

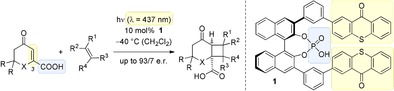
Abstract
A chiral phosphoric acid with a 2,2’‐binaphthol core was prepared that displays two thioxanthone moieties at the 3,3’‐position as light‐harvesting antennas. Despite its relatively low triplet energy, the phosphoric acid was found to be an efficient catalyst for the enantioselective intermolecular [2+2] photocycloaddition of β‐carboxyl‐substituted cyclic enones (e.r. up to 93:7). Binding of the carboxylic acid to the sensitizer is suggested by NMR studies and by DFT calculations to occur by means of two hydrogen bonds. The binding event not only enables an enantioface differentiation but also modulates the triplet energy of the substrates.
Keywords: enantioselectivity, hydrogen bonds, organocatalysis, photochemistry, sensitizers
Bind me tender: Cyclic enone‐3‐carboxylic acids (X=CH2, O) display a binding motif (in blue) for an assembly with chiral acid 1. The acid acts as photocatalyst by transferring the energy of visible photons to the substrate via its thioxanthone substituents (in yellow) enabling an enantioselective [2+2] photocycloaddition (see scheme).
Recent interest in visible light‐mediated reactions has triggered a large number of studies towards the synthesis of new chromophores and chiral catalysts.1 The long known thioxanthone chromophore2 has been revisited in the context of triplet sensitization3 and it has been attached to chiral scaffolds for applications in enantioselective photochemistry.4 The most frequently used chiral modification is represented by compound 1 (Figure 1)4a in which a thioxanthone is attached via an oxazole to position C7 of 1,5,7‐trimethyl‐3‐azabicyclo[3.3.1]nonan‐2‐one. The latter device serves as a hydrogen bonding site5 and allows to process lactams in [2+2] photocycloaddition4a, 6 and deracemization7 reactions. By attachment to a chiral bisoxazoline, thioxanthone can be part of a bifunctional chiral metal catalyst and ligand 2 has been successfully employed in the Ni‐catalyzed oxygenation of β‐ketoesters.4b Chiral imidazolinone‐based organocatalysts have recently been reported for enamine catalysis in which the thioxanthone acts via single electron transfer.4d However, not any binding motif known from thermal reactions will automatically lead to a successful chiral catalyst. Thiourea‐linked thioxanthones such as 3, for example, did not display the expected enantioselectivity in photochemical reactions.4c
Figure 1.
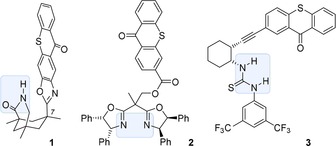
Structure of chiral thioxanthones 1–3 with a substrate or metal binding site (in gray).
Nonetheless, given the limited number of substrates that can yet be processed by chiral thioxanthones, it seems desirable to further investigate possible binding motifs to which a thioxanthone entity can be attached. In this communication, we describe the synthesis of a C 2‐symmetric chiral phosphoric acid with two thioxanthone units and report on preliminary studies as to its mode of action in photochemical [2+2] cycloaddition reactions.
Since their initial use in organocatalysis,8 chiral phosphoric acids have emerged as a highly efficient class of compounds for a plethora of applications.9 Most phosphoric acids display aryl groups in positions C3 and C3’ of the 2,2’‐binaphthol core and we envisaged that these positions would serve as suitable points of attachment for a thioxanthone unit. However, the direct linkage of a 9‐oxo‐9H‐thioxanthen‐3‐yl group to positions C3 and C3’ led to a phosphoric acid which did not induce any enantioselectivity in photochemical test reactions (see Supporting Information for further details). Upon further screening, a more promising catalyst 4 (Scheme 1) was discovered which was prepared by Suzuki cross‐coupling of aryl bromide 7 and known diboronic acid 8.10 The former component was obtained by a dehydrogenative condensation11 of 3‐bromobiphenyl (5) and thiosalicylic acid (6). After demethylation and phosphorylation the free acid was liberated by treatment with water. A major asset of phosphoric acid 4 as compared to thioxanthones 1 and 3 is its C 2 symmetry which invites the coordination of substrates with a symmetric binding motif. In this context, carboxylic acids RCOOH seemed particularly interesting to us because studies by List and co‐workers had established that they can be activated in thermal reactions by coordination to chiral phosphoric acids.12
Scheme 1.
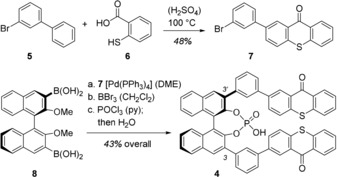
Synthesis of phosphoric acid 4 from boronic acid 8 and aryl bromide 7. Conditions and yields for the individual steps a–c: (a) 2.5 equiv 7, 10 mol % Pd(PPh3)4, 10 equiv 2 m Na2CO3 (aq), 85 °C, 16 h (DME), 96 %; (b) 6.0 equiv BBr3, 0 °C→rt, 2 h (CH2Cl2), 83 %; (c) 2.0 equiv POCl3, 95 °C, 12 h (py), then H2O, 95 °C, 54 %.
Preliminary experiments commenced with 3,4‐dihydro‐2,2‐dimethyl‐4‐oxo‐2H‐pyran‐6‐carboxylic acid (9, Scheme 2) which had been previously employed in intermolecular [2+2] photocycloaddition reactions upon UV irradiation (λ=366 nm).13 To our delight, we found that the previously reported reaction with cyclopentene to racemic products rac‐10 could be conducted with visible light in the presence of parent thioxanthone (thioxanthen‐9‐one, see Supporting Information for further details). With catalytic quantities of acid 4, the reaction proceeded at λ=437 nm with improved chemoselectivity (vide infra) and the desired product was isolated as a mixture of two diastereoisomers with the cis‐anti‐cis diastereoisomer 10 a prevailing over the cis‐syn‐cis diastereoisomer (d.r.=69/31).
Scheme 2.
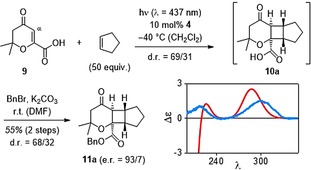
[2+2] Photocycloaddition of cyclopentene to acid 9 catalyzed by thioxanthone 4 and subsequent benzylation; schematic CD spectrum (calculated in red, vs. measured in blue) of acid 10 (for full details, see Supporting Information).
Although isolation of products 10 was possible (61 % yield), the enantiomeric ratio (e.r.) could not be determined by chiral HPLC analysis. Derivatization to the UV active benzyl ester was feasible and esters 11 were formed in 55 % yield over two steps. Benzyl ester 11 a displayed a remarkable e.r. of 93/7 which suggests a high enantioface differentiation in the [2+2] photocycloaddition to product 10 a. The absolute configuration of the major enantiomer was determined by comparison of the measured and calculated CD spectra of compound 10 a. Formally, the absolute configuration corresponds to a Si face approach onto the α‐carbon atom in the α,β‐unsaturated carbonyl compound 9.
Cyclohex‐2‐enone‐3‐carboxylic acid (12) had been previously involved in an enantioselective [2+2] photocycloaddition14, 15 employing a chiral amine as chiral template. Enantioselectivities of up to 24 % ee (e.r.=62/38) had been achieved employing five equivalents of the template (λ>320 nm). Products had not been isolated and the enantioselectivity had been determined by GLC upon derivatization. In our case, it was possible to perform the reaction with visible light (λ=437 nm) employing only 10 mol % of catalyst 4 but the separation of the polar regioisomeric products 13 and 14 was not feasible. The benzylation turned out to be sluggish and yields of products 15 and 16 were low (Scheme 3).
Scheme 3.
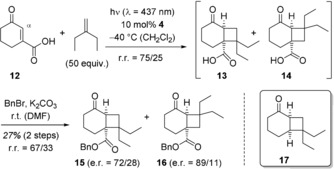
Catalyzed enantioselective [2+2] photocycloaddition of 2‐cyclohexenone‐3‐carboxylic acid (12) and 2‐ethylbutene and subsequent benzylation; inset: structure of the decarboxylation product 17 obtained from intermediate 13.
Like for product 11 a a significant enantioface differentation was recorded with the higher e.r. (89/11) being found for the minor regioisomer 16. The absolute configuration of chiral product 13 was assessed by decarboxylation16 to known cyclobutane 17.17 The direction of attack at the α‐carbon atom of enone 12 is identical to the direction of attack for compound 9 (Si face). The assignment of the absolute configuration for all other major enantiomers in the [2+2] photocycloaddition of 9 and 12 was based on analogy. Indeed, it was established that other olefins also react successfully under sensitized conditions in the presence of chiral phosphoric acid 4. After benzylation, products 18–21 were isolated with high to moderate enantioselectivities (Figure 2). However, the yield for the two‐step protocol remained relatively low, presumably due to the non‐optimized benzylation protocol (26–42 %, see Supporting Information for further details).
Figure 2.
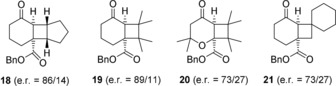
Structure and enantiomeric ratio of [2+2] photocycloaddition products 18–21 obtained from acids 9 and 12 via energy transfer from chiral phosphoric acid 4.
A notable feature of thioxanthone 4 as compared to the parent compound (thioxanthen‐9‐one) was the fact that undesired decarboxylation reactions were suppressed and that the photocycloaddition reaction was more efficient (see Supporting Information for further details). Luminescence measurements in dichloromethane (Figure 3) revealed that the triplet energy (E T) of compound 4 is surprisingly low. From the phosphorescence emission (77 K, dichloromethane) at the long wavelength shoulder the energy was calculated as E T=235±2 kJ mol−1 which is much lower than the reported triplet energy of thioxanthen‐9‐one (E T=272 kJ mol−1).18 Luminescence spectra of the two carboxylic acids showed the typical signature of α,β‐unsaturated enones19 with a readily detectable 0‐0 transition. The triplet energy calculated from this transition was E T=288±2 kJ mol−1 for compound 9 and E T=287±2 kJ mol−1 for compound 12 (77 K, pentane/isopentane).
Figure 3.
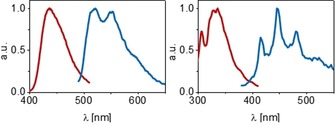
Fluorescence (red) and phosphorescence (blue) spectra recorded at r.t. (fluorescence) and 77 K (phosphorescence) for catalyst 4 (left) in a dichloromethane matrix and for acid 9 (right) in a pentane/isopentane matrix (for futher details, see the Supporting Information).
Combined with the results obtained from the thioxanthen‐9‐one irradiation experiments, the data suggest a preferred energy transfer within a complex between the carboxylic acids and compound 4 while thioxanthen‐9‐one leads to partial oxidation of the acid. Given the high energy difference of the triplet energies in a non‐complexed situation it is also likely that the phosphoric acid lowers the triplet energy of the carboxylic acids upon association. Although it is known that coordination of Lewis acids to carbonyl groups alters the triplet state energy1, 20 we are not aware of this phenomenon having been observed for phosphoric acid/carboxylic acid combinations.
At the reaction temperature (−40 °C) binding of carboxylic acid 9 to phosphoric acid 4 was proven by NMR studies in CD2Cl2. The identification of two NOE contacts between 4 and 9 as well as Diffusion Ordered Spectroscopy (DOSY) experiments validated 4⋅9 complex formation, the latter showing a significant increase (≈1.7 Å) of the hydrodynamic radius of carboxylic acid 9 in the presence of catalyst 4 (see Supporting Information for details and spectra).12a Significant line broadening and a slight highfield shifting (≈0.1 ppm) of the signals of 9 in the presence of 4 demonstrate a fast exchange on the NMR timescale between complex 4⋅9 and the separated molecules 4 and 9 at this temperature (see Supporting Information for spectra).
Next, NMR measurements between −40 °C and −93 °C were applied to gain information about the hydrogen bond situation in this system. Indeed, specific signals of hydrogen bonds could be detected, which is to our knowledge the first time for complexes between chiral phosphoric acids and carboxylic acids. The hydrogen bond region21 of 4 showed various minor populated signals summarized as HX (blue region in Figure 4) and two distinct signals H2 and H3 (green region). The temperature coefficients of H2 and H3 are very small (−0.9 ppb K−1) indicating that these protons are effectively sequestered from solvent interactions. This is typical for molecules featuring strong internal H‐bonds or stable complexes with internal H‐bonds, for example, dimers (or higher aggregates) of 4.22 The minimum temperature coefficient of the signals labelled HX is significantly higher (−9.6 ppb K−1) than that for H2 and H3, but still by far smaller than the one of carboxylic acid 9 (−38.6 ppb K−1). This indicates a partial sequestration of the HX protons from the solvent, which would be rational for different rotational isomers of monomeric 4. Indeed, signals HX of the catalyst collapse to one signal H1 in the presence of 9, while signals H2 and H3 are unaffected.23 The respective 31P NMR spectra show a similar behaviour (see the Supporting Information) and thus support the assignment of HX and H1 as monomeric, and H2 and H3 as dimeric (or oligomeric) catalyst species. The averaged proton signal H1 indicates very low rotation barriers of and within the 3,3’‐substituent in the 4⋅9 complex, which is in accordance with the theoretical calculations (vide infra). In contrast, in the absence of 9 the various proton signals HX indicate higher rotation barriers in monomeric 4 despite reduced absolute steric hindrance. The combination of a medium temperature coefficient, higher rotation barriers and significant spreading of the chemical shifts of HX protons over ≈3 ppm hints at various internal hydrogen bonds of the phosphoric acid to the aromatic moieties of the 3,3’‐substituent (see the Supporting Information for further discussion), which stabilize different rotational isomers of the free catalyst. To the best of our knowledge, this feature has not yet been observed for chiral phosphoric acids and is potentially useful as a concept for pre‐organization.
Figure 4.
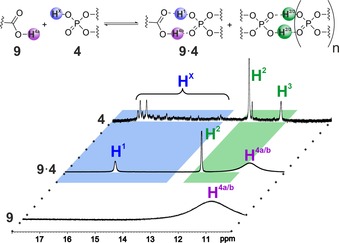
Hydrogen bond region of the 1H‐NMR spectra of 4, 9 and an equimolar solution of 4 and 9 at 600 MHz and −40° C in CD2Cl2, showing the presence of different species of the catalyst.
Preliminary DFT calculations revealed a relatively shallow energy hypersurface for the 1:1 complex of catalyst 4 with carboxylic acid 12. There are several local minima detected upon rotation around the indicated bonds (for more details, see the Supporting Information). Due to the free rotation around the thioxanthone‐phenyl bond (Figure 5, left) the C 2 symmetry of the catalyst is lost in most rotamers which complicates the analysis. The energetically lowest diastereomeric conformation is according to the calculation 3.5 kJ mol−1 more stable than the shown conformation (Figure 5, right). While the former conformation does not account for the correct product configuration, the latter arrangement illustrates the observed preferential approach of the olefin from the Si face of the α‐carbon atom. The extent of the enantioface differentiation depends on the exact trajectory of the olefin and on the position of initial attack.
Figure 5.
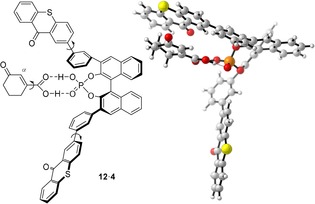
Models for the complex of cyclohex‐2‐enone‐3‐carboxylic acid (12) with chiral phosphoric acid 4: Rotatable single bonds influencing the reactive conformation (left), optimized structure of a 1:1 complex in which the Si face at the α‐carbon atom of acid 12 is accessible (right). DFT calculations were performed using the M06‐2X functional24 with the def2‐TZVP25 basis set for all atoms employing Gaussian0926 with D3 dispersion27 and low‐frequency entropy28 corrections by Grimme.
In conclusion, the first member of a new class of bifunctional photocatalysts has been synthesized and screened in [2+2] photocycloaddition reactions. The catalyst appears to lower the triplet energy of the substrates by hydrogen bonding and enables a notable enantioface differentiation. Different catalyst species (monomeric rotational isomers and higher aggregates) were observed by NMR, but only the monomeric catalyst participates in the complexation to the substrate, while higher aggregates are not affected by the substrate. The formation of a catalyst substrate complex was proven by NMR studies and a potential structure of the 1:1 complex was revealed by DFT calculations. A better understanding of the binding mode and of the interaction between the sensitizing unit and the substrate is expected to facilitate the design of modified catalysts.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement No 665951–ELICOS) is gratefully acknowledged. We thank Dr. J. D. Jolliffe for his help in the preparation of catalyst 4 and M. Eder for his assistance with the CD measurements. J. G. thanks the Fonds der Chemischen Industrie for funding (Kekulé fellowship), the SPP 1807 (dispersion) for intellectual support and Stephan Reichl for his help in preparing water‐free NMR‐samples.
F. Pecho, Y.-Q. Zou, J. Gramüller, T. Mori, S. M. Huber, A. Bauer, R. M. Gschwind, T. Bach, Chem. Eur. J. 2020, 26, 5190.
Contributor Information
Franziska Pecho, http://www.oc1.ch.tum.de/home_en/.
Prof. Dr. Thorsten Bach, Email: thorsten.bach@ch.tum.de.
References
- 1.Reviews:
- 1a. Silvi M., Melchiorre P., Nature 2018, 554, 41–49; [DOI] [PubMed] [Google Scholar]
- 1b. Garrido-Castro A. F., Maestro M. C., Alemán J., Tetrahedron Lett. 2018, 59, 1286–1294; [Google Scholar]
- 1c. Brenninger C., Jolliffe J. D., Bach T., Angew. Chem. Int. Ed. 2018, 57, 14338–14349; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14536–14547. [Google Scholar]
- 2.Examples:
- 2a. DeBoer C. D., Schlessinger R. H., J. Am. Chem. Soc. 1972, 94, 655–656; [Google Scholar]
- 2b. Yates S. F., Schuster G. B., J. Org. Chem. 1984, 49, 3349–3356; [Google Scholar]
- 2c. Meier K., Zweifel H., J. Photochem. 1986, 35, 353–366. [Google Scholar]
- 3.Reviews:
- 3a. Strieth-Kalthoff F., James M. J., Teders M., Pitzer L., Glorius F., Chem. Soc. Rev. 2018, 47, 7190–7202; [DOI] [PubMed] [Google Scholar]
- 3b. Zhou Q.-Q., Zou Y.-Q., Lu L.-Q., Xiao W.-J., Angew. Chem. Int. Ed. 2019, 58, 1586–1604; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1600–1619. [Google Scholar]
- 4.
- 4a. Alonso R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 4368–4371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4457–4460; [Google Scholar]
- 4b. Ding W., Lu L.-Q., Zhou Q.-Q., Wei Y., Chen J.-R., Xiao W.-J., J. Am. Chem. Soc. 2017, 139, 63–66; [DOI] [PubMed] [Google Scholar]
- 4c. Mayr F., Mohr L.-M., Rodriguez E., Bach T., Synthesis 2017, 49, 5238–5250; [Google Scholar]
- 4d. Rigotti T., Casado-Sánchez A., Cabrera S., Pérez-Ruiz R., Liras M., de la Peña O‘Shea V. A., Alemán J., ACS Catal. 2018, 8, 5928–5940. [Google Scholar]
- 5. Burg F., Bach T., J. Org. Chem. 2019, 84, 8815–8836. [DOI] [PubMed] [Google Scholar]
- 6. Tröster A., Alonso R., Bauer A., Bach T., J. Am. Chem. Soc. 2016, 138, 7808–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Hölzl-Hobmeier A., Bauer A., Silva A. V., Huber S. M., Bannwarth C., Bach T., Nature 2018, 564, 240–243; [DOI] [PubMed] [Google Scholar]
- 7b. Tröster A., Bauer A., Jandl C., Bach T., Angew. Chem. Int. Ed. 2019, 58, 3538–3541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3576–3579. [Google Scholar]
- 8.Reviews:
- 8a. Akiyama T., Chem. Rev. 2007, 107, 5744–5758; [DOI] [PubMed] [Google Scholar]
- 8b. Terada M., Synthesis 2010, 1929–1962; [Google Scholar]
- 8c. Parmar D., Sugiono E., Raja S., Rueping M., Chem. Rev. 2014, 114, 9047–9153; [DOI] [PubMed] [Google Scholar]
- 8d. Maji R., Mallojjala S. C., Wheeler S. E., Chem. Soc. Rev. 2018, 47, 1142–1158. [DOI] [PubMed] [Google Scholar]
- 9.Recent examples:
- 9a. Li J. Q., Grosslight S., Miller S. J., Sigman M. S., Toste F. D., ACS Catal. ACS Cat. 2019, 9, 9794–9799; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Wang L., Zhong J. L., Li X. F., Angew. Chem. Int. Ed. 2019, 58, 15824–15828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15971–15975; [Google Scholar]
- 9c. Sun M., Ma C., Zhou S.-J., Lou S. F., Xiao J., Jiao Y. C., Shi F., Angew. Chem. Int. Ed. 2019, 58, 8703–8708; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8795–8800; [Google Scholar]
- 9d. Lin J.-S., Li T.-T., Liu J.-R., Jiao G.-Y., Gu Q.-S., Cheng J.-T., Guo Y.-L., Hong X., Liu X.-Y., J. Am. Chem. Soc. 2019, 141, 1074–1083; [DOI] [PubMed] [Google Scholar]
- 9e. Suneja A., Schneider C., Org. Lett. 2018, 20, 7576–7580. [DOI] [PubMed] [Google Scholar]
- 10. Hatano M., Horibe T., Ishihara K., J. Am. Chem. Soc. 2010, 132, 56–57. [DOI] [PubMed] [Google Scholar]
- 11. Davis E. G., Smiles S., J. Chem. Soc. Trans. 1910, 97, 1290–1299. [Google Scholar]
- 12.
- 12a. Monaco M. R., Poladura B., Diaz de Los Bernardos M., Leutzsch M., Goddard R., List B., Angew. Chem. Int. Ed. 2014, 53, 7063–7067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7183–7187; [Google Scholar]
- 12b. Monaco M. R., Prévost S., List B., Angew. Chem. Int. Ed. 2014, 53, 8142–8145; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8280–8283; [Google Scholar]
- 12c. Monaco M. R., Fazzi D., Tsuji N., Leutzsch M., Liao S., Thiel W., List B., J. Am. Chem. Soc. 2016, 138, 14740–14749. [DOI] [PubMed] [Google Scholar]
- 13. Piva-Le Blanc S., Hénon S., Piva O., Tetrahedron Lett. 1998, 39, 9683–9684. [Google Scholar]
- 14. Yanagisawa Y., Nishiyama Y., Tanimoto H., Morimoto T., Kakiuchi K., Tetrahedron Lett. 2014, 55, 2123–2126. [Google Scholar]
- 15.For racemic photocycloaddition reactions of substrate 12, see:
- 15a. Agostas W. C., Lowrance W. W., Tetrahedron Lett. 1969, 10, 3053–3054; [Google Scholar]
- 15b. Agosta W. C., Lowrance W. W., J. Org. Chem. 1970, 35, 3851–3856; [Google Scholar]
- 15c. Lange G. L., Decicco C., Tan S. L., Chamberlain G., Tetrahedron Lett. 1985, 26, 4707–4710; [Google Scholar]
- 15d. Webster F. X., Silverstein R. M., J. Org. Chem. 1986, 51, 5226–5231; [Google Scholar]
- 15e.ref. [13];
- 15f. Tsutsumi K., Nakano H., Furutani A., Endou K., Merpuge A., Shintani T., Morimoto T., Kakiuchi K., J. Org. Chem. 2004, 69, 785–789. [DOI] [PubMed] [Google Scholar]
- 16. Griffin J. D., Zeller M. A., Nicewicz D. A., J. Am. Chem. Soc. 2015, 137, 11340–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poplata S., Bach T., J. Am. Chem. Soc. 2018, 140, 3228–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Determined at 77 K in methylcyclohexane: Iyer A., Clay A., Jockusch S., Sivaguru J., J. Phys. Org. Chem. 2017, 30, e3738. [Google Scholar]
- 19.Examples:
- 19a. Magnifico M., O'Connell E. J., Fratini A. V., Shaw C. M., J. Chem. Soc. Chem. Commun. 1972, 1095–1096; [Google Scholar]
- 19b. Blok P. M. L., Dekkers H. P. J. M., Photochem. Photobiol. 1991, 53, 421–429; [Google Scholar]
- 19c. Blok P. M. L., Jacobs H. J. C., Dekkers H. P., J. Am. Chem. Soc. 1991, 113, 794–801; [Google Scholar]
- 19d. Kelly J. F. D., Doyle M. E., Guha M., Kavanagh P. V., Kelly J. M., McMurry T. B. H., J. Chem. Soc. Perkin Trans. 2 1998, 1635–1642; [Google Scholar]
- 19e. Bagnicha S. A., Khropika N. N., Knyukshtoa V. N., Mikhalchukb A. L., Rubinovb D. B., Spectrochim. Acta Part A 2005, 61, 2161–2167. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Blum T. R., Miller Z. D., Bates D. M., Guzei I. A., Yoon T. P., Science 2016, 354, 1391–1395; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Daub M. E., Jung H., Lee B. J., Won J., Baik M.-H., Yoon T. P., J. Am. Chem. Soc. 2019, 141, 9543–9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Steiner T., Angew. Chem. Int. Ed. 2002, 41, 48–76; [Google Scholar]; Angew. Chem. 2002, 114, 50–80; [Google Scholar]
- 21b. Sorgenfrei N., Hioe J., Greindl J., Rothermel K., Morana F., Lokesh N., Gschwind R. M., J. Am. Chem. Soc. 2016, 138, 16345–16354; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Melikian M., Gramüller J., Hioe J., Greindl J., Gschwind R. M., Chem. Sci. 2019, 10, 5226–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andersen N. H., Neidigh J. W., Harris S. M., Lee G. M., Liu Z., Tong T., J. Am. Chem. Soc. 1997, 119, 8547–8561. [Google Scholar]
- 23.In the presence of 9, proton H3 is overlapped by proton H4 However, at 180 K, the detection of H3 is feasible and its chemical shift remains unchanged in presence or absence of 4. (see Supporting Information for spectra).
- 24. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 25. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 26.M. J. Frisch et al. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009.
- 27. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104-1-154104-19. [DOI] [PubMed] [Google Scholar]
- 28. Grimme S., Chem. Eur. J. 2012, 18, 9955–9964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary