

Abstract
Tomosyn, a protein encoded by syntaxin‐1‐binding protein 5 (STXBP5) gene, has a well‐established presynaptic role in the inhibition of neurotransmitter release and the reduction of synaptic transmission by its canonical interaction with the soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor machinery. However, the postsynaptic role of tomosyn in dendritic arborization, spine stability, and trafficking of ionotropic glutamate receptors remains to be elucidated. We used short hairpin RNA to knock down tomosyn in mouse primary neurons to evaluate the postsynaptic cellular function and molecular signaling regulated by tomosyn. Knockdown of tomosyn led to an increase of RhoA GTPase activity accompanied by compromised dendritic arborization, loss of dendritic spines, decreased surface expression of AMPA receptors, and reduced miniature excitatory postsynaptic current frequency. Inhibiting RhoA signaling was sufficient to rescue the abnormal dendritic morphology and the surface expression of AMPA receptors. The function of tomosyn regulating RhoA is mediated through the N‐terminal WD40 motif, where two variants each carrying a single nucleotide mutation in this region were found in individuals with autism spectrum disorder (ASD). We demonstrated that these variants displayed loss‐of‐function phenotypes. Unlike the wild‐type tomosyn, these two variants failed to restore the reduced dendritic complexity, spine density, as well as decreased surface expression of AMPA receptors in tomosyn knockdown neurons. This study uncovers a novel role of tomosyn in maintaining neuronal function by inhibiting RhoA activity. Further analysis of tomosyn variants also provides a potential mechanism for explaining cellular pathology in ASD.
Keywords: autism, dendrite, RRID:AB_10563941, RRID:AB_10622025, RRID:AB_11042324, RRID:AB_221568, RRID:AB_2278725, RRID:AB_2336064, RRID:AB_2619878, RRID:AB_2629468, RRID:AB_325403, RRID:AB_476692, RRID:AB_477256, RRID:AB_887844, RRID:CVCL_0470, SNARE, STXBP5, WD40 motif
Tomosyn inhibits RhoA activity via WD40 motif to regulate dendritic stability. Silence or autism spectrum disorder‐associated variants of tomosyn result in reduced dendritic complexity and surface expression of AMPA receptors.
Significance.
This study unveils a postsynaptic role of tomosyn in the maintenance of neuronal morphology and glutamate receptor trafficking via the regulation of the Rho signaling pathway. The WD40 domain of tomosyn is necessary to conduct this Rho regulation, and two autism‐associated variants localized at this region perturb this function. This study reveals a novel molecular link between dendritic stability and synaptic function, which could advance a greater understanding of the cellular pathologies involved in neurodevelopmental disorders, such as autism.
1. INTRODUCTION
Formation and maintenance of neuronal connections are crucial for promoting proper brain function. Failure of these processes is often observed in neurological disorders accompanied by detrimental behaviors. Excitatory neurons are major projection cells in the brain and extend complex dendritic arbors to establish extensive connections with other neurons. Mature neurons have actin‐rich spines that protrude from dendrites and are the major structures harboring synaptic components. Cytoskeleton rearrangement by Rho signaling pathways is the driving force that regulates neuronal stability and synaptic plasticity (Govek et al., 2004; Lin & Koleske, 2010; Nakayama, Harms, & Luo, 2000). Ionotropic glutamate receptors, including AMPA and NMDA receptors, cluster primarily on dendritic spines and are responsible for excitatory synaptic transmission and synaptic plasticity (Diering & Huganir, 2018; Lau & Zukin, 2007; Reiner & Levitz, 2018). Understanding the mechanistic regulations between the cytoskeletal machinery and glutamate receptors trafficking will allow us to better understand the underlying mechanisms that promote neuronal stability and function.
Tomosyn was first identified as a protein encoded by the syntaxin‐1‐binding protein 5 (STXBP5) gene in rats (Fujita et al., 1998). Structurally, tomosyn contains an N‐terminal WD40 repeats domain and a C‐terminal coiled‐coil motif (Masuda, Huang, Fisher, Luo, & Scheller, 1998). The R‐SNARE‐like structure at the C terminus allows tomosyn to inhibit the formation of the SNARE complex by competing with Munc18 for binding to syntaxin‐1, thereby negatively regulating neurotransmitter release (Ben‐Simon et al., 2015; Fujita et al., 1998; Sakisaka et al., 2008). Mice overexpressing tomosyn in the hippocampus showed impaired spatial learning and memory, potentially via reduced synaptic transmission in mossy fiber (MF)‐CA3 synapses (Barak et al., 2013). Conversely, tomosyn knockout mice exhibited accelerated kindling behaviors due to increased glutamate release in the hippocampal dentate gyrus (Batten et al., 2017). Selectively knocking down tomosyn in MF‐CA3 synapses also impaired facilitation, long‐term potentiation (LTP), and PKA‐induced potentiation (Ben‐Simon et al., 2015). While there is accumulating evidence for the presynaptic roles of tomosyn, relatively few studies have focused on its role in postsynaptic compartments. One study has shown that tomosyn regulates neurite outgrowth in immature neurons by strongly binding to Rho‐associated serine/threonine kinase (ROCK)‐phosphorylated syntaxin‐1 (Sakisaka et al., 2004). Tomosyn has also been shown to regulate dendritic morphology via the ubiquitin‐proteasome system (Saldate, Shiau, Cazares, & Stuenkel, 2018). Here, we provide evidence showing that tomosyn regulates RhoA signaling to maintain the structural stability of neurons.
Mechanisms that regulate neuronal stability and synaptic function are often vulnerable targets in neurodevelopmental disorders, such as autism spectrum disorder (ASD; Forrest, Parnell, & Penzes, 2018; Hampson & Blatt, 2015; Subramanian et al., 2017). Individuals with ASD commonly exhibit difficulties in social communication and interaction, repetitive behaviors, and restricted interests (American Psychiatric Association, 2013). Although ASD exhibits a high degree of genetic heterogeneity, many identified risk genes converge on similar cellular pathways, including those that regulate neurite outgrowth, spine stability, synaptic plasticity, excitatory/inhibitory balance, and trafficking of glutamate receptors (Bridi, Park, & Huang, 2017; Chahrour et al., 2016; Iossifov et al., 2014; Lin, Frei, Kilander, Shen, & Blatt, 2016; Oblak, Gibbs, & Blatt, 2010, 2011; Sanders et al., 2015; Short et al., 2018; Volk, Chiu, Sharma, & Huganir, 2015). Several human genetic studies, including genome‐wide association studies and whole exome sequencing, have linked the deletion, and two tomosyn variants (L412V and Y502C) to ASD (Cukier et al., 2014; Davis et al., 2009; Hussman et al., 2011; Matsunami et al., 2013). In this study, we first examined the postsynaptic role of tomosyn. Reductions of dendritic arborization and spine density accompanied by enhanced RhoA activity and decreased surface expression of glutamate receptors were observed in tomosyn knockdown neurons. We then further elucidate whether autism‐related tomosyn variants affect the tomosyn function. ASD‐associated tomosyn L412V and Y502C variants displayed a loss‐of‐function effect and failed to restore the cellular defects in tomosyn knockdown neurons. Our study reveals a pivotal postsynaptic function of tomosyn, which mediates a potential pathway for maintaining the structural stability of neurons, and may be altered in ASD.
2. MATERIALS AND METHODS
Key materials and reagents, and Primary antibodies are summarized in Table S1 and Table S2, respectively.
2.1. Animals
C57BL/6J mice were obtained from the University of Maryland School of Medicine Program in Comparative Medicine. Mice were housed and cared for by the AAALAC accredited program of the University of Maryland School of Medicine. Female mice were group‐housed and male mice were singly housed with ad libitum food and water accessibility under a standard 12‐hr light/dark cycle. Neonatal mice of both sexes were sacrificed for neuronal culture preparation. Animals of both sexes were used for biochemistry as well to match the mixed‐gender condition in cultures. All experiments performed were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) of the University of Maryland School of Medicine and the Hussman Institute for Autism.
2.2. Plasmids
The pcDNA™4/myc‐His‐m‐tomosyn plasmid was a gift from Dr. Sushant Bhatnagar, University of Alabama. The tomosyn‐GFP was engineered by adding a green fluorescent protein (GFP) tag at the 3′ end of the pcDNA™4/myc‐His‐m‐tomosyn. Short hairpin RNA (shRNA) constructs targeting tomosyn were generated by inserting the anti‐tomosyn RNAi (shRNA482 also referred to as shTomosyn‐GCACTGAGCGAGGAAACTAC, shRNA1083‐GGAACCATATGCTGTGGTTGT) or a scrambled shRNA control (Scramble: CAGGAACGCATAGACGCATGA) into the third generation pLL3.7‐RFP vector using the HpaI and XhoI restriction sites as previously described (Ben‐Simon et al., 2015). L412V or Y502C point mutation was introduced by site‐directed mutagenesis (QuikChange II XL Site‐Directed Mutagenesis Kit, Agilent Technologies) using pcDNA™4/myc‐His‐m‐tomosyn‐GFP plasmid as a template. The shRNA‐resistant tomosyn (wild‐type WTr‐Tom‐GFP, L412Vr‐Tom‐GFP, and Y502Cr ‐Tom‐GFP) constructs were made by mutating three nucleotides in the shTomosyn sequences with two PCR primers: Forward 5′‐AGTGGCTCTATGTGGGCACGGAACGCGGAAACATACACATTGTTA‐3′ and Reverse 5′‐TAACAATGTGTATGTTTCCGCGTTCCGTGCCCACATAGAGCCACT‐3′. shTomosyn‐WTr‐Tom‐RFP, shTomosyn‐L412Vr‐Tom‐RFP, and shTomosyn‐Y502Cr‐Tom‐RFP were generated by inserting WTr‐Tom, L412Vr‐Tom, or Y502Cr‐Tom into the shTomosyn vector. pcDNA3‐EGFP‐RhoA‐WT (Addgene, plasmid # 12965) and pcDNA3‐EGFP‐RhoA‐T19N (Addgene, plasmid # 12967) were gifts from Gary Bokoch (Subauste et al., 2000). pCI‐SEP‐GluR1 (Addgene, plasmid #24000) was a gift from Robert Malinow (Kopec, Li, Wei, Boehm, & Malinow, 2006). pTriEx‐RhoA FLARE.sc Biosensor WT (Addgene, plasmid # 12150) was a gift from Klaus Hahn (Pertz, Hodgson, Klemke, & Hahn, 2006). pCAGIG (IRES‐GFP; Addgene, plasmid # 11159) was a gift from Connie Cepko (Matsuda & Cepko, 2004). pRFP‐N1 was constructed by replacing GFP in the pEGFP‐N1 (Clontech, Catalog # 6085‐1) with RFP. Full‐length (FL) tomosyn and domain‐mutant tomosyn with BamHI and EcoRI restriction sites were subcloned into the pRFP‐N1 vector. Primer sequences for FL‐Tom‐RFP: Forward 5′‐ATATGAATTCGCCACCATGAGGAAATTCAACATCAG‐3′ and Reverse 5′‐ATATGGATCCATGAACTGGTACCACTTCTTATCTTTG‐3′; Tom‐ΔC‐RFP: Forward 5′‐ATATGAATTCGCCACCATGAGGAAATTCAACATCAG‐3′ and Reverse 5′‐ATATGGATCCATGCCCGGGATGTGTTGTG‐3′; and Tom‐ΔN‐RFP: Forward 5′‐ATATGAATTCGCCACCATGCCTGGTGGGATC‐3′ and Reverse 5′‐ATATGGATCCATTTTGTATTTCAGCATCATCTCATGAG‐3′.
2.3. Cell culture and transfections
Neuro‐2a (N2a, a mouse neuroblastoma cell line; ATCC) cells were maintained in high‐glucose DMEM (Invitrogen) growth media supplemented with 1% penicillin/streptomycin (Invitrogen), 2 mM l‐glutamine (Invitrogen), and 10% fetal bovine serum (Sigma‐Aldrich). Primary hippocampal cultures were prepared from postnatal day 0 (P0) newborn mice (8–10 animals per culture), and plated at a density of 3 × 105 cells/well for morphometric analysis, electrophysiology, biochemistry, and Förster resonance energy transfer (FRET) experiments, or 3 × 104 cells/well for surface staining on 12‐mm coverslips in 24‐well plates coated with 20 μg/ml poly‐(d)‐lysine (Millipore Sigma). Neurons were maintained in serum‐free neural basal media (Invitrogen) containing 1% pen/strep, 2 mM l‐glutamine, and 2% B27‐supplement. Lipofectamine 3000 reagent (Invitrogen) was used to transfect N2a cells according to the manufacturer's protocol. Primary hippocampal neurons were transfected using Lipofectamine 3000 at 4 days in vitro (DIV) followed by fixation at 7 DIV for quantifying the dendritic arborization, and calcium‐phosphate transfection method at 11–12 DIV followed by fixation at 15 DIV for spine quantification. For inhibiting RhoA activity, 24 hr after transfection, neurons were either treated with 1.0 µg/ml of RhoA inhibitor C3 transferase (Cytoskeleton) for 16 hr or 10 µl of ddH2O as a control for 16 hr prior to fixation. For electrophysiological recordings, primary hippocampal neurons were transfected at 9 DIV using Lipofectamine 2000 (Invitrogen) and incubated until 14–16 DIV. All cultures were replicated with the same experimental condition at least three times for each experiment.
2.4. Synaptic fractionation
Forebrains from P21 mice (two pooled forebrains per sample, six animals) were obtained for synaptic fraction analyses. Synaptic fractionation was prepared as described previously (Bermejo, Milenkovic, Salahpour, & Ramsey, 2014). Briefly, forebrains were homogenized in 4 ml of 0.32 M HEPES‐buffered sucrose solution (0.32 M sucrose, 4 mM HEPES, 0.25 mM PMSF, and protease and phosphatase inhibitors) at 4°C by using a motor‐driven glass‐Teflon tissue homogenizer. Homogenates (total) were centrifuged at 900g for 10 min at 4°C to generate the nuclear fraction pellet (P1) and the supernatant (cytosol/membranes, S1). S1 was centrifuged at 10,000g for 15 min at 4°C to obtain the crude synaptosomes (P2) and the supernatant (cytosol/light membranes, S2). P2 was washed with 0.32 M HEPES‐buffered sucrose solution and re‐centrifuged at 10,000g for 15 min at 4°C. P2′, obtained from P2, was lysed in 4 ml ddH2O by hypoosmotic shock and was adjusted back to 4 mM HEPES. P2′ lysate was centrifuged at 25,000g for 20 min at 4°C to isolate the synaptosomal membrane fraction (synaptosomes, P3) in the pellet and the crude vesicular fraction in the supernatant (synaptic vesicles, S3). P3 in 1 ml of 0.32 M HEPES‐buffered sucrose solution was loaded on discontinuous sucrose gradient containing 3.0 ml of 0.8 M sucrose, 3.0 ml of 1 M sucrose, and 3.5 ml of 1.2 M sucrose in 4 mM HEPES‐buffered solution, and re‐centrifuged at 150,000g for 2 hr at 4°C to obtain synaptic plasma membranes (SPM). SPM was re‐suspended in Triton X‐100/HEPES/EDTA solution (0.54% Triton X‐100, 50 mM HEPES, 2 mM EDTA) and centrifuged at 32,000g for 20 min at 4°C to obtain the postsynaptic density (PSD) fraction. A quantity of 10 µg protein from each fractionation was loaded for Western blot analysis.
2.5. Western blot analysis
Hippocampi and whole brains from mice (two pooled hippocampus and one whole brain per age group) of both sexes at P0, P7, P14, and P21 were dissected for Western blot analysis of the temporal expression (six animals for hippocampus and three animals for whole brain per age‐point). Cortex, hippocampus, striatum, and cerebellum from four P14 mice (two to three pooled brain areas per sample, 8–12 animals) of both sexes were prepared for Western blot analysis of the spatial expression. Results from at least three independent cultures of N2a cells were collected for each experiment. Brain tissues or N2a cells transfected with plasmids were harvested in RIPA buffer containing 20 mM Tris HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP‐40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM beta‐glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin, 1 mM PMSF, and a phosphatase and protease inhibitor cocktail (Millipore Sigma). Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). About 50 µg of total protein from N2a cell lysates was used to determine the knockdown efficiency of tomosyn shRNAs and to validate tomosyn domain mutants. Ten micrograms of total protein from different brain regions was used to examine tomosyn expression. Protein samples were run on 8% SDS‐PAGE gels and transferred to a PVDF membrane. After blocking the membrane with 5% skim milk in TBST (0.1% Tween 20 in TBS) for 1 hr at room temperature, the membrane was immunoblotted with the following primary antibodies: rabbit anti‐tomosyn (Synaptic Systems), mouse anti‐β‐Actin (Sigma‐Aldrich), mouse anti‐syntaxin‐1 (Synaptic Systems), mouse anti‐syntaxin‐4 (Synaptic Systems), mouse anti‐PSD‐95 (UC Davis/NIH NeuroMab Facility), rabbit anti‐RFP (Thermo Fisher Scientific), and rabbit anti‐GAPDH (Cell Signaling Technology). The membrane was washed three times with TBST for 10 min and incubated with the HRP‐conjugated secondary antibodies for 60 min at room temperature. The membrane was then washed five times with TBST for 10 min at room temperature. The chemiluminescent reaction was induced by incubating the membrane with Clarity™ Western ECL substrate (Bio‐Rad). The chemiluminescent blots were then imaged with the ChemiDoc™ Touch Imaging System (Bio‐Rad). The densitometry of the protein signals was analyzed using Image Lab™ software (Bio‐Rad).
2.6. Immunocytochemistry and morphometric analysis
Cultured neurons were fixed at 7 or 15 DIV with 4% paraformaldehyde in phosphate buffer (PB; 320 mM Na2HPO4, 76 mM NaH2PO4) for 15 min and permeabilized and blocked in TBS containing 0.1% Triton X‐100, 3% BSA, and 1% donkey serum. Cells were immunostained with the following primary antibodies: mouse anti‐GFP (Molecular Probes), rat anti‐RFP (Chromotek), rabbit anti‐tomosyn (Synaptic Systems), mouse anti‐MAP2 (Millipore Sigma), mouse anti‐CaMKIIα (Thermo Fisher Scientific), and mouse anti‐GAD67 (Millipore Sigma) followed by Alexa Fluor 488, 568, 594‐conjugated secondary antibodies (Thermo Fisher Scientific). For dendrite analysis, z‐stack images were taken using a confocal laser scanning microscope (Zeiss LSM 780) with a 40 X/1.3 oil DIC objective. Original images of neurons were blinded and then used to perform dendrites tracing using Fiji (NIH) with NeuronJ plugin (Meijering et al., 2004) for further analysis. Only dendrites that are longer than 10 μm are included for the quantification of branch number and total dendrite length. Traced neurons were further analyzed using Fiji with Sholl analysis plugin (Ferreira et al., 2014). Concentric circles having 10 µm increments in radius were defined from the center of the cell body. The number of traced dendrites crossing each circle was counted. Dendrite complexity was quantified by the area under the Sholl curve. For spine analysis, two to three dendrite segments per neuron were selected randomly from secondary dendrites. Z‐stack images were taken using Zeiss LSM 780 with a 63 ×/1.4 oil DIC objective and four zooms at 512 × 512 resolution. Images were blinded prior to the analysis and the number of dendritic spines, which are ≤2 µm in length, per dendritic segment measured was quantified using Fiji. In addition, images of neurons co‐immunostained for either CaMKIIα or GAD67 and tomosyn were taken using the EVOS FL Auto2 imaging system with a 40×/0.95 NA air objective (Thermo Fisher Scientific).
2.7. Electrophysiology
Neurons at 14–16 DIV were used for whole‐cell electrophysiological recordings following either scrambled shRNA or shTomosyn transfection at 9 DIV. Coverslips were transferred from a 24‐well plate into a recording chamber superfused with a buffer containing the following (in mM): 125 NaCl, 3 KCl, 10 HEPES, 5 dextrose, 1 MgCl2, and 2 CaCl2. The recording buffer was adjusted to ~260 mOsm and pH 7.2, and the bath was maintained at a temperature of 30°C. Glass pipettes pulled to a resistance of 3–7 MΩ were filled with internal solution containing the following (in mM): 134 KMeSO4, 3 KCl, 10 HEPES, 1 MgCl2, 4 MgATP, 0.5 Na2GTP, 5 K2‐creatine phosphate, and 5 Na2‐creatine phosphate. The internal solution was adjusted to ~250 mOsm and pH 7.4. Only RFP‐expressing cells with a series resistance ≤30 MΩ and a resting membrane potential ≤−50 mV were accepted for final analysis. Whole‐cell parameters were monitored throughout the recording with a 100 ms, −10 mV step delivered every 30 s. Recordings were made using an Axon MultiClamp 700B amplifier (Molecular Devices). Data were filtered at 2 kHz and digitized at 10 kHz with a National Instruments digital–analog converter under the control of Igor Pro software (WaveMetrics). Miniature EPSCs were recorded in voltage mode (Vh = −70 mV) in the presence of 50 µM AP5, 1 µM tetrodotoxin (TTX), and 10 µM gabazine (GBZ). The frequency, amplitude, and kinetics of miniature excitatory postsynaptic currents (mEPSCs) were analyzed with Mini Analysis software (Synaptosoft), with a threshold of 3 × RMS noise for event discrimination. At least 50 events per cell were used in these calculations. Transfection conditions were blinded prior to recording.
2.8. FRET analysis
Hippocampal neurons were co‐transfected with pTriEx‐RhoA FLARE.sc Biosensor WT and either scrambled shRNA or shTomosyn at 4 DIV. Cells were fixed with 4% paraformaldehyde in PB and mounted at 7 DIV. Imaging was performed with an upright confocal microscope (Zeiss LSM 780) and a 40× EC Plan‐NeoFluar/1.3 oil‐immersion DIC objective lens. For sensitized emission, FRET excitation was performed using the 458 nm laser, and emission filter bands were set to 440–510 nm for CFP and 517–606 nm for YFP. The 561 nm laser was used for excitation of RFP to identify shRNA‐transfected neurons. Single plane images were blinded and exported to Fiji (NIH) for analysis. All images were corrected for shading (uneven illumination in the field of view) and the RFP channel image was used as a reference to create a whole‐cell ROI (region‐of‐interest), which was subsequently overlaid on CFP and YFP channel images and mean fluorescence intensity (I) was measured within the ROI and thereafter background‐corrected. As the RhoA FLARE is an intramolecular FRET biosensor, the FRET efficiency is calculated by simple ratiometry of the acceptor (YFP) over the donor (CFP) intensity: FRET efficiency = I YFP/I CFP. Acceptor photobleaching FRET was done by placing a circular bleach ROI (r = 2.04 μm) over secondary or tertiary parts of either apical or basal dendritic compartments of transfected neurons. Acceptor bleaching was executed over 50 iterations with the 514 nm laser at maximum power. Changes in fluorescence intensities in the donor (CFP) and acceptor (YFP) channels were recorded using low power (2%) excitation with the 458 and 514 nm lasers, respectively, and a total of 10 scans were acquired: 5 before (pre) and 5 after (post) the single bleaching event. The mean fluorescence intensity values (I) of ROIs were exported to Excel. The donor channel bleach ROI data were background‐corrected and normalized to unbleached background ROIs and reference ROIs, respectively. FRET efficiency in percentage was calculated according to: FRET efficiency = ((Idonorpost − Idonorpre)/Idonorpost) × 100. All calculated FRET efficiency data were exported to GraphPad Prism 8 for statistical analysis.
2.9. Immunoprecipitation
Immunoprecipitations of endogenous binding partners of tomosyn or syntaxin‐4 were performed using lysates of cultured cortical neurons. Cultured cortical neurons were lysed in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X‐100) containing phosphatase and protease inhibitor cocktail (Thermo Fisher Scientific). Lysates were precleared with protein A/G agarose beads (Thermo Fisher Scientific) for 1 hr. Two micrograms of rabbit anti‐tomosyn or mouse anti‐syntaxin‐4 antibody was added to the lysate followed by rotation overnight at 4°C. About 20 µl of protein A/G agarose was added to the immune complex and incubated for 1 hr at 4°C. The beads were washed five times with TBST. Bound proteins were eluted in 2 × Laemmli sample buffer (Bio‐Rad) containing 5% 2‐mercaptoethanol by heating at 95°C for 5 min. Immunoprecipitations were repeated three times from three independent cultures to confirm the results.
2.10. Surface staining
Neurons were co‐transfected with pCI‐SEP‐pHluorin‐GluR1 and scrambled shRNA or shTomosyn. The surface staining of pHluorin‐GluR1 was performed as previously described (Gu et al., 2016; Noel et al., 1999). Briefly, transfected neurons were incubated with mouse anti‐GFP antibody in prechilled artificial cerebrospinal fluid (aCSF; 124 mM NaCl, 5 mM KCl, 1.23 mM NaH2PO4, 26 mM NaHCO3, 1 mM MgCl2, 2 mM CaCl2) containing 10% BSA for 1 hr at room temperature. Cells were washed three times with aCSF, incubated with donkey anti‐mouse Alexa Fluor 647‐conjugated secondary antibody (Thermo Fisher Scientific) for 1 hr at room temperature, and again washed three times with aCSF. For total pHluorin‐GluR1 staining, cells were fixed with 4% paraformaldehyde in PB for 15 min, permeabilized, and blocked in blocking solution. Cells were immunostained with rabbit anti‐GFP (Antibodies‐Online) and rat anti‐RFP antibodies (Chromotek). Cells were washed three times followed by incubation with goat anti‐rat Alexa Fluor 568‐ and donkey anti‐rabbit Alexa Fluor 488‐conjugated secondary antibody (Thermo Fisher Technologies). Coverslips were mounted on the glass slides with Prolong Diamond Antifade Mountant (Molecular Probes) prior to imaging. Three dendrite segments per neuron were selected randomly from secondary branches on the apical dendrite. Identical acquisition settings were applied to each image within all experiments. For surface GluR1 expression quantification, both whole dendritic segments and individual spines were chosen as ROI. Surface GluR1/total GluR1 was calculated as surface/total = (mean intensity of surface − background intensity)/(mean intensity of total − background intensity). Images were blinded prior to analysis.
2.11. Statistical analysis
Sample sizes were predetermined using a priori power analysis that was adequate for power of 0.8 and statistically significant differences of p < 0.05 for each experiment. Values were expressed as mean ± SD (standard deviation) and n represents the samples analyzed for statistical differences. All statistical comparisons were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA). Student's t‐test was performed in comparing the differences between two samples. Variable comparisons among three and more groups were performed using either one‐way ANOVA with Dunnett's post hoc test or two‐way ANOVA with Tukey's or Sidak's multiple comparisons test. All statistical results are summarized in Table S3.
3. RESULTS
3.1. Tomosyn is expressed at pre and postsynaptic areas
Protein expression of tomosyn in developing mouse brains was first determined using Western blot analysis. The expression of tomosyn in the hippocampus was readily detectable from P0 and increased temporally during development (Figure 1a). Tomosyn levels reached a fourfold elevation by P14, suggesting an increasing role during neuronal maturation. Further examination in the whole brain, however, did not show significant differences at different developmental time points (Figure S1a). Spatial analysis at P14 revealed a similar expression profile of tomosyn in different brain regions (Figure S1b). In cultured cortical neurons, tomosyn also showed a gradual increase of expression with PSD‐95, although not to the same degree, suggesting a role in synaptogenesis (Figure S1c). To determine the subcellular localization of tomosyn, we first performed immunocytochemistry on cultured hippocampal neurons. Endogenous tomosyn was shown to localize in both the soma and in MAP2‐positive dendrites (Figure 1b). Overexpressed tomosyn‐GFP also localized to dendritic compartments, including dendritic spines (Figure 1c). The expression level of tomosyn in dendritic spines was similar to that in dendritic shafts (Figure 1d). Furthermore, partial tomosyn colocalized with PSD‐95 (Figure 1e). In addition, synaptic fractionation of mouse forebrain was performed (Figure 1f). Tomosyn was enriched in presynaptic compartments including syntaxin‐1‐rich SPM and synaptic vesicles (S3). However, tomosyn was also detected in the PSD, indicated by the strong presence of PSD‐95. In addition, immunofluorescence labeling of cultured hippocampal neurons revealed tomosyn expression in both CaMKIIα‐positive excitatory neurons and GAD67‐positive inhibitory neurons (Figure S1d). These data establish the spatiotemporal expression pattern of tomosyn in the brain and suggest a potential postsynaptic role in dendritic development.
FIGURE 1.
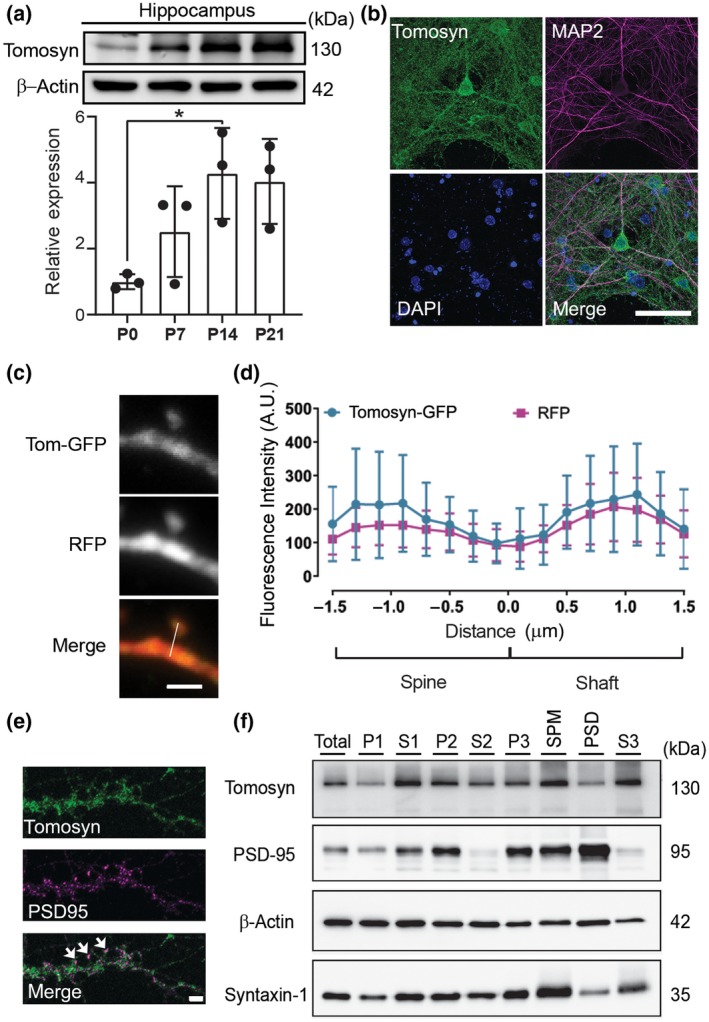
Tomosyn is localized in both pre and postsynaptic compartments. (a) Western blot analysis shows the developmental expression pattern of tomosyn protein in the hippocampus at P0, P7, P14, and P21; n = 3. (b) Confocal immunofluorescence images showing tomosyn (green), MAP2 (red), and DAPI (blue) from a cultured hippocampal neuron at 15 DIV. Scale bar, 50 μm. (c) Confocal images of a dendritic spine of a cultured neuron transfected with RFP and tomosyn‐GFP. Scale bar, 2 µm. (d) Fluorescence intensity of tomosyn‐GFP and RFP in the dendritic spine versus the dendritic shaft was quantified by line scan (shown in c); n = 29. (e) Confocal images of a dendritic segment of a cultured neuron immunostained with tomosyn and PSD‐95 antibodies. Arrows indicate the colocalization of tomosyn and PSD‐95. Scale bar, 2 μm. (f) Synaptic fractionation of forebrain lysates (total) shows the subcellular localization of tomosyn in P21 mouse brains. P1, nuclei; S1, cytosol/membranes; P2, crude synaptosomes; S2, cytosol/light membranes; P3, synaptosomes; SPM, synaptic plasma membranes; PSD, postsynaptic density; S3, synaptic vesicles. Brightness and contrast were adjusted in b and e for better visualization
3.2. Knockdown of tomosyn decreases dendritic complexity and dendritic spine density
To examine the effect of tomosyn on neuronal morphology, a shRNA expression system was used to knock down tomosyn in cultured hippocampal neurons. The knockdown efficiency of two shRNAs targeting tomosyn, shRNA482 and shRNA1083, was first determined in N2a cells. shRNA482 and shRNA1083 resulted in 87.9% and 79.3% reduction, respectively, in the amount of overexpressing GFP‐tagged tomosyn protein compared to scrambled shRNA (Figure S2a). shRNA482, which most effectively decreased tomosyn expression, was chosen for all subsequent experiments and is referred to as shTomosyn hereafter. To further evaluate the knockdown specificity of shTomosyn, a shRNA‐resistant form of tomosyn was engineered as Tomr‐GFP (tomosyn rescue). Co‐expressing Tomr‐GFP with shTomosyn effectively restored the protein level of tomosyn back to control levels (Figure S2b). shTomosyn was then used to knock down tomosyn expression in cultured hippocampal neurons, and their dendritic arborization was examined. Reconstructions of dendritic trees (Figure 2a) and Sholl analysis (Figure 2b) showed that the overall size of the dendritic arbors was smaller in tomosyn knockdown neurons. Co‐expression of Tomr‐GFP restored dendritic complexity in knockdown neurons. Further quantification showed that the branch number (Figure 2c; scramble: 26.19 ± 7.521, shTomosyn: 17.68 ± 4.930, Tomosyn rescue: 23.93 ± 5.434) and total dendrite length (Figure 2d; scramble: 1,613 ± 379.2, shTomosyn: 844.7 ± 231.0, Tomosyn rescue: 1,317 ± 292.0 µm) were significantly restored by Tomr‐GFP expression. Knockdown of tomosyn also resulted in overall decreased spine density at different branches (Primary—scramble: 0.46 ± 0.092, shTomosyn: 0.34 ± 0.078 µm−1; Secondary—scramble: 0.41 ± 0.12, shTomosyn: 0.25 ± 0.075 µm−1; Tertiary—0.33 ± 0.064, shTomosyn: 0.22 ± 0.056 µm−1), while re‐expressing Tomr‐GFP (Primary—0.41 ± 0.082, Secondary—0.38 ± 0.085, Tertiary—0.31 ± 0.07 µm−1) successfully rescued dendritic spine loss (Figure 2e,f). Since the secondary branches showed the most significant differences in the knockdown condition, spine density on the secondary branches was selectively examined in the following experiments. Together, these data suggest that tomosyn is required for dendritic arborization and dendritic spine formation.
FIGURE 2.
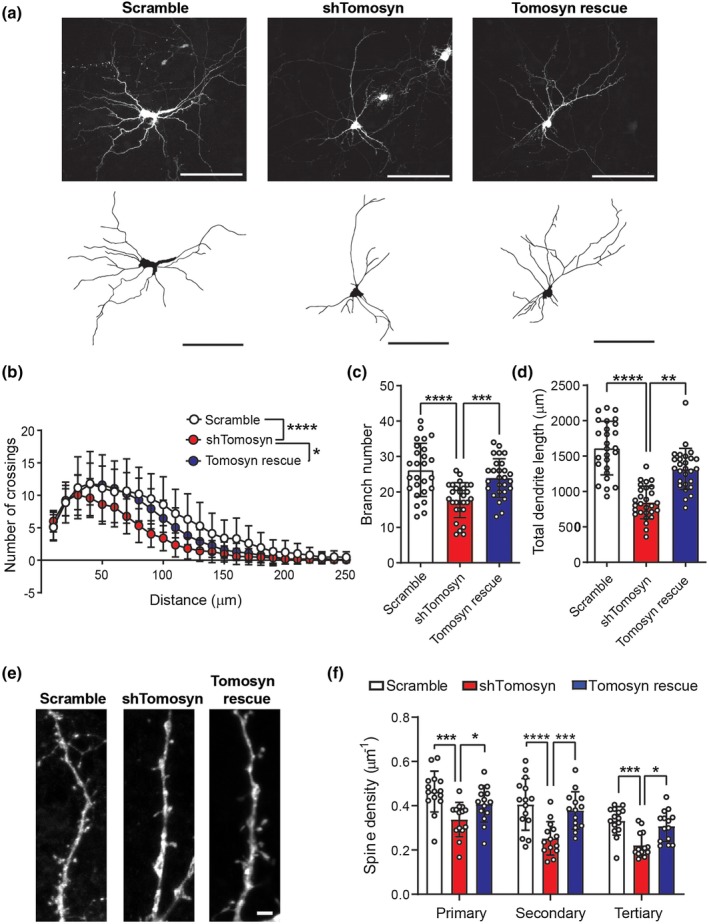
Knockdown of tomosyn reduces dendritic complexity and dendritic spine density. (a) Representative images and reconstructed dendritic trees from mouse hippocampal neurons transfected with scrambled shRNA + GFP or shTomosyn + GFP or shTomosyn + Tomr‐GFP. Scale bar, 100 µm. (b) Sholl analysis, (c) branch number, and (d) total dendrite length of reconstructed neurons. General dendrite complexity was quantified by the area under the Sholl curve. Neurons expressing shTomosyn exhibited compromised dendritic complexity, whereas expression of Tomr‐GFP rescued simplified dendritic complexity in shTomosyn‐expressing neurons compared to scramble controls. *p < 0.05, ****p < 0.0001, one‐way ANOVA with Tukey's test. n = 26 scramble, 28 shTomosyn, and 27 Tomosyn rescue. (e) Confocal images of dendritic spines from neurons transfected with scrambled shRNA + GFP, shTomosyn + GFP, or shTomosyn + Tomr‐GFP for 72 hr. Scale bar, 2 µm. (f) Mean spine density was decreased in tomosyn knockdown neurons at primary, secondary, and tertiary dendrites. Spine density was restored in Tomr‐GFP rescue and shTomosyn co‐expressing neurons compared to shTomosyn + GFP. *p < 0.05, ***p < 0.001, ****p < 0.0001 by two‐way ANOVA with Tukey's test multiple comparisons test. n = 15 scramble, 14 shTomosyn, and 15 Tomosyn rescue [Color figure can be viewed at https://www.wileyonlinelibrary.com]
3.3. Tomosyn deficiency leads to reduced frequency of mEPSCs
Because tomosyn deficiency results in abnormal dendritic arborization and spinogenesis, we asked whether tomosyn plays a role in maintaining basal excitatory synaptic transmission. To answer this question, the effect of tomosyn knockdown on AMPA receptor (AMPAR)‐mediated mEPSCs in cultured hippocampal neurons was examined. mEPSC frequency was significantly reduced in tomosyn‐knockdown cells compared to that in controls (Figure 3a,c, scramble: 5.187 ± 3.699, shTomosyn: 1.461 ± 1.669 Hz). Charge (Figure 3e, scramble: 42.41 ± 16.94, shTomosyn: 32.89 ± 10.22 fC), but not amplitude (Figure 3b,d, scramble: 16.36 ± 6.887, shTomosyn: 14.55 ± 4.628 pA), was decreased in tomosyn knockdown neurons compared to scrambled shRNA‐transfected neurons. Analysis of mEPSC kinetics revealed that the decay time constant was significantly smaller in tomosyn knockdown neurons than in the scramble controls (Figure 3f, scramble: 3.71 ± 1.202, shTomosyn: 2.81 ± 0.972 ms). These data, showing a reduction of mEPSC frequency, support the spine loss phenotype observed in tomosyn knockdown neurons, while the attenuation in the decay time constant may indicate a change in AMPAR subunit composition at the synapse in shTomosyn neurons.
FIGURE 3.
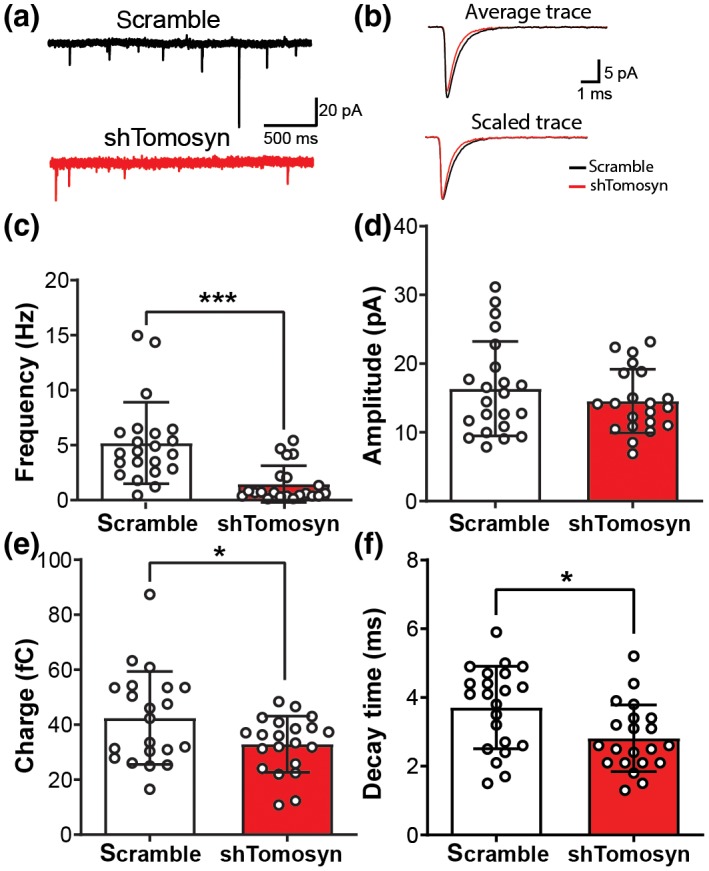
Frequency of miniature EPSCs is decreased in tomosyn knockdown neurons. Cultured hippocampal neurons were transfected with either scrambled shRNA or shTomosyn. (a) An example of current traces collected from whole‐cell voltage‐clamp of hippocampal neurons. (b) Examples of average (top) and scaled (bottom) mEPSC traces from scrambled shRNA and shTomosyn transfected neurons. Summary graphs show (c) frequency, (d) amplitude, (e) charge, and (f) decay time constant of recorded mEPSCs. *p < 0.05, ****p < 0.0001, Student's t‐test. n = 22 scramble and 21 shTomosyn [Color figure can be viewed at https://www.wileyonlinelibrary.com]
3.4. Tomosyn knockdown leads to elevated RhoA GTPase activity
Decreased dendritic arborization and destabilization of dendritic spines on pyramidal neurons have previously been linked to the increased activity of RhoA GTPase (Nakayama et al., 2000). Thus, we investigated whether the simplification of dendritic arbors and loss of dendritic spines observed in neurons with tomosyn knockdown were the result of elevated RhoA activity. Hippocampal neurons were co‐transfected with either scrambled shRNA or shTomosyn, as well as an intramolecular FRET RhoA biosensor, which contains the CFP (donor)/YFP (acceptor) FRET pair flanked by FL RhoA on one side, and the Rho‐binding domain of the effector rhotekin (RBD) on the other side (Pertz et al., 2006). Upon activation of RhoA, the RBD element associates with RhoA‐GTP, thereby causing the biosensor molecule to change its conformation into a FRET‐favorable state. Hence, RhoA activity can be monitored by measuring the FRET occurring between the CFP and YFP fluorophores (Aoki & Matsuda, 2009; Pertz et al., 2006). FRET efficiency was recorded by two well‐established techniques: sensitized emission (Figure 4a,b) and acceptor photobleaching (Figure S3). Whole‐cell RhoA biosensor FRET signal recorded by sensitized emission revealed that knockdown of tomosyn resulted in a significantly higher FRET ratio compared to scramble controls (Figure 4a,b, scramble: 1.17 ± 0.206, shTomosyn: 1.50 ± 0.116). Similarly, acceptor (YFP) photobleaching FRET (pbFRET) performed at randomly selected regions along apical and basal dendrites showed that overall FRET was greater in tomosyn knockdown neurons compared to scramble controls (apical—scramble: 14.27 ± 5.119, shTomosyn: 26.26 ± 5.016%; basal—scramble: 15.76 ± 4.248, shTomosyn: 23.35 ± 6.881%). However, within each sample group (scramble and shTomosyn) no significant differences in EFRET were observed between the specified dendritic compartments (Figure S3c). Collectively, these data show that RhoA activity is increased in tomosyn knockdown neurons, indicating that tomosyn might play a role in the maintenance of dendritic morphology by inhibiting the activity of RhoA GTPase.
FIGURE 4.
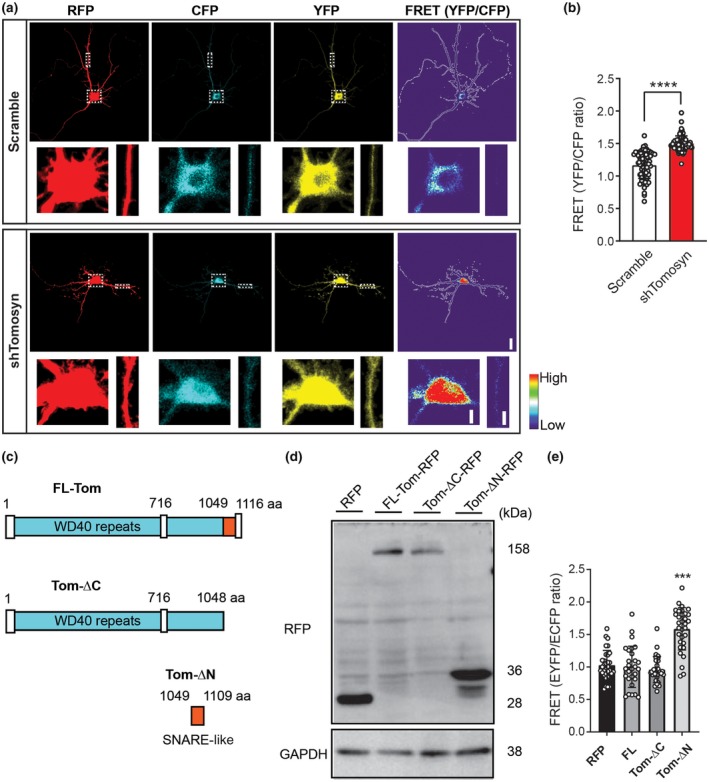
Tomosyn knockdown results in increased RhoA activity in hippocampal neurons. Hippocampal neurons were co‐transfected with a RhoA biosensor and either scrambled shRNA or shTomosyn. (a) Representative images show the neurons in the intensity‐modulated display mode. Higher magnification of the soma and a dendritic segment in dashed box areas are enlarged for better visualization. Scale bar, 20 and 5 µm. (b) FRET efficiency was determined as a ratio of YFP/CFP. ****p < 0.0001, unpaired Student's t‐test. n = 72 scramble and 69 shTomosyn. (c) Schematic structure of domain mutant tomosyn. FL‐Tom, full‐length mouse tomosyn (1‐1166 aa); Tom‐ΔC, containing an N‐terminal domain with WD40 repeats (1‐1048 aa); Tom‐ΔN, containing a SNARE coil‐coiled domain (1049‐1109 aa). (d) Western blot shows the expression of domain mutant tomosyn (Tom‐ΔC‐RFP and Tom‐ΔN‐RFP), FL‐Tom‐RFP, and RFP control in N2a cells. (e) Summary graph showing FRET efficiency in domain mutant tomosyn‐expressing neurons. Neurons with Tom‐ΔN‐RFP showed increased FRET efficiency. ***p < 0.001 compared to RFP, FL‐tomosyn, and ΔC‐tomosyn, one‐way ANOVA with Dunnett's multiple comparisons test. n = 32 RFP, 28 FL‐Tom‐RFP, 29 Tom‐ΔC‐RFP, and 32 Tom‐ΔN‐RFP
To further investigate which part of tomosyn is essential for inhibiting RhoA activity, two domain mutants were constructed: Tom‐ΔC‐RFP, which contains the N‐terminal WD40 domain, and Tom‐ΔN‐RFP, which contains only the R‐SNARE domain of tomosyn (Figure 4c). Western blot analysis showed that both Tom‐ΔC‐RFP and Tom‐ΔN‐RFP migrated as shorter versions of tomosyn at the predicted size (Figure 4d). FRET analysis showed that RhoA activity in neurons expressing Tom‐ΔC‐RFP is comparable to that of RFP control and FL tomosyn. However, EFRET was significantly increased in neurons expressing Tom‐ΔN‐RFP (Figure 4e, RFP: 1.03 ± 0.228, FL: 1.00 ± 0.318, ΔC: 0.96 ± 0.201, ΔN: 1.59 ± 0.325), suggesting the lack of the WD40 domain of tomosyn is sufficient to increase RhoA activity. Taken together, these data suggest that tomosyn likely functions as a negative regulator for RhoA activity via the WD40 domain in neurons.
3.5. Inhibition of RhoA signaling restores dendritic arborization and spine density in tomosyn‐deficient neurons
To determine whether inhibiting RhoA activity is sufficient to rescue the aberrant dendritic phenotypes in tomosyn knockdown neurons, dominant‐negative RhoA (T19N‐RhoA) was co‐transfected in scramble control‐ and shTomosyn‐expressing neurons (Figure 5). In agreement with previous findings, WT‐RhoA or T19N‐RhoA did not affect dendritic branching and spinogenesis in scramble control neurons due to the overall inactive RhoA state (Nakayama et al., 2000; Figure 5c–e). As shown earlier, tomosyn knockdown neurons exhibited less dendritic complexity measured by total dendrite length (scramble: 1,092.62 ± 341.772 µm, shTomosyn: 742.84 ± 305.527 µm), branch number (scramble: 23.41 ± 8.049, shTomosyn: 16.19 ± 6.333), and spine density (scramble: 0.46 ± 0.118, shTomosyn: 0.34 ± 0.093 µm−1). When T19N‐RhoA was co‐expressed, tomosyn knockdown neurons showed similar total dendrite length (scramble: 1,163.67 ± 232.799, shTomosyn: 1,067.38 ± 295.038 µm), branch number (scramble: 21.53 ± 5.690, shTomosyn: 20.00 ± 6.485) and spine density (scramble: 0.47 ± 0.141, shTomosyn: 0.53 ± 0.117 µm−1) compared to control neurons (Figure 5c–e). In contrast, overexpression of WT‐RhoA did not affect the compromised dendritic morphology of tomosyn knockdown neurons (total dendrite length—scramble: 1,169.43 ± 383.284, shTomosyn: 774.95 ± 288.905 µm; branch number—scramble: 20.74 ± 6.464, shTomosyn: 15.29 ± 4.396; spine density—scramble: 0.48 ± 0.113, shTomosyn: 0.34 ± 0.107 µm−1; Figure 5c–e). Treating cultured neurons with C3T, a RhoA inhibitor, also fully rescued the dendritic phenotypes in tomosyn knockdown neurons (Figure S4; total dendrite length—scramble: 938.83 ± 235.331, shTomosyn: 901.65 ± 282.320 µm; branch number—scramble: 19.69 ± 6.260, shTomosyn: 19.53 ± 7.238; spine density—scramble: 0.53 ± 0.151, shTomosyn: 0.52 ± 0.128 µm−1). These data further suggest that tomosyn regulates dendritic arborization and the stability of dendritic spines through RhoA inhibition.
FIGURE 5.
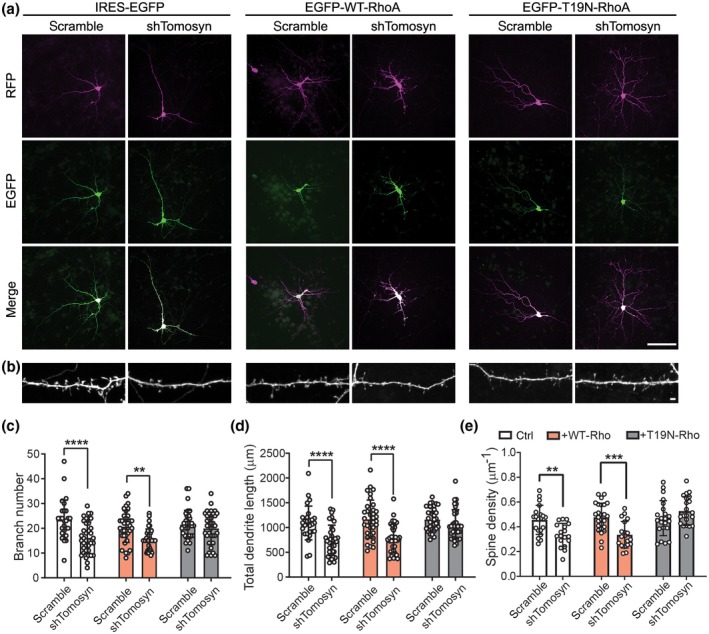
Inhibition of the Rho signaling pathway restores altered dendritic structures in tomosyn knockdown neurons. Representative images show (a) dendritic morphology at DIV7 and (b) spine morphology at DIV15. Transfected neurons were co‐expressed by scramble shRNA or IRES‐EGFP together with IRES‐EGFP (left), EGFP‐WT‐RhoA (middle), and EGFP‐T19N‐RhoA (right). Scale bar, 100 µm in a and 2 µm in b. Quantification of (c) branch number, (d) total dendrite length, and (e) spine density of neurons co‐transfected neurons. Co‐expression of T19N‐RhoA resulted in similar levels of dendritic complexity and spine density between scramble control‐ and shTomosyn‐expressing neurons. **p < 0.01, ***p < 0.001, ****p < 0.0001, two‐way ANOVA with Sidak's multiple comparisons test. n = 27 scramble + ctrl, 35 scramble + WT‐RhoA, 34 scramble + T19N‐RhoA, 37 shTomosyn + ctrl, 35 shTomosyn + WT‐RhoA, and 36 shTomosyn + T19N‐RhoA in c and d; n = 20 scramble + ctrl, 26 scramble + WT‐RhoA, 22 scramble + T19N‐RhoA, 18 shTomosyn + ctrl, 19 shTomosyn + WT‐RhoA, 21 shTomosyn + T19N‐RhoA in e
3.6. Knockdown of tomosyn leads to altered surface‐expressed glutamate receptors
Altered mEPSC kinetics in tomosyn knockdown neurons suggested that the composition of AMPA receptors may be affected (Figure 3). Previous studies have shown that in addition to syntaxin‐1, tomosyn can form a complex with syntaxin‐4 (Madera‐Salcedo et al., 2018; Widberg, Bryant, Girotti, Rea, & James, 2003; Zhu et al., 2014), a postsynaptic t‐SNARE that regulates AMPAR trafficking during neuronal plasticity (Kennedy, Davison, Robinson, & Ehlers, 2010). Indeed, co‐immunoprecipitation of endogenous tomosyn and syntaxin‐4 in cultured cortical neurons showed that these two proteins interact with each other in neurons (Figure 6a). We questioned whether tomosyn could regulate AMPAR trafficking by affecting SNARE formation. We first overexpressed and knocked down tomosyn and then measured the surface expression of AMPA receptors by quantifying the fluorescence of pHluorin‐GluR1 on the surface of dendritic spines. Overexpression or knockdown of tomosyn did not show any difference of the GluR1 surface expression on dendritic spines (Figure 6b; OE: 0.77 ± 0.169, scramble: 0.69 ± 0.201, shTomosyn: 0.62 ± 0.204). However, when we measured the whole dendritic segment, knocking down tomosyn significantly reduced the surface expression of GluR1 (Figure 6c,d; OE: 0.63 ± 0.123, scramble: 0.56 ± 0.211, shTomosyn: 0.41 ± 0.131). This prompted us to hypothesize that the reduced surface expression of AMPA receptors was due to the loss of dendritic spines, where most AMPA receptors are inserted. As previously shown, we could rescue the spine loss in tomosyn knockdown neurons with C3T (Figure S4e). C3T treatment significantly restored the surface expression of GluR1 on dendritic segments in tomosyn knockdown neurons back to the control levels (Figure 6c,d; shTomosyn + C3T: 0.62 ± 0.199). These data suggested that tomosyn likely regulates the surface expression of AMPA receptors through the regulation of RhoA signaling pathway rather than affecting SNARE machinery.
FIGURE 6.
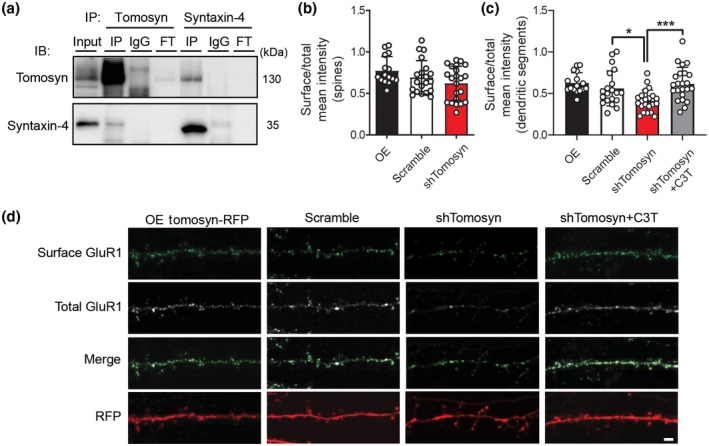
Surface expression of GluR1 subunits is reduced in tomosyn knockdown neurons. (a) Western blot analysis of co‐immunoprecipitation experiments in cultured cortical neurons. Cultured cortical neuron lysates at 15 DIV were immunoprecipitated with either anti‐tomosyn antibody, rabbit IgG control, anti‐syntaxin‐4 antibody, or mouse IgG control. Immunoblot was probed with tomosyn and syntaxin‐4 antibodies. FT = flow through. Quantification of the surface expression of pHluorin‐GluR1 in (b) dendritic spines and (c) dendritic segments. Cultured neurons were co‐transfected with either Tomosyn‐RFP (OE), scrambled shRNA, or shTomosyn and pHluorin‐GluR1. Tomosyn knockdown neurons were treated with the RhoA inhibitor, C3T. *p < 0.05, ***p < 0.001, one‐way ANOVA with Dunnett's multiple comparisons test. n = 16 OE, 20 scramble, 22 shTomosyn, and 23 shTomosyn + C3T. (d) Representative confocal images showing surface staining for pHluorin‐GluR1 in cultured hippocampal neurons at 15 DIV. Scale bar, 2 µm
3.7. Autism‐associated tomosyn variants fail to restore dendritic complexity and the surface expression of GluR1
We have shown that the N‐terminal WD40 domain of tomosyn likely inhibits RhoA activity (Figure 4e). Two tomosyn variants harboring missense mutations (L412V and Y502C) in the WD40 domain have been identified in individuals with autism (Cukier et al., 2014; Figure 7a). To evaluate potential alterations from these two proteins, shRNA‐resistant tomosyn variants, L412Vr‐Tom‐GFP and Y502Cr‐Tom‐GFP, were first engineered. When co‐expressed with shTomosyn, WTr‐Tom‐GFP, L412Vr‐Tom‐GFP, and Y502Cr‐Tom‐GFP all effectively rescued the protein level of tomosyn (Figure S5a). RhoA activity was then determined using FRET biosensor analysis. Consistent with the previous experiment, tomosyn knockdown neurons exhibited higher FRET ratio (1.43 ± 0.278) compared to control neurons (0.98 ± 0.133). A significant increase in FRET signal ratio was detected in neurons co‐expressing the RFP‐tagged and shTomosyn‐resistant L412Vr‐Tom‐RFP (1.26 ± 0.354) or Y502Cr‐Tom‐RFP (1.27 ± 0.268) variant compared to those expressing WTr‐Tom‐RFP (0.98 ± 0.158; Figure S5b). Neuronal morphology was further examined to determine the contributions of the shRNA‐resistant tomosyn variants, L412Vr‐Tom‐GFP and Y502Cr‐Tom‐GFP, in tomosyn knockdown neurons. As previously shown, knockdown of tomosyn caused a reduction in dendritic arbor total length (scramble: 1,348.6 ± 574.3, shTomosyn: 771.4 ± 270.5 µm) and branch number (scramble: 24.33 ± 8.640, shTomosyn: 17.10 ± 7.222) as measured by Sholl analysis in comparison with scramble controls (Figure 7b–e). WTr‐Tom‐GFP rescued the decrease in total dendrite length (1,252 ± 306.5 µm) and branch number (22.85 ± 6.136), whereas cells co‐expressing Y502Cr‐Tom‐GFP still showed simplified dendritic morphology with reduced branching (18.53 ± 7.615) and dendrite length (865.0 ± 290.9 µm) compared to scramble controls. Co‐expression of L412Vr‐Tom‐GFP, however, partially rescued branch number (21.38 ± 7.540) with no significant difference compared to the scramble or shTomosyn conditions, but did not rescue total dendrite length in tomosyn knockdown neurons (960.7 ± 299.2 µm). Similarly, a decrease in dendritic spine density was observed as previously shown in tomosyn knockdown neurons (0.37 ± 0.095 µm−1) compared to control neurons (0.61 ± 0.097 µm−1; Figure 7f,g). Co‐expression of WTr‐Tom‐GFP (0.54 ± 0.110 µm−1) rescued spine loss compared to tomosyn knockdown neurons. Co‐expression of Y502Cr‐Tom‐GFP (0.47 ± 0.130 µm−1) also showed partial spine rescue when compared to the knockdown condition. However, co‐expression of L412Vr‐Tom‐GFP (0.38 ± 0.105 µm−1) or Y502Cr‐Tom‐GFP both failed to rescue the spine loss to the control levels. Similarly, the reduced surface expression of GluR1 was restored by WTr‐Tom‐GFP, but neither by L412Vr‐Tom‐GFP nor by Y502Cr‐Tom‐GFP (Figures 7h and S5c; scramble: 0.61 ± 0.273, shTomosyn: 0.48 ± 0.155, WT: 0.66 ± 0.227, L412V: 0.48 ± 0.103, Y502C: 0.56 ± 0.136). Collectively, our findings support a model wherein tomosyn or ASD‐associated tomosyn variants affect dendritic arborization, spinogenesis, and AMPA receptor trafficking via the RhoA signaling pathway, leading to alterations in neuronal activity (Figure 8).
FIGURE 7.
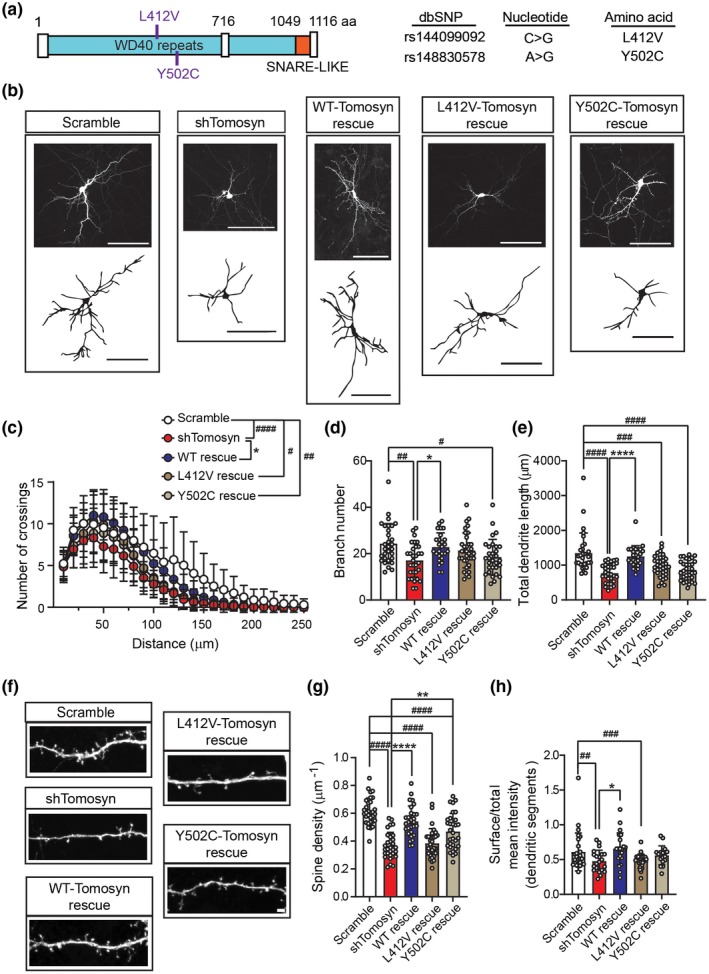
Autism‐associated mutant tomosyn fails to restore dendritic arborization and spine loss in tomosyn knockdown neurons. (a) An illustration of the location of L412V and Y502C mutations in tomosyn. (b) Representative images of reconstructed dendritic trees and (c) Sholl analysis, (d) branch number, and (e) total dendrite length from mouse hippocampal neurons transfected with scrambled shRNA + GFP, shTomosyn + GFP, shTomosyn + WTr‐Tom‐GFP, shTomosyn + L412Vr‐Tom‐GFP, or shTomosyn + Y502Cr‐Tom‐GFP. Scale bar, 100 µm. Both L412V and Y502C mutant tomosyn failed to rescue total dendrite length compared to scrambled shRNA. Only Y502C failed to rescue branch number compared to scramble. # p < 0.05, ## p < 0.01, ### p < 0.001, #### p < 0.0001 when compared to scramble; *p < 0.05, **p < 0.01, ****p < 0.0001 when compared to shTomosyn, one‐way ANOVA with Tukey multiple comparisons test. n = 30 scramble, 31 shTomosyn, 27 WT rescue, 33 L412V rescue, and 31 Y502C rescue. (f) Representative confocal images and (g) the quantification of spine density in cultured hippocampal neurons transfected with different tomosyn constructs as described in b, c, d, and e. Scale bar, 2 µm. #### p < 0.0001 when compared to scramble; ****p < 0.0001 when compared to shTomosyn, one‐way ANOVA with Dunnett's multiple comparison tests. n = 31 scramble, 30 shTomosyn, 29 WT rescue, 31 L412V rescue, and 31 Y502C rescue. (h) A reduction of GluR1 surface expression in dendritic segments in neurons co‐expressing shTomosyn‐L412Vr‐Tom‐RFP, compared to shTomosyn‐WTr‐Tom‐RFP, was observed. **p < 0.01 when compared to shTomosyn; ## p < 0.01, ### p < 0.001 when compared to shTomosyn‐L412Vr‐Tom‐RFP by one‐way ANOVA with Tukey's multiple comparisons test. n = 28 scramble, 21 shTomosyn, 22 WT rescue, 24 L412V rescue, 20 Y502C rescue
FIGURE 8.
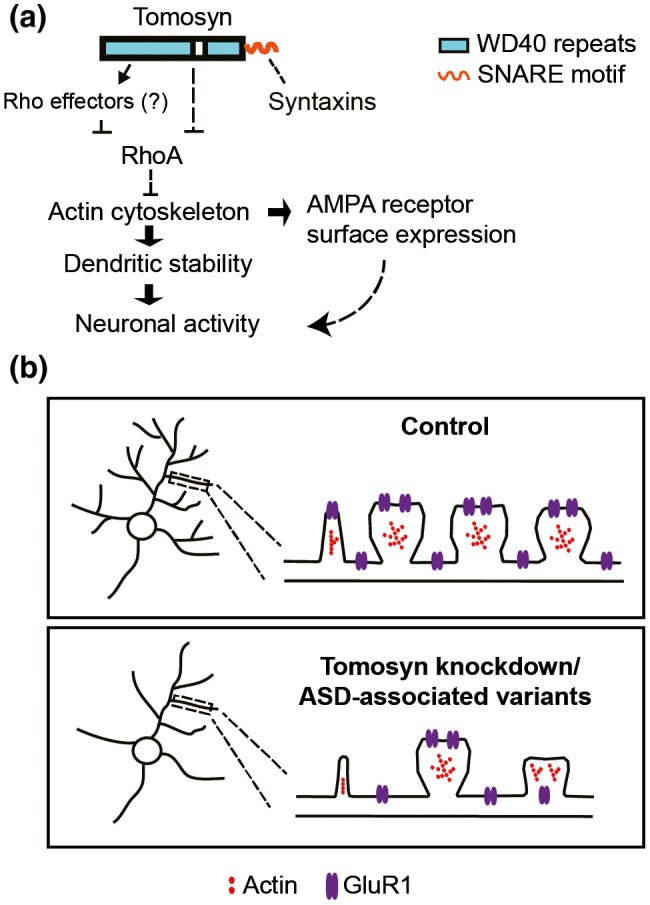
Working model of tomosyn function in regulating dendritic stability. (a) Mechanistic regulation of tomosyn in the maintenance of dendritic stability and AMPA receptor trafficking. Tomosyn regulates dendritic stability and AMPA receptor trafficking via inhibition of the RhoA signaling pathway. (b) Schematic model of spine loss, GluR1 trafficking impairment in tomosyn knockdown or autism‐associated variants rescued neurons [Color figure can be viewed at https://www.wileyonlinelibrary.com]
4. DISCUSSION
The role of tomosyn in regulating neurotransmitter release and exocytosis has been extensively studied (Ashery, Bielopolski, Barak, & Yizhar, 2009; Batten et al., 2017; Ye et al., 2014; Zhu et al., 2014). Because tomosyn is also distributed throughout dendrites (Barak et al., 2010; Fujita et al., 1998; Madera‐Salcedo et al., 2018), an undiscovered role in postsynaptic function is likely present. Here, we described the presence and function of tomosyn in postsynaptic areas. We have shown that tomosyn is a molecular link between neuronal stability and transmission, which acts as an inhibitory regulator of RhoA GTPase and plays a positive role in AMPA receptor trafficking in neurons. Autism‐associated tomosyn variants L412V and Y502C showed a loss‐of‐function effect, resulting in a failed rescue of spine loss and simplified dendritic arborization accompanied by decreased GluR1 surface expression in tomosyn knockdown neurons.
Using biochemical approaches, we revealed that tomosyn is widely expressed in multiple brain regions, including the cerebral cortex, hippocampus, thalamus, striatum, and cerebellum, as well as diverse neuronal types, including CaMKIIα‐positive excitatory neurons and GAD67‐positive inhibitory neurons. Consistent with previous studies examining mRNA expression, the protein level of tomosyn is developmentally regulated and peaks at P14 in the mouse brain, which is equivalent to early childhood in humans (Groffen, Jacobsen, Schut, & Verhage, 2005; Li et al., 2017). These findings suggest that tomosyn may play an important role in brain development. Our further subcellular examinations demonstrated that tomosyn is detected in both PSD fractions and in presynaptic compartments. Moreover, this study, leveraging the sparsely transfected neurons that receive unchanged presynaptic inputs, has revealed the postsynaptic role of tomosyn in neuronal morphogenesis and activity. The changes in neuronal morphology and neurotransmission upon tomosyn knockdown also showed that tomosyn exhibits a cell‐autonomous effect on dendritic structures and synaptic activity. Our findings demonstrate the detailed expression profile of tomosyn in the developing brain and indicate an essential role for tomosyn in the maintenance of dendritic arborization, spine density, and synaptic strength.
Previous studies have indicated that tomosyn, containing WD40 repeats, shows significant sequence similarity to lethal (2) giant larvae (L2gl) and yeast protein Sro7p (Lehman, Rossi, Adamo, & Brennwald, 1999; Mechler, McGinnis, & Gehring, 1985). The WD40 domain is a scaffolding structure involved in small GTPase‐mediated protein–protein interaction (Schapira, Tyers, Torrent, & Arrowsmith, 2017). Interaction of Sro7p and Sec4p Rab GTPase is implicated in Rab GTPase‐dependent vesicle clustering and tethering (Rossi, Watson, Demonch, Temple, & Brennwald, 2015; Rossi, Watson, Kennedy, & Brennwald, 2018). Sro7p has also been reported to disrupt cell polarity via suppression of a small GTPase Rho3p (Kagami, Toh‐e, & Matsui, 1998). Similar to the yeast homolog, tomosyn has recently been shown to interact with the GTP‐bound state of Rab3A to partition synaptic vesicle pools (Cazares et al., 2016). Together, this evidence suggests that tomosyn may interact with the small Rho GTPase RhoA via the WD40 domain. In the current study, FRET analysis revealed an increase in RhoA activity in tomosyn knockdown neurons. Domain analysis further indicated that the N terminus of tomosyn with its WD40 repeats is likely responsible for mediating this RhoA inhibition. However, more sophisticated biochemical studies are required for further understanding of this inter‐molecular regulation.
The dendritic SNARE fusion machinery has been implicated in the regulation of constitutive and activity‐dependent glutamate receptor trafficking. Syntaxin‐4 has previously been shown to regulate AMPAR trafficking in LTP, and surface expression of GluR2 and GluN1 in basal neurotransmission (Bin et al., 2018; Kennedy et al., 2010). Synaptosomal‐associated proteins (SNAPs) are members of the plasma membrane‐associated SNARE family required for glutamate receptor insertion. SNAP‐25, heavily studied for its role in neurotransmitter release, has recently been shown to be involved in AMPAR insertion via the formation of SNAP‐25‐syntaxin1A/B‐VAMP2 complexes (Gu et al., 2016). Tomosyn was first identified as a syntaxin‐1‐binding protein that negatively regulated neurotransmitter release and exocytosis via interaction with SNAP‐25 and syntaxin‐1 (Hatsuzawa, Lang, Fasshauer, Bruns, & Jahn, 2003; Sakisaka et al., 2008), while later experiments in nonneuronal cells also showed that tomosyn regulates mast cell degranulation by switching interaction between syntaxin‐4 and syntaxin‐3 (Madera‐Salcedo et al., 2018). This evidence suggests that tomosyn may play a role in glutamate receptor trafficking in hippocampal neurons via interaction with syntaxins and SNAPs. However, we found decreased GluR1 surface expression on dendritic segments but not dendritic spines in tomosyn knockdown neurons. Therefore, the reduced surface GluR1 expression in tomosyn knockdown neurons may simply result from a decreased number of dendritic spines. A stable cytoskeletal structure is tightly modulated by depolymerization and polymerization of actin, providing scaffolds to facilitate AMPAR trafficking along actin filaments (Allison, Gelfand, Spector, & Craig, 1998; Kim & Lisman, 1999; Zhou, Xiao, & Nicoll, 2001). The increased RhoA activity in tomosyn knockdown neurons likely collapses the actin cytoskeleton, thereby disrupting the structure that supports AMPAR trafficking. Therefore, C3T treatment restores spine stability and consequently rescues surface expression of AMPA receptors. Our data suggest that the reduced surface expression of AMPA receptors is likely secondary to the increase of RhoA activity in tomosyn knockdown neurons.
Although STXBP5 is not a prominent autism‐risk gene, variants have been found in individuals with ASD (Cukier et al., 2014). In the present study, we examined two variants that carry single nucleotide mutations (L412V and Y502C) on the WD40 domain of tomosyn. ASD often features cellular characteristics involving altered dendritic structures and neuronal activity (Forrest et al., 2018; Moretto, Murru, Martano, Sassone, & Passafaro, 2018). Since the WD40 domain of tomosyn likely mediates regulation of RhoA activity, L412V and Y502C variants may affect the dendritic structures of neurons by interrupting RhoA signaling. FRET analysis confirmed that neither L412V nor Y502C variant sufficiently suppressed the increased RhoA activity in tomosyn knockdown neurons. Both variants failed to completely restore total dendrite length and spine density following tomosyn knockdown and subsequently failed to restore the decreased GluR1 surface expression. All these data suggest that L412V and Y502C mutations result in loss‐of‐function of tomosyn and abrogate normal RhoA signaling. It is important to note that while our strategy of examining tomosyn variants in tomosyn knockdown neurons directly reveals the functional alterations caused by the mutations relative to wild‐type tomosyn, this does not mimic the likely biology of ASD. High genetic heterogeneity is a key feature for ASD, as most individuals suffer from multiple heterozygous genetic mutations. Whether L412V and Y502C variants contribute to pathological phenotypes in a more in vivo‐relevant environment still requires further investigation. Additionally, the potential contribution of sex differences was not able to be examined here due to the nature of the culture system. Nonetheless, the signaling mechanism that we described here may potentially underlie the phenotype of disrupted neuronal structures often observed in ASD brains (Figure 8b).
Taken together, this study uncovers a crucial role of tomosyn in the maintenance of neuronal morphology and neuronal transmission via inhibition of RhoA activity. Our findings provide insights into the function of tomosyn as a regulator for the maintenance of neuronal stability and activity and suggest how this mechanism may be disrupted in ASD.
DECLARATION OF TRANSPARENCY
The authors, reviewers, and editors affirm that in accordance with the policies set by the Journal of Neuroscience Research, this manuscript presents an accurate and transparent account of the study being reported and that all critical details describing the methods and results are present.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interests.
AUTHOR CONTRIBUTIONS
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, W.S. and Y.L.; Methodology, W.S., M.K., M.B., J.F., S.H., and Y.L.; Validation, W.S., M.K., J.F., R.N., and Y.L.; Investigation, W.S., M.K., M.B., J.F., R.N., and Y.L.; Formal Analysis, W.S., M.K., M.B., and Y.L.; Resources, W.S. and M.K.; Writing – Original Draft, W.S. and Y.L.; Writing – Review & Editing, W.S., M.K., M.B., J.F., R.N., S.H., and Y.L.; Visualization, W.S., M.K., and Y.L.; Supervision, S.H. and Y.L.; Project Administration, Y.L.; Funding Acquisition, S.H. and Y.L.
Supporting information
FIGURE S1 Spatial and temporal expression patterns of tomosyn during different developmental stages. (a) Temporal expression pattern of tomosyn in whole brain at P0, P7, P14, and P21; n = 4. (b) Spatial expression of tomosyn in different brain regions at P14. CB, cerebellum; CX, cortex; HC, hippocampus; Th/St, thalamus and striatum; n = 4. (c) Temporal expression of tomosyn and PSD‐95 in cultured cortical neurons at 1, 4, 7, 10, and 14 DIV. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to 1 DIV, one‐way ANOVA with Dunnett’s multiple comparisons test; n = 3. (d) Fluorescence images showing either CaMKIIα or GAD67 and tomosyn from cultured hippocampal neurons at 15 DIV. Scale bar, 20 μm
FIGURE S2 Silencing effect of shRNA on tomosyn and rescuing effect of shRNA‐resistant tomosyn. (a) Western blot analysis shows the knockdown efficiency of shRNAs against tomosyn. Tomosyn‐GFP was co‐transfected with scrambled shRNA, shRNA482, or shRNA1083 in N2a cells. shRNA482 resulted in the most effective reduction of tomosyn protein level and is shown as shTomosyn in subsequent experiments. ****p < 0.0001 compared to scramble control, one‐way ANOVA with Dunnett’s multiple comparisons test; n = 3. (b) Western blot analysis shows the protein level of Tomr‐GFP, a shRNA‐resistant form of tomosyn, in tomosyn‐knockdown N2a cells. While shTomosyn reduced the expression of Tom‐GFP, the protein level of Tomr‐GFP was not affected by shTomosyn knockdown. **p < 0.01 when compared with shTomosyn + Tom‐GFP, one‐way ANOVA with Dunnett’s multiple comparisons test; n = 3
FIGURE S3 Tomosyn knockdown leads to increased RhoA activity in different dendritic areas. (a) Representative images of dendritic segments taken immediately before and after photobleaching of acceptor (YFP). Photobleaching FRET (pbFRET) was used to measure RhoA activity in different dendritic areas. Bleached area is highlighted as a white‐dashed circle (ROI). Scale bar, 2 µm. (b) A representative graph of a pbFRET experiment showing changes in fluorescence intensities within bleach ROI over the course of 10 frames. Photobleaching was performed after the fifth frame. (c) FRET efficiency was determined by calculating the percentage (%) of the increase in donor (CFP) fluorescence intensity after acceptor photobleaching. Hippocampal neurons were co‐transfected with a RhoA Biosensor and either scrambled shRNA or shTomosyn. Tomosyn knockdown neuron showed a higher FRET efficiency. No significant increase in FRET efficiency was found between apical dendrites (a) and basal dendrites (b) within each group. p = 0.92. n = 14 scramble and 17 shTomosyn. ****p < 0.0001 when compared to scramble, two‐way ANOVA with Tukey’s multiple comparisons test
FIGURE S4 Inhibition of the Rho signaling pathway restores altered dendritic structures in tomosyn knockdown neurons. Transfected neurons expressing scramble shRNA or shTomosyn were treated with control (ddH2O) and C3T. (a) Representative images show dendritic morphology at 7 DIV. Scale bar, 100 µm. (b) Representative images show spine morphology at 15 DIV. Scale bar, 2 µm. Tomosyn knockdown neurons treated with C3T rescued the (c) branch number, (d) total dendrite length, and (e) spine density. *p < 0.05, two‐way ANOVA with Sidak’s multiple comparisons test. n = 23 scramble + ctrl, 32 scramble + C3T, 23 shTomosyn + ctrl, and 32 shTomosyn + C3T in c and d. n = 23 scramble + ctrl, 26 scramble + C3T, 25 shTomosyn + ctrl, and 30 shTomosyn + C3T in (e)
FIGURE S5 shRNA‐resistant autism‐associated tomosyn variants result in increased RhoA activity in tomosyn knockdown neurons. (a) Representative Western blot and densitometry graph show protein levels of shRNA‐resistant WTr‐Tom‐GFP, L412Vr‐Tom‐GFP, and Y502Cr‐Tom‐GFP in N2a cells co‐transfected with shTomosyn. **p < 0.01, ***p < 0.001 by one‐way ANOVA with Dunnett’s multiple comparisons test. n = 3. (b) Summary graph showing FRET efficiency in tomosyn knockdown neurons rescued with WTr, L412Vr, or Y502Cr tomosyn constructs. Neurons were co‐transfected with a RhoA biosensor and scrambled shRNA, shTomosyn, shTomosyn‐WTr‐Tom‐RFP, shTomosyn‐L412Vr‐Tom‐RFP, or shTomosyn‐Y502Cr‐Tom‐RFP. ***p < 0.001 compared to scrambled shRNA; # p < 0.05, ### p < 0.001 compared to shTomosyn‐WTr‐Tom‐RFP, one‐way ANOVA with Dunnett’s multiple comparisons test. n = 42 scramble, 39 shTomosyn, 34 WT rescue, 31 L412V rescue, and 42 Y502C rescue. (c) Example confocal images show the surface expression of pHluorin‐GluR1 in neurons expressing scrambled shRNA, shTomosyn, WT‐tomosyn rescue, L412V‐tomosyn rescue, and Y502C‐tomosyn rescue. Scale bar, 2 µm
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
Tables S1‐S3
TABLE S1 Key reagents table
TABLE S2 Primary antibodies
TABLE S3 Statistical results
ACKNOWLEDGMENTS
We thank Drs. Louis DeTolla and Turhan Coksaygan at the University of Maryland School of Medicine for providing veterinary service and consultation. The authors gratefully acknowledge Elizabeth Benevides and Drs. Gene J. Blatt and John P. Hussman for their edits and helpful comments on the manuscript.
Shen W, Kilander MBC, Bridi MS, et al. Tomosyn regulates the small RhoA GTPase to control the dendritic stability of neurons and the surface expression of AMPA receptors. J Neuro Res. 2020;98:1213–1231. 10.1002/jnr.24608
Edited by Nathalie Dehorter. Reviewed by Monika Bijata and Patrick Brennwald.
The peer review history for this article is available at https://publons.com/publon/10.1002/jnr.24608.
Wenjuan Shen and Michaela B. C. Kilander should be considered joint first author.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Allison, D. W. , Gelfand, V. I. , Spector, I. , & Craig, A. M. (1998). Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: Differential attachment of NMDA versus AMPA receptors. Journal of Neuroscience, 18(7), 2423–2436. 10.1523/JNEUROSCI.18-07-02423.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association . (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Arlington, VA: American Psychiatric Publishing: American Psychiatric Press, Inc. [Google Scholar]
- Aoki, K. , & Matsuda, M. (2009). Visualization of small GTPase activity with fluorescence resonance energy transfer‐based biosensors. Nature Protocols, 4, 1623–1631. 10.1038/nprot.2009.175 [DOI] [PubMed] [Google Scholar]
- Ashery, U. , Bielopolski, N. , Barak, B. , & Yizhar, O. (2009). Friends and foes in synaptic transmission: The role of tomosyn in vesicle priming. Trends in Neurosciences, 32(5), 275–282. 10.1016/j.tins.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak, B. , Okun, E. , Ben‐Simon, Y. , Lavi, A. , Shapira, R. , Madar, R. , … Ashery, U. (2013). Neuron‐specific expression of tomosyn1 in the mouse hippocampal dentate gyrus impairs spatial learning and memory. Neuromolecular Medicine, 15(2), 351–363. 10.1007/s12017-013-8223-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak, B. , Williams, A. , Bielopolski, N. , Gottfried, I. , Okun, E. , Brown, M. A. , … Ashery, U. (2010). Tomosyn expression pattern in the mouse hippocampus suggests both presynaptic and postsynaptic functions. Frontiers in Neuroanatomy, 4, 149 10.3389/fnana.2010.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten, S. R. , Matveeva, E. A. , Whiteheart, S. W. , Vanaman, T. C. , Gerhardt, G. A. , & Slevin, J. T. (2017). Linking kindling to increased glutamate release in the dentate gyrus of the hippocampus through the STXBP5/tomosyn‐1 gene. Brain and Behavior, 7(9), e00795 10.1002/brb3.795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Simon, Y. , Rodenas‐Ruano, A. , Alviña, K. , Lam, A. D. , Stuenkel, E. L. , Castillo, P. E. , & Ashery, U. (2015). A combined optogenetic‐knockdown strategy reveals a major role of tomosyn in mossy fiber synaptic plasticity. Cell Reports, 12(3), 396–404. 10.1016/j.celrep.2015.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo, M. K. , Milenkovic, M. , Salahpour, A. , & Ramsey, A. J. (2014). Preparation of synaptic plasma membrane and postsynaptic density proteins using a discontinuous sucrose gradient. Journal of Visualized Experiments (91), e51896 10.3791/51896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin, N.‐R. , Ma, K. E. , Harada, H. , Tien, C.‐W. , Bergin, F. , Sugita, K. , … Sugita, S. (2018). Crucial role of postsynaptic syntaxin 4 in mediating basal neurotransmission and synaptic plasticity in hippocampal CA1 neurons. Cell Reports, 23(10), 2955–2966. 10.1016/j.celrep.2018.05.026 [DOI] [PubMed] [Google Scholar]
- Bridi, M. S. , Park, S. M. , & Huang, S. (2017). Developmental disruption of GABA(A)R‐meditated inhibition in Cntnap2 KO mice. eNeuro, 4(5), ENEURO.0162‐0117. 2017 10.1523/ENEURO.0162-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazares, V. A. , Njus, M. M. , Manly, A. , Saldate, J. J. , Subramani, A. , Ben‐Simon, Y. , … Stuenkel, E. L. (2016). Dynamic partitioning of synaptic vesicle pools by the SNARE‐binding protein tomosyn. Journal of Neuroscience, 36(44), 11208–11222. 10.1523/JNEUROSCI.1297-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour, M. , O'Roak, B. J. , Santini, E. , Samaco, R. C. , Kleiman, R. J. , & Manzini, M. C. (2016). Current perspectives in autism spectrum disorder: From genes to therapy. Journal of Neuroscience, 36(45), 11402–11410. 10.1523/jneurosci.2335-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukier, H. N. , Dueker, N. D. , Slifer, S. H. , Lee, J. M. , Whitehead, P. L. , Lalanne, E. , … Pericak‐Vance, M. A. (2014). Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Molecular Autism, 5(1), 1 10.1186/2040-2392-5-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, L. K. , Meyer, K. J. , Rudd, D. S. , Librant, A. L. , Epping, E. A. , Sheffield, V. C. , & Wassink, T. H. (2009). Novel copy number variants in children with autism and additional developmental anomalies. Journal of Neurodevelopmental Disorders, 1(4), 292–301. 10.1007/s11689-009-9013-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering, G. H. , & Huganir, R. L. (2018). The AMPA receptor code of synaptic plasticity. Neuron, 100(2), 314–329. 10.1016/j.neuron.2018.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, T. A. , Blackman, A. V. , Oyrer, J. , Jayabal, S. , Chung, A. J. , Watt, A. J. , … van Meyel, D. J. (2014). Neuronal morphometry directly from bitmap images. Nature Methods, 11(10), 982–984. 10.1038/nmeth.3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest, M. P. , Parnell, E. , & Penzes, P. (2018). Dendritic structural plasticity and neuropsychiatric disease. Nature Reviews Neuroscience, 19, 215–234. 10.1038/nrn.2018.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, Y. , Shirataki, H. , Sakisaka, T. , Asakura, T. , Ohya, T. , Kotani, H. , … Takai, Y. (1998). Tomosyn: A syntaxin‐1‐binding protein that forms a novel complex in the neurotransmitter release process. Neuron, 20(5), 905–915. [DOI] [PubMed] [Google Scholar]
- Govek, E. E. , Newey, S. E. , Akerman, C. J. , Cross, J. R. , Van der Veken, L. , & Van Aelst, L. (2004). The X‐linked mental retardation protein oligophrenin‐1 is required for dendritic spine morphogenesis. Nature Neuroscience, 7(4), 364–372. 10.1038/nn1210 [DOI] [PubMed] [Google Scholar]
- Groffen, A. J. , Jacobsen, L. , Schut, D. , & Verhage, M. (2005). Two distinct genes drive expression of seven tomosyn isoforms in the mammalian brain, sharing a conserved structure with a unique variable domain. Journal of Neurochemistry, 92(3), 554–568. 10.1111/j.1471-4159.2004.02890.x [DOI] [PubMed] [Google Scholar]
- Gu, Y. , Chiu, S. L. , Liu, B. , Wu, P. H. , Delannoy, M. , Lin, D. T. , … Huganir, R. L. (2016). Differential vesicular sorting of AMPA and GABAA receptors. Proceedings of the National Academy of Sciences of the United States of America, 113(7), E922–E931. 10.1073/pnas.1525726113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson, D. R. , & Blatt, G. J. (2015). Autism spectrum disorders and neuropathology of the cerebellum. Frontiers in Neuroscience, 9, 420 10.3389/fnins.2015.00420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsuzawa, K. , Lang, T. , Fasshauer, D. , Bruns, D. , & Jahn, R. (2003). The R‐SNARE motif of tomosyn forms SNARE core complexes with syntaxin 1 and SNAP‐25 and down‐regulates exocytosis. Journal of Biological Chemistry, 278(33), 31159–31166. 10.1074/jbc.M305500200 [DOI] [PubMed] [Google Scholar]
- Hussman, J. P. , Chung, R.‐H. , Griswold, A. J. , Jaworski, J. M. , Salyakina, D. , Ma, D. , … Pericak‐Vance, M. A. (2011). A noise‐reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Molecular Autism, 2(1), 1 10.1186/2040-2392-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov, I. , O’Roak, B. J. , Sanders, S. J. , Ronemus, M. , Krumm, N. , Levy, D. , … Wigler, M. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515(7526), 216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami, M. , Toh‐e, A. , & Matsui, Y. (1998). Sro7p, a Saccharomyces cerevisiae counterpart of the tumor suppressor l(2)gl protein, is related to myosins in function. Genetics, 149(4), 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, M. J. , Davison, I. G. , Robinson, C. G. , & Ehlers, M. D. (2010). Syntaxin‐4 defines a domain for activity‐dependent exocytosis in dendritic spines. Cell, 141(3), 524–535. 10.1016/j.cell.2010.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, C.‐H. , & Lisman, J. E. (1999). A role of actin filament in synaptic transmission and long‐term potentiation. Journal of Neuroscience, 19(11), 4314–4324. 10.1523/JNEUROSCI.19-11-04314.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec, C. D. , Li, B. , Wei, W. , Boehm, J. , & Malinow, R. (2006). Glutamate receptor exocytosis and spine enlargement during chemically induced long‐term potentiation. Journal of Neuroscience, 26(7), 2000–2009. 10.1523/JNEUROSCI.3918-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, C. G. , & Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nature Reviews Neuroscience, 8, 413 10.1038/nrn2153 [DOI] [PubMed] [Google Scholar]
- Lehman, K. , Rossi, G. , Adamo, J. E. , & Brennwald, P. (1999). Yeast homologues of tomosyn and lethal giant larvae function in exocytosis and are associated with the plasma membrane SNARE, Sec9. Journal of Cell Biology, 146(1), 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Zhang, W. , Yang, H. , Howrigan, D. P. , Wilkinson, B. , Souaiaia, T. , … Coba, M. P. (2017). Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nature Neuroscience, 20(8), 1150–1161. 10.1038/nn.4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. C. , Frei, J. A. , Kilander, M. B. , Shen, W. , & Blatt, G. J. (2016). A subset of autism‐associated genes regulate the structural stability of neurons. Frontiers in Cellular Neuroscience, 10, 263 10.3389/fncel.2016.00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. C. , & Koleske, A. J. (2010). Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annual Review of Neuroscience, 33, 349–378. 10.1146/annurev-neuro-060909-153204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madera‐Salcedo, I. K. , Danelli, L. , Tiwari, N. , Dema, B. , Pacreau, E. , Vibhushan, S. , … Blank, U. (2018). Tomosyn functions as a PKCδ‐regulated fusion clamp in mast cell degranulation. Science Signalling, 11(537), eaan4350 10.1126/scisignal.aan4350 [DOI] [PubMed] [Google Scholar]
- Masuda, E. S. , Huang, B. C. , Fisher, J. M. , Luo, Y. , & Scheller, R. H. (1998). Tomosyn binds t‐SNARE proteins via a VAMP‐like coiled coil. Neuron, 21(3), 479–480. [DOI] [PubMed] [Google Scholar]
- Matsuda, T. , & Cepko, C. L. (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proceedings of the National Academy of Sciences of the United States of America, 101(1), 16–22. 10.1073/pnas.2235688100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami, N. , Hadley, D. , Hensel, C. H. , Christensen, G. B. , Kim, C. , Frackelton, E. , … Hakonarson, H. (2013). Identification of rare recurrent copy number variants in high‐risk autism families and their prevalence in a large ASD population. PLoS ONE, 8(1), e52239 10.1371/journal.pone.0052239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechler, B. M. , McGinnis, W. , & Gehring, W. J. (1985). Molecular cloning of lethal(2)giant larvae, a recessive oncogene of Drosophila melanogaster. The EMBO Journal, 4(6), 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering, E. , Jacob, M. , Sarria, J. C. , Steiner, P. , Hirling, H. , & Unser, M. (2004). Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A, 58(2), 167–176. 10.1002/cyto.a.20022 [DOI] [PubMed] [Google Scholar]
- Moretto, E. , Murru, L. , Martano, G. , Sassone, J. , & Passafaro, M. (2018). Glutamatergic synapses in neurodevelopmental disorders. Progress in Neuro‐Psychopharmacology and Biological Psychiatry, 84, 328–342. 10.1016/j.pnpbp.2017.09.014 [DOI] [PubMed] [Google Scholar]
- Nakayama, A. Y. , Harms, M. B. , & Luo, L. (2000). Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. Journal of Neuroscience, 20(14), 5329–5338. 10.1523/jneurosci.20-14-05329.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel, J. , Ralph, G. S. , Pickard, L. , Williams, J. , Molnar, E. , Uney, J. B. , … Henley, J. M. (1999). Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF‐dependent mechanism. Neuron, 23(2), 365–376. 10.1016/S0896-6273(00)80786-2 [DOI] [PubMed] [Google Scholar]
- Oblak, A. L. , Gibbs, T. T. , & Blatt, G. J. (2010). Decreased GABA(B) receptors in the cingulate cortex and fusiform gyrus in autism. Journal of Neurochemistry, 114(5), 1414–1423. 10.1111/j.1471-4159.2010.06858.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oblak, A. L. , Gibbs, T. T. , & Blatt, G. J. (2011). Reduced GABAA receptors and benzodiazepine binding sites in the posterior cingulate cortex and fusiform gyrus in autism. Brain Research, 1380, 218–228. 10.1016/j.brainres.2010.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertz, O. , Hodgson, L. , Klemke, R. L. , & Hahn, K. M. (2006). Spatiotemporal dynamics of RhoA activity in migrating cells. Nature, 440, 1069–1072. 10.1038/nature04665 [DOI] [PubMed] [Google Scholar]
- Reiner, A. , & Levitz, J. (2018). Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron, 98(6), 1080–1098. 10.1016/j.neuron.2018.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, G. , Watson, K. , Demonch, M. , Temple, B. , & Brennwald, P. (2015). In vitro reconstitution of Rab GTPase‐dependent vesicle clustering by the yeast lethal giant larvae/tomosyn homolog, Sro7. Journal of Biological Chemistry, 290(1), 612–624. 10.1074/jbc.M114.595892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, G. , Watson, K. , Kennedy, W. , & Brennwald, P. (2018). The tomosyn homologue, Sro7, is a direct effector of the Rab GTPase, Sec4, in post‐Golgi vesicle tethering. Molecular Biology of the Cell, 29(12), 1476–1486. 10.1091/mbc.E18-02-0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakisaka, T. , Baba, T. , Tanaka, S. , Izumi, G. , Yasumi, M. , & Takai, Y. (2004). Regulation of SNAREs by tomosyn and ROCK: Implication in extension and retraction of neurites. Journal of Cell Biology, 166(1), 17–25. 10.1083/jcb.200405002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakisaka, T. , Yamamoto, Y. , Mochida, S. , Nakamura, M. , Nishikawa, K. , Ishizaki, H. , … Takai, Y. (2008). Dual inhibition of SNARE complex formation by tomosyn ensures controlled neurotransmitter release. Journal of Cell Biology, 183(2), 323–337. 10.1083/jcb.200805150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldate, J. J. , Shiau, J. , Cazares, V. A. , & Stuenkel, E. L. (2018). The ubiquitin‐proteasome system functionally links neuronal Tomosyn‐1 to dendritic morphology. Journal of Biological Chemistry, 293(7), 2232–2246. 10.1074/jbc.M117.815514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, S. J. , He, X. , Willsey, A. J. , Ercan‐Sencicek, A. G. , Samocha, K. E. , Cicek, A. E. , … State, M. W. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron, 87(6), 1215–1233. 10.1016/j.neuron.2015.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira, M. , Tyers, M. , Torrent, M. , & Arrowsmith, C. H. (2017). WD40 repeat domain proteins: A novel target class? Nature Reviews Drug Discovery, 16(11), 773–786. 10.1038/nrd.2017.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short, P. J. , McRae, J. F. , Gallone, G. , Sifrim, A. , Won, H. , Geschwind, D. H. , … Hurles, M. E. (2018). De novo mutations in regulatory elements in neurodevelopmental disorders. Nature, 555(7698), 611–616. 10.1038/nature25983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste, M. C. , Von Herrath, M. , Benard, V. , Chamberlain, C. E. , Chuang, T.‐H. , Chu, K. , … Hahn, K. M. (2000). Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. Journal of Biological Chemistry, 275(13), 9725–9733. [DOI] [PubMed] [Google Scholar]
- Subramanian, K. , Brandenburg, C. , Orsati, F. , Soghomonian, J.‐J. , Hussman, J. P. , & Blatt, G. J. (2017). Basal ganglia and autism—A translational perspective. Autism Research, 10(11), 1751–1775. 10.1002/aur.1837 [DOI] [PubMed] [Google Scholar]
- Volk, L. , Chiu, S. L. , Sharma, K. , & Huganir, R. L. (2015). Glutamate synapses in human cognitive disorders. Annual Review of Neuroscience, 38, 127–149. 10.1146/annurev-neuro-071714-033821 [DOI] [PubMed] [Google Scholar]
- Widberg, C. H. , Bryant, N. J. , Girotti, M. , Rea, S. , & James, D. E. (2003). Tomosyn interacts with the t‐SNAREs syntaxin4 and SNAP23 and plays a role in insulin‐stimulated GLUT4 translocation. Journal of Biological Chemistry, 278(37), 35093–35101. 10.1074/jbc.M304261200 [DOI] [PubMed] [Google Scholar]
- Ye, S. , Huang, Y. , Joshi, S. , Zhang, J. , Yang, F. , Zhang, G. , … Whiteheart, S. W. (2014). Platelet secretion and hemostasis require syntaxin‐binding protein STXBP5. Journal of Clinical Investigation, 124(10), 4517–4528. 10.1172/JCI75572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q. , Xiao, M. , & Nicoll, R. A. (2001). Contribution of cytoskeleton to the internalization of AMPA receptors. Proceedings of the National Academy of Sciences of the United States of America, 98(3), 1261–1266. 10.1073/pnas.031573798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Q. , Yamakuchi, M. , Ture, S. , de la Luz Garcia‐Hernandez, M. , Ko, K. A. , Modjeski, K. L. , … Lowenstein, C. J. (2014). Syntaxin‐binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion. Journal of Clinical Investigation, 124(10), 4503–4516. 10.1172/JCI71245 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Spatial and temporal expression patterns of tomosyn during different developmental stages. (a) Temporal expression pattern of tomosyn in whole brain at P0, P7, P14, and P21; n = 4. (b) Spatial expression of tomosyn in different brain regions at P14. CB, cerebellum; CX, cortex; HC, hippocampus; Th/St, thalamus and striatum; n = 4. (c) Temporal expression of tomosyn and PSD‐95 in cultured cortical neurons at 1, 4, 7, 10, and 14 DIV. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to 1 DIV, one‐way ANOVA with Dunnett’s multiple comparisons test; n = 3. (d) Fluorescence images showing either CaMKIIα or GAD67 and tomosyn from cultured hippocampal neurons at 15 DIV. Scale bar, 20 μm
FIGURE S2 Silencing effect of shRNA on tomosyn and rescuing effect of shRNA‐resistant tomosyn. (a) Western blot analysis shows the knockdown efficiency of shRNAs against tomosyn. Tomosyn‐GFP was co‐transfected with scrambled shRNA, shRNA482, or shRNA1083 in N2a cells. shRNA482 resulted in the most effective reduction of tomosyn protein level and is shown as shTomosyn in subsequent experiments. ****p < 0.0001 compared to scramble control, one‐way ANOVA with Dunnett’s multiple comparisons test; n = 3. (b) Western blot analysis shows the protein level of Tomr‐GFP, a shRNA‐resistant form of tomosyn, in tomosyn‐knockdown N2a cells. While shTomosyn reduced the expression of Tom‐GFP, the protein level of Tomr‐GFP was not affected by shTomosyn knockdown. **p < 0.01 when compared with shTomosyn + Tom‐GFP, one‐way ANOVA with Dunnett’s multiple comparisons test; n = 3
FIGURE S3 Tomosyn knockdown leads to increased RhoA activity in different dendritic areas. (a) Representative images of dendritic segments taken immediately before and after photobleaching of acceptor (YFP). Photobleaching FRET (pbFRET) was used to measure RhoA activity in different dendritic areas. Bleached area is highlighted as a white‐dashed circle (ROI). Scale bar, 2 µm. (b) A representative graph of a pbFRET experiment showing changes in fluorescence intensities within bleach ROI over the course of 10 frames. Photobleaching was performed after the fifth frame. (c) FRET efficiency was determined by calculating the percentage (%) of the increase in donor (CFP) fluorescence intensity after acceptor photobleaching. Hippocampal neurons were co‐transfected with a RhoA Biosensor and either scrambled shRNA or shTomosyn. Tomosyn knockdown neuron showed a higher FRET efficiency. No significant increase in FRET efficiency was found between apical dendrites (a) and basal dendrites (b) within each group. p = 0.92. n = 14 scramble and 17 shTomosyn. ****p < 0.0001 when compared to scramble, two‐way ANOVA with Tukey’s multiple comparisons test
FIGURE S4 Inhibition of the Rho signaling pathway restores altered dendritic structures in tomosyn knockdown neurons. Transfected neurons expressing scramble shRNA or shTomosyn were treated with control (ddH2O) and C3T. (a) Representative images show dendritic morphology at 7 DIV. Scale bar, 100 µm. (b) Representative images show spine morphology at 15 DIV. Scale bar, 2 µm. Tomosyn knockdown neurons treated with C3T rescued the (c) branch number, (d) total dendrite length, and (e) spine density. *p < 0.05, two‐way ANOVA with Sidak’s multiple comparisons test. n = 23 scramble + ctrl, 32 scramble + C3T, 23 shTomosyn + ctrl, and 32 shTomosyn + C3T in c and d. n = 23 scramble + ctrl, 26 scramble + C3T, 25 shTomosyn + ctrl, and 30 shTomosyn + C3T in (e)
FIGURE S5 shRNA‐resistant autism‐associated tomosyn variants result in increased RhoA activity in tomosyn knockdown neurons. (a) Representative Western blot and densitometry graph show protein levels of shRNA‐resistant WTr‐Tom‐GFP, L412Vr‐Tom‐GFP, and Y502Cr‐Tom‐GFP in N2a cells co‐transfected with shTomosyn. **p < 0.01, ***p < 0.001 by one‐way ANOVA with Dunnett’s multiple comparisons test. n = 3. (b) Summary graph showing FRET efficiency in tomosyn knockdown neurons rescued with WTr, L412Vr, or Y502Cr tomosyn constructs. Neurons were co‐transfected with a RhoA biosensor and scrambled shRNA, shTomosyn, shTomosyn‐WTr‐Tom‐RFP, shTomosyn‐L412Vr‐Tom‐RFP, or shTomosyn‐Y502Cr‐Tom‐RFP. ***p < 0.001 compared to scrambled shRNA; # p < 0.05, ### p < 0.001 compared to shTomosyn‐WTr‐Tom‐RFP, one‐way ANOVA with Dunnett’s multiple comparisons test. n = 42 scramble, 39 shTomosyn, 34 WT rescue, 31 L412V rescue, and 42 Y502C rescue. (c) Example confocal images show the surface expression of pHluorin‐GluR1 in neurons expressing scrambled shRNA, shTomosyn, WT‐tomosyn rescue, L412V‐tomosyn rescue, and Y502C‐tomosyn rescue. Scale bar, 2 µm
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
Tables S1‐S3
TABLE S1 Key reagents table
TABLE S2 Primary antibodies
TABLE S3 Statistical results
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.