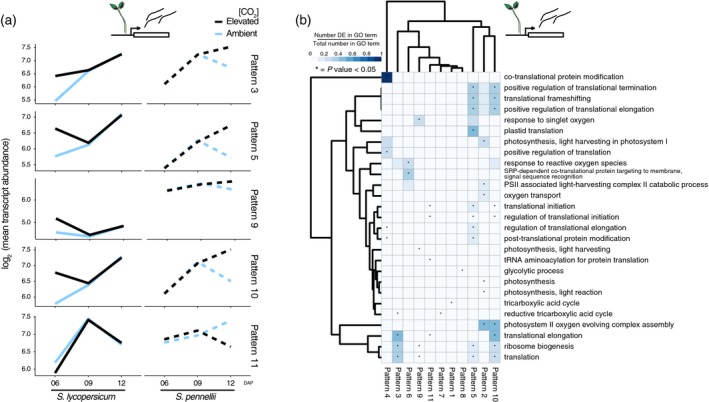
Elevated CO2 perturbs regulatory modules associated with translation.
(a) Representative line plots of gene expression patterns associated with translation. Lines represent the average log2 normalized transcript abundance (total RNA‐seq) of all genes assigned to a dominant pattern; blue = ambient CO2; black = elevated CO2.
(b) Enriched GO categories associated with dominant patterns. Scale = ratio between the number of significant genes in the category and the total number of genes assigned to the same category (*P < 0.05).
Divergent transcriptional changes between species in response to eCO2 have not been previously reported, neither have changes in translation. We thus hypothesized that there may be changes in which transcripts are associated with ribosomes in the shoot in response to eCO2. These transcriptional changes represent averaged changes across organs and we anticipate an increased complexity of changes within and across individual cell types.
Preferential transcript loading on ribosomes
To test if changes in transcript–ribosome association occur in response to eCO2, we characterized these ribosome‐associated RNA populations in the shoots of both species at 6 and 12 DAP using Translating Ribosome Affinity Purification (TRAP)‐Seq from 35S:RPL18b‐His6‐FLAG‐GFP of S. lycopersicum cv M82 and S. pennellii. These time points were chosen as transcript accumulation related to translation initiation and elongation was enriched in the shoot and divergent at 12 DAP. In order to identify transcripts that are differentially loaded onto ribosomes (translational regulation), we selected 2599 upregulated or downregulated genes that showed differences in expression in response to eCO2 between both species in the transcriptome data or in the translatome data (Figure S5a,b; Experimental procedures). These 2599 genes could be further divided into 11 co‐regulated groups or ‘dominant patterns’ (Figure 5a). As an example, genes in Pattern 2 are decreased in transcript abundance in the ribosome‐associated fraction at 12 DAP in S. lycopersicum in response to eCO2, (while they typically increase over development time under ambient CO2) and show little to no difference in their abundance in the transcriptome data (Figure 5b). In contrast, these same genes in Pattern 2 are increased in their transcript abundance in response to eCO2 in S. pennellii in both ambient and in eCO2 (Figure 5b). To further characterize the biological processes associated with these differences between species in translationally regulated genes, we queried enriched GO categories (Figure 5c). GO terms related to transcriptional regulation and transcription factor activity were uniquely enriched in Pattern 2, a marked contrast with the translational regulatory pathways observed in the transcriptional profiling. Such enrichment for entire categories related to transcriptional regulation have not previously been identified in response to abiotic stresses (Mustroph et al., 2009; Ribeiro et al., 2012; Juntawong and Bailey‐Serres, 2012; Yángüez et al., 2013; Aubry et al., 2014; Lin et al., 2014; Cheng et al., 2015; Vragović et al., 2015; Basbouss‐Serhal et al., 2015; Zoschke et al., 2016; Meteignier et al., 2017; Xu et al., 2017; Li et al., 2018; Chen et al., 2018; Wu et al., 2019; Tian et al., 2019). Other patterns had few and general terms associated with them (patterns 3, 6, 9, 10, and 11; Figure S6; Table S4).
Figure 5.
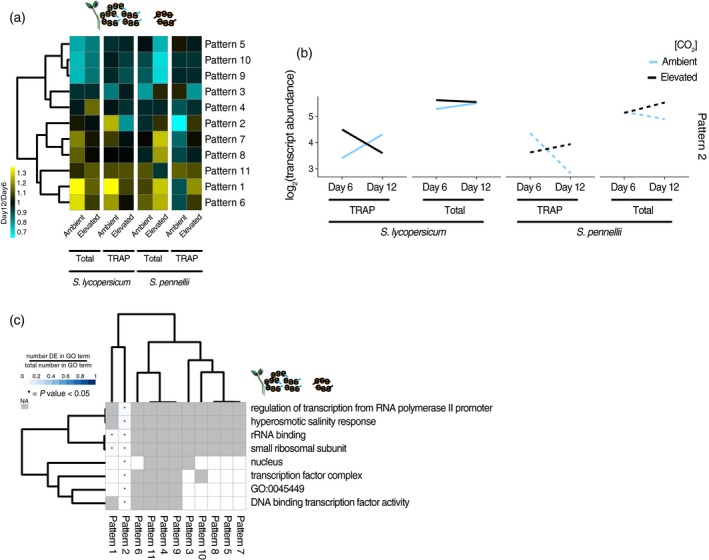
Genes with translational profiles that respond to eCO2 in a species‐specific manner are enriched in GO categories related to transcription.
(a) Dominant patterns representing uniquely (upregulated or downregulated) differentially expressed genes that differ between total RNA and TRAP RNA. The colour scale represents temporal changes (12 DAP/6 DAP).
(b) Genes that change expression between species in the ribosome‐associated RNA population. Lines represent the mean expression (as measured from transcript abundance in total RNA‐seq or ribosome loading in TRAP) of the genes (n = 140) assigned to Pattern 2. Blue lines = transcript abundance under ambient CO2; black lines = elevated CO2. Solid lines = Solanum lycopersicum, dashed lines = S. pennellii.
(c) GO terms enriched in Pattern 2. Scale = ratio between the number of significant genes in the category and the size of the category. Asterisk = significant enrichment (GoSeq; P < 0.05). Cells in grey (NA) represent categories without enough genes to be considered in the analysis.
The group of genes associated with Pattern 2 demonstrates feedback to transcriptional regulation in response to eCO2, which differs between species (Figure 5b and Table S4). Genes in this co‐regulated group comprise members of the Mediator complex (MED11, 18, and 19B), involved in transcription initiation (Seizl et al., 2011; Buendía‐Monreal and Gillmor, 2016) and transcriptional repression (Dolan and Chapple, 2016), as well as 15 transcription factors. Subunits of the Mediator complex play roles in transcriptional regulation and in the abiotic stress response (Liao et al., 2016). To the best of our knowledge, these have never been shown to be translationally regulated. Among the 15 transcription factors in this group, some have been associated with abscisic acid (ABA) and drought including Nuclear Factor Y (NF‐Y) (Warpeha et al., 2007; Nelson et al., 2007; Li et al., 2008b), DEAR5/RAP2.9 (Agarwal et al., 2006; Fujita et al., 2011), and TCTP/P23 (Kim et al., 2012). Together, these results suggested a translational regulatory mechanism in which ribosomal loading of transcripts associated with transcriptional initiation were selectively increased in S. pennellii and decreased in S. lycopersicum, thus providing transcriptional feedback. Similar to the transcriptome analyses, we anticipate a greater complexity of changes in translational abundance within and across individual cell types within both the shoot and the root.
Overall, our results highlight the importance of a systematic understanding of the whole plant's response to eCO2 as well as the importance of molecular variation as a potential resource for breeding and can provide insight into the different means by which plant species can respond to elevated atmospheric CO2. Figure 6 offers an interpretation of how the molecular findings in this study underlie distinct global regulatory mechanisms that can lead to the same physiological response to eCO2. Our results point to the translational coordination of specific transcriptional machinery – the Mediator complex and other transcription factors – in a species‐specific manner. Further characterization of the potential adaptive role of development stage‐specific widening of xylem vessel diameter as well as of specific shoot and root metabolites identified here will be a valuable tool to help enable efforts to prepare for the upcoming increase in eCO2 and to sustainably prepare for the resulting changes to global agriculture.
Figure 6.
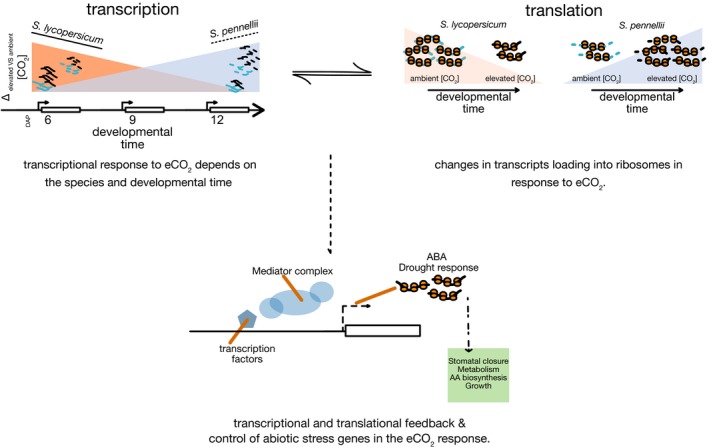
Model of the effects of eCO2 on transcription and translation in Solanum species.
The transcriptional response to elevated CO2 has elements of species‐specificity, this depends on developmental stage and influences translation. A subset of genes is translationally regulated (Pattern 2, Figure 5b) in a species‐specific manner in response to elevated CO2. These genes include transcriptional regulators (members of the Mediator complex as well as other transcription factors) and genes associated with abscisic acid (ABA) response. We hypothesize that co‐ordinated feedback between transcriptional and translational regulation differs between these species, and yet gives rise to similar changes in metabolism, growth, and stomatal closure.
Experimental procedures
Plants and growth conditions
Seeds of S. lycopersicum (cv M82) and S. pennellii (LA0716) were obtained from the C. Rick Tomato Genetics Resource Centre, sterilized, and sown in Profile Calcined Clay medium (MVP Products, Buffalo Grove, IL; www.turface.com) in 4‐inch pots with a modified removable bottom. Plants were grown in Conviron PGR15 growth chambers (Controlled Environments Ltd, Winnipeg, Manitoba, Canada; http://www.conviron.com). Chambers were set at either ambient (400 ppm) or eCO2 (700 ppm), 325 μmol m−2 sec−1 PPFD, 70% relative humidity, 16 h:8 h light:dark schedule, and 22°C at the Controlled Environment Facility at the University of California, Davis, USA. Carbon dioxide levels were monitored continuously with a monitor that immediately reported any major deviations in CO2 levels. No major deviations were recorded over the time period of the experiment. Plants were watered every 2–3 days with 40% Long‐Ashton solution containing 6 mm NH4NO3. The location of the flats within each chamber and the location of plants within each flat were rotated upon every watering, and CO2 treatment assignments were rotated between chambers at every experimental replication to avoid any experimental artefacts caused by differences between chambers.
Shoots and roots were harvested 28–35 DAP for shoot and root biomass. Twenty‐four plants (24 biological replicates) were measured for M82 and S. pennellii, respectively, for elevated and ambient carbon dioxide treatments. Shoots and roots were oven dried (at least 70°C), and dry biomass was recorded. Biomass measurements and the associated scripts to analyze these data can be found online at https://github.com/rodriguezmDNA/Gray_RodriguezMedina_CO2Supplemental.
Measurement of photosynthetic rate and stomatal conductance
In situ leaf‐level photosynthetic rate (A) and stomatal conductance (gsc) were measured using the LiCOR 6400 system (LI‐COR; https://www.licor.com). Raw LiCOR measurements can be found at https://github.com/rodriguezmDNA/Gray_RodriguezMedina_CO2Supplemental. Measurements were taken consecutively over 3 days for S. lycopersicum and S. pennellii plants under ambient or eCO2 conditions in the youngest fully expanded leaf in 12‐week‐old plants. Measurements were taken from this leaf for three to five biological replicates per CO2 condition (ambient or elevated) for each species. The anova() function from R package lmerTest (R Core Team, 2013) (Kuznetsova et al., 2017) was used to test the main effects of CO2 and species and their interaction in a linear model, treating the date of measurement as a random effect (Experimental procedures). The experiments were repeated four times. Measurements were taken between 15:00 and 18:00, consecutively over 3 days.
Measurement of root anatomy and morphology
Whole root systems were collected at 7, 10, and 13 DAP, submerged in deionized water in a shallow, clear dish, and scanned on a flatbed scanner (Epson Perfection V700 Photo Scanner; Epson America, Long Beach, CA, USA; https://epson.com/usa). Images were analyzed using the ImageJ plugin SmartRoot (Lobet et al., 2011) to provide data on total root system length, number of lateral roots, and root diameter. Following scanning, 1 cm samples were collected at 2–3 cm from the root tip, embedded in agarose and preserved in FAA for later cross‐sections. A linear model in R (R Core Team, 2013) was used to test for significant main effects of CO2, species, and developmental time, and their two‐way and three‐way interactions.
Cellular anatomy measurements of roots
Root tissue samples that had been preserved in FAA were rehydrated using a stepwise series of ethanol concentrations and sectioned into 200‐µm thick sections using a Vibratome Series 1000 sectioning system. Sections were stained with toluidine blue, de‐stained in water, and mounted in 50% glycerol. Sections were imaged using an Olympus Vanox microscope (www.olympus-lifescience.com), and analyzed using ImageJ for cross‐sectional diameter, and diameter of the largest metaxylem vessel. Cross‐sections were prepared, imaged, and analyzed from root tissue collected from S. lycopersicum and S. pennellii plants at four time points: 7, 10, 13 DAP, and at 12 weeks old. Raw data for root anatomical measurements can be found at https://github.com/rodriguezmDNA/Gray_RodriguezMedina_CO2Supplemental. Four biological replicates were analyzed for each time‐point (7, 10, and 13 DAP) in either ambient or elevated carbon dioxide in each species. When measuring xylem vessel diameter in mature roots, 11–12 biological replicates (representing sections from different plants) were analyzed from lateral roots and primary roots, for each carbon dioxide concentration and for each species. Only xylem vessels larger than 20 µm in diameter were considered, following the method in Rincon et al. (2003). A linear model in R was used to test for significant main effects of CO2, species, and developmental time, and their two‐way and three‐way interactions. Outliers were either substituted by their group average between replicates (for young xylem measurements) or removed (for mature metaxylem measurements).
Tissue collection for RNA‐seq and metabolite profiling
Whole root and whole shoot samples were collected for transcriptomic and metabolomic profiling at 6, 9, and 12 DAP. To collect each sample, the entire pot was lowered into deionized water and then the pot was slowly lifted up and away from the removable bottom of the pot, allowing the medium to gently float away from the root system. The plant was then moved to a secondary container of deionized water to ensure that the roots were free of medium. Then, the roots and shoots were severed using a clean razor blade, and the tissue was flash‐frozen in liquid nitrogen. From submergence of the root system to placement of samples in liquid nitrogen, a maximum of c. 75 sec elapsed.
For transcriptome profiling, root tissue from three individual plants was pooled together to yield one root sample per treatment‐species combination day–1 on each of the three sampling days, and shoot tissue from the same three plants was pooled together to yield one shoot sample per treatment‐species combination day–1 on each of the three sampling days. The same sampling scheme and chambers were used for metabolite and physiological profiling. The experiments were repeated four times for all traits measured.
RNA‐seq library preparation
RNA‐seq libraries were prepared using the method described by Kumar et al. (2012). Briefly, for each library, the pooled root or shoot samples were homogenized in lysis and binding buffer containing B‐mercaptoethanol. Tissue was disrupted using a mortar and pestle and a Mini‐beadbeater‐96 high‐throughput cell disruptor (BioSpec Products, Bartlesville, OK, USA; www.biospec.com). Oligo(dT)25‐coated Dynabeads (Ambion, Foster City, CA, USA; www.thermofisher.com) were then added to the lysate, and mixed for 10 min to allow oligo(dT) beads to hybridize to poly‐adenylated mRNA. cDNA was then synthesized and digested to c. 300 bp fragments using NEBNext DNA fragmentase enzyme mix (NEB, Beverly, MA, USA; www.neb.com). Library fragments greater than 300 bp were purified using AMPure XP solid phase reverse immobilization (SPRI) magnetic beads (Agencourt Bioscience, Beverly, MA, USA). For each library, one of the 96 unique barcoded adapters described in Kumar et al. (2012) was ligated to each library, enabling multiplexing. cDNA was quantified using a SYBR green‐based method on a plate reader, libraries were pooled, and fragment size and quality was checked using a BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA; www.agilent.com).
Translating ribosome affinity purification
To determine the effects of eCO2 on the translatome of shoot tissue, transgenic S. lycopersicum and S. pennellii expressing a FLAG epitope on the ribosomal L18 (RPL18) under the 35S promoter (Ron et al., 2013) were grown in ambient and elevated CO2 under the same conditions and chambers as described above. At 6 and 12 DAP, tissue was collected for the TRAP protocol. Because the biomass of plant tissues in young seedlings was small, and because the TRAP populations were segregating, we pooled multiple plants (up to 15) together to ensure that adequate mRNA for library preparation could be collected via the ribosomal pulldown.
The TRAP procedure has been described previously by Zanetti et al. (2005). Briefly, tissue from transgenic plants expressing the FLAG epitope was ground in liquid nitrogen using a mortar and pestle, and homogenized in polysome extraction buffer. Samples were clarified by centrifugation and filtration through a Miracloth, and the clarified extract was added to magnetic beads that were coated in anti‐Flag antibody. Beads and extract were incubated at 4°C using a nutator for 2 h, and washed six or seven times with wash buffer. Polysomes bound to magnetic beads were then frozen at −80°C until RNA‐seq library preparation.
RNA‐seq libraries were prepared from TRAP polysomes using the method described by Townsley et al. (2015). Lysis and binding buffers containing B‐mercaptoethanol were added to anti‐Flag magnetic beads and bound ribosomes, allowing elution of mRNA. mRNA was then hybridized to biotin–20nt‐dT oligos, allowing pulldown of mRNA out of solution with the use of magnetic streptavidin beads. mRNA was then washed with three successive buffers, eluted off the beads at 80°C, and a secondary wash was performed by adding more biotin–20nt‐dT oligos to the mRNA, repeating the bead pulldown and the buffer washes. mRNA was then heat fragmented, and first and second strand cDNA was synthesized. The optimal fragment size was selected using AMPure beads, and libraries were enriched using PCR.
Library sequencing and bioinformatics pipeline
Pooled and barcoded libraries were submitted to the Vincent J. Coates Genomic Sequencing Facility at UC Berkeley (http://qb3.berkeley.edu/qb3/gsl/index.cfm) and were sequenced on an Illumina HiSeq 2000 instrument (https://www.illumina.com). A custom Perl script was used to split sequence reads into individual libraries based on barcode sequences (Toal et al., 2018), and FastQC (Andrews, 2010) was used to plot read quality. We used the bowtie 1.2.2 tool (Langmead et al., 2009) to align RNA‐seq reads to the S. lycopersicum reference transcriptome (ITAG 3.2, http://www.solgenomics.net). Mapped reads were aggregated into a counts table using a custom R script and used in downstream expression and clustering analysis. Raw total and TRAP sequencing data can be obtained from the Gene Expression Omnibus (GEO) under the accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126430. Normalized CPM data are found in Table S5.
Differential expression analysis
Counts were normalized using edgeR (Robinson et al., 2009) and limma‐voom (Ritchie et al., 2015) Bioconductor packages in R. A linear model of the main and interaction effects between CO2, species, and developmental time was used to test for differentially expressed genes. Data from roots or shoots were analyzed separately. False discovery rates were calculated using the ashR (Stephens, 2016) package in R. Normalized CPM data for total and TRAP RNA‐Seq is available in Table S5. Correlation between all samples is also in Table S5. Only samples present in these tables were considered for the analyses.
Clustering analysis
Voom‐normalized counts were calculated using the voom() function from the limma package. These values were used for clustering analysis, and selecting for the top varying genes. This analysis was carried out using the algorithm described by Orlando et al. (2009). Genes assigned to each dominant pattern are listed in Table S4.
Functional enrichment analysis
Enrichment of GO categories was analyzed using GOseq (Young et al., 2010). For custom gene lists, Fisher's Exact Tests were used to test for enrichment. The glycolysis and TCA cycle‐related genes that are eCO2‐responsive were obtained from the list described in Markelz et al. (2014a). Multiple hypothesis testing was not performed for GO categories due to the graph structure of GO terms. It has been discussed that enrichment tests for different GO categories can be correlated, and therefore multiple testing corrections in GO analyses cannot be simply addressed by calculating ordinary false discovery rates (Mi et al., 2012) or that the correction can be too conservative and yield few or no significant terms (Alexa and Rahnenfuhrer, 2007).
Motif enrichment
We used AME (McLeay and Bailey, 2010) to identify enriched transcription factor binding motifs in the 1 kb region upstream from the transcription start site of genes assigned to dominant patterns 3, 5, 9, 10, and 11. The parameters were set as follows: ‐‐verbose 1 ‐‐scoring avg ‐‐method fisher ‐‐hit‐lo‐fraction 0.25 ‐‐evalue‐report‐threshold 10.0 ‐‐control ‐‐shuffle‐‐ ‐‐kmer 2. The Arabidopsis DAP‐Seq (O'Malley et al., 2016) database was used as a reference.
Untargeted metabolite profiling
Tissues were collected for metabolite profiling at the same time and using the same method as the tissue collection for the RNA‐seq experiment. Root and shoot tissue were separately pooled for three individuals of each species and each CO2 treatment at 6, 9, or 12 DAP (four biological replicates each). Samples were lyophilized, ground, and extracted in a chilled mixture of methanol, chloroform, and water (5:2:2). Samples were then vortexed, centrifuged, and the supernatant was dried (Labconco Centrivap cold trap concentrator), and resuspended in degassed acetonitrile and water (1:1) to clean the sample of membrane lipids and triglycerides. Samples were again dried, Fatty Acid Methyl Esters (FAMEs) of eight to 30 carbons in length were added as internal standards, and samples were then derivatized using methoxyamine hydrochloride in pyridine followed by N‐methyl‐N‐(trimethylsilyl)trifluoroacetamide. Data were acquired using gas chromatography and time‐of‐flight mass spectrometry as described by Fiehn et al. (2008). Briefly, a volume of 0.50 μl was injected for each sample on an rtx5Sil‐MS column (Restek Corporation; Bellefonte, PA). The temperature gradient was as follows: 50°C for 1 min, increasing 20°C min−1 up to 330°C, and held constant at 330°C for 5 min. Compound identification was accomplished via mass spectrometry (Leco Pegasus IV mass spectrometer; Leco Corporation, St. Joseph, MI). Here, 191 compounds were identified in the samples from this experiment. Sample extraction, derivatization, and data acquisition were all completed at the West Coast Metabolomics Facility (http://metabolomics.ucdavis.edu/). Data for each metabolite were first log2 transformed and then a linear model was fit to test for main effects of CO2, species and developmental time, and their interactions using the R function lm(). Multiple testing correction was performed using the ashR package in R. Roots were analyzed separately from shoots. Log2 transformed data and results from the linear model and P‐value correction are in Table S3. Data can also be found at https://github.com/rodriguezmDNA/Gray_RodriguezMedina_CO2Supplemental.
Selection of a gene set with unique transcriptional and translational profiles
To select uniquely upregulated or downregulated genes at the transcriptional or translational level, we performed pairwise comparisons to test for the effect of CO2 between samples of the total or TRAP, species (S. lycopersicum or S. pennellii), and day (6 or 12 DAP), with eight contrasts. Differentially expressed genes (q‐value < 0.01) were separated on the basis of their direction of change: upregulated (logFC > 1) or downregulated (logFC < 1). For each list of upregulated or downregulated genes, we performed a set analysis between samples of the same RNA source on both days (6 and 12 DAP) and species. To identify genes uniquely regulated in each species, we took the symmetric difference between species regardless of development time response.
Conflict of interest
The authors declare no affiliations with or involvement in any organization or entity with any financial interest, or non‐financial interest in the subject matter or materials discussed in this manuscript.
Author contributions
SBG and SMB conceived the original research and experiment design; SBG performed the experiments; SBG and JRM performed analysis of the data.; SBG, JRM, and SMB interpreted the results and wrote the article. KK performed TRAP sequencing experiments. SR and TWT provided technical assistance to SBG; DR provided significant guidance on data analysis. SMB agrees to serve as the author responsible for contact and ensures communication.
Supporting information
Figure S1. Whole root to metaxylem ratios.
Figure S2. Elevated CO2 has no significant effects on mature metaxylem in either species.
Figure S3. Number of significantly differentially expressed genes in each of the linear model terms in shoots or roots.
Figure S4. Clustering of genes that change in a (CO2 × Species × Day 12) interaction manner reveals genetic modules with dominant patterns.
Figure S5. Strategy to identify groups of genes with unique transcriptional or translational patterns.
Figure S6. Number of GO terms enriched per dominant pattern in total and TRAP RNA‐Seq data.
Table S1. anova tables from root area and diameter analyses
Table S2. anova tables from shoot or root biomass, photosynthesis, and stomatal conductance analyses
Table S3. Log2 transformed data and results of the metabolomic analysis
Table S4. Genes assigned to dominant patterns and motif enrichment analysis
Table S5. CPM normalized data from total and TRAP RNA‐Seq
Acknowledgments
This research was supported by NSF‐IOS‐1052395 and NSF‐PGRP‐1238243. SMB was partially supported by a Howard Hughes Medical Institute Faculty Scholar Fellowship. SBG was supported by an NSF‐PGRP Post‐doctoral Fellowship. JRM was partially supported by the University of California Institute for Mexico and the United States (UCMEXUS) and Consejo Nacional de Ciencia y Tecnología (CONACYT). KK was supported by a Finnish Cultural Foundation post‐doctoral fellowship. DR was supported by a United States Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA), Hatch project 1010469. Many thanks to Cody Markelz, Julia Bailey‐Serres, Elizabeth Georgian and Andrew Leakey for their gracious critiques of the manuscript.
Data Availability Statement
Raw total and TRAP sequencing data can be obtained from GEO under the accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126430. Normalized CPM data for total and TRAP RNA‐Seq are provided in Table S5. Raw LiCOR, root anatomy and morphology and associated scripts can be found at https://github.com/rodriguezmDNA/Gray_RodriguezMedina_CO2Supplemental.
References
- Agarwal, P.K. , Agarwal, P. , Reddy, M.K. and Sopory, S.K. (2006) Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep. 25, 1263–1274. [DOI] [PubMed] [Google Scholar]
- Ainsworth, E.A. and Long, S.P. (2004) What have we learned from 15 years of free‐air CO2 enrichment (FACE)? A meta‐analytic review of the responses of photosynthesis, canopy properties and plant production to rising CO2: Tansley review. New Phytol. 165, 351–372. [DOI] [PubMed] [Google Scholar]
- Alexa, A. and Rahnenfuhrer, J. (2007) Gene Set Enrichment Analysis with topGO. https://bioconductor.riken.jp/packages/3.7/bioc/vignettes/topGO/inst/doc/topGO.pdf [Google Scholar]
- Andrews, S. (2010) FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Aubry, S. , Smith‐Unna, R.D. , Boursnell, C.M. , Kopriva, S. and Hibberd, J.M. (2014) Transcript residency on ribosomes reveals a key role for the Arabidopsis thaliana bundle sheath in sulfur and glucosinolate metabolism. Plant J. 78, 659–673. [DOI] [PubMed] [Google Scholar]
- Basbouss‐Serhal, I. , Soubigou‐Taconnat, L. , Bailly, C. and Leymarie, J. (2015) Germination potential of dormant and nondormant Arabidopsis seeds is driven by distinct recruitment of messenger RNAs to polysomes. Plant Physiol. 168, 1049–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin, J. , Baerenfaller, K. , Gosai, S.J. , Gregory, B.D. , Crespi, M. and Bailey‐Serres, J. (2017) Global analysis of ribosome‐associated noncoding RNAs unveils new modes of translational regulation. Proc. Natl Acad. Sci. USA, 114, E10018–E10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, K.A. , Leakey, A.D.B. and Ainsworth, E.A. (2014) How seasonal temperature or water inputs affect the relative response of C3 crops to elevated [CO2]: a global analysis of open top chamber and free air CO2 enrichment studies. Food Energy Secur. 3, 33–45. [Google Scholar]
- Buendía‐Monreal, M. and Gillmor, C.S. (2016) Mediator: a key regulator of plant development. Dev. Biol. 419, 7–18. [DOI] [PubMed] [Google Scholar]
- Chen, G.‐H. , Liu, M.‐J. , Xiong, Y. , Sheen, J. and Wu, S.‐H. (2018) TOR and RPS6 transmit light signals to enhance protein translation in deetiolating Arabidopsis seedlings. Proc. Natl Acad. Sci. USA, 115, 12823–12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, M.‐C. , Ko, K. , Chang, W.‐L. , Kuo, W.‐C. , Chen, G.‐H. and Lin, T.‐P. (2015) Increased glutathione contributes to stress tolerance and global translational changes in Arabidopsis. Plant J. 83, 926–939. [DOI] [PubMed] [Google Scholar]
- Cohen, I. , Rapaport, T. , Berger, R.T. and Rachmilevitch, S. (2018) The effects of elevated CO2 and nitrogen nutrition on root dynamics. Plant Sci. 272, 294–300. [DOI] [PubMed] [Google Scholar]
- Crookshanks, M. , Taylor, G. and Dolan, L. (1998) A model system to study the effects of elevated CO2 on the developmental physiology of roots: the use of Arabidopsis thaliana . J. Exp. Bot. 49, 593–597. [Google Scholar]
- Dehan, K. and Tal, M. (1978) Salt tolerance in the wild relatives of the cultivated tomato: responses of Solanum pennelii to high salinity. Irrigation Sci. 1, 71–76. [Google Scholar]
- Dermody, O. , Long, S.P. and DeLucia, E.H. (2006) How does elevated CO2 or ozone affect the leaf‐area index of soybean when applied independently? New Phytol. 169, 145–155. [DOI] [PubMed] [Google Scholar]
- Dolan, W.L. and Chapple, C. (2016) Conservation and divergence of mediator structure and function: insights from plants. Plant Cell Physiol. 58, 4–21. [DOI] [PubMed] [Google Scholar]
- Drake, B.G. , Gonzàlez‐Meler, M.A. and Long, S.P. (1997) MORE EFFICIENT PLANTS: a consequence of rising atmospheric CO2? Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 609–639. [DOI] [PubMed] [Google Scholar]
- Easlon, H.M. and Richards, J.H. (2009) Drought response in self‐compatible species of tomato (Solanaceae). Am. J. Bot. 96, 605–611. [DOI] [PubMed] [Google Scholar]
- Fiehn, O. , Wohlgemuth, G. , Scholz, M. , Kind, T. , Lee, D.Y. , Lu, Y. , Moon, S. and Nikolau, B. (2008) Quality control for plant metabolomics: reporting MSI‐compliant studies: quality control in metabolomics. Plant J. 53, 691–704. [DOI] [PubMed] [Google Scholar]
- Frary, A. , Göl, D. , Keleş, D. , Ökmen, B. , Pınar, H. , Şığva, H.Ö. , Yemenicioğlu, A. and Doğanlar, S. (2010) Salt tolerance in Solanum pennellii: antioxidant response and related QTL. BMC Plant Biol. 10, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frary, A. , Keleş, D. , Pinar, H. , Göl, D. and Doğanlar, S. (2011) NaCl tolerance in Lycopersicon pennellii introgression lines: QTL related to physiological responses. Biol. Plant. 55, 461–468. [Google Scholar]
- Fujita, Y. , Fujita, M. , Shinozaki, K. and Yamaguchi‐Shinozaki, K. (2011) ABA‐mediated transcriptional regulation in response to osmotic stress in plants. J. Plant. Res. 124, 509–525. [DOI] [PubMed] [Google Scholar]
- Gong, P. , Zhang, J. , Li, H. et al . (2010) Transcriptional profiles of drought‐responsive genes in modulating transcription signal transduction, and biochemical pathways in tomato. J. Exp. Bot. 61, 3563–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, S.B. and Brady, S.M. (2016) Plant developmental responses to climate change. Dev. Biol. 419, 64–77. [DOI] [PubMed] [Google Scholar]
- Gray, S.B. , Strellner, R.S. , Puthuval, K.K. , Ng, C. , Shulman, R.E. , Siebers, M.H. , Rogers, A. and Leakey, A.D.B. (2012) Minirhizotron imaging reveals that nodulation of field‐grown soybean is enhanced by free‐air CO2 enrichment only when combined with drought stress. Funct. Plant Biol. 40, 137. [DOI] [PubMed] [Google Scholar]
- Huang, J. , Sun, S. , Xu, D. et al . (2012) A TFIIIA‐type zinc finger protein confers multiple abiotic stress tolerances in transgenic rice (Oryza sativa L.). Plant Mol. Biol. 80, 337–350. [DOI] [PubMed] [Google Scholar]
- Hussain, M.Z. , VanLoocke, A. , Siebers, M.H. , Ruiz‐Vera, U.M. , Cody Markelz, R.J. , Leakey, A.D.B. , Ort, D.R. and Bernacchi, C.J. (2013) Future carbon dioxide concentration decreases canopy evapotranspiration and soil water depletion by field‐grown maize. Glob. Change Biol. 19, 1572–1584. [DOI] [PubMed] [Google Scholar]
- IPCC (2013) Summary for policymakers In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (Stocker T. F., Qin D., Plattner G.‐K. et al., Cambridge and New York, NY: Cambridge University Press, pp. 1–30. Available at: http://www.climatechange2013.org [Google Scholar]
- Jauregui, I. , Aparicio‐Tejo, P.M. , Avila, C. , Rueda‐López, M. and Aranjuelo, I. (2015) Root and shoot performance of Arabidopsis thaliana exposed to elevated CO2: a physiologic, metabolic and transcriptomic response. J. Plant Physiol. 189, 65–76. [DOI] [PubMed] [Google Scholar]
- Jauregui, I. , Aparicio‐Tejo, P.M. , Avila, C. , Cañas, R. , Sakalauskiene, S. and Aranjuelo, I. (2016) Root‐shoot interactions explain the reduction of leaf mineral content in Arabidopsis plants grown under elevated [CO2] conditions. Physiol. Plant. 158, 65–79. [DOI] [PubMed] [Google Scholar]
- Juntawong, P. and Bailey‐Serres, J. (2012) Dynamic light regulation of translation status in Arabidopsis thaliana . Front. Plant Sci. 3 Available at: http://journal.frontiersin.org/article/10.3389/fpls.2012.00066/abstract [Accessed August 23, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, F. , Zhao, W. , Richards, J.T. , Wheeler, R.M. , Guy, C.L. and Levine, L.H. (2012) Transcriptional and metabolic insights into the differential physiological responses of Arabidopsis to optimal and supraoptimal atmospheric CO2 . PLoS ONE, 7, e43583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi, R. , Girke, T. , Bray, E.A. and Bailey‐Serres, J. (2004) Differential mRNA translation contributes to gene regulation under non‐stress and dehydration stress conditions in Arabidopsis thaliana . Plant J. 38, 823–839. [DOI] [PubMed] [Google Scholar]
- Kim, Y.‐M. , Han, Y.‐J. , Hwang, O.‐J. , Lee, S.‐S. , Shin, A.‐Y. , Kim, S.Y. and Kim, J.‐I. (2012) Overexpression of Arabidopsis translationally controlled tumor protein gene AtTCTP enhances drought tolerance with rapid ABA‐induced stomatal closure. Mol. Cells, 33, 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koca, H. , Ozdemir, F. and Turkan, I. (2006) Effect of salt stress on lipid peroxidation and superoxide dismutase and peroxidase activities of Lycopersicon esculentum and L. pennellii . Biol. Plant. 50, 745–748. [Google Scholar]
- Koenig, D. , Jimenez‐Gomez, J.M. , Kimura, S. et al . (2013) Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc. Natl Acad. Sci. USA, 110, E2655–E2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, R. , Ichihashi, Y. , Kimura, S. , Chitwood, D.H. , Headland, L.R. , Peng, J. , Maloof, J.N. and Sinha, N.R. (2012) A high‐throughput method for illumina RNA‐Seq library preparation. Front. Plant Sci. 3 10.3389/fpls.2012.00202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova, A. , Brockhoff, P.B. and Christensen, R.H.B. (2017) lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26. [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. and Salzberg, S.L. (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layat, E. , Probst, A.V. and Tourmente, S. (2013) Structure, function and regulation of transcription factor IIIA: from Xenopus to Arabidopsis. Biochim. Biophys. Acta, 1829, 274–282. [DOI] [PubMed] [Google Scholar]
- Leakey, A.D.B. and Lau, J.A. (2012) Evolutionary context for understanding and manipulating plant responses to past, present and future atmospheric [CO2]. Philo. Trans. Royal Soc. B, 367, 613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leakey, Andrew D.B. , Ainsworth, E.A. , Bernacchi, C.J. , Rogers, A. , Long, S.P. and Ort, D.R. (2009a) Elevated CO2 effects on plant carbon, nitrogen, and water relations: six important lessons from FACE. J. Exp. Bot. 60, 2859–2876. [DOI] [PubMed] [Google Scholar]
- Leakey, A.D.B. , Xu, F. , Gillespie, K.M. , McGrath, J.M. , Ainsworth, E.A. and Ort, D.R. (2009b) Genomic basis for stimulated respiration by plants growing under elevated carbon dioxide. Proc. Natl Acad. Sci. USA, 106, 3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P. , Ainsworth, E.A. , Leakey, A.D.B. , Ulanov, A. , Lozovaya, V. , Ort, D.R. and Bohnert, H.J. (2008a) Arabidopsis transcript and metabolite profiles: ecotype‐specific responses to open‐air elevated [CO2]. Plant Cell Environ. 31, 1673–1687. [DOI] [PubMed] [Google Scholar]
- Li, W.‐X. , Oono, Y. , Zhu, J. et al . (2008b) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell, 20, 2238–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y.‐F. , Zheng, Y. , Vemireddy, L.R. et al. (2018) Comparative transcriptome and translatome analysis in contrasting rice genotypes reveals differential mRNA translation in salt‐tolerant Pokkali under salt stress. BMC Genom., 19 Available at: https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-5279-4 [Accessed August 23, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, C.‐J. , Lai, Z. , Lee, S. , Yun, D.J. and Mengiste, T. (2016) Arabidopsis HOOKLESS1 regulates responses to pathogens and abscisic acid through interaction with MED18 and acetylation of WRKY33 and ABI5 chromatin. Plant Cell, 28, 1662–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.‐Y. , Chen, P.‐W. , Chuang, M.‐H. , Juntawong, P. , Bailey‐Serres, J. and Jauh, G.‐Y. (2014) Profiling of translatomes of in vivo‐grown pollen tubes reveals genes with roles in micropylar guidance during pollination in Arabidopsis . Plant Cell, 26, 602–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, M.‐J. , Wu, Szu‐Hsien , Wu, J.‐F. , Lin, W.‐D. , Wu, Y.‐C. , Tsai, T.‐Y. , Tsai, H.‐L. and Wu, Shu‐Hsing (2013) Translational landscape of photomorphogenic Arabidopsis . Plant Cell, 25, 3699–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobet, G. , Pagès, L. and Draye, X. (2011) A novel image‐analysis toolbox enabling quantitative analysis of root system architecture. Plant Physiol. 157, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoszek, R. , Feist, P. and Ignatova, Z. (2016) Insights into the adaptive response of Arabidopsis thaliana to prolonged thermal stress by ribosomal profiling and RNA‐Seq. BMC Plant Biol. 16, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markelz, R.J.C. , Lai, L.X. , Vosseler, L.N. and Leakey, A.D.B. (2014a) Transcriptional reprogramming and stimulation of leaf respiration by elevated CO2 concentration is diminished, but not eliminated, under limiting nitrogen supply: leaf respiration response to CO2 and N supply. Plant Cell Environ. 37, 886–898. [DOI] [PubMed] [Google Scholar]
- Markelz, R.J.C. , Vosseller, L.N. and Leakey, A.D.B. (2014b) Developmental stage specificity of transcriptional, biochemical and CO2 efflux responses of leaf dark respiration to growth of Arabidopsis thaliana at elevated [CO2]: respiration over leaf development under high CO2 . Plant Cell Environ. 37, 2542–2552. [DOI] [PubMed] [Google Scholar]
- McLeay, R.C. and Bailey, T.L. (2010) Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics, 11, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medek, D.E. , Schwartz, J. and Myers, S.S. (2017) Estimated effects of future atmospheric CO2 concentrations on protein intake and the risk of protein deficiency by country and region. Environ. Health Perspect. 125, 087002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meteignier, L.‐V. , El Oirdi, M. , Cohen, M. et al . (2017) Translatome analysis of an NB‐LRR immune response identifies important contributors to plant immunity in Arabidopsis. J. Exp. Bot. 68, 2333–2344. [DOI] [PubMed] [Google Scholar]
- Mi, G. , Di, Y. , Emerson, S. , Cumbie, J.S. and Chang, J.H. (2012) Length bias correction in gene ontology enrichment analysis using logistic regression. PLoS ONE, 7, e46128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustroph, A. , Zanetti, M.E. , Jang, C.J.H. , Holtan, H.E. , Repetti, P.P. , Galbraith, D.W. , Girke, T. and Bailey‐Serres, J. (2009) Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc. Natl Acad. Sci. USA, 106, 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, D.E. , Repetti, P.P. , Adams, T.R. et al . (2007) Plant nuclear factor Y (NF‐Y) B subunits confer drought tolerance and lead to improved corn yields on water‐limited acres. Proc. Natl Acad. Sci. USA, 104, 16450–16455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley, R.C. , Huang, S.C. , Song, L. , Lewsey, M.G. , Bartlett, A. , Nery, J.R. , Galli, M. , Gallavotti, A. and Ecker, J.R. (2016) Cistrome and epicistrome features shape the regulatory DNA landscape. Cell, 165, 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando, D.A. , Brady, S.M. , Koch, J.D. , Dinneny, J.R. and Benfey, P.N. (2009) Manipulating large‐scale Arabidopsis microarray expression data: identifying dominant expression patterns and biological process enrichment In Plant Systems Biology (Belostotsky D. A., ed.). Totowa, NJ: Humana Press, pp. 57–77. Available at: http://link.springer.com/10.1007/978-1-60327-563-7_4 [Accessed August 24, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013) R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; Available at: http://www.R-project.org/ [Google Scholar]
- Ribeiro, D.M. , Araújo, W.L. , Fernie, A.R. , Schippers, J.H.M. and Mueller‐Roeber, B. (2012) Translatome and metabolome effects triggered by gibberellins during rosette growth in Arabidopsis. J. Exp. Bot. 63, 2769–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon, C.A. , Raper, C.D. Jr and Patterson, R.P. (2003) Genotypic differences in root anatomy affecting water movement through roots of soybean. Int. J. Plant Sci. 164, 543–551. [Google Scholar]
- Ritchie, M.E. , Phipson, B. , Wu, D. , Hu, Y. , Law, C.W. , Shi, W. and Smyth, G.K. (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M.D. , McCarthy, D.J. and Smyth, G.K. (2009) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, H.H. , Runion, G.B. and Krupa, S.V. (1994) Plant responses to atmospheric CO2 enrichment with emphasis on roots and the rhizosphere. Environ. Pollut. 83, 155–189. [DOI] [PubMed] [Google Scholar]
- Ron, M. , Dorrity, M.W. , de Lucas, M. , Toal, T. , Hernandez, R.I. , Little, S.A. , Maloof, J.N. , Kliebenstein, D.J. and Brady, S.M. (2013) Identification of novel loci regulating interspecific variation in root morphology and cellular development in tomato. Plant Physiol. 162, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seizl, M. , Larivière, L. , Pfaffeneder, T. , Wenzeck, L. and Cramer, P. (2011) Mediator head subcomplex Med11/22 contains a common helix bundle building block with a specific function in transcription initiation complex stabilization. Nucleic Acids Res. 39, 6291–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18, 275–294. 10.1093/biostatistics/kxw041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, G. (2003) Spatial and temporal effects of free‐air CO2 enrichment (POPFACE) on leaf growth, cell expansion, and cell production in a closed canopy of poplar. Plant Physiol. 131, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, C. , Wang, Y. , Yu, H. et al. (2019) A gene expression map of shoot domains reveals regulatory mechanisms. Nat. Commun., 10 Available at: http://www.nature.com/articles/s41467-018-08083-z [Accessed August 23, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toal, T.W. , Ron, M. , Gibson, D. et al. (2018) Regulation of root angle and gravitropism. G3: Genes, Genomes, Genetics, 8, 3841–3855. 10.1534/g3.118.200540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley, B.T. , Covington, M.F. , Ichihashi, Y. , Zumstein, K. and Sinha, N.R. (2015) BrAD‐seq: Breath Adapter Directional sequencing: a streamlined, ultra‐simple and fast library preparation protocol for strand specific mRNA library construction. Front. Plant Sci. 6 Available at: http://www.frontiersin.org/Plant_Evolution_and_Development/10.3389/fpls.2015.00366/abstract [Accessed December 5, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente, R. , Pérez, P. , Martínez‐Carrasco, R. et al . (2016) Metabolic and transcriptional analysis of durum wheat responses to elevated CO2 at low and high nitrate supply. Plant Cell Physiol. 57, 2133–2146. [DOI] [PubMed] [Google Scholar]
- Vicente, R. , Martínez‐Carrasco, R. , Pérez, P. and Morcuende, R. (2018) New insights into the impacts of elevated CO2, nitrogen, and temperature levels on the regulation of C and N metabolism in durum wheat using network analysis. New Biotechnol. 40, 192–199. [DOI] [PubMed] [Google Scholar]
- Vragović, K. , Sela, A. , Friedlander‐Shani, L. , Fridman, Y. , Hacham, Y. , Holland, N. , Bartom, E. , Mockler, T.C. and Savaldi‐Goldstein, S. (2015) Translatome analyses capture of opposing tissue‐specific brassinosteroid signals orchestrating root meristem differentiation. Proc. Natl Acad. Sci. USA, 112, 923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warpeha, K.M. , Upadhyay, S. , Yeh, J. , Adamiak, J. , Hawkins, S.I. , Lapik, Y.R. , Anderson, M.B. and Kaufman, L.S. (2007) The GCR1, GPA1, PRN1, NF‐Y signal chain mediates both blue light and abscisic acid responses in Arabidopsis. Plant Physiol. 143, 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, C.K. , Sato, S. , Yanagisawa, S. , Uesono, Y. , Terashima, I. and Noguchi, K. (2014) Effects of elevated CO2 on levels of primary metabolites and transcripts of genes encoding respiratory enzymes and their diurnal patterns in Arabidopsis thaliana: possible relationships with respiratory rates. Plant Cell Physiol. 55, 341–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H.‐Y.L. , Song, G. , Walley, J.W. and Hsu, P.Y. (2019) The tomato translational landscape revealed by transcriptome assembly and ribosome profiling. Plant Physiol. 181, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, G. , Greene, G.H. , Yoo, H. , Liu, L. , Marqués, J. , Motley, J. and Dong, X. (2017) Global translational reprogramming is a fundamental layer of immune regulation in plants. Nature, 545, 487–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yángüez, E. , Castro‐Sanz, A.B. , Fernández‐Bautista, N. , Oliveros, J.C. and Castellano, M.M. (2013) Analysis of genome‐wide changes in the translatome of Arabidopsis seedlings subjected to heat stress. PLoS ONE, 8, e71425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, M.D. , Wakefield, M.J. , Smyth, G.K. and Oshlack, A. (2010) Gene ontology analysis for RNA‐seq: accounting for selection bias. Genome Biol. 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak, D.R. , Pregitzer, K.S. , King, J.S. and Holmes, W.E. (2000) Elevated atmospheric CO2, fine roots and the response of soil microorganisms: a review and hypothesis: REVIEW elevated CO2 and soil microorganisms. New Phytol. 147, 201–222. [Google Scholar]
- Zanetti, M.E. , Chang, I.F. , Gong, F. , Galbraith, D.W. and Bailey‐Serres, J. (2005) Immunopurification of polyribosomal complexes of Arabidopsis for global analysis of gene expression. Plant Physiol. 138, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , Zhang, T. , Liu, J. , Zhang, J. and He, C. (2018) Comprehensive analysis of differentially expressed genes reveals the molecular response to elevated CO2 levels in two sea buckthorn cultivars. Gene, 660, 120–127. [DOI] [PubMed] [Google Scholar]
- Zoschke, R. , Watkins, K.P. , Miranda, R.G. and Barkan, A. (2016) The PPR‐SMR protein PPR53 enhances the stability and translation of specific chloroplast RNAs in maize. Plant J. 85, 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Whole root to metaxylem ratios.
Figure S2. Elevated CO2 has no significant effects on mature metaxylem in either species.
Figure S3. Number of significantly differentially expressed genes in each of the linear model terms in shoots or roots.
Figure S4. Clustering of genes that change in a (CO2 × Species × Day 12) interaction manner reveals genetic modules with dominant patterns.
Figure S5. Strategy to identify groups of genes with unique transcriptional or translational patterns.
Figure S6. Number of GO terms enriched per dominant pattern in total and TRAP RNA‐Seq data.
Table S1. anova tables from root area and diameter analyses
Table S2. anova tables from shoot or root biomass, photosynthesis, and stomatal conductance analyses
Table S3. Log2 transformed data and results of the metabolomic analysis
Table S4. Genes assigned to dominant patterns and motif enrichment analysis
Table S5. CPM normalized data from total and TRAP RNA‐Seq
Data Availability Statement
Raw total and TRAP sequencing data can be obtained from GEO under the accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126430. Normalized CPM data for total and TRAP RNA‐Seq are provided in Table S5. Raw LiCOR, root anatomy and morphology and associated scripts can be found at https://github.com/rodriguezmDNA/Gray_RodriguezMedina_CO2Supplemental.