

Abstract
Understanding mechanisms that determine the behavior of human hematopoietic stem cells (HSCs) is essential for developing novel strategies to expand ex vivo the number of fully functional HSCs. In this review, we focus on the complex interplay between intrinsic mechanisms regulated by transcriptional and mitochondrial networks and extrinsic signals imposed by the bone marrow microenvironment, which in concert regulate the balance between HSC self‐renewal and differentiation. Such integrated signaling mechanisms that dictate the fate of HSCs in vivo must be recapitulated ex vivo to achieve successful expansion of clinically relevant HSCs. We also highlight some of the most recent ex vivo HSC expansion strategies that have currently entered clinical development. Finally, based on the evidence reviewed here and lessons learned from ex vivo HSC expansion, we raise some critical questions regarding HSC fate and the cellular plasticity of hematopoietic cells that challenge the unidirectional model of human hematopoiesis.
Keywords: HSCs, ex vivo expansion, mitochondrial network, BM niches
In this review, we focus on the complex interplay between intrinsic mechanisms regulated by transcriptional and mitochondrial networks and extrinsic signals imposed by the bone marrow microenvironment, which in concert regulate the balance between HSC self‐renewal and differentiation. We also highlight some of the most recent ex vivo HSC expansion strategies that have currently entered clinical development and raise some critical questions regarding HSC fate and the cellular plasticity of hematopoietic cells that challenge the unidirectional model of human hematopoiesis.
Introduction
Hematopoiesis relies on the preservation of the integrity of a unique pool of long‐term hematopoietic stem cells (HSCs). Long‐term HSCs occupy the apex of the hierarchy of hematopoietic cells and possess both self‐renewal and multipotent differentiation capacities. Such HSCs differentiate into a full spectrum of mature blood cells via intermediate progenitor stages, whereas their long‐term self‐renewal potential allows for sustaining the primitive pool of HSCs throughout life.1, 2 The balance between HSC self‐renewal and differentiation is central to homeostasis and is regulated by a complex interplay of both cell‐intrinsic and ‐extrinsic signaling networks.
Primitive long‐term HSCs are predominantly quiescent and rely heavily on anaerobic glycolysis for energy production.3 They reside in specialized bone marrow (BM) niches, where they maintain their undifferentiated state and a markedly low metabolic activity.4 Upon hematopoietic stress, HSCs receive signals from cellular components that comprise the HSC niches and sense changes occurring in the microenvironment causing them to exit quiescence and undergo either HSC self‐renewal or differentiation.5 The classical unidirectional model of hematopoiesis implies that upon initiation of commitment, a fraction of HSCs gradually lose their self‐renewal potential and become multipotent progenitors, which then give rise to more lineage‐committed progenitors and eventually differentiated hematopoietic cells (Fig. 1A). A metabolic switch from glycolysis toward mitochondrial metabolism accompanies the transition of HSCs from quiescence to an active cell cycling state. This switch is complex and is regulated not only by extrinsic cues imposed by the HSC niches, but also by a well‐coordinated hub of intrinsic signaling and mechanisms, which rely on the remarkable plasticity of the mitochondrial network. In fact, mitochondrial activity and metabolism are critical determinants of HSC fate decisions.
Figure 1.
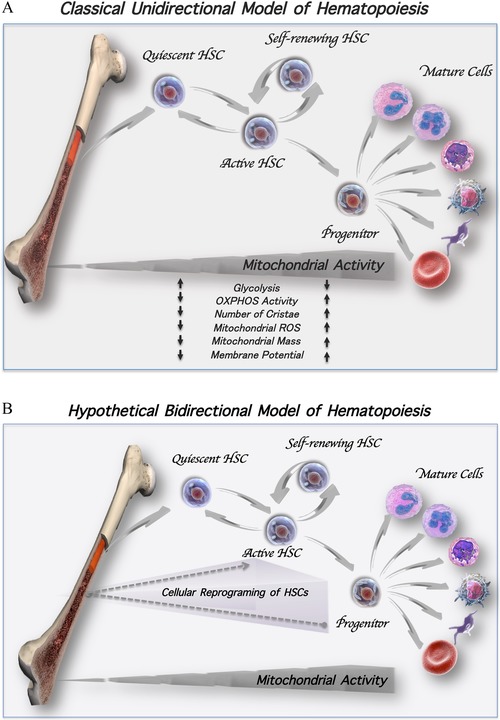
Models of hematopoiesis. (A) Classical unidirectional model of hematopoiesis. Quiescent HSCs reside in the BM niches and rely mainly on glycolysis for their energy production. Upon hematopoietic stress, HSCs exit the quiescent state and become activated into slowly dividing HSCs, giving rise to a daughter cell that may remain in the cell cycle or to a daughter cell that may return back into a quiescent state to maintain the pool of primitive HSCs. Frequent and rapid cell divisions lead to transiently amplifying progenitors, which in turn give rise to more differentiated effector hematopoietic cells. A metabolic switch associated with an increased level of mitochondrial OXPHOS activity, ROS generation, and mass occurs during the course of differentiation. (B) Hypothetical bidirectional model of hematopoiesis. Under hematopoietic stress, progenitor cells might be reprogramed and reacquire a stem‐like fate to replenish and sustain the pool of dividing HSCs with self‐renewal potential (dotted arrows).
Defining the characteristics and understanding the mechanisms underlying the self‐renewal of primitive HSCs holds the key for the development of innovative approaches aimed at ex vivo expansion of clinically relevant human HSCs. In this regard, umbilical cord blood units (UCBs) provide an excellent alternative source of HSCs and hematopoietic progenitor cells (HPCs) for patients who require allogeneic stem cell transplantation but lack a matched donor. However, the limited number of both HPCs and HSCs present within individual UCB units is a major limitation for their use as grafts for adult recipients. This limitation can theoretically be overcome by the ex vivo expansion of UCB‐derived CD34+ cells (UCB‐CD34+). Expansion of the number of functional HPCs, and most importantly, HSCs, which exhibit transcriptomic and metabolic profiles that closely resemble primary HSCs, is the goal of a growing number of ex vivo expansion strategies. A successful approach would provide sufficient numbers of human progenitor cells capable of establishing short‐term multilineage engraftment and accelerating the time to neutrophil and platelet recovery. Such a successful strategy would also boost the number of primitive HSCs capable of achieving robust long‐term multilineage engraftment in a myeloablated adult host.2, 6
In this review, we will highlight several recent ex vivo strategies that have been used to generate increased numbers of functional progenitor and stem cells. We will also focus on the characteristics of HSCs and mechanisms that determine their long‐term repopulating capacity. Efforts to clarify such mechanisms might help to identify new cellular pathways that can be targeted and then utilized for more effective ex vivo strategies.
Moreover, we present the recent evidence that might challenge the classical unidirectional hierarchical organization of the human hematopoietic system. This new evidence implies the existence of a bidirectional model of hematopoiesis and points to the possibility that cellular plasticity drives the reverse transition of committed progenitors into primitive HSCs.
Lessons learned from ex vivo simulating niche regulation of HSC self‐renewal and differentiation
The balance between HSC self‐renewal and differentiation in vivo relies on the complex and dynamic environment of HSC niches.7, 8, 9, 10, 11, 12, 13, 14, 15 Recapitulating such an environment has been thought to be critical to optimizing ex vivo cultures and achieving successful HSC expansion.
HSC niches are composed of multiple types of cells with specialized functions. These cells secrete specific cytokines and chemokines and provide physical interactions, which are essential for the retention of HSC function.16, 17, 18, 19, 20 These niche factors, including thrombopoietin, stem cell factor, Flt3 ligand, interleukin‐6, and granulocyte colony‐stimulating factor, have been used in ex vivo cultures to influence HSC proliferation. Initial clinical studies utilizing either cytokine‐driven static cultures21 or cultures of UCB‐CD34+ cells with continuous perfusion with cytokines22 have resulted in limited clinical success since the expanded product failed to improve the time to neutrophil and platelet engraftment.23 Recently, however, a phase I clinical trial using ex vivo−expanded cells with a new cytokine combination that recapitulates a “hypoxia effect” has shown promising results.24, 25
Mimicking ex vivo the complexity of the stem cell “niches” for HSC expansion has proven to be a difficult task. However, preclinical studies have revealed that direct interaction between mesenchymal stromal cells (MSCs) and either unpurified UCB cells26 or purified UCB‐CD34+ cells in ex vivo cultures results in extensive expansion of the total numbers of nucleated cells.27, 28 BM endothelial cells have also been shown to be important for the generation of HSCs in ex vivo cultures.29 Activation of naturally occurring pathways within the BM endothelium, including the Notch pathway, and immobilization of its delta‐1 ligand have been reported to effectively increase ex vivo the number of UCB‐CD34+cells. These expanded cells shortened the time of neutrophil recovery in transplant recipients in a phase I clinical trial.30, 31, 32, 33 However, the neutrophil recovery proved to be transient, indicating the lack of long‐term HSCs and the dominance of short‐term repopulating HPCs within the expanded cell product.26, 30, 34 It is possible that the lack of in vivo persistence in these transplanted patients could either be due to loss of stem cell self‐renewal capacity during culture, or to immune‐mediated rejection.23, 30
Together, this current evidence supports the notion that simulating the BM niches alone might not be sufficient to regulate HSC self‐renewal potential in ex vivo cultures. Importantly, it indicates that loss of HSC function in ex vivo cultures is likely to occur as a consequence of the silencing of the gene expression and metabolic programs that define HSCs. Accordingly, additional key factors, including HSC plasticity and HSC metabolic and transcriptome profiles, should be taken into consideration and adjusted to increase the capacity of ex vivo cultures to expand the numbers of HSCs with characteristics that closely resemble primary HSCs.
Mitochondrial activity is a critical determinant of HSC self‐renewal and differentiation potential
HSCs display a primitive mitochondrial network characterized by low mitochondrial activity and metabolism, which reflects their low energy demands. Indeed, HSCs predominantly depend on glycolysis for energy production. Low mitochondrial mass, membrane potential, and reactive oxygen species (ROS) generation, as well as functionally immature mitochondria with globular shapes and poorly developed cristae, mark self‐renewing HSCs35, 36, 37, 38, 39, 40, 41, 42, 43 (Fig. 1A). Upon commitment, HSCs rapidly switch to mitochondrial OXPHOS activity to meet the robust energy demands imposed by differentiation.44, 45 Enhanced mitochondrial mass, membrane potential, and ROS generation accompany the metabolic switch during the course of HSC differentiation.42, 46, 47 In fact, mitochondrial activity and metabolism are not simply passive hallmarks of HSCs, but critical drivers of their fate.
Mitochondrial mass and activity in HSCs is controlled by the tightly synchronized processes of mitochondrial biogenesis and turnover. HSCs exhibit a limited capacity to boost mitochondrial biogenesis, and the suppression of both mitochondrial biogenesis and metabolism preserves the functionality of primitive HSCs.48 Although the amount of mitochondrial content in HSCs still remains a matter of debate,49 the emerging view underlines that self‐renewing HSCs rely on autophagy/mitophagy activation, which acts as a primary gatekeeper of their metabolic activity. Autophagy activation limits the number of active mitochondria, mitigates the deleterious effects of cellular stress, and thus enables the retention of HSC primitive metabolic state.50, 51, 52, 53
Quiescent, self‐renewing, and differentiating HSCs have distinct metabolic profiles. Low mitochondrial activity offers protective advantages for resting long‐term HSCs due to the generation of low levels of ROS.36, 41 A limited elevation in ROS levels and mitochondrial activity drives the quiescence HSCs to enter an active but slowly cell cycling phase (Fig. 1A). Progressive increments in ROS levels, however, result in frequent cell divisions and HSC differentiation.54, 55 When ROS levels exceed the capacity of the cellular antioxidant defense, HSCs undergo senescence or cell death.56 Thus, ROS act as rheostats that dictate HSC fate.
To preserve their self‐renewal potential and reduce ROS generation, HSCs utilize complex regulatory pathways, including the sirtuin family proteins (SIRTs), forkhead box O3 (FOXO3A), AMP protein kinase (AMPK), and p53 signaling pathways.56, 57, 58, 59, 60, 61, 62, 63, 64, 65 Notably, each of these diverse pathways converges to preserve the mitochondrial network, which, in turn, engages and coordinates them to mount an antioxidant defense and regulate HSC redox status. Activation of such pathways is tightly coupled to redox sensors, including thioredoxin‐interacting protein (TXNIP) and magnesium superoxide dismutase (MnSOD).63 Intriguingly, upon transient and mild oxidative stress, TXNIP switches the function of p53 from acting as a prooxidant to an antioxidant.66 In turn, limited activation of p53 mediates the expression of ROS scavengers required to suppress ROS levels and, therefore, retain the regenerative capacity of HSCs.59, 63, 66
Mitochondrial activity in close interaction with BM milieu modulates the fine balance between HSC quiescence, self‐renewal, and differentiation
The role of mitochondria as critical determinants of HSC fate decision cannot be envisioned as being separated but rather as coupled to the signals received from the BM microenvironment. Emerging evidence indicates that mitochondria do not act alone but in concert with HSC niches to maintain a low metabolic state, which characterize primitive HSCs.
Primitive HSCs reside not only in extremely hypoxic BM niches, but also in periarteriolar and highly vascular areas of the BM where the oxygen tension is higher.9, 67 Thus, it appears that hypoxia might not be the sole mechanism by which the BM niche contributes to primitive HSCs. Indeed, it has been reported that arterial BM endothelial cells, which are exposed to the highest levels of physiological oxygen tension, create a barrier for the quiescent HSCs.9 The integrity of this barrier has been attributed to the ability of endothelial cells to enhance their antioxidant defense mechanisms and scavenge excessive ROS. In addition, endothelial cells in periarteriolar regions of the BM rely mainly on glycolysis to avoid ROS generation by mitochondrial OXPHOS activity. Accordingly, this barrier of endothelial cells provides the ROSlow microenvironment that is essential for HSC maintenance.9
Mitochondria and ROS, in fact, can be transferred to other stem cell compartments within the BM milieu. In vivo, following myeloablation, connexin‐43 (Cx43), which is a component of gap junctional channels, mediates the transfer of ROS from HSCs to MSCs resulting in reduced ROS levels in HSCs.68 Alternatively, Cx43 might exert its protective role on HSCs by facilitating the transfer and scavenging of mitochondria by the BM compartments. While this intriguing evidence is preliminary and merits further investigation, it clearly indicates the complexity of signaling networks that regulate the metabolic state of HSCs. The role of the signaling interplay between the mitochondria of HSCs and the BM has been recently reinforced by a report indicating that HSCs sense extracellular adenosine within the BM microenvironment to suppress Ca2+ influx and mitochondrial activity, leading to the retention of their primitive pool.69
Together, this evidence highlights the ability of HSCs to employ and integrate extrinsic signals imposed by the BM with intrinsic mechanisms and redox signaling regulated by mitochondria. This ongoing and highly dynamic relationship is decisive in determining HSC fate decisions and the balance between quiescence, self‐renewal, and differentiation. Accordingly, controlling and manipulating the dynamics of this interrelationship would be undeniably beneficial for establishing strategies to effectively expand ex vivo the numbers of functional HSCs for clinical use.
Ex vivo expansion strategies and clinical outcomes
Several ex vivo strategies to expand the numbers of functional human hematopoietic cells have been based upon high‐throughput unbiased screenings of libraries of small molecules and chemical compounds. These efforts have resulted in the identification of several small molecules that are now being evaluated in the clinic to promote the expansion of hematopoietic cells from UCB‐CD34+ cells in ex vivo cultures. In this review, we will focus on a few of these new strategies, which currently hold high expectations in transplantation settings.
Ex vivo expansion of UCB‐CD34+ cells with the aryl hydrocarbon receptor antagonist SR1
Screening of a large library of 100,000 small molecules led to the identification of the aryl hydrocarbon receptor (AhR) antagonist, StemReginin1 (SR1). SR1 treatment combined with a serum‐free expansion media and a cytokine cocktail increased by 50‐fold the number of UCB‐CD34+ cells and 17‐fold the number of HSCs that were capable of establishing long‐term engraftment in immunodeficient mice.70 Mechanistic insights underlying ex vivo expansion by inhibiting the AhR pathway with SR1 treatment have yet to be determined. Intriguingly, AhR activation by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) compromises the functional activity of HSCs in murine models,71 whereas the complete loss of AhR is associated with enhanced oxidative stress, premature HSC exhaustion, and hematopoietic stress.72 Although this evidence clearly implicates the AhR pathway in HSC metabolism and hematopoiesis, additional studies are needed to precisely understand the role of the AhR in the self‐renewal of human HSCs. The characterization of this pathway indeed becomes more complex given the evidence that the role of AhR pathway in self‐renewal is cell context and differentiation stage specific.73, 74
A phase I/II clinical study performed with one unmanipulated UCB unit and the progeny of the CD34+ cells from another UCB unit treated for 15 days ex vivo with SR1 demonstrated rapid neutrophil and platelet recovery. Importantly, this recovery was significantly more rapid than in patients receiving only the unmanipulated UCB.75 Only one‐third of the patients receiving the expanded grafts showed mixed myeloid chimerism associated with rapid neutrophil recovery derived from both fractions. Although promising, it remains to be determined whether SR1‐generated grafts from UCB‐CD34+ cells engraft without the additional presence of the second unmanipulated UCB unit.
Ex vivo expansion of UCB‐CD34+ cells with UM171
UM171, a pyrimidoindole derivative, is another potent small molecule that triggers a robust ex vivo expansion of human HSCs with a remarkable marrow repopulating capacity. Although the mechanism responsible for this expansion is unknown, UM171 enhances the pool of primitive HSCs independently of the AhR pathway. Gene expression signatures of cells expanded with UM171 are distinct from those expanded with SR1. UM171 is a highly potent agonist of HSC renewal and leads to a higher degree of expansion of more primitive human HSCs than SR1.76 Interestingly, both SR1 and UM171 do not affect ex vivo expansion of murine HSCs.70, 76
Notably, the UM171‐expanded pool of human cells enriched for HSCs with high engraftment potential in myeloablated murine models exhibit increased expression of the endothelial protein C receptor (EPCR). Originally identified as a key component of the endothelial barrier in the BM, EPCR is also expressed by murine HSCs.76, 77 EPCR promotes the retention of murine HSCs in the BM78, 79 by binding to the anticoagulant protease aPC and activating protease‐activated receptor‐1 signaling.
Recently, EPCR has been recognized as a reliable marker for the identification of human HSCs with extensive self‐renewal potential. Only the subpopulation of EPCR+CD34+ cells express CD90 and CD133, both of which mark the most primitive human HSCs.77 HSCs enriched for EPCR expression display a transcriptome that matches the transcriptome of freshly isolated HSCs.77
Single UCB expanded grafts with UM171 are currently being tested in a clinical trial.80 Preliminary results are promising and indicate that UCB‐CD34+ cells expanded for 7 days with UM171 establish rapid engraftment and full donor chimerism and provide clinical benefits associated with low infection and transplant‐related mortality.80 Only the future will demonstrate whether transplants with ex vivo UM171‐expanded cells from single UCB units are feasible.
Ex vivo expansion of UCB‐CD34+ cells with valproic acid
Unlike SR1 and UM171, which were found in unbiased screens of small molecules, several decades of research have revealed that epigenetic modifiers can be used to expand the pool of primitive and clinically relevant HSCs. Such modifiers, including various histone deacetylase (HDAC) inhibitors, alter the epigenetic landscape leading to a substantially increased number of both murine and human HSCs.81, 82, 83, 84, 85 Studies from our group have revealed that the deacetylase inhibitor valproic acid (VPA) triggers cellular reprogramming of UCB‐CD34+ cells leading to the ex vivo expansion of the numbers of functional primitive HSCs. The expanded HSCs are capable of establishing multilineage hematopoiesis in both primary and secondary NSG mice.83, 86 Notably, this reprogramming is accompanied by the acquisition and retention of a transcriptome and a primitive mitochondrial profile, which closely resembles that of primary functional human HSCs.84
Ex vivo culture conditions have been reported to induce stress leading to rapid HSC proliferation and loss of the primitive characteristics of primary HSCs. However, ex vivo VPA‐expanded cultures are highly enriched for cells with trancriptome profiles that are reminiscent of those previously reported to characterize both long‐term human and nonhuman primary HSCs. Key regulators that govern self‐renewal capacity and quiescence of HSCs are strongly augmented by VPA treatment. Expression profiles of both single and bulk expanded HSCs are highly enriched for long‐term HSCs phenotypic markers, including CD90 and EPCR84 (and unpublished data). Importantly, these transcriptomic alterations occur promptly, indicating that the expansion of the HSC pool with VPA is not merely a result of the rapid proliferation of the existing stem cells. Instead, such an increase in the HSC number is a result of the acquisition and retention of a primitive transcriptomic and metabolic profile combined with a limited number of cell divisions.84
Similar to primary HSCs, VPA‐expanded cells exhibit a low metabolic profile, which is characterized by enhanced glycolysis and diminished mitochondrial OXPHOS activity. The cellular reprogramming of primitive HSCs from UCB‐CD34+ cells with VPA treatment is accompanied by metabolic rewiring, which is tightly linked to a remodeled primitive mitochondrial network. This remodeling is concomitant with reduced mitochondrial ROS levels, membrane potential, and mass. Not surprisingly, the subpopulation of cells expressing high levels of long‐term HSC phenotypic markers display reduced mitochondrial ROS levels and content as opposed to HSCs that express low levels of these markers. Notably, the restructured mitochondrial network is composed of globular mitochondria with a low number of deformed cristae, which reflect their limited mitochondrial activity. Such mitochondrial morphology and characteristics resemble the mitochondrial network of primary long‐term HSCs. It has yet to be determined whether these alterations are consequences of a direct effect of VPA on mitochondria or downstream events of the epigenetic reprogramming occurring in the nucleus. It is also possible that VPA treatment reduces mitochondrial mass and activity by influencing both mitophagy activation and mitochondrial biogenesis.
Cellular reprogramming of HSCs is in fact reversible and requires the continuous presence of VPA during ex vivo culture.84 Despite this, expanded HSCs are capable of undergoing serial engraftment in NSG mice without additional exposure to VPA.83 Although unknown, such sustained self‐renewal capacity of HSCs and retention of their epigenetic and primitive mitochondrial profiles is possibly due to the metabolic and signaling cues imposed by the BM milieu. Nevertheless, these findings together with other recent reports36, 41, 87, 88 support the emerging view that transplantation of HSCs selected based on their mitochondrial status along with phenotypic markers might prove clinically beneficial and support long‐term hematopoiesis in humans.
A restructured primitive mitochondrial network accompanied by low activity is critical for HSC fate. However, alone, remodeling of the mitochondrial network is not sufficient for full and successful ex vivo reprogramming of UCB‐CD34+ cells into more primitive HSCs. It appears that cellular reprogramming with VPA also requires a limited increase in p53 activity. Although p53 activity does not affect mitochondrial mass, its enhanced activity is necessary to further suppress ROS levels. It is conceivable that p53 might exert its antioxidant effects to cope with the oxidative stress imposed by both cellular reprogramming and ex vivo culture conditions. Such antioxidant activity of p53, which is required for the activation of MnSOD, might also be needed to impede the frequent and rapid cycling of HSCs that can lead to their exhaustion.
Several reports have revealed the multifaceted roles of p53 as a pro‐ and antioxidant and its implications in hematopoiesis. A limited activation of p53 is essential for the repopulating potential of HSCs, whereas highly activated p53 can lead to senescence and death of HSCs.89 Despite these observations, its role in cellular reprogramming triggered by VPA treatment was unexpected since p53 activity impairs iPSC reprogramming from fibroblast cells.90, 91, 92 Therefore, our findings imply a unique role for p53 in cellular reprogramming and regulation of the HSC fate. Simultaneously with our report, a study from another group has shown that upon suppression of intracellular Ca2+ levels, dividing murine HSCs retain a slow rate of cycling and their self‐renewal potential through the upregulation of p53‐related genes.69
The remarkable effect of VPA on cellular reprogramming is evident not only in human and murine ex vivo expansion cultures, but also upon the generation of iPS cells from human fibroblasts. In fact, the combination of only two reprogramming factors, octamer‐binding transcription factor 4 (OCT4‐4) and SRY (sex‐determining region y)‐box 2 (SOX2), with VPA is sufficient to achieve reprogramming of human fibroblasts with similar efficiency as those achieved with the combination of three reprogramming factors. Importantly, the reprogramed iPS cells display both pluripotency and gene expression profiles that resemble those of human ES cells, thus supporting a central role for VPA as a chromatin modifier in cellular reprogramming.93 This evidence is consistent with an unbiased in vivo screening of 550 compounds in a transgenic zebrafish embryo model. Notably, this screen identified HDAC inhibitors and particularly VPA as a potent agonist of HSC expansion in vivo.94
Ex vivo HSC expansion with VPA represents a very attractive and novel approach due to the high degree of expansion of the numbers of both short‐term and more importantly, long‐term HSCs with transcriptomic and mitochondrial profiles that are reminiscent of primary primitive HSCs. HSCs ex vivo expansion with VPA occurs rapidly and requires only 7 days of culture, which limits the degree of differentiation, the risk of genetic instability, and bacterial contamination. While the numbers and primitive characteristic are potent predictors of HSC functionality, only the ongoing clinical trial will validate the engraftment potential of the expanded cell product and its efficacy in humans.
Concluding remarks and perspective
Evidence outlined in this review highlights the feasibility of effective ex vivo HSC expansion and its eventual transition to the clinic. Clearly, the progress achieved in this area is due to the advanced understanding of the complex mechanisms that govern the HSC fate. Recent evidence has added new insights into the plasticity of both the BM niches and mitochondrial networks, which together dictate HSC fate. Yet, these new findings have also introduced new layers of complexity, making it difficult to determine the drivers and sensors of this highly dynamic interrelationship.
Understanding such integrated signaling mechanisms that define the balance between quiescence and self‐renewal in vivo is challenging due in part to HSC heterogeneity. Despite major methodological developments, including new clonal tracking strategies and cellular barcoding, our ability to trace HSC fate decisions within BM niches is limited.7 Greater insights into instructions provided by the BM and on/off signals that switch the mitochondrial metabolic rewiring and alter HSC fates within the BM niches will markedly benefit the development of future ex vivo HSC expansion systems.
The classical model of hematopoiesis suggests that within stem and progenitor cell hierarchies, commitment and differentiation are unidirectional (Fig. 1A). This current model implies that quiescent HSCs, which are extremely rare, become activated into slowly dividing cells. In turn, each HSC gives rise to at least one identical daughter HSC that might return to a quiescent state to maintain a pool of primitive HSCs. The other daughter cell continues to cycle and transiently commits to a multipotent progenitor cell, which further differentiates into mature blood cells. It has been proposed that HSCs shift between quiescence and the cycling state. Yet, such persistent activation of HSCs out of quiescence might result in exhaustion and loss of their functional identity. On the other hand, it has been shown that the self‐renewal ability of an HSC both in vitro and in vivo depends on the number of cell divisions. Indeed, each single cell division impacts the regenerative capacity of HSCs.95, 96 Together, these behaviors might compromise the integrity of the primitive pool of HSCs and theoretically could lead to the exhaustion of their potential to support long‐term hematopoiesis. However, BM failure is rarely observed even among the most elderly individuals. These observations, therefore, raise questions regarding our current understanding of HSC self‐renewal behavior and whether in vivo, the HSC fate decisions within each hierarchy are truly irreversible.
Based on the evidence obtained from the ex vivo HSC expansion strategies described by our laboratory, it is possible that in vivo, the renewal of the HSC pool might be in part due to the propensity of more committed progenitor cells to reacquire stem cell properties. Similar to VPA, small molecules that act as epigenetic modifiers might exist in the BM milieu in vivo, leading to epigenetic reprogramming and reacquisition of HSC properties by committed progenitor cells (Fig. 1B, dotted arrow). In support of this hypothesis is a recent report that revealed the presence of a short‐chain fatty acid molecule pentanoate, which exerts an HDAC‐inhibitory activity in the intestinal lumen.97 Strikingly, this small molecule that is produced by bacteria, induces metabolic and epigenetic reprogramming of lymphocytes leading to the suppression of aberrant immune cell activation in both the gut and central nervous system. In addition to pentanoate, other short‐chain fatty acids, such as acetate, propionate, and butyrate, are also generated in the intestinal tract. Interestingly, all of these small molecules have been reported to have varying potency as HDAC inhibitors. Whether and which of the naturally occurring short‐chain fatty acid(s) might be responsible for HSC reprogramming in man remains to be revealed.
Evidence indicates that in intestinal tissues where the presence of the short‐chain fatty acids is evident, the secretory progenitor cells can revert back and reacquire a stem cell phenotype to facilitate tissue repair in vivo.98, 99 However, intestinal tissues, which are the fastest self‐renewing tissues in mammals, are not the only tissues where such plasticity of progenitor cells has been observed. A similar process of dedifferentiation has been reported in multiple other epithelial tissues, including the lung and kidney,100, 101, 102 implying that more committed and differentiated cells can pursue an alternative fate and replenish the number of stem cells following recovery from tissue damage. It is possible that such cellular plasticity reflects alterations in the chromatin status.
Clearly, future research will be required to address the plasticity of hematopoietic committed progenitors and their potential to reacquire a stem cell fate, which might contribute to the maintenance of the self‐renewing pool of HSCs throughout a normal life span. Presently, however, we can use our knowledge of the coordination of epigenetic and metabolic events obtained from current in vivo and ex vivo studies to define conditions required to efficiently expand the number of clinically relevant potent human HSCs.
Competing interests
The authors declare no competing interests.
Acknowledgments
This work was supported by NYSTEM grants C030136 and C030159 to R.H. We would like to thank Bartek Jablonski for his feedback and revision of the manuscript.
Contributor Information
Luena Papa, Email: Luena.Papa@mssm.edu.
Ronald Hoffman, Email: Ronald.Hoffman@mssm.edu.
References
- 1. Warren, L.A. & Rossi D.J.. 2009. Stem cells and aging in the hematopoietic system. Mech. Ageing Dev. 130: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jacobsen, S.E.W. & Nerlov C.. 2019. Haematopoiesis in the era of advanced single‐cell technologies. Nat. Cell Biol. 21: 2–8. [DOI] [PubMed] [Google Scholar]
- 3. Simsek, T. , Kocabas F., Zheng J., et al 2010. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7: 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheung, T.H. & Rando T.A.. 2013. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 14: 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park, D. , Sykes D.B. & Scadden D.T.. 2012. The hematopoietic stem cell niche. Front. Biosci. (Landmark Ed). 17: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Radtke, S. , Adair J.E., Giese M.A., et al 2017. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci. Transl. Med. 9 10.1126/scitranslmed.aan1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Copley, M.R. , Beer P.A. & Eaves C.J.. 2012. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell 10: 690–697. [DOI] [PubMed] [Google Scholar]
- 8. Birbrair, A. & Frenette P.S.. 2016. Niche heterogeneity in the bone marrow. Ann. N.Y. Acad. Sci. 1370: 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Itkin, T. , Gur‐Cohen S., Spencer J.A., et al 2016. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532: 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boulais, P.E. & Frenette P.S.. 2015. Making sense of hematopoietic stem cell niches. Blood 125: 2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei, Q. & Frenette P.S.. 2018. Niches for hematopoietic stem cells and their progeny. Immunity 48: 632–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ding, L. & Morrison S.J.. 2013. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495: 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang, J. , Niu C., Ye L., et al 2003. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425: 836–841. [DOI] [PubMed] [Google Scholar]
- 14. Mendez‐Ferrer, S. , Michurina T.V., Ferraro F., et al 2010. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466: 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kiel, M.J. , Yilmaz O.H., Iwashita T., et al 2005. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121: 1109–1121. [DOI] [PubMed] [Google Scholar]
- 16. Qian, H. , Buza‐Vidas N., Hyland C.D., et al 2007. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell 1: 671–684. [DOI] [PubMed] [Google Scholar]
- 17. Nakamura‐Ishizu, A. , Takubo K., Fujioka M. & Suda T.. 2014. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem. Biophys. Res. Commun. 454: 353–357. [DOI] [PubMed] [Google Scholar]
- 18. Lennartsson, J. & Ronnstrand L.. 2012. Stem cell factor receptor/c‐Kit: from basic science to clinical implications. Physiol. Rev. 92: 1619–1649. [DOI] [PubMed] [Google Scholar]
- 19. Rappold, I. , Watt S.M., Kusadasi N., et al 1999. Gp130‐signaling synergizes with FL and TPO for the long‐term expansion of cord blood progenitors. Leukemia 13: 2036–2048. [DOI] [PubMed] [Google Scholar]
- 20. Li, T. & Wu Y.. 2011. Paracrine molecules of mesenchymal stem cells for hematopoietic stem cell niche. Bone Marrow Res. 2011 10.1155/2011/353878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shpall, E.J. , Quinones R., Giller R., et al 2002. Transplantation of ex vivo expanded cord blood. Biol. Blood Marrow Transplant. 8: 368–376. [DOI] [PubMed] [Google Scholar]
- 22. Jaroscak, J. , Goltry K., Smith A., et al 2003. Augmentation of umbilical cord blood (UCB) transplantation with ex vivo‐expanded UCB cells: results of a phase 1 trial using the AastromReplicell System. Blood 101: 5061–5067. [DOI] [PubMed] [Google Scholar]
- 23. Dahlberg, A. , Delaney C. & Bernstein I.D.. 2011. Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood 117: 6083–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Forcade, E. , Cognet C., Sicard X., et al 2015. Comparable immune reconstitution between ex vivo amplification and un‐manipulated umbilical cord blood transplantation. Blood 126: 1936. [Google Scholar]
- 25. Duchez, P. , Chevaleyre J., Vlaski M., et al 2012. Definitive setup of clinical scale procedure for ex vivo expansion of cord blood hematopoietic cells for transplantation. Cell Transplant. 21: 2517–2521. [DOI] [PubMed] [Google Scholar]
- 26. de Lima, M. , McNiece I., Robinson S.N., et al 2012. Cord‐blood engraftment with ex vivo mesenchymal‐cell coculture. N. Engl. J. Med. 367: 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wagner, W. , Roderburg C., Wein F., et al 2007. Molecular and secretory profiles of human mesenchymal stromal cells and their abilities to maintain primitive hematopoietic progenitors. Stem Cells 25: 2638–2647. [DOI] [PubMed] [Google Scholar]
- 28. Flores‐Guzman, P. , Flores‐Figueroa E., Montesinos J.J., et al 2009. Individual and combined effects of mesenchymal stromal cells and recombinant stimulatory cytokines on the in vitro growth of primitive hematopoietic cells from human umbilical cord blood. Cytotherapy 11: 886–896. [DOI] [PubMed] [Google Scholar]
- 29. Lewis, I.D. , Almeida‐Porada G., Du J., et al 2001. Umbilical cord blood cells capable of engrafting in primary, secondary, and tertiary xenogeneic hosts are preserved after ex vivo culture in a noncontact system. Blood 97: 3441–3449. [DOI] [PubMed] [Google Scholar]
- 30. Delaney, C. , Heimfeld S., Brashem‐Stein C., et al 2010. Notch‐mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat. Med. 16: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Delaney, C. , Varnum‐Finney B., Aoyama K., et al 2005. Dose‐dependent effects of the Notch ligand Delta1 on ex vivo differentiation and in vivo marrow repopulating ability of cord blood cells. Blood 106: 2693–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Butler, J.M. , Nolan D.J., Vertes E.L., et al 2010. Endothelial cells are essential for the self‐renewal and repopulation of Notch‐dependent hematopoietic stem cells. Cell Stem Cell 6: 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohishi, K. , Varnum‐Finney B. & Bernstein I.D.. 2002. Delta‐1 enhances marrow and thymus repopulating ability of human CD34(+)CD38(–) cord blood cells. J. Clin. Invest. 110: 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mehta, R.S. , Rezvani K., Olson A., et al 2015. Novel techniques for ex vivo expansion of cord blood: clinical trials. Front. Med. (Lausanne) 2: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maryanovich, M. , Zaltsman Y., Ruggiero A., et al 2015. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun. 6: 7901. [DOI] [PubMed] [Google Scholar]
- 36. Vannini, N. , Girotra M., Naveiras O., et al 2016. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 7: 13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Romero‐Moya, D. , Bueno C., Montes R., et al 2013. Cord blood‐derived CD34+ hematopoietic cells with low mitochondrial mass are enriched in hematopoietic repopulating stem cell function. Haematologica 98: 1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Warr, M.R. & Passegue E.. 2013. Metabolic makeover for HSCs. Cell Stem Cell 12: 1–3. [DOI] [PubMed] [Google Scholar]
- 39. Chen, C. , Liu Y., Liu R., et al 2008. TSC‐mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 205: 2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Piccoli, C. , Ria R., Scrima R., et al 2005. Characterization of mitochondrial and extra‐mitochondrial oxygen consuming reactions in human hematopoietic stem cells. Novel evidence of the occurrence of NAD(P)H oxidase activity. J. Biol. Chem. 280: 26467–26476. [DOI] [PubMed] [Google Scholar]
- 41. Jang, Y.Y. & Sharkis S.J.. 2007. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low‐oxygenic niche. Blood 110: 3056–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du, J. , Li Q., Tang F., et al 2014. Cited2 is required for the maintenance of glycolytic metabolism in adult hematopoietic stem cells. Stem Cells Dev. 23: 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu, M. , Zeng H., Chen S., et al 2018. SRC‐3 is involved in maintaining hematopoietic stem cell quiescence by regulation of mitochondrial metabolism in mice. Blood 132: 911–923. [DOI] [PubMed] [Google Scholar]
- 44. Yu, W.M. , Liu X., Shen J., et al 2013. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 12: 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takubo, K. , Nagamatsu G., Kobayashi C.I., et al 2013. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maryanovich, M. & Gross A.. 2013. A ROS rheostat for cell fate regulation. Trends Cell Biol. 23: 129–134. [DOI] [PubMed] [Google Scholar]
- 47. Maryanovich, M. , Oberkovitz G., Niv H., et al 2012. The ATM‐BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 14: 535–541. [DOI] [PubMed] [Google Scholar]
- 48. Qian, P. , He X.C., Paulson A., et al 2016. The Dlk1‐Gtl2 locus preserves LT‐HSC function by inhibiting the PI3K‐mTOR pathway to restrict mitochondrial metabolism. Cell Stem Cell 18: 214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Almeida, M.J. , Luchsinger L.L., Corrigan D.J., et al 2017. Dye‐independent methods reveal elevated mitochondrial mass in hematopoietic stem cells. Cell Stem Cell 21: 725–729.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Warr, M.R. , Binnewies M., Flach J., et al 2013. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494: 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ito, K. , Turcotte R., Cui J., et al 2016. Self‐renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354: 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ho, T.T. , Warr M.R., Adelman E.R., et al 2017. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543: 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nguyen‐McCarty, M. & Klein P.S.. 2017. Autophagy is a signature of a signaling network that maintains hematopoietic stem cells. PLoS One 12: e0177054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Suda, T. , Takubo K. & Semenza G.L.. 2011. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9: 298–310. [DOI] [PubMed] [Google Scholar]
- 55. Ito, K. , Bonora M. & Ito K.. 2019. Metabolism as master of hematopoietic stem cell fate. Int. J. Hematol. 109: 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ito, K. , Hirao A., Arai F., et al 2006. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 12: 446–451. [DOI] [PubMed] [Google Scholar]
- 57. Tothova, Z. & Gilliland D.G.. 2007. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 1: 140–152. [DOI] [PubMed] [Google Scholar]
- 58. Tothova, Z. , Kollipara R., Huntly B.J., et al 2007. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128: 325–339. [DOI] [PubMed] [Google Scholar]
- 59. Jung, H. , Kim D.O., Byun J.E., et al 2016. Thioredoxin‐interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat. Commun. 7 10.1038/ncomms13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matsui, K. , Ezoe S., Oritani K., et al 2012. NAD‐dependent histone deacetylase, SIRT1, plays essential roles in the maintenance of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 418: 811–817. [DOI] [PubMed] [Google Scholar]
- 61. Zhang, H. , Menzies K.J. & Auwerx J.. 2018. The role of mitochondria in stem cell fate and aging. Development 145 https://10.1242/dev.143420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nakada, D. , Saunders T.L. & Morrison S.J.. 2010. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brown, K. , Xie S., Qiu X., et al 2013. SIRT3 reverses aging‐associated degeneration. Cell Rep. 3: 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen, C. , Liu Y., Liu Y. & Zheng P.. 2009. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2: ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen, C. , Liu Y., Liu Y. & Zheng P.. 2009. The axis of mTOR–mitochondria–ROS and stemness of the hematopoietic stem cells. Cell Cycle 8: 1158–1160. [DOI] [PubMed] [Google Scholar]
- 66. Jung, H. , Kim M.J., Kim D.O., et al 2013. TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 18: 75–85. [DOI] [PubMed] [Google Scholar]
- 67. Crane, G.M. , Jeffery E. & Morrison S.J.. 2017. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 17: 573–590. [DOI] [PubMed] [Google Scholar]
- 68. Taniguchi Ishikawa, E. , Gonzalez‐Nieto D., Ghiaur G., et al 2012. Connexin‐43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc. Natl. Acad. Sci. USA 109: 9071–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Umemoto, T. , Hashimoto M., Matsumura T., et al 2018. Ca(2+)–mitochondria axis drives cell division in hematopoietic stem cells. J. Exp. Med. 215: 2097–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Boitano, A.E. , Wang J., Romeo R., et al 2010. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329: 1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Singh, K.P. , Wyman A., Casado F.L., et al 2009. Treatment of mice with the Ah receptor agonist and human carcinogen dioxin results in altered numbers and function of hematopoietic stem cells. Carcinogenesis 30: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Singh, K.P. , Bennett J.A., Casado F.L., et al 2014. Loss of aryl hydrocarbon receptor promotes gene changes associated with premature hematopoietic stem cell exhaustion and development of a myeloproliferative disorder in aging mice. Stem Cells Dev. 23: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jackson, C.S. , Durandt C., Janse van Rensburg I., et al 2017. Targeting the aryl hydrocarbon receptor nuclear translocator complex with DMOG and Stemregenin 1 improves primitive hematopoietic stem cell expansion. Stem Cell Res. 21: 124–131. [DOI] [PubMed] [Google Scholar]
- 74. Casado, F.L. 2016. The Aryl hydrocarbon receptor relays metabolic signals to promote cellular regeneration. Stem Cells Int. 2016 10.1155/2016/4389802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wagner, J.E., Jr. , Brunstein C.G., Boitano A.E., et al 2016. Phase I/II trial of StemRegenin‐1 expanded umbilical cord blood hematopoietic stem cells supports testing as a stand‐alone graft. Cell Stem Cell 18: 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fares, I. , Chagraoui J., Gareau Y., et al 2014. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self‐renewal. Science 345: 1509–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fares, I. , Chagraoui J., Lehnertz B., et al 2017. EPCR expression marks UM171‐expanded CD34(+) cord blood stem cells. Blood 129: 3344–3351. [DOI] [PubMed] [Google Scholar]
- 78. Gur‐Cohen, S. , Itkin T., Chakrabarty S., et al 2015. PAR1 signaling regulates the retention and recruitment of EPCR‐expressing bone marrow hematopoietic stem cells. Nat. Med. 21: 1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gur‐Cohen, S. , Kollet O., Graf C., et al 2016. Regulation of long‐term repopulating hematopoietic stem cells by EPCR/PAR1 signaling. Ann. N.Y. Acad. Sci. 1370: 65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sandra Cohen, J.R. , Lachance S., Marinier A., et al 2017. Single UM171 expanded cord blood transplant is feasible, safe, and permits transplantation of better HLA matched cords with very low transplant related mortality. Blood 130: 658. [Google Scholar]
- 81. Milhem, M. , Mahmud N., Lavelle D., et al 2004. Modification of hematopoietic stem cell fate by 5aza 2′deoxycytidine and trichostatin A. Blood 103: 4102–4110. [DOI] [PubMed] [Google Scholar]
- 82. De Felice, L. , Tatarelli C., Mascolo M.G., et al 2005. Histone deacetylase inhibitor valproic acid enhances the cytokine‐induced expansion of human hematopoietic stem cells. Cancer Res. 65: 1505–1513. [DOI] [PubMed] [Google Scholar]
- 83. Chaurasia, P. , Gajzer D.C., Schaniel C., et al 2014. Epigenetic reprogramming induces the expansion of cord blood stem cells. J. Clin. Invest. 124: 2378–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Papa, L. , Zimran E., Djedaini M., et al 2018. Ex vivo human HSC expansion requires coordination of cellular reprogramming with mitochondrial remodeling and p53 activation. Blood Adv. 2: 2766–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Addison, C.L. , Nor J.E., Zhao H., et al 2005. The response of VEGF‐stimulated endothelial cells to angiostatic molecules is substrate‐dependent. BMC Cell Biol. 6: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Iancu‐Rubin, C. & Hoffman R.. 2015. Role of epigenetic reprogramming in hematopoietic stem cell function. Curr. Opin. Hematol. 22: 279–285. [DOI] [PubMed] [Google Scholar]
- 87. Mantel, C.R. , O'Leary H.A., Chitteti B.R., et al 2015. Enhancing hematopoietic stem cell transplantation efficacy by mitigating oxygen shock. Cell 161: 1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Notta, F. , Doulatov S., Laurenti E., et al 2011. Isolation of single human hematopoietic stem cells capable of long‐term multilineage engraftment. Science 333: 218–221. [DOI] [PubMed] [Google Scholar]
- 89. Pant, V. , Quintas‐Cardama A. & Lozano G.. 2012. The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood 120: 5118–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hong, H. , Takahashi K., Ichisaka T., et al 2009. Suppression of induced pluripotent stem cell generation by the p53‐p21 pathway. Nature 460: 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kawamura, T. , Suzuki J., Wang Y.V., et al 2009. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460: 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Krizhanovsky, V. & Lowe S.W.. 2009. Stem cells: the promises and perils of p53. Nature 460: 1085–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Huangfu, D. , Osafune K., Maehr R., et al 2008. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 26: 1269–1275. [DOI] [PubMed] [Google Scholar]
- 94. Arulmozhivarman, G. , Krater M., Wobus M., et al 2017. Zebrafish in‐vivo screening for compounds amplifying hematopoietic stem and progenitor cells: ‐ preclinical validation in human CD34+ stem and progenitor cells. Sci. Rep. 7: 12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bernitz, J.M. , Kim H.S., MacArthur B., et al 2016. Hematopoietic stem cells count and remember self‐renewal divisions. Cell 167: 1296–1309.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Qiu, J. , Papatsenko D., Niu X., et al 2014. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep. 2: 473–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Luu, M. , Pautz S., Kohl V., et al 2019. The short‐chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic‐epigenetic crosstalk in lymphocytes. Nat. Commun. 10: 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. van Es, J.H. , Sato T., van de Wetering M., et al 2012. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat. Cell Biol. 14: 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Visvader, J.E. & Clevers H.. 2016. Tissue‐specific designs of stem cell hierarchies. Nat. Cell Biol. 18: 349–355. [DOI] [PubMed] [Google Scholar]
- 100. Rock, J.R. , Onaitis M.W., Rawlins E.L., et al 2009. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 106: 12771–12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tata, P.R. , Mou H., Pardo‐Saganta A., et al 2013. Dedifferentiation of committed epithelial cells into stem cells in vivo . Nature 503: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Witzgall, R. , Brown D., Schwarz C. & Bonventre J.V.. 1994. Localization of proliferating cell nuclear antigen, vimentin, c‐Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Invest. 93: 2175–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]