

Abstract
Ene‐reductases allow regio‐ and stereoselective reduction of activated C=C double bonds at the expense of nicotinamide adenine dinucleotide cofactors [NAD(P)H]. Biological NAD(P)H can be replaced by synthetic mimics to facilitate enzyme screening and process optimization. The ene‐reductase FOYE‐1, originating from an acidophilic iron oxidizer, has been described as a promising candidate and is now being explored for applied biocatalysis. Biological and synthetic nicotinamide cofactors were evaluated to fuel FOYE‐1 to produce valuable compounds. A maximum activity of (319.7±3.2) U mg−1 with NADPH or of (206.7±3.4) U mg−1 with 1‐benzyl‐1,4‐dihydronicotinamide (BNAH) for the reduction of N‐methylmaleimide was observed at 30 °C. Notably, BNAH was found to be a promising reductant but exhibits poor solubility in water. Different organic solvents were therefore assayed: FOYE‐1 showed excellent performance in most systems with up to 20 vol% solvent and at temperatures up to 40 °C. Purification and application strategies were evaluated on a small scale to optimize the process. Finally, a 200 mL biotransformation of 750 mg (R)‐carvone afforded 495 mg of (2R,5R)‐dihydrocarvone (>95 % ee), demonstrating the simplicity of handling and application of FOYE‐1.
Keywords: biocatalysis, biotransformations, cofactor mimics, Old Yellow Enzymes, oxidoreductases, solvent stability
Enantioselective hydrogenation: Ene‐reductases (ERs, OYEs) are versatile biocatalysts for selective hydrogenation of activated C=C double bonds. FOYE‐1 was characterized with respect to solvent stability for the conversion of (R)‐carvone into (2R,5R)‐dihydrocarvone at the expense of the synthetic nicotinamide cofactor 1‐benzyl‐1,4‐dihydronicotinamide, the activity of which is compared with that of the natural electron donor NAD(P)H.

Introduction
The monoterpenoid (2R,5R)‐dihydrocarvone serves as a chiral building block for the synthesis of natural products such as sesquiterpenes or striatenic acid, copolymers, and antimalarial drugs.1, 2, 3 It is naturally present in dill oil and caraway seeds, but cannot be obtained through extraction.4 On a large scale, it is produced either by carvone hydrogenation or by limonene oxidation.3 Because of its industrial relevance, its production through several strategies has been investigated.2, 3, 5 Among those strategies, the use of biocatalysts has been described, and here the ene‐reductases (ERs) acting on (R)‐carvone are preferred (Scheme 1).6 Despite the catalytic power and selectivity of those enzymes, the employment of ERs as biocatalysts poses several challenges that need to be addressed.5b, 6 Firstly, these enzymes require reducing power that is naturally obtained from NAD(P)H. This cofactor can be recycled in situ in whole‐cell and cell‐free systems2, 7 or can be replaced by nicotinamide cofactor mimics in cell‐free systems.6d, 8 Secondly, in this case the substrate and product (carvone and dihydrocarvone) are poorly soluble in water and so cosolvents are necessary and need to be implemented in the system used.9 These cosolvents act as a substrate reservoir and as an extraction solvent for in situ product removal. Thirdly, carvones are reported to have toxic or antimicrobial activity,10 and this limits the application of whole‐cell biotransformation without further process engineering.2, 9 Finally, ER‐based whole‐cell biotransformation can result in by‐products caused by racemization.2 Therefore, cell‐free ER approaches in combination with a simple source of reducing power might be favored for small‐scale studies in order to obtain highly pure products rapidly. Here the cost‐effective 1‐benzyl‐1,4‐dihydronicotinamide (BNAH) has been used.
Scheme 1.

Stereoselective reduction of (R)‐carvone through the action of an ene‐reductase (ER). The nicotinamide (biological or synthetic, NA) acts as an electron donor to reduce the flavin cofactor FMN of the ER; this subsequently allows the transfer of a hydride to Cα of the unsaturated substrate. A proton from a conserved Tyr residue in the ER is added to Cβ to yield (2R,5R)‐dihydrocarvone.2, 6
Ene‐reductases of the Old Yellow Enzyme family (OYEs) are flavin‐dependent enzymes that employ nicotinamide cofactors for an initial flavin reduction followed by a direct trans‐hydrogenation of the substrate. A classic example would be the reduction of (R)‐carvone to (2R,5R)‐dihydrocarvone (Scheme 1).2, 6, 9, 11, 12 The substrates of ERs typically need to contain an activated C=C double bond that can be hydrogenated, generating up to two stereogenic centers.6 To study and evaluate ERs, maleimides were employed first because these are cost‐effective and soluble in buffer and do not interfere with the cosubstrate or cofactor. However, it was shown that they can form covalent adducts with the cysteine residues frequently found in the active sites of ERs.13 This leads to a rapid inactivation and can be circumvented either by site‐directed mutagenesis or by avoiding enzyme incubation in the presence of those maleimides.
Thermo‐ and solvent stability are important factors to consider if an enzyme is to be employed in industrial biocatalysis.6, 9, 13, 14 Here, the above‐mentioned substrate‐based inactivation due to covalent modification should be avoided. Additionally, the enzyme must be stable over time to allow high turnover numbers and increased rates through the use of higher reaction temperatures and/or the possibility of adding cosolvents. The use of cosolvents is furthermore important for solubilization of the (co)substrates carvone and 1‐benzyl‐1,4‐dihydronicotinamide (BNAH), for example.
In this study we describe the recently identified thermostable ene‐reductase FOYE‐114b with respect to applied biocatalysis in order to show its potential to produce an optically pure compound of industrial interest: (2R,5R)‐dihydrocarvone.1, 2, 3, 4, 5, 9, 10, 11 This procedure provides access to chiral and highly pure ER products, as well as to simple‐to‐handle and scalable biotransformations. The activity of FOYE‐1 with efficient usage of BNAH is described for the first time. In addition, its enzymatic activity and stability in the presence of various organic solvents were investigated.
Results and Discussion
Enzyme production and specific activity
The protein FOYE‐1 was produced in a yield similar to that described previously: 4.2 mg per L fermentation broth in an unoptimized approach based on gene expression.14b Despite indications of the formation of inclusion bodies (Figure S1 in the Supporting Information), the total amount (8.4 mg from 2 L expression culture) allowed catalytic properties and stability issues to be investigated, as well as various biotransformations to be set up.
Initially, the protein was purified by Ni‐affinity chromatography and total activity was determined as described previously.14b The specific activity of the enzyme [U mg−1] is by convention given as the rate either of substrate consumption or of corresponding product formation [μmol min−1] per mg enzyme. It amounted to a specific activity of 60.8 U mg−1 under standard conditions (50 mm KH2PO4/Na2HPO4 buffer, pH 7.1, 1 mm maleimide (1), 200 μm NADPH; NADPH consumption) with respect to the total protein amount of the preparation. This is in congruence with earlier investigations (65.4 U mg−1) and showed the reproducibility of FOYE‐1 production. The chromatographically enriched protein was found to be only partially saturated with the flavin mononucleotide (FMN) cofactor (46.5 % of monomer loaded with FMN for the batch used for most experiments presented herein). Protein production was repeated three times and the obtained purified protein showed FMN saturations in the range of 30 to 50 %, respectively. Loading the remaining apoprotein by addition of excess FMN, followed by incubation and washing steps to remove surplus free FMN, was partially successful. In cases of excess FMN over total protein, the reaction rate could be increased by a factor of 1.3 to 1.7 depending on the protein batch. Accordingly, a maximum FMN load of 60 to 80 % of purified FOYE‐1 was reached while the specific activity remained stable. Thus, only a fraction of the total protein pool participated in catalysis, and specific activity calculations in this study are therefore generally based on the amount of protein with FMN‐saturated active sites. In addition, it needs to be mentioned that surplus FMN in solution can be reduced by the BNAH employed for biocatalysis. This might lead to side reactions such as unproductive hydrogen peroxide formation or FMNH2–protein interaction and make any kinetic data analysis or turnover calculations complicated.
FMN‐saturated FOYE‐1 (holoprotein) had an observed specific activity of 130.7 U mg−1 on maleimide (1) with NADPH as electron donor (Table 1). This calculation procedure was used for all subsequent activity data. The kinetic data fit to a clear kinetic profile according to the Michaelis–Menten model as is typical for ERs.6, 8a, 15 A number of potential substrates was screened and compared with the standard (Table 1, Figure S3). It was shown that maleimides are preferred substrates of class III ERs,6d, 14 with N‐methylmaleimide (2) in particular being converted efficiently.
Table 1.
Observed activity of FOYE‐1 on various substrates (Figure S3).
|
|
Substrate |
T [°C][a] |
Observed activity |
|---|---|---|---|
|
|
|
|
[U mg−1][b] |
|
1 |
maleimide |
20 |
130.7±1.6 |
|
2 |
N‐methylmaleimide |
20 |
144.7±3.7 |
|
|
|
22.5 |
172.0±18.0 |
|
|
|
30 |
264.1±2.2 |
|
3 |
N‐(2,4,6‐trichlorophenyl)maleimide |
22.5 |
11.6±0.3 |
|
4 |
indole‐2‐carboxylic acid |
22.5 |
n.d. |
|
5 |
3‐hydroxy‐2‐methylpyran‐4‐one |
22.5 |
n.d. |
|
6 |
mesaconic acid |
22.5 |
0.7±0.1 |
[a] Temperature was set to ambient conditions and kept constant during the assays. [b] Each assay mixture consisted of 50 mm KH2PO4/Na2HPO4 buffer (pH 7.1), 1 mm (compounds 1–3) or 10 mm (compounds 4–6) substrate, 200 μm NADPH, and 30 nm purified FOYE‐1, no additional FMN. The activity was calculated on the basis of FMN‐saturated FOYE‐1. n.d.=not detected.
FOYE‐1 was prepared and purified under different conditions to establish a simple preparation for biotransformation studies (Table S1, Figure S2). It became clear that either the direct use of crude extract or purified protein is preferable: without substantial purification the specific activity of FOYE‐1 is already roughly 62 % of the maximum reachable activity achieved by affinity chromatography, thus making this a potentially economic preparation for scaled‐up work.
With a partial FMN saturation of 46.5 %, the remaining apo‐FOYE‐1 in the cell‐free crude extract could be saturated with FMN and added to the pool of functional protein. To check this, the assays were repeated in the presence of additional FMN (10 to 70 μm). This resulted in an increase in units of active enzyme while the high specific activity was kept. The maximum achievable FMN saturation of about 80 % was already reached in the presence of 10 μm FMN in the relevant assays. With maleimide (1) as substrate, the observed activity increased from 64.5 mU to 109.7 mU (specific activity of 172 U mg−1) at 22 °C and from 99.0 mU to 168.3 mU (264 U mg−1) at 30 °C. It can thus be stated that FOYE‐1 is one of the most active OYEs reported for this substrate so far.6 Nevertheless, it needs to be mentioned that the surplus FMN was most likely not tightly bound to the protein, because a maximum saturation of only about 50 % FMN could be determined in the purified FOYE‐1 (cf. above).
Application of various electron donors
ERs are known to accept a variety of electron donors to reduce FMN, such as natural and synthetic nicotinamide cofactors (Scheme 1).6d, 8, 17 FOYE‐1 accepts both NADH and NADPH, with a clear preference for the phosphorylated form (Table 2).14b
Table 2.
Performance of FOYE‐1 with natural and artificial nicotinamide cofactors.
|
Electron donor [a] |
T [°C] |
Observed activity [U mg−1] [b] |
||
|---|---|---|---|---|
|
Conc. of electron donor [μm] |
200 |
300 |
1000 |
|
|
NADH |
20 |
7.3±0.7 |
8.3±0.3 |
8.6±0.2 |
|
NADPH |
20 |
140.5±0.4 |
141.5±2 |
163.1±2.4 |
|
BNAH |
20 |
76.0±1.2 |
93.6±1.6 |
174.1±2.7 |
|
NADH |
30 |
11.4±0.5 |
11.6±0.6 |
12.7±1.0 |
|
NADPH |
30 |
264.1±2.2 |
282.2±2.9 |
319.7±3.2 |
|
BNAH |
30 |
87.9±2.2 |
108.9±1.3 |
206.7±3.4 |
[a] The electron donor served as the substrate to initiate the reaction. [b] Each assay mixture consisted of 50 mm KH2PO4/Na2HPO4 buffer (pH 7.1), 1 mm 2, 200/300/1000 μm of the appropriate electron donor, and 8.6 nm FOYE‐1 (holoprotein).
With respect to the cost factor, the traditional synthetic cofactor mimic BNAH is an attractive electron donor6b, 8 because it can easily be obtained at comparably low cost [approximately €12.50 per g at 95 % purity (09/2019), in comparison with NADPH at about €615.00 per g at 95 % purity (09/2019)]. We thus studied the kinetics and solvent stability of FOYE‐1 with BNAH because this cofactor mimic is only partially soluble in aqueous buffers. In a first standardized test, BNAH was compared with NADH and NADPH at 20 and 30 °C (Table 2). NADPH was found to be the best electron donor, especially at elevated temperatures. About three to five times more BNAH is required in order to achieve similar activities.
The enzyme kinetics relating to BNAH as the electron donor and N‐methylmaleimide (2) as main substrate (Figure 1, Table 3) confirmed our assumption: more than 1 mm BNAH is needed to saturate the active site of FOYE‐1, due to the high K m value of about 600 μm. On comparing the kinetic parameters from the experiments varying either electron donor or substrate concentration while keeping the other constant, it became obvious that FOYE‐1 suffers from nonproductive uncoupling: the enzyme can employ only about 63 % of the reducing equivalents of BNAH to reduce N‐methylmaleimide, which is efficiently bound by the enzyme and converted at a high activity of about 188.6 U mg−1 under standard conditions (pH 7.1 and 22 °C). Interestingly, the catalytic turnover frequencies observed with mimics are the same as those seen with NADPH, or even higher, and only the ER XenA showed a comparable catalytic efficiency.6d, 14b, 17
Figure 1.

Michaelis–Menten kinetic analysis of FOYE‐1 with BNAH as cosubstrate. The standard enzyme assay was performed as described in the Experimental Section, in KH2PO4/Na2HPO4 buffer at 22.5 °C. FOYE‐1 (8.6 nm, 0.375 μg mL−1, holoprotein) was used without the addition of extra FMN. The concentration of A) BNAH, or B) substrate 2 was varied (BNAH 0–1200 μm, 2 0–200 μm), while the other was kept in excess. Data were analyzed by nonlinear fitting of the Michaelis–Menten equation with the aid of the KaleidaGraph software package (Table 3).
Table 3.
Kinetic parameters of FOYE‐1 with respect to BNAH and N‐methylmaleimide (2).
|
Substrate[a]/ |
K m |
V max |
k cat |
k cat/K m |
|---|---|---|---|---|
|
conditions |
[μm] |
[U mg−1] |
[s−1] |
[s−1 mm −1] |
|
BNAH |
600±19 |
298±5 |
216±4 |
360 |
|
BNAH/20 % acetone[b] |
1133±80 |
59.8±2.7 |
43.4±2 |
38 |
|
N‐methylmaleimide (2) |
7.3±0.7 |
188.6±3.5 |
136.7±2.5 |
18 726 |
The standard enzyme assay as described in the Experimental Section was performed in KH2PO4/Na2HPO4 buffer at 22.5 °C; 8.6 nm (0.375 μg mL‐1, holoprotein) FOYE‐1 was used without the addition of extra FMN. [a] The substrate concentration was varied (BNAH 0–1200 μm and 2 0–200 μm) while the other one was kept in excess accordingly. [b] The same experiment was repeated in the presence of 20 vol% acetone.
Knaus et al. determined the steady‐state kinetics for three ERs: PETNR, TOYE, and XenA.17 In that work the enzyme performance with the natural nicotinamide electron donors in comparison with synthetic mimics was determined under substrate saturation (cyclohex‐2‐en‐1‐one) conditions at 30 °C. Similar experiments were performed with OYERo2a and use of N‐methylmaleimide (2) as the substrate at 25 °C.13a From these data sets, the following order of catalytic efficiency can be deduced (under substrate saturation conditions and with BNAH as electron donor): XenA (≈1860 s−1 mm −1)>FOYE‐1 (≈20 %)>OYERo2a (≈17 %)>PETNR≈TOYE (4 %). With respect to turnover frequency, FOYE‐1 is the most powerful ER of the set (k app four to 40 times higher than those of the other ERs listed).13a, 17
Upon addition of 20 vol% acetone during kinetic experiments to enhance BNAH solubility (Table 3), the activity decreased by a factor of 5 and also the affinity for the cosubstrate was lowered significantly (factor of 2). It might be reasoned that the cosolvent affects the protein structure or even its stability, thus limiting its applicability. The effect of organic solvents on the performance of FOYE‐1 needed to be further elucidated (see below).
To evaluate the cosubstrate and its effect on biocatalysis, small‐scale biotransformations were performed. A number of substrates was tested to demonstrate the scope and selectivity with respect to various electron donors (Figure S3, Table S2). Varying the electron donor did not influence the enzyme's selectivity: in all cases the same enantiomers or diastereomers were obtained, mostly with similar ee values. The determined ee values were generally acceptable, with substrates 2‐methyl‐N‐phenylmaleimide (9), (R)‐carvone (12), (S)‐carvone (13), dimethylcitraconic acid (14), and dimethylitaconic acid (16) being hydrogenated with ee values above 90 % (for substrate structures see Figure S3). Products obtained at higher turnover frequencies can racemize in solution over time and thus gave moderate (compound 10) to low (compound 7) ee values with the natural electron donor and afforded slightly lower enantiopurity with the synthetic electron donors, possibly due to even higher turnover frequencies.17 With respect to conversion, no significant difference was observed. It seems that, in general, mimics can efficiently replace the natural electron donor (NADPH) for FOYE‐1. Strikingly, (R)‐carvone (12) was converted in a highly selective manner (97 % optical purity, 14–18 % conversion) and thus became the model substrate for further investigations.
Activity and stability of FOYE‐1 in the presence of organic solvents
When employing unnatural nicotinamide mimics and various organic substrates, the use of cosolvents in order to bring both to the active site of the enzyme in a sufficiently high concentration while keeping the enzyme at work is essential.6, 8 FOYE‐1 was reported to be stable at elevated temperatures,14b and this often correlates with higher general stability.18, 19 We thus investigated its enzymatic activity and stability towards various organic solvents.
Typical solvents used in biocatalysis were analyzed in terms of compatibility with the enzymatic reaction over a concentration range of 0–60 vol%, with both natural and mimic cosubstrates (Figure 2). For the NADPH data set (Figure 2 A), most solvent candidates, including ethanol, methanol, acetone, isopropanol, DMSO, and THF, caused no significant loss in activity when supplemented to a final volume of 20 % of the assay. Increases of up to 120 % relative activity were even found in few cases; similar observations have been reported previously for other ERs.14a, 17, 18 Even at 40 vol% solvent concentration the enzyme showed more than 50 % relative activity for most additives. This is outstanding in comparison with other ERs. Only the presence of acetonitrile led to significant enzyme inactivation even at low concentrations. It can thus be concluded that FOYE‐1 is stable against a variety of useful cosolvents and can even become more active under those conditions. In particular, ethanol, acetone, or isopropanol are promising candidates because they each increase the relative enzymatic activity to a certain extent and can be used up to 30–40 vol% to support substrate and electron donor solubility. With respect to the use of NAD(P)H as source of electrons for ERs (in vitro), dehydrogenases are necessary to recycle these electron donors, and these enzymes accept ethanol or isopropanol.20 Thus the application of those cosolvents not only supported the activity of FOYE‐1 but could also provide access to efficient NAD(P)H recycling.
Figure 2.
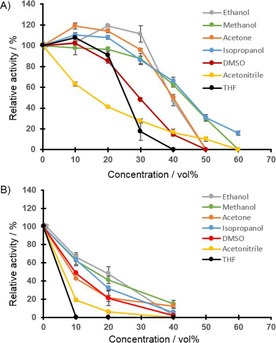
FOYE‐1 activity in presence of cosolvents. The standard enzyme assay was performed while the concentrations of solvents were varied, initial rates were determined. A) NADPH (200 μm), or B) BNAH (1000 μm) served as electron donor; N‐methylmaleimide (2, 1 mm) was used as substrate. Data are shown as values relative to an enzyme assay without cosolvents [A) 100 %=140 U mg−1, B) 100 %=170 U mg−1].
We furthermore analyzed FOYE‐1 activity in the presence of organic solvents and BNAH as the electron donor (Figure 2 B). In all cases the enzymatic activity dropped. THF and acetonitrile in particular led to reduced enzyme performance in terms of initial activity. Still, ethanol and methanol can be used up to 20 vol%, with 50 % relative activity being retained. The other tested solvents—isopropanol, acetone, and DMSO—allowed moderate enzyme performance between 10 and 20 vol% added solvent. At this point it is questionable why an exchange of electron donor led to this reduction in enzymatic activity. It should be noted that the enzyme's stability appears to be unaffected, because with NADPH as source of reducing power a much better performance was found (see also below and Figure S4). Thus, the combination of organic solvents and BNAH must cause this loss of activity and this remains to be investigated in more detail. For this study, it was crucial to show that FOYE‐1 can work under those conditions and allows the selective conversion of a desired substrate.
For the next step, we studied the storability of the enzyme for more than 24 h in the presence of cosolvents (20 vol%, Figure S4). The enzyme is stable in 20 vol% ethanol, acetone, or isopropanol for more than 24 h when kept on ice. No loss in activity was determined; in the first 2 h an increase of activity was even observed. Furthermore, the enzyme activity was studied at various temperatures (Figure S4), and results similar to those published previously were obtained.14b Up to 40 °C the enzyme is highly active, but at higher temperatures the activity decreases significantly.
The observation that FOYE‐1 and other ER enzyme activity can be enhanced by adding solvents might have two explanations: 1) increased solubility, and thus availability, of (co)substrates, and/or 2) increased flexibility of the enzyme itself. We believe that the second factor is most important, because the employed (co)substrates, except for BNAH, in these experiments were generally soluble in buffer (50 mm phosphate buffer). We thus conclude that, on the one hand, the presence of a cosolvent (up to 30 % ethanol, acetone, or isopropanol) can lead to beneficial structural changes or to more mobility in the protein, most likely due to altered solvent accessibility or a changed hydrogen bond network in the protein (backbone). On the other hand, this introduced flexibility might hamper the binding of nicotinamide mimics such as BNAH that display per se weaker binding to the active site, as is obvious from the K m values of 72 μm NADPH versus 600 μm BNAH.14b In the presence of acetone the K m value for BNAH is even doubled (Table 3), which supports this hypothesis. Therefore, we can conclude that FOYE‐1 is stable towards various organic solvents but that these affect the performance of the enzyme in a manner dependent on the electron donor employed.
Biotransformation of (R)‐carvone
The conversion of (R)‐carvone (12) through the action of ene‐reductases is known and was therefore used here as a model reaction for an industrially relevant substrate.2, 6 In order to rule out the possibility that the applied cosubstrates NADPH or BNAH promote the direct reduction of (R)‐carvone, the corresponding standard assays were performed in the absence of FOYE‐1. Neither (R)‐carvone reduction nor dihydrocarvone production was determined by GC‐FID (data not shown).
To provide a first view of the enzymatic performance in a scaled‐up process, the consumption of BNAH over time was assayed (Figure S5). Here, crude extract and purified enzyme preparations were compared (each about 1 μm FOYE‐1 as holoprotein). It was determined that 10 mm BNAH were rapidly consumed and mostly converted within 2 h. Both preparations performed similarly, with the crude extract preparation being slightly slower. However, (R)‐carvone (12) was not fully converted at this stage (not shown). This indicates that BNAH might need to be added stepwise and a fed‐batch‐like biotransformation established.
This information was used to set up two 10 mL biotransformations of (R)‐carvone (12), one with chromatographically enriched FOYE‐1 obtained from Ni‐affinity chromatography and one containing a crude extract preparation, each with about 1 μm holoprotein. The concentration of BNAH was set to 10 mm at the beginning, and then addition of solid BNAH, corresponding to 7.5 mm, was carried out hourly to achieve maximum substrate conversion into the desired product (Figure 3, Table S3). After 2 h, 21 % conversion of 12 into (2R,5R)‐dihydrocarvone was achieved with >99 % ee in the case of the chromatographically enriched protein preparation. The level of conversion reached a maximum of 23 % (>97 % ee of the product) after 8 h. Surprisingly, on employment of the cell‐free crude extract preparation, 42 % conversion (>98 % ee of the product) were already reached within 2 h. This indicates that a cell‐free crude extract containing the ER is sufficient to produce large quantities of product quickly and in decent purity. If necessary, however, the one‐step purification method allows slightly higher product enantiopurity to be achieved at the price of lower conversion rates.
Figure 3.

Biotransformation of (R)‐carvone (12) through the action of FOYE‐1 with different protein preparations, 10 mL scale. For comparison, A) a chromatographically enriched, or B) a crude extract preparation was employed; 1 μm enzyme was applied to convert 5 mm (7.5 mg) substrate 12 while the electron donor BNAH was fed stepwise (initial 10 mm+7.5 mm h−1 in solid form). Substrate and products were analyzed by chiral GC.
Finally, an experiment was performed at 750 mg scale to show the scalability and accessibility of ER products. Thus, 751 mg (R)‐carvone (12) in a 200 mL reaction volume, corresponding to 5 mmol, were converted under optimal conditions for FOYE‐1: 15 vol% acetone in phosphate buffer at 30 °C. The enzyme was applied as a crude extract preparation, comprising about 1.5 μm FMN‐loaded FOYE‐1. BNAH was provided from the beginning and fed over time to achieve high substrate conversion. BNAH was completely consumed at the end of the reaction. The reaction was stopped after 8 h and the mixture was extracted with n‐pentane (>99 % extraction yield, 750 mg dried product/substrate mix). The actual yield and purity were determined by NMR (Figures S7 and S8) and GC‐FID. This allowed differentiation between substrate and product (both possible diastereomers). Thus, a final extracted product in the form of 495 mg (2R,5R)‐dihydrocarvone, corresponding to a 65 % yield, was obtained. The optical purity achieved was 95 % in the final preparation and thus comparable to that of the small‐scale biotransformation (Table S3). This shows that scale‐up and changing the conditions (higher solvent concentration and use of a BNAH feeding strategy) does not significantly affect the selectivity in FOYE‐1 biocatalysis. Moreover, the bioconversion could be performed without adding an organic cosolvent: by employing pure (R)‐carvone as second phase, conveniently serving as a reservoir for BNAH, a substrate load of 100 g L−1 (10 %, v/v in overall volume) was achieved, resulting in conversion rates similar to those before (data not shown).
Conclusion
FOYE‐1 production is reproducible and simple to achieve. The protein can be applied as a crude preparation in semisynthetic approaches so chromatographic enrichments or even purification by sophisticated chromatographic methods are avoided. The enzyme is oxygen‐insensitive and accepts various nicotinamide cofactor mimics as electron donors. It is stable at elevated temperatures (30 to 40 °C) even in the presence of various organic solvents such as acetone or isopropanol (up to 30 vol%), allowing efficient in situ NAD(P)H regeneration by alcohol dehydrogenases (not shown).20 With NADPH employed as reductant, FOYE‐1 has one of the highest ER activities reported: 264 to 320 U mg−1 (here on maleimides) at 30 °C. Even with BNAH an activity of 88 to 207 U mg−1 for the conversion of 2 was observed. The use of BNAH as artificial cosubstrate somewhat limits the applicability of organic solvents due to overall lower reaction rates. Nevertheless, for experiments at various scales BNAH is still favored because a cost reduction by a factor of 50 can be achieved just by using mimetic reducing equivalents. In the case of the 200 mL biotransformation performed here this amounted to a cost of about €130 for BNAH whereas NADPH would have cost €6400 from commercial suppliers. The costs can be further reduced by synthesizing BNAH freshly, as was done here. Because FOYE‐1 showed long lifetimes in the solvents used, the reaction time can be increased to compensate lower turnover rates. Of the tested substrates, (R)‐carvone seemed a promising candidate for conversion into a valuable product because the production of (2R,5R)‐dihydrocarvone—our model compound here—was achieved at high conversion rates (up to 65 % extracted product in 8 h with use of a crude preparation) and with high optical purity (>95 %).
To sum up, it was shown for the first time that FOYE‐1 acts efficiently with BNAH as electron donor and is stable and active in various organic solvents. Even though the addition of cosolvents is necessary and lowers the enzyme activity when BNAH is used, this approach allows simple conversion of selected substrates for biochemists, chemists, and others. FOYE‐1 can thus be considered a valuable and easily accessible addition to the toolbox for selective hydrogenation reactions of C=C double bonds.
Experimental Section
Chemicals: NADPH and NADH were purchased from Prozomix. BNAH was freshly synthesized as described previously and assessed for stability by UV/Vis spectroscopy prior to each application.13a All other chemicals were purchased from Sigma–Aldrich or TCI Europe (Belgium or Germany) at highest purity available.
Enzyme production: The previously described expression vector pET_FOYE_01 was checked by sequencing with pET primers and freshly transformed into Escherichia coli BL21(DE3) for gene expression.14b, 21 A single colony was used to inoculate precultures (50 mL) consisting of lysogeny broth (LB) medium [tryptone (10 g L−1), yeast extract (5 g L−1), NaCl (10 g L−1)] and appropriate antibiotics [ampicillin (100 μg mL−1) and chloramphenicol (50 μg mL−1)]. These were incubated at 37 °C overnight and used to inoculate main cultures (each 500 mL) of LBNB medium [tryptone (10 g L−1), yeast extract (5 g L−1), NaCl (29.2 g L−1), glucose (2 g L−1), as well as betaine (1 mm)] and antibiotics in same final concentrations as described above. The cultivation was routinely monitored by measuring the optical density (OD) at 600 nm against a medium blank. The incubation was maintained at 37 °C until an OD600 of about 0.4 was reached, the mixture was then allowed to cool to 22 °C, and gene expression was induced at an OD600 of about 0.6 by addition of isopropyl β‐d‐1‐thiogalactopyranoside (IPTG, 0.1 mm final concentration). The protein production was continued overnight at these conditions, and cells were finally harvested by centrifugation (4200 g, 40 min). The cell pellet obtained was resuspended in phosphate buffer (50 mm Na2HPO4/ KH2PO4, pH 7.1) and stored in aliquots at −20 °C until further processing.
Enzyme purification: The cell pellet was thawed and treated with an ultrasonic probe on ice (10 cycles of 30 s treatment at 50 % power and output by means of a probe, 60 s pause between each treatment to cool the sample, Branson Sonifier 250). DNase I (grade II, 1 mg mL−1, 5 μL) was added to degrade the DNA prior to further processing. Various routes to prepare different purification grades of the biocatalyst were then followed: 1) crude extract, 2) enrichment by affinity chromatographically, and 3) ammonium sulfate precipitation (cf. Supporting Information). The purity of protein samples was analyzed by means of SDS‐PAGE as reported earlier.14
1) The crude extract sample was prepared as follows: after sonication and centrifugation (17 000 g for 40 min at 4 °C) the sample was aliquoted in fractions (1 mL) and flash‐frozen with liquid nitrogen. The crude extract preparation was then stored at −20 °C.
2) The chromatographically enriched sample was prepared by means of Ni‐affinity chromatography from crude extract samples as follows: centrifugation of cell debris at 17 000 g for 40 min at 4 °C gave a cell‐free crude extract. The supernatant was then filtered with a syringe filter (0.2 μm pore size). Protein purification was achieved with a HisTrap HP column (5 mL) and use of an ÄKTA device (both GE Healthcare) as described earlier.14b During this procedure, the removal of nonspecific proteins was achieved by first rinsing with binding buffer (10 mm Tris⋅HCl, 500 mm NaCl). A washing step was performed with 50 mm imidazole containing buffer (10 % of elution buffer; 10 mm Tris⋅HCl, 500 mm NaCl, 500 mm imidazole). Elution of the target protein FOYE‐1 was achieved with a linear imidazole gradient from 50 to 500 mm over eight column volumes. Fractions containing the enzyme (monitored by the yellow color due to the presence of the flavin cofactor; can be determined at 460 nm; protein elution was also followed at 280 nm) were collected, pooled, and subsequently concentrated by use of an ultrafiltration device with a molecular weight cut‐off of 30 kDa. Protein aliquots were stored at −20 °C in phosphate buffer (Na2HPO4/KH2PO4, 50 mm, pH 7.1) containing glycerol (50 vol%).
Protein, cofactor, and activity determination: The protein concentration was measured by the Bradford method22 with use of bovine serum albumin (BSA) as standard to generate a calibration curve. Absorbance of standard and FOYE‐1 samples was measured at 595 nm with a plate reader (Bio‐Tek Instruments μQuant) against a blank of the corresponding buffer.
The loading of FOYE‐1 with the cofactor FMN was determined by FMN quantification directly from the protein samples. A known protein dilution was prepared in a quartz cuvette (1 mL) and the absorbance was measured at 460 nm (extinction coefficient 12.5 mm −1 cm−1)14a, 17, 23 Saturation of the protein with FMN was calculated by comparing the protein concentration and amount of FMN in the samples.
Loading of FOYE‐1 with additional FMN was tried by various approaches. Firstly, FMN was provided either prior to or after Ni‐affinity chromatography in excess over estimated FOYE‐1 concentration. Surplus FMN was removed by ultrafiltration as described above. However, this did not yield a fully FMN‐saturated protein pool and was therefore not further investigated. In addition, the purified FOYE‐1 was assayed for NADPH or BNAH consumption with excess FMN (10 to 70 μm, see below for assay details). The increase in rate was used as a factor representing the amount of participating active sites. Also in this case, only partial saturation was achieved.
The ene‐reductase activity assay for FOYE‐1 was performed as described previously.14 The assay was carried out in quartz cuvettes (1 mL) and the consumption of NADPH was measured spectrophotometrically at 340 nm (extinction coefficient 6.22 mm −1 cm−1, Figure S6, example shown for different protein samples). In the case of BNAH the same procedure was followed. For kinetic experiments BNAH stocks were prepared in methanol, whereas for biotransformation studies it was used in solid form. Extinction was measured at 340 nm (extinction coefficient 4.75 mm −1 cm−1) in a concentration range from 0 to 350 μm; above this concentration of BNAH, measurement was carried out at 395 nm (extinction coefficient 1.70 mm −1 cm−1) to avoid detector saturation. The absorbance maximum of BNAH in the applied buffer was determined to be 358 nm (extinction coefficient 6.18 mm −1 cm−1). The assay mixtures contained final concentrations of KH2PO4/Na2HPO4 buffer (50 mm, pH 7.1), 1 mm substrate [mostly N‐methylmaleimide (2) or (R)‐carvone (12) used as standard], and 200 μm cosubstrate NADPH if not otherwise stated. Typically, the assay was performed at 22.5 °C, with all components being preheated prior to measurements. A final concentration of 8.6 nm (0.375 μg mL−1) holoprotein FOYE‐1 was added to start the reaction.
The enzyme activity is expressed in U mg−1, defined as follows: 1 unit represents the conversion of 1 μmol substrate per minute. This was correlated to the amount of FMN‐saturated FOYE (holoprotein) in mg.
Biotransformation: Small‐scale biotransformations were performed at 1 mL reaction volume in 2 mL microreaction tubes. The reactions were set up with KH2PO4/Na2HPO4 buffer (25 mm), holoenzyme (1.875 μm), the appropriate electron donor (NAD(P)H or mimic, 10 mm) and substrate (10 mm). Control experiments were performed with (R)‐carvone (12) as substrate in absence of protein. Tubes were shaken for 4 h at 30 °C at 800 rpm (thermoblock). Reactions were stopped by addition of ethyl acetate [500 μL, containing dodecane (5 mm) as an internal standard] and vortexing for 30 s. Afterwards, the tubes were centrifuged for 5 min at 10 800 g. The organic supernatant was isolated, dried with anhydrous magnesium sulfate, centrifuged, and transferred into GC vials for analysis.14a
The biotransformation of (R)‐carvone was performed in a final volume of 10 mL [solvent: KH2PO4/Na2HPO4 buffer (50 mm, pH 7.1) containing acetone (15 vol%)]. Substrate (5 mm, 7.5 mg) and cosubstrate (BNAH, initial concentration of 10 mm) were added. To start the reaction, FOYE‐1 (1 μm) was added to the preheated solution and the mixture was gently shaken at 30 °C. The BNAH concentration was increased every hour by addition of 7.5 mm in solid form. The formation of product was measured hourly by sampling and chiral GC‐FID analysis (see below). The samples drawn (300 μL each) were treated as described above before analysis by GC. The experiment was performed for 10 h. The final product could also be extracted by using n‐pentane and a subsequent concentration step under reduced pressure to obtain an enriched product fraction.
The same procedure was repeated at higher scale. A reaction volume of 200 mL in a round‐bottomed flask (250 mL) was prepared as described above in phosphate buffer: acetone (15 vol%), (R)‐carvone (25 mm, 751 mg), BNAH (25 mm initially + 7.5 mm hourly), and FOYE‐1 (1.5 μm). The reaction was started by adding the enzyme (1.5 μm FMN‐loaded FOYE‐1) and incubation was performed at 30 °C (with gentle shaking) until the conversion stopped after about 8 h.
Product identification: In cases of small‐scale biotransformations, the products and purity were identified by GC‐FID or HPLC‐UV/Vis analyses as described previously.14 Otherwise, substrate and product concentration were determined by GC‐FID with a Shimadzu 2010 GC system containing a Hydrodex β‐6TBDM column (Macherey–Nagel, Germany). Each sample was extracted with an equal volume of ethyl acetate containing octan‐1‐ol as internal standard. After extraction, the organic phase was dried with anhydrous magnesium sulfate and the supernatant was placed in an analytic vial for further measurement. The column temperature was kept constant at 120 °C and the measurement was performed for 10 min. Retention times were determined with the aid of authentic standards as follows: octan‐1‐ol 3.5 min, dihydrocarvone (R and S enantiomers) 5.2/5.7 min, and (R)‐carvone 7.7 min.
In addition, 1H NMR spectra (Bruker, Rheinstetten, Germany, DPX‐200 NMR) were recorded for the product obtained from the 200 mL experiment. For this, the reaction mixture was extracted with pentane; this gave a mixture of products and substrate suitable for 1H NMR analysis with BNAH left unextracted in the reaction mixture.
(R)‐Carvone: 1H NMR (200 MHz, [D6]DMSO): δ=7.00–6.67 (m, 1 H), 4.88–4.61 (m, 2 H), 2.76–2.54 (m, 1 H), 2.51–2.19 (m, 6 H), 1.78–1.63 ppm (m, 6 H).
(2R,5R)‐Dihydrocarvone: 1H NMR (200 MHz, [D6]DMSO): δ=4.88–4.51 (m, 2 H), 2.47–2.13 (m, 4 H), 2.13–1.95 (m, 1 H), 1.92–1.51 (m, 5 H), 1.40–1.15 (m, 1 H), 0.99–0.77 ppm (m, 3 H).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Saxonian Ministry of Science and Fine Arts and the European Union (EU) in the framework of the European Social Fund (ESF; project numbers 100101363 and 100236458). D.T., A.G.B., and C.M. were supported by the Federal Ministry for Innovation, Science and Research of North Rhine–Westphalia (PtJ‐TRI/1411ng006)—ChemBioCat.
D. Tischler, E. Gädke, D. Eggerichs, A. Gomez Baraibar, C. Mügge, A. Scholtissek, C. E. Paul, ChemBioChem 2020, 21, 1217.
References
- 1.
- 1a. Alarcon J., Lamilla C., Cespedes C. L., Ind. Crop. Prod. 2013, 42, 268–272; [Google Scholar]
- 1b. Aubin Y., Audran G., Monti H., Tetrahedron Lett. 2006, 47, 3669–3671; [Google Scholar]
- 1c. de Rouville H. P. J., Vives G., Tur E., Rapenne G., Crassous J., New J. Chem. 2009, 33, 293–299; [Google Scholar]
- 1d. Dong Y., McCullough K. J., Wittlin S., Chollet J., Vennerstrom J. L., Bioorg. Med. Chem. Lett. 2010, 20, 6359–6361; [DOI] [PubMed] [Google Scholar]
- 1e. Krawczyk H., Sliwinski M., Kedzia J., Wojciechowski J., Wolf W. M., Tetrahedron: Asymmetry 2007, 18, 2712–2718. [Google Scholar]
- 2. Castiglione K., Fu Y., Polte I., Leupold S., Meo A., Weuster-Botz D., Biochem. Eng. J. 2017, 117, 102–111. [Google Scholar]
- 3. Lowe J. R., Tolman W. B., Hillmyer M. A., Biomacromolecules 2009, 10, 2003–2008. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Gildemeister R., Hoffman F., The Volatile Oils, Vol. 1, 2nd ed. (Ed.: E. Kremers), Wiley, New York, 1913; [Google Scholar]
- 4b. Verghese J., Vihar A., Perfum. Flavor. 1980, 5, 23–26. [Google Scholar]
- 5.
- 5a. Raucher S., Hwang K.-J., Synth. Commun. 1980, 10, 133–137; [Google Scholar]
- 5b. Zhang B., Zheng L., Lin J., Wei D., Biotechnol. Lett. 2016, 38, 1527–1534. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Issa I. S., Toogood H. S., Johannissen L. O., Raferty J., Scrutton N. S., Gardiner J. M., Chem. Eur. J. 2019, 25, 2983–2988; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Knaus T., Toogood H. S., Scrutton N. S. in Green Biocatalysis (Ed.: R. N. Patel), Wiley, Hoboken, 2016, pp. 473–488; [Google Scholar]
- 6c. Mähler C., Burger C., Kratzl F., Weuster-Botz D., Castiglione K., Molecules 2019, 24, E2550, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Scholtissek A., Tischler D., Westphal A. H., van Berkel W. J. H., Paul C. E., Catalysts 2017, 7, 130; [Google Scholar]
- 6e. Toogood H. S., Scrutton N. S., Curr. Opin. Chem. Biol. 2014, 19, 107–115; [DOI] [PubMed] [Google Scholar]
- 6f. Toogood H. S., Scrutton N. S., ACS Catal. 2018, 8, 3532–3549; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6g. Winkler C. K., Faber K., Hall M., Curr. Opin. Chem. Biol. 2018, 43, 97–105. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Han L., Liang B., World J. Microbiol. Biotechnol. 2018, 34, 141; [DOI] [PubMed] [Google Scholar]
- 7b. Wang X., Saba T., Yiu H. H. P., Howe R. F., Anderson J. A., Shi J., Chem. Eur. J. 2017, 2, 621–654. [Google Scholar]
- 8.
- 8a. Falcone N., She Z., Syed J., Lough A., Kraatz H.-B., ChemBioChem 2019, 20, 838–845; [DOI] [PubMed] [Google Scholar]
- 8b. Guarneri A., van Berkel W. J. H., Paul C. E., Curr. Opin. Biotechnol. 2019, 60, 63–71; [DOI] [PubMed] [Google Scholar]
- 8c. Paul C. E., Hollmann F., Appl. Microbiol. Biotechnol. 2016, 100, 4773–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown G., Moody T. S., Smyth M., Taylor S. J. C. in Biocatalysis: An Industrial Perspective (Eds.: G. de Gonzalo, P. D. de María), 2017, The Royal Society of Chemistry, London, pp. 229–256. [Google Scholar]
- 10. de Carvalho C., da Fonseca M., Food Chem. 2006, 95, 413–422. [Google Scholar]
- 11.
- 11a. Peters C., Frasson D., Sievers M., Buller R. M. U., ChemBioChem 2019, 20, 1569–1577; [DOI] [PubMed] [Google Scholar]
- 11b. Stuermer R., Hauer B., Hall M., Faber K., Curr. Opin. Chem. Biol. 2007, 11, 203–213; [DOI] [PubMed] [Google Scholar]
- 11c. Winkler C. K., Tasnádi G., Clay D., Hall M., Faber K., J. Biotechnol. 2012, 162, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Haas E., Biochem. Z. 1938, 298, 369–390; [Google Scholar]
- 12b. Warburg O., Christian W., Naturwissenschaften 1932, 20, 688-688. [Google Scholar]
- 13.
- 13a. Scholtissek A., Gädke E., Paul C. E., Westphal A. H., van Berkel W. J. H., Tischler D., Front. Microbiol. 2018, 9, 2410; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Spiegelhauer O., Mende S., Dickert F., Knauer S. H., Ullmann G. M., Dobbek H., J. Mol. Biol. 2010, 398, 66–82. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Riedel A., Mehnert M., Paul C. E., Westphal A. H., van Berkel W. J. H., Tischler D., Front. Genet. 2015, 6, 1073; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Scholtissek A., Ullrich S. R., Mühling M., Schlömann M., Paul C. E., Tischler D., Appl. Microbiol. Biotechnol. 2017, 101, 609–619. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Chen B.-S., Médici R., van der Helm M. P., van Zwet Y., Gjonaj L., van der Geest R., Otten L. G., Hanefeld U., Appl. Microbiol. Biotechnol. 2018, 102, 5545–5556; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Mansell D. J., Toogood H. S., Waller J., Hughes J. M., Levy C. W., Gardiner J. M., Scrutton N. S., ACS Catal. 2013, 3, 370–379; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Steinkellner G., Gruber C. C., Pavkov-Keller T., Binter A., Steiner K., Winkler C., Łyskowski A., Schwamberger O., Oberer M., Schwab H., Faber K., Macheroux P., Gruber K., Nat. Commun. 2014, 5, 4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kitzing K., Fitzpatrick T. B., Wilken C., Sawa J., Bourenkov G. P., Macheroux P., Clausen T., J. Biol. Chem. 2005, 280, 27904–27913. [DOI] [PubMed] [Google Scholar]
- 17. Knaus T., Paul C. E., Levy C. W., de Vries S., Mutti F. G., Hollmann F., Scrutton N. S., J. Am. Chem. Soc. 2016, 138, 1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Clay D., Winkler C. K., Tasnádi G., Faber K., Biotechnol. Lett. 2014, 36, 1329–1333; [DOI] [PubMed] [Google Scholar]
- 18b. Fu Y., Hoelsch K., Weuster-Botz D., Process Biochem. 2012, 47, 1988–1997. [Google Scholar]
- 19.
- 19a. Demirjian D. C., Shah P. C., Morís-Varas F. in Biocatalysis: From Discovery to Application (Ed.: D.-D. Fessner), Springer, Berlin, 1999, pp. 1–29; [Google Scholar]
- 19b. Gupta M. N., Eur. J. Biochem. 1992, 203, 25–32. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Dascier D., Kambourakis S., Hua L., Rozzell J. D., Stewart J. D., Org. Process Res. Dev. 2014, 18, 793–800; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Han L., Liang B., World J. Microbiol. Biotechnol. 2018, 34, 141. [DOI] [PubMed] [Google Scholar]
- 21. Qi J., Schlömann M., Tischler D., J. Mol. Catal. B 2016, 130, 9–17. [Google Scholar]
- 22. Bradford M. M., Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 23. Macheroux P. in Methods in Molecular Biology, Vol. 131: Flavoprotein Protocols (Eds.: S. K. Chapman, G. A. Reid), Humana, Totowa, 1999, pp. 1–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary