

Summary
Introgression from one species in a specific environment to another may facilitate colonization of the environment by the recipient species. However, such environment‐dependent introgression has been clarified in limited plant taxa.
In northern Japan, there are two interfertile oak species: Quercus dentata (Qd) in coastal areas and Q. mongolica var. crispula (Qc) in inland areas. However, at higher latitudes where Qd is rare, a coastal Qc ecotype with Qd‐like traits is distributed in the coastal areas. We distinguished inland Qc, coastal Qc, and coastal Qd populations based on genome‐wide genotypes and multitrait phenotypes and verified introgression from coastal Qd to coastal Qc using reduced library sequencing.
Genotypes and phenotypes differed among the populations, and coastal Qc was intermediate between inland Qc and coastal Qd. The ABBA–BABA test showed introgression from coastal Qd to coastal Qc. In coastal Qc, we found various stages of introgression after the first generation of backcross but detected no genomic regions where introgression was enhanced.
Overall, we show evidence for introgression from a coastal species to an ecotype of an inland species, which has colonized the coastal environment. It remains unclear whether introgressed alleles are selected in the coastal environment.
Keywords: ancestry proportion, coastal stress, double‐digest restriction‐site‐associated DNA (ddRAD) sequencing, heterozygosity, morphological traits, Patterson's D, principal component analysis, Weir–Cockerham's FST
Short abstract
See also the Editorial on this article by https://doi.org/10.1111/nph.16560.
Introduction
Introgression is the transfer of genes from one taxon to a genomic background of another taxon through hybridization and recurrent backcrossing (Anderson, 1953; Goulet et al., 2017). Recombination during backcrossing reorganizes the genome of the recipient taxon with introgressed genes from the donor taxon. The introgressed alleles add new genetic variation to the recipient taxon (Anderson & Stebbins, 1954; Suarez‐Gonzalez et al., 2018b). In comparison with other sources of genetic variation, such as standing variation and novel mutation, introgression may be advantageous because the introgressed alleles have been maintained in the environment of the donor taxon (Anderson, 1948). Thus, introgression potentially allows the recipient taxon to rapidly colonize a new habitat that the donor taxon has inhabited (Arnold & Kunte, 2017). Such environment‐dependent introgression has been found in some plants, resulting in the expansion of their habitats and the creation of different ecotypes (Rieseberg et al., 2007). Such phenomena have been clarified in limited plant taxa (Whitney et al., 2010, 2015; Arnold et al., 2016) but have been investigated recently in various plants, including trees (Suarez‐Gonzalez et al., 2016, 2018a; Khodwekar & Gailing, 2017).
Oaks are dominant tree species of temperate forests in the northern hemisphere (Denk et al., 2017) and are often interfertile among species with ambiguous species boundaries (Petit et al., 2004). Introgression between oak taxa, even between deeply divergent lineages (McVay et al., 2017), has been found frequently (Guichoux et al., 2013; Eaton et al., 2015; Sork et al., 2016; Kim et al., 2018; Ortego et al., 2018). Thus, oaks are models for investigating evolutionary consequences of introgression. Oak trees inhabit various types of environment between more stressful habitats, such as coastal, arid, and volcanic areas, and less stressful habitats in mild and productive conditions (Cavender‐Bares, 2019). Therefore, environment‐dependent introgression in oaks has great potential to allow the expansion of their habitats and the creation of different ecotypes.
In northern Japan, there are two species, Quercus dentata Thunberg (Qd) in coastal areas and Quercus mongolica Fischer ex Ledebour var. crispula (Blume) H. Ohashi (Qc) in inland areas (Matsumoto et al., 2009). In northern Hokkaido, the northernmost part of Japan, Qd trees are rare because this region is beyond the northern distributional limit of Qd. In the region, a coastal Qc ecotype with unique traits, which are similar to Qd phenotypes and are associated with tolerance to coastal stress, occurs in the coastal area (Aizawa et al., 2018). Some taxonomists regard the coastal Qc ecotype as a putative hybrid between Qc and Qd (Ohba, 2006). Nuclear microsatellites demonstrate that an admixture with Qd characterizes the genetic background of the coastal Qc ecotype (Nagamitsu et al., 2019). Thus, environment‐dependent introgression from Qd to the coastal Qc ecotype is expected but has not yet been confirmed because incomplete lineage sorting of ancestral polymorphism also produces a similar admixture pattern (Muir & Schlötterer, 2004; Lexer et al., 2006; Pease & Hahn, 2015).
Genomic studies are effective to distinguish introgression from incomplete lineage sorting of ancestral polymorphism (Goulet et al., 2017) and to detect genomic regions where introgression patterns deviate from expectations of neutral genetic processes (Gompert et al., 2017; Suarez‐Gonzalez et al., 2018b). A whole‐genome sequence of Quercus robur Linnaeus is available (Plomion et al., 2018), which facilitates genomic studies on oaks. Restriction‐site‐associated DNA (RAD) sequencing is useful for mapping polymorphic sites to the oak reference sequence and determining genotypes at genome‐wide loci (Baird et al., 2008). We used double‐digest RAD (ddRAD) sequencing to obtain genome‐wide genotypes (Peterson et al., 2012).
Here, we aimed to verify introgression from Qd to Qc in a coastal environment. Before the verification, we assigned inland Qc, coastal Qc, and coastal Qd populations as nonintrogressed, putatively introgressed, and donor populations, respectively. We used genome‐wide genotypes, multitrait phenotypes, and coastal stress to distinguish these populations. To verify the introgression, we performed the ABBA–BABA test with Patterson's D statistic (Durand et al., 2011) for the three populations and the outgroup, Q. robur, of which the whole‐genome sequence was used as the reference for genotyping. After the verification, we described introgression patterns among individuals and among loci. We expected that nonneutral processes, such as selection in coastal environment, during sufficient backcrossing with Qc resulted in genomic heterogeneity in introgression. We examined introgression stages using both ancestry proportion and interancestry heterozygosity (Fitzpatrick, 2013) and genomic heterogeneity using F ST outliers (Foll & Gaggiotti, 2008).
Materials and Methods
Sampling
We collected 16 samples each from the inland Qc, coastal Qc, and coastal Qd trees in northern Hokkaido (Fig. 1). The two species, Qc and Qd, can hybridize with each other, and their F1 hybrids are fertile (Ubukata et al., 1999). Though most of the morphological traits of leaves and shoots are continuous between the two species (Ishida et al., 2003; Ito, 2009), we identified samples with hairy shoots as Qd and those with hairless shoots as Qc (Fig. 1a–c). We sampled coastal Qc and coastal Qd trees from the shore side of oak forests on coastal dunes or cliffs and inland Qc trees from broadleaf forests at the foot of mountains (Fig. 1d). The northern limits of the distributional range of Qd are located at 44.8°N on the western side and at 44.5°N on the eastern side of Hokkaido (Nagamitsu et al., 2019). We sampled coastal Qd trees from 44.0°N to the northern limits on both sides of Hokkaido and coastal Qc trees from the northern limits to the northernmost part of Hokkaido (Fig. 1d).
Figure 1.
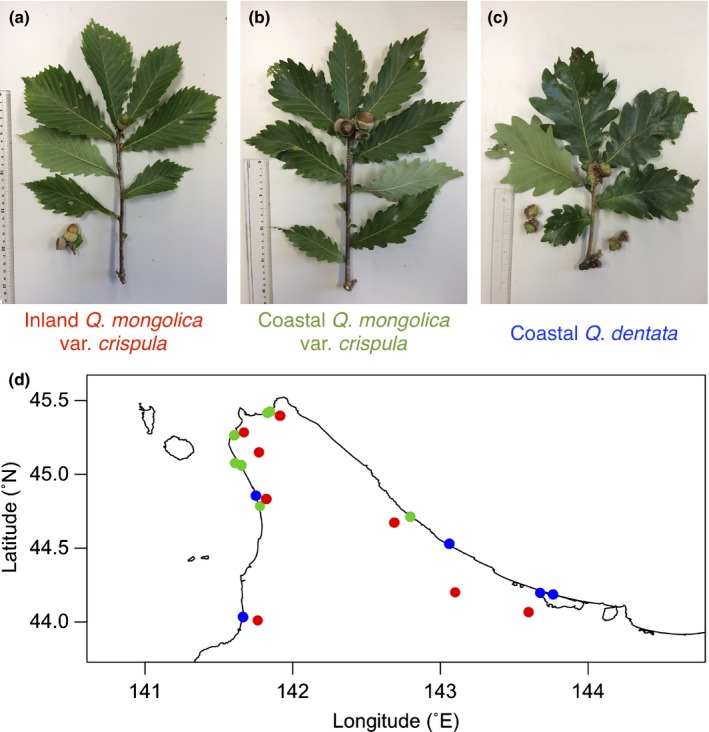
Current‐year shoots and acorns of (a) inland Quercus mongolica var. crispula (Qc), (b) coastal Qc, and (c) coastal Quercus dentata (Qd). (d) Sampling locations (circles) of inland Qc (red), coastal Qc (green), and coastal Qd (blue) in northern Hokkaido.
In the forests, we sampled trees > 20 m apart from each other to avoid sampling closely related trees. The locations of sampled trees were recorded using GPSMAP 64 (Garmin, Olathe, KS, USA). Two or three branches were collected from sampled trees. Leaves and shoots of collected branches were dried for morphological measurement and preserved in the Hokkaido Research Center, Forestry and Forest Products Research Institute. Fresh leaves were stored at −20°C until DNA extraction.
Phenotypes and coastal stress
To investigate phenotypes of collected samples, we selected 11 morphological traits of leaves and shoots (Table 1), which are thought to be distinctive between the two species and to be associated with the tolerance to coastal stress (Nagamitsu et al., 2019). We measured the morphological traits of sampled trees as described in Supporting Information Notes S1 and Fig. S1.
Table 1.
Mean values of morphological traits in inland Quercus mongolica var. crispula (Qc), coastal Qc, and coastal Quercus dentata (Qd) samples.
| Trait | Mean value in population | Loading | ||
|---|---|---|---|---|
| Inland Qc | Coastal Qc | Coastal Qd | to first PC | |
| Relative leaf width (leaf width/leaf length) | 0.519 | 0.590 | 0.595 | 0.271 |
| Relative petiole length (petiole length/leaf length) | 0.0253 | 0.0319 | 0.0303 | 0.066 |
| Lateral vain interval (mm) (leaf length/(number of lateral veins + 1)) | 8.84 | 11.92 | 12.98 | 0.365 |
| Tooth apex angle (°) of serrations | 77.7 | 134.9 | 156.0 | 0.393 |
| Density (mm−2) of stellate hairs | 3.22 | 7.95 | 9.06 | 0.224 |
| Length (mm) of radial filaments of stellate hairs | 0.102 | 0.120 | 0.286 | 0.312 |
| Leaf mass per area (mg mm−2) | 0.757 | 0.940 | 0.932 | 0.292 |
| Shoot diameter (mm) | 3.49 | 5.45 | 5.57 | 0.394 |
| Number of axillary buds at leaf scars | 7.72 | 8.98 | 9.38 | 0.283 |
| Number of axillary buds at lower stipule scars | 3.15 | 3.60 | 3.50 | 0.171 |
| Number of axillary buds at bud scale scars | 0.821 | 2.511 | 3.271 | 0.370 |
Loadings of the traits to the first principal component (PC) of phenotypes are shown.
To describe the leaf shape, four traits were recorded: relative leaf width, relative petiole length, lateral vein interval, and tooth apex angle of serrations. Among the four traits, the two latter traits are important to identify the two species (Ohba, 2006). Stellate hairs on the lower leaf surface vary between the two species, although their functions for the tolerance to coastal stress are unknown (Ishida et al., 2003). Both the density and size of stellate hairs were measured. Leaf mass per area and shoot diameter may have physiological functions for adaptation to coastal environment, such as intense light, severe drought, and strong wind (Poorter et al., 2009). Salt spray and harsh wind in winter cause the mortality of buds, particularly those in the upper part of shoots (Asai et al., 1986). Bud production in the lower part of shoots seems to convey the tolerance to coastal stress because more buds can compensate for the bud mortality. Thus, the number of axillary buds was recorded at leaf scars, at lower stipule scars, and at bud scale scars.
As already mentioned, the bud mortality pattern in shoots reflects coastal stress (Nagamitsu et al., 2019). To evaluate coastal stress, we calculated the proportion of flushing buds in the lower part of shoots, n/(m + n), where m and n are the numbers of flushing buds in the upper and lower parts of a shoot, respectively (Fig. S1; Notes S1).
Genotypes
DNA was extracted from stored fresh leaves of each sampled tree using the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). A ddRAD library was prepared from the 48 samples in the modified methods of Peterson et al. (2012). Extracted DNA was digested with PstI and Sau3AI restriction enzymes, ligated with Y‐shaped adaptors, and amplified by PCR with the KAPA HiFi polymerase (Kapa Biosystems, Woburn, MA, USA). After PCR amplification with adapter‐specific primer pairs (Access Array Barcode Library for Illumina; Fluidigm, South San Francisco, CA, USA), an equal amount of DNA from each sample was mixed and size‐selected with the BluePippin agarose gel (Sage Science, Beverly, MA, USA). Approx. 450 bp library fragments were retrieved. The library quality was checked using a 2100 Bioanalyzer with a high‐sensitivity DNA chip (Agilent Technologies, Waldbronn, Germany). The detailed methods of library preparation are described in Notes S2. The library was sequenced using an Illumina MiSeq to generate paired‐end reads with a 75 bp length.
The reads obtained were mapped to the reference sequences, 12 pseudomolecules (chromosomes) of oak genome assembly PM1N, of Q. robur (Plomion et al., 2018). The read mapping and variant calling were conducted using ddocent (Puritz et al., 2014). The subsequent filtering procedures are explained in Notes S3. From the variant loci (sites) with various types of polymorphism, we selected sites that were biallelic without indels in both the reference and samples and polymorphic in the samples, with higher sequencing quality and fewer missing genotypes, using VCftools (Danecek et al., 2011). We removed sites mapped to the regions of transposable elements determined in Q. robur (Plomion et al., 2018). In addition, we removed sites that deviated extremely from the Hardy–Weinberg equilibrium within populations using VCftools and sites that potentially had null alleles using GBstools (Cooke et al., 2016). Finally, we thinned sites that were tightly linked at < 1 kb intervals.
Populations
To summarize both phenotypic and genetic variations of the samples obtained, we performed principal component analysis (PCA) using the function prcomp of R 3.1.3 (R Core Team, 2016). Some traits, the density and size of stellate hairs and the number of axillary buds at bud scale scars, were skewed. Thus, values of the size, the density + 1, and the number + 1 were log‐transformed. Genotypes at selected ddRAD sites were coded as 0, 1, and 2, which were homozygotes of an allele the same as the reference, heterozygotes of the reference and nonreference (different from the reference) alleles, and homozygotes of the nonreference allele, respectively. The phenotypic values of 11 observed traits and the genotypic values at selected ddRAD sites without missing values were standardized (mean: 0; standard deviation: 1) and applied to the PCA. The contributions of principal components (PCs) to the total phenotypic or genetic variation were obtained. In the PCA of phenotypes, the loadings of the 11 traits to the first PC were calculated.
We distinguished inland Qc, coastal Qc, and coastal Qd populations based on the phenotypic and genetic variations and coastal stress. The samples were plotted on the coordinates of the PCs of genotypes and phenotypes and the proportion of flushing buds in the lower part of shoots. Based on the distribution of the samples, we selected samples representing distinct populations of inland Qc, coastal Qc, and coastal Qd.
Detecting introgression
The ABBA–BABA test with the Patterson's D statistic is useful to detect introgression as distinguishing from incomplete lineage sorting of ancestral polymorphism (Durand et al., 2011; Goulet et al., 2017). We assumed a phylogenetic relationship (((inland Qc, coastal Qc), coastal Qd), Q. robur) according to phylogenetic studies of white oaks (the section Quercus) using multiple nuclear loci (Hubert et al., 2014; McVay et al., 2017; Hipp et al., 2018). We calculated the genome‐wide or chromosomal D values in this phylogeny as follows:
(p i1, the frequency of a derived allele at site i in inland Qc (nonintrogressed population); p i2, the frequency of a derived allele at site i in coastal Qc (putatively introgressed recipient population); p i3, the frequency of a derived allele at site i in coastal Qd (donor population); p i4, the frequency of a derived allele at site i in Q. robur (outgroup population); n, the number of sites in the whole genome or each chromosome; Eaton et al., 2015). A positive D value shows that introgression occurred from the donor population to the putatively introgressed recipient population.
The selected samples representing the three populations were used for the ABBA–BABA test. It was ambiguous whether a nonreference allele was a derived one or not because a single reference sequence of Q. robur was available. Thus, ambiguous derived alleles were incorporated into the calculation of D values using p i4 randomly obtained from a probability distribution (Notes S4).
To obtain genome‐wide and chromosomal D estimates, we performed a 1k random sampling of p i4 and then a 1k bootstrap sampling of sites for each sampled p i4 (Notes S4). We obtained the median values and 95% ranges of the 1M bootstrap samples as the estimates and credible intervals, respectively, using the Two functions sample and quantile in R 3.1.3. The probabilities that the genome‐wide D values of bootstrap samples were negative and that the chromosomal D values of bootstrap samples were less than the genome‐wide estimate were calculated. The probabilities for 12 chromosomes were adjusted to q values of the false discovery rate (FDR) in 12 multiple tests using the function p.adjust in R 3.1.3. A significantly positive genome‐wide D value rejects a null hypothesis that no introgression from coastal Qd to coastal Qc populations has occurred. A chromosomal D value significantly higher than the genome‐wide estimate rejects a null hypothesis that the introgression level of each chromosome is the same as the genome‐wide level.
Introgression patterns
We described introgression patterns among samples and among sites. We inferred introgression stages using both ancestry proportion S (also called hybrid index) and interancestry heterozygosity H (Fitzpatrick, 2013) and evaluated genomic heterogeneity in introgression using F ST outliers (Foll & Gaggiotti, 2008). The selected samples representing inland Qc, coastal Qc, and coastal Qd populations were used for describing the introgression patterns.
The introgression stages were depicted on the coordinates of S and H, where S is the proportion of alleles descending from the coastal Qd population and H is the proportion of heterozygous loci with both alleles descending from the inland Qc and coastal Qd populations (Fitzpatrick, 2013). We estimated S and H of the samples using the function HIest with the options, method = “SANN”, iterations = 10 000, in the package hiest (Fitzpatrick, 2013) in R 3.1.3. The allele frequencies at every site in inland Qc and coastal Qd populations were obtained from the selected samples representing these populations. To obtain simulated samples in various introgression stages, we created 100 random genotypes at all sites from each of the inland Qc and coastal Qd populations with the allele frequencies. We simulated random mating between these populations (Qc and Qd) and obtained 100 simulated samples of their F1 hybrids. Similarly, we obtained subsequent generations, F2 hybrids and the first generation of backcross with Qc (BC1 Qc) and with Qd (BC1 Qd), and further generations from mating between BC1 Qc and F1, between BC1 Qc and F2, within BC1 Qc, and between BC1 Qc and Qc. In the simulated samples, S and H were estimated using HIest in the same method with iterations = 1000. The simulated and observed samples were plotted on the coordinates of S and H to infer the introgression stages in the coastal Qc population.
Wright's F ST, indicating a difference in allele frequency between populations, represents an indirect measure of gene flow between them. To investigate patterns of introgression across genome‐wide loci, we calculated Weir and Cockerham's estimator of F ST at each site in every pair of the three populations using the option ‐weir‐fst‐pop in VCftools 0.1.14 (Danecek et al., 2011). To detect F ST outlier sites, we surveyed sites with F ST values that significantly deviated from an expectation of a neutral genetic process based on the FDR q values using bayescan 2.1 (Foll & Gaggiotti, 2008). Outlier sites with high F ST values between inland Qc and coastal Qc populations and low F ST values between coastal Qc and coastal Qd populations indicate enhanced introgression. On the other hand, outlier sites with the opposite combination of F ST values indicate restricted introgression. To illustrate patterns of introgression in individual chromosomes, we plotted the three F ST values between inland Qc and coastal Qd populations, between inland Qc and coastal Qc populations, and between coastal Qc and coastal Qd populations at individual sites along sequence positions in every chromosome.
Results
Genetic and phenotypic variations and coastal stress
We obtained genome‐wide genotypes of 13 inland Qc, 15 coastal Qc, and 16 coastal Qd samples because we omitted three samples of inland Qc and one sample of coastal Qc with > 30% missing genotypes. Based on ddRAD reads, genotypes at 2772 selected sites in 12 chromosomes were obtained from the 44 samples (Fig. S2a). The intervals of these sites along individual chromosomes varied from 1 kb to 3.2 Mb, and the mean and median intervals were 0.26 Mb and 0.14 Mb, respectively (Fig. S2b). Among the sites, 1347 sites without missing genotypes were used for PCA. The PCA of genotypes resulted in much higher standard deviations of the first PC, which contributed to 12.0% of the total genetic variation, than those of subsequent PCs (< 3.0%; Fig. S3a). The first PC discriminated inland Qc, coastal Qc, and coastal Qd samples, although the subsequent PCs did not (Fig. S3b–f).
In the 44 samples, mean values of 11 measured traits tended to be higher in coastal Qc and coastal Qd samples than in inland Qc samples (Table 1). The PCA of phenotypes of the 11 traits resulted in much higher standard deviations of the first PC (which contributed 40.8% of the total phenotypic variation) than those of subsequent PCs (< 13.6%; Fig. S4a). The first PC separated inland Qc samples from coastal Qc and coastal Qd samples, although the subsequent PCs did not discriminate the three groups of samples (Fig. S4b–f). The lateral vein interval, tooth apex angle, shoot diameter, and number of axillary buds at bud scale scars had high (> 0.35) loadings to the first PC of phenotypes (Table 1).
The proportion of flushing buds in the lower part of shoots was higher in coastal Qc and coastal Qd samples than in inland Qc samples (Kruskal–Wallis test, P < 0.001; Fig. 2a), indicating that stress was higher in the coastal environment. Two coastal Qd samples had lower values of the first PC of genotypes than the other coastal Qd samples (Fig. 2b,d). One inland Qc sample had higher values of the first PC of phenotypes than the other inland Qc samples (Fig. 2c,d). These three samples showed intermediate coastal stress between inland and coastal environments and similar phenotypes to coastal Qc samples (Fig. 2c). On the coordinates of the first PCs of genotypes and phenotypes, inland Qc, coastal Qc, and coastal Qd samples were clearly separated except for the three samples (Fig. 2d). Thus, we omitted the three samples and selected 12 inland Qc, 15 coastal Qc, and 14 coastal Qd samples representing distinct populations. The first PCs of genotypes and phenotypes differed among these populations (Kruskal–Wallis test, P < 0.001), and the first PCs of coastal Qc population were intermediate between those of inland Qc and coastal Qd populations (Fig. 2d). The coastal Qc population was distinct from the coastal Qd population in genotypes and distinct from the inland Qc population in phenotypes. (Fig. 2d). The coastal Qc population differed from the coastal Qd population phenotypically and differed from the inland Qc population genetically (Wilcoxon test, P < 0.001), but these differences were relatively small (Fig. 2d).
Figure 2.
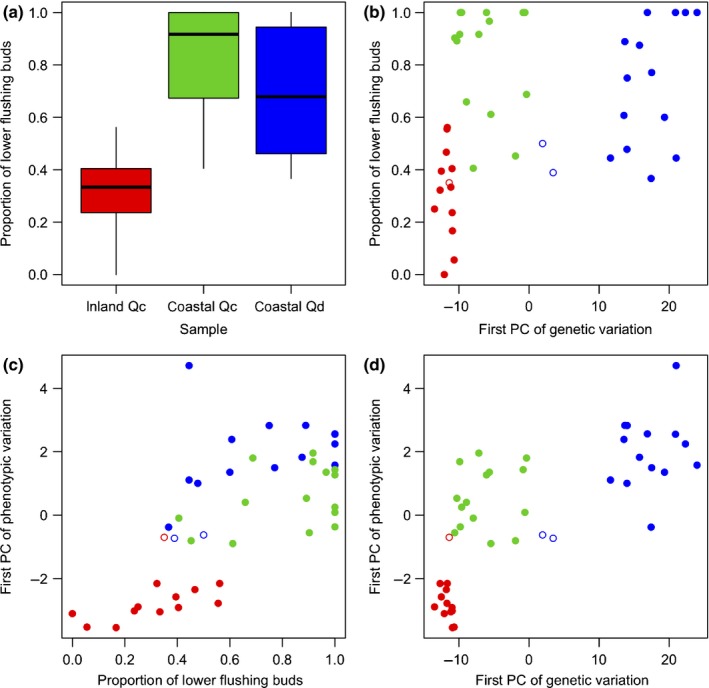
Genetic and phenotypic variations and coastal stress. (a) Box plots of the proportion of lower flushing buds, an index of coastal stress, in inland Quercus mongolica var. crispula (iQc), coastal Qc (cQc), and coastal Quercus dentata (cQd) samples. Boxes indicate the first and third quartiles, and whiskers indicate ranges. (b) Relationship between the coastal stress index and the first principal component (PC) values of genotypes. (c) Relationship between the coastal stress index and the first PC values of phenotypes. (d) Relationship between the first PCs of genotypes and phenotypes. Circles indicate iQc (red), cQc (green), and cQd (blue) samples (b–d). Closed circles indicate samples representing iQc, cQc and cQd populations, which are selected for the following analyses, and open circles are removed from the following analyses (b–d).
Introgression
The Patterson's D statistic, estimated from allele frequencies at the 2772 ddRAD sites in the 41 selected samples, was significantly positive (D = 0.044, bootstrap P < 0.001), showing introgression from coastal Qd to coastal Qc populations at genome‐wide loci (Table 2). In the 12 chromosomes, the D estimates were positive (≥ 0.007) but were not significantly different from the genome‐wide estimate (FDR q > 0.186; Table 2).
Table 2.
Genome‐wide and chromosomal estimates of the Patterson's D statistic with 95% ranges of bootstrap samples.
| Estimate | Bootstrap sample | P‐value | ||
|---|---|---|---|---|
| 2.5 percentile | 97.5 percentile | |||
| Genome‐wide | 0.044 | 0.026 | 0.062 | < 0.001 |
| Chromosome | ||||
| 1 | 0.038 | −0.023 | 0.099 | 0.568 |
| 2 | 0.092 | 0.049 | 0.135 | 0.015 |
| 3 | 0.052 | −0.013 | 0.115 | 0.392 |
| 4 | 0.007 | −0.068 | 0.079 | 0.836 |
| 5 | 0.049 | −0.006 | 0.104 | 0.452 |
| 6 | 0.048 | −0.019 | 0.115 | 0.455 |
| 7 | 0.042 | −0.013 | 0.099 | 0.544 |
| 8 | 0.020 | −0.043 | 0.084 | 0.783 |
| 9 | 0.014 | −0.057 | 0.083 | 0.801 |
| 10 | 0.046 | −0.017 | 0.111 | 0.468 |
| 11 | 0.019 | −0.049 | 0.088 | 0.774 |
| 12 | 0.039 | −0.040 | 0.117 | 0.557 |
P‐values of the ABBA–BABA tests that the genome‐wide D value is negative and that the chromosomal D values are lower than the genome‐wide estimate are also shown.
The Qd‐ancestry proportion S and the interancestry heterozygosity H were estimated from genotypes at the 2772 sites in 40 of the 41 selected samples because the estimation failed in one coastal Qc sample due to an extremely low maximum log‐likelihood. On the coordinate of S and H, observed samples of inland Qc were located at S = 0 and H = 0, and those of coastal Qd were located near S = 1 and H = 0, where simulated samples of their populations (Qc and Qd) were located (Fig. 3a). Simulated samples were distributed around the expected locations of F1 hybrids (S = 0.5, H = 1), F2 hybrids (S = 0.5, H = 0.5), and the first generations of backcross with Qc (BC1 Qc: S = 0.25, H = 0.5) and with Qd (BC1 Qd: S = 0.75, H = 0.5; Fig. 3a). Observed samples of coastal Qc were located in a wide range on the coordinates (0.06 ≤ S ≤ 0.44, 0.00 ≤ H ≤ 0.69; Fig. 3). This range was different from the ranges of simulated samples of F2 and BC1 Qc (Fig. 3a) but was overlapped with those of simulated samples of further generations from mating between BC1 Qc and F1, between BC1 Qc and F2, within BC1 Qc, and between BC1 Qc and Qc (Fig. 3b). Most of the coastal Qc samples were located around the expectation, H = 2S(1 − S), from random mating between Qc and Qd populations (Fig. 3).
Figure 3.
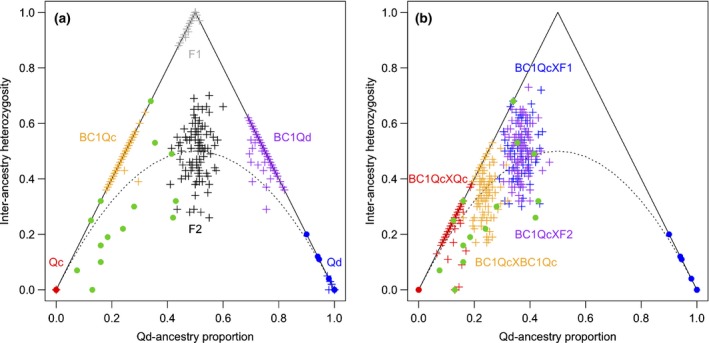
Scatterplots of relationship between the Quercus dentata (Qd)‐ancestry proportion and the interancestry heterozygosity in observed (circles) and simulated (crosses) samples. Red, green, and blue circles indicate observed samples of inland Quercus mongolica var. crispula (Qc), coastal Qc, and coastal Qd, respectively. A dotted line indicates an expectation from random mating between inland Qc and coastal Qd populations. A solid line indicates an expectation from recurrent backcrossing (a) Red, blue, grey, black, orange, and purple crosses indicate simulated samples of inland Qc and coastal Qd populations, their F1 and F2 hybrids, and the first generations of backcross with Qc (BC1 Qc) and Qd (BC1 Qd), respectively. (b) Red, orange, blue, and purple crosses indicate simulated samples from mating between BC1 Qc and Qc, within BC1 Qc, between BC1 Qc and F1, and between BC1 Qc and F2, respectively.
The Weir and Cockerham estimator of Wright's F ST in every pair of the three populations consisting of the 41 selected samples was obtained from 2633 ddRAD sites, at which F ST values were calculated in every pair of the three populations. These F ST values were highest between the inland Qc and coastal Qd populations, intermediate between the coastal Qc and coastal Qd populations, and lowest between the inland Qc and coastal Qc populations, in spite of their large variances (Kruskal–Wallis test, P < 0.001; Fig. 4a). No outlier sites were detected in F ST between the inland Qc and coastal Qd populations (FDR q ≥ 0.074; Fig. S5a), between the inland Qc and coastal Qc populations (FDR q ≥ 0.128; Fig. S5b), or between the coastal Qc and coastal Qd populations (FDR q ≥ 0.164; Fig. S5c). Nine sites with relatively high (> 0.25) F ST values between the inland Qc and coastal Qc populations (0.251 ≤ F ST ≤ 0.408) and 11 sites with relatively high (> 0.65) F ST values between the coastal Qc and coastal Qd populations (0.656 ≤ F ST ≤ 0.763) were plotted on the F ST distributions (Figs 4b–d, S5). The nine sites showing high genetic differentiation between environments in Qc had relatively low F ST values (−0.034 ≤ F ST ≤ 0.269) between the coastal Qc and coastal Qd populations (Fig. 4b,c). The 11 sites showing high genetic differentiation between species in the coastal environment had relatively low F ST values (−0.055 ≤ F ST ≤ 0.064) between the inland Qc and coastal Qc populations (Fig. 4c,d). Thus, introgression from Qd to Qc in the coastal environment tended to be enhanced at the nine former sites but restricted at the 11 latter sites, although these sites were not regarded as outliers. The F ST values at the 2633 sites in the 12 chromosomes are illustrated in Fig. S6. Three of the nine sites indicating enhanced introgression were aggregated in a genomic region (position 51–53 Mb) of the eighth chromosome (Fig. S6).
Figure 4.
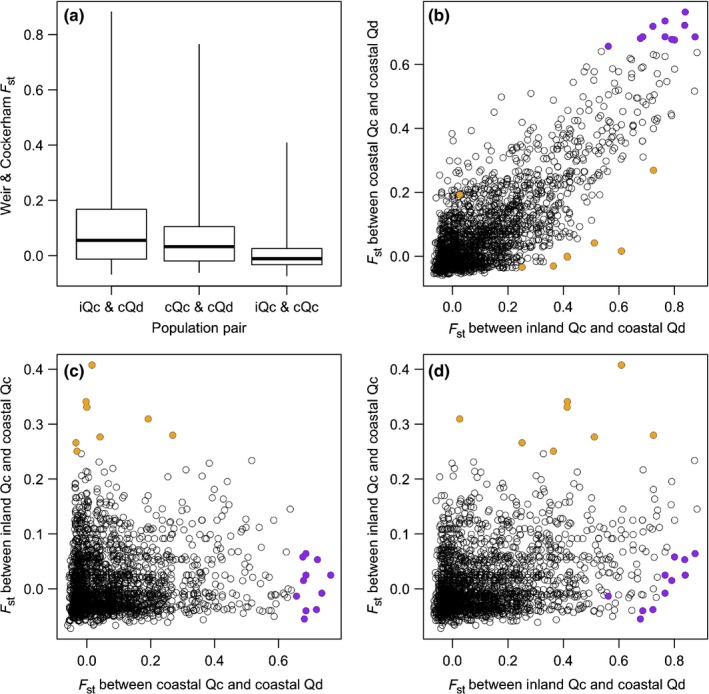
Weir and Cockerham's F ST at double‐digest restriction‐site‐associated DNA sites in pairs of three populations, inland Quercus mongolica var. crispula (Qc), coastal Qc, and coastal Quercus dentata (Qd). (a) Box plots of F ST values in three pairs of inland Qc (iQc), coastal Qc (cQc), and coastal Qd (cQd) populations. Boxes indicate the first and third quartiles, and whiskers indicate ranges. (b–d) Relationships in F ST values between two of the three populations. Orange circles indicate sites with relatively high (> 0.25) F ST values between iQc and cQc populations. Purple circles indicate sites with relatively high (> 0.65) F ST values between cQc and cQd populations.
Discussion
The coastal Qc ecotype is recognized as a Qc population in the coastal environment in northern Hokkaido, which is genetically admixed with Qd and is phenotypically similar to Qd (Ohba, 2006; Aizawa et al., 2018; Nagamitsu et al., 2019). This study confirms these properties quantitatively based on genome‐wide genotypes, multitrait phenotypes, and coastal stress. Oaks in the coastal environment are under stress from various factors, such as wind, salinity, drought, and heat (Ciccarelli, 2015). Among these factors, strong wind and salt spray in winter are critical factors that damage terminal buds and inhibit shoot elongation, affecting tree shape and canopy height (Asai et al., 1986). Thus, we measured the flushing pattern of buds in the upper and lower parts of shoots as an index of coastal stress. This study demonstrates that the coastal Qc ecotype is under coastal stress similar to that of Qd in the coastal environment and different from that of Qc in the inland environment. Because the flushing pattern of buds is a response of oak traits to environment, not only environmental conditions but also genotypes and phenotypes are involved in the difference in the coastal stress index. As expected from the properties of the coastal Qc ecotype, its genotypes and phenotypes were intermediate between those of inland Qc and coastal Qd. This study reveals that the coastal Qc ecotype is distinct from the coastal Qd in genotypes and distinct from the inland Qc in phenotypes. On the other hand, the coastal Qc ecotype shows relatively small differences from the coastal Qd in phenotypes and from the inland Qc in genotypes.
The relatively large phenotypic variation between inland Qc and coastal Qc in spite of the relatively small genetic variation between them may be partly due to phenotypic plasticity responding to their different environments. Oaks are known to have phenotypic plasticity in response to various environmental conditions related to coastal stress, such as temperature, drought, and light (Ramirez‐Valiente et al., 2010; Cavender‐Bares, 2019). The observed phenotypic variation was mainly associated with leaf shape, shoot diameter, and bud production. Our previous study shows that these traits depend not only on the proportion of admixture with Qd estimated from nuclear microsatellites but also on the proportion of lower flushing buds, which is the same as measured in this study (Nagamitsu et al., 2019). Thus, the findings suggest both genetic (admixture with Qd) and environmental (coastal stress) effects on the phenotypes of the coastal Qc ecotype. The previous study also shows interactions between the genetic and environmental effects, suggesting different reaction norms to a coastal environment among trees with different admixture proportions (Nagamitsu et al., 2019). To discriminate the genetic and environmental effects and their interactions, comparison of traits of trees planted in common gardens is necessary (Cavender‐Bares & Ramírez‐Valiente, 2017). In such an experiment, it is required that trees are sampled from inland Qc, coastal Qc, and coastal Qc populations, and that common gardens are located in inland and coastal environments. In the experiment, survival, growth, and reproduction of the trees can be evaluated. These approaches are essential to elucidate the adaptation of the coastal Qc ecotype to the coastal environment (Hedrick, 2013; Suarez‐Gonzalez et al., 2018b), however, which is beyond the scope of this study.
Although we distinguished the three populations, we found some samples with intermediate genotypes and phenotypes among the populations. The findings suggest that genetic and phenotypic variations in the fields are more continuous than those in the samples of this study, forming clines among the populations. A cline from one taxon to another through their hybrids is called a hybrid zone (Harrison, 1990). Hybrid zones are often accompanied by environmental gradients, and the parental taxa of the hybrids inhabit different environments (Barton & Hewitt, 1985). Fundamental theories and useful tools have been developed to investigate hybrid zones (Gompert et al., 2017). Coastal oak forests that include both Qc and Qd trees in northern Japan are known to form hybrid zones between Qd on the coastal side and Qc on the inland side (Matsumoto et al., 2009). However, the coastal Qc ecotype in northern Hokkaido is in a system different from a typical hybrid zone because Qd is rare on the coastal side at higher latitudes in this region. The northern distributional limit of Qd spatially separates the coastal Qc ecotype from Qd, which may result in the observed genetic gap between them. Because of these features, population‐based analyses are more suitable for the samples of this study than common tools for hybrid zones, such as genomic cline analyses (Gompert & Buerkle, 2009, 2011). Thus, we defined inland Qc, coastal Qc, and coastal Qd populations as nonintrogressed, putatively introgressed, and donor populations, respectively, to investigate introgression.
Allele sharing between taxa results from both introgression between them and incomplete lineage sorting of polymorphism in their ancestral lineage (Muir & Schlötterer, 2004; Lexer et al., 2006; Goulet et al., 2017). In the former, alleles that emerged in either taxon transfer between the taxa after their divergence. In the latter, both taxa inherit alleles that emerged in the ancestral lineage before the divergence. We tried to detect introgression as discriminating from incomplete lineage sorting using the ABBA–BABA test with a European white oak species, Q. robur, as an outgroup (Durand et al., 2011; Pease & Hahn, 2015). Both Qc and Qd belong to white oaks (Denk et al., 2017), and their comprehensive phylogeny has been reconstructed (Hubert et al., 2014; McVay et al., 2017; Hipp et al., 2018). Hipp et al. (2018) estimated that East Asian white oaks (the ancestral lineage of Qc and Qd) and European white oaks (the sister group) diverged 15–20 Ma and that Q. mongolica (the species including Qc) and the clade including Qd diverged c. 10 Ma. These estimates indicate similar durations when derived alleles accumulated in Qd and the ancestral lineage. Thus, it is feasible that Qc and Qd share derived alleles by means of introgression as well as incomplete lineage sorting of ancestral polymorphism. The ABBA–BABA test of this study shows introgression from coastal Qd to the coastal Qc ecotype at genome‐wide loci. Because the ABBA–BABA test assumes that alleles derived from an ancestral lineage are randomly sorted to descendant populations, nonneutral processes that bias this sorting can lead to false‐positive introgression (Martin et al., 2015). However, it is unlikely that nonneutral processes, such as selection in a coastal environment, affect genome‐wide loci of the coastal Qc ecotype.
Introgression begins with hybridization and proceeds during recurrent backcrossing (Anderson, 1953; Goulet et al., 2017). We estimated stages of this process using two parameters: the ancestry proportion and interancestry heterozygosity (Fitzpatrick, 2013). We found various introgression stages after the first generation of backcross (BC1) with Qc in the coastal Qc ecotype. Because subsequent hybrid generations created from random mating are expected to have similar parameters, it is difficult to discriminate different generations after F2 hybrids and BC1 (Gompert & Buerkle, 2016). Thus, it is unclear how many generations after BC1 have passed in the coastal Qc ecotype. Not only current hybridization but also ancient introgression is known in white oaks (Sork et al., 2016; Kim et al., 2018). These examples suggest a possibility that the coastal Qc ecotype originated from ancient introgression. Drift or selection decreases the interancestry heterozygosity, leading to fixation of introgressed alleles in the genomic background of either ancestry (Gompert et al., 2014). Such fixation was rare in the coastal Qc ecotype, and most of the samples were expected to derive from random mating between Qc and Qd ancestry. These findings do not support that selection of introgressed alleles in a coastal environment and/or drift due to demographic bottlenecks alter the genomic composition of the coastal Qc ecotype.
Genomic heterogeneity in introgression was found in European white oaks that experienced secondary contacts (Guichoux et al., 2013; Leroy et al., 2017). A locus showing enhanced introgression was identified in North American red oaks (the section Lobatae) along a soil moisture gradient, suggesting that dry environment selected introgressed alleles at the locus (Lind‐Riehl et al., 2014; Khodwekar & Gailing, 2017). In this study, we detected neither chromosomes nor loci deviated from a neutral expectation of introgression. However, we found some loci with a combination of high genetic differentiation within Qc between inland and coastal environments and low genetic differentiation within the coastal environment between Qc and Qd, suggesting enhanced introgression. We also found some loci with the opposite combination, suggesting restricted introgression. Some of the loci showing enhanced introgression were aggregated in a genomic region. These findings imply genomic heterogeneity in introgression, although this study failed to detect outliers. Insufficient numbers of loci and samples in this study may be responsible for the failure. First, it is difficult to detect loci at which introgressed alleles have been sufficiently selected due to their narrow blocks of linkage disequilibrium using a small number of loci; this is because sufficient recombination during the selection reduces the length of the blocks (Gompert et al., 2017). Second, it is difficult to detect loci at which introgressed alleles have been insufficiently selected due to their weak signature of selection using a small number of samples.
In conclusion, this study shows introgression from the coastal oak species Qd to an ecotype of the inland oak species Qc, which colonized the coastal environment outside the distributional range of Qd. This system is a typical example of environment‐dependent introgression resulting in the expansion of habitats and the creation of ecotypes. The coastal Qc ecotype is a mixture of individuals in various introgression stages after the first generation of backcross. Although genomic heterogeneity in introgression is expected, this study detects no genomic regions deviated from a neutral expectation of introgression. Thus, it remains unclear that introgressed Qd alleles are selected in the coastal environment. Further studies are necessary to show whether the introgression in the coastal Qc ecotype leads to the adaptation to the colonizing environment.
Author contributions
TN and HS designed the study. TN and AN collected the samples. KU, AI and TN analyzed the data. TN wrote the manuscript.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Morphological measurements of leaf and shoot traits and coastal stress.
Fig. S2 Distributions of selected ddRAD sites.
Fig. S3 Principal component analysis of genotypes.
Fig. S4 Principal component analysis of phenotypes.
Fig. S5 Relationships between q values of the false discovery rate (FDR) and the Weir and Cockerham's F ST between populations.
Fig. S6 Weir and Cockerham's F ST among populations at ddRAD sites in individual chromosomes.
Notes S1 Morphological measurements.
Notes S2 Preparation of ddRAD library.
Notes S3 Variant calling and site filtering.
Notes S4 Patterson's D with ambiguous derived alleles.
Acknowledgements
We thank the towns of Obira and Omu, as well as the Hokkaido Regional Forest Offices and the Soya and Kitami District Forest Offices, for permission to collect the samples, Akiko Takazawa for assistance in the laboratory work, Saneyoshi Ueno for computational arrangement, and Mineaki Aizawa and James Worth for suggestions for improving the manuscript. This study was supported by JSPS KAKENHI grant no. 17K07859 to TN and Research grant no. 201610 of the Forestry and Forest Products Research Institute to AN.
See also the Editorial on this article by https://doi.org/10.1111/nph.16560.
References
- Aizawa M, Maekawa K, Mochizuki H, Saito H, Harada K, Kadomatsu M, Iizuka K, Ohkubo T. 2018. Unveiling the origin of Quercus serrata subsp. mongolicoides found in Honshu, Japan, by using genetic and morphological analyses. Plant Species Biology 33: 174–190. [Google Scholar]
- Anderson E. 1948. Hybridization of the habitat. Evolution 2: 1–9. [Google Scholar]
- Anderson E. 1953. Introgressive hybridization. Biological Reviews of the Cambridge Philosophical Society 28: 280–307. [Google Scholar]
- Anderson E, Stebbins GL. 1954. Hybridization as an evolutionary stimulus. Evolution 8: 378–388. [Google Scholar]
- Arnold ML, Kunte K. 2017. Adaptive genetic exchange: a tangled history of admixture and evolutionary innovation. Trends in Ecology & Evolution 32: 601–611. [DOI] [PubMed] [Google Scholar]
- Arnold BJ, Lahner B, DaCosta JM, Weisman CM, Hollister JD, Salt DE, Bomblies K, Yant L. 2016. Borrowed alleles and convergence in serpentine adaptation. Proceedings of the National Academy of Sciences, USA 113: 8320–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Shinmura Y, Usui G. 1986. Mortality factors of the winter buds of Quercus dentata and Quercus mongolica var. grosseserrata in natural coastal forests of northern Hokkaido. Journal of Japanese Forest Society 68: 368–374. [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. 2008. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3: e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH, Hewitt GM. 1985. Analysis of hybrid zones. Annual Review of Ecology and Systematics 16: 113–148. [Google Scholar]
- Cavender‐Bares J. 2019. Diversification, adaptation, and community assembly of the American oaks (Quercus), a model clade for integrating ecology and evolution. New Phytologist 221: 669–692. [DOI] [PubMed] [Google Scholar]
- Cavender‐Bares J, Ramírez‐Valiente JA. 2017. Physiological evidence from common garden experiments for local adaptation and adaptive plasticity to climate in American live oaks (Quercus section Virentes): implications for conservation under global change In: Gil‐Pelegrín E, Peguero‐Pina J, Sancho‐Knapik D, eds. Oaks physiological ecology. Exploring the functional diversity of genus Quercus L. Tree physiology, vol.7 Cham, Switzerland: Springer, 107–135. [Google Scholar]
- Ciccarelli D. 2015. Mediterranean coastal dune vegetation: are disturbance and stress the key selective forces that drive the psammophilous succession? Estuarine, Coastal and Shelf Science 165: 247–253. [Google Scholar]
- Cooke TF, Yee M‐C, Muzzio M, Sockell A, Bell R, Cornejo OE, Kelley JL, Bailliet G, Bravi CM, Bustamante CD et al 2016. GBstools: a statistical method for estimating allelic dropout in reduced representation sequencing data. PLoS Genetics 12: e1005631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST et al 2011. The variant call format and VCftools . Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk T, Grimm GW, Manos PS, Deng M, Hipp AL. 2017. An updated infrageneric classification of the oaks: review of previous taxonomic schemes and synthesis of evolutionary patterns In: Gil‐Pelegrín E, Peguero‐Pina J, Sancho‐Knapik D, eds. Oaks physiological ecology. Exploring the functional diversity of genus Quercus L. Tree physiology, vol. 7 Cham, Switzerland: Springer, 13–38. [Google Scholar]
- Durand EY, Patterson N, Reich D, Slatkin M. 2011. Testing for ancient admixture between closely related populations. Molecular Biology and Evolution 28: 2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DAR, Hipp AL, González‐Rodríguez A, Cavender‐Bares J. 2015. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69: 2587–2601. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick BM. 2013. Alternative forms for genomic clines. Ecology and Evolution 3: 1951–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M, Gaggiotti O. 2008. A genome‐scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert Z, Buerkle CA. 2009. A powerful regression‐based method for admixture mapping of isolation across the genome of hybrids. Molecular Ecology 18: 1207–1224. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Buerkle CA. 2011. Bayesian estimation of genomic clines. Molecular Ecology 20: 2111–2127. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Buerkle CA. 2016. What, if anything, are hybrids: enduring truths and challenges associated with population structure and gene flow. Evolutionary Applications 9: 909–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert Z, Lucas LK, Buerkle CA, Forister ML, Fordyce JA, Nice CC. 2014. Admixture and the organization of genetic diversity in a butterfly species complex revealed through common and rare genetic variants. Molecular Ecology 23: 4555–4573. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Mandeville EG, Buerkle CA. 2017. Analysis of population genomic data from hybrid zones. Annual Review of Ecology, Evolution, and Systematics 48: 207–229. [Google Scholar]
- Goulet BE, Roda F, Hopkins R. 2017. Hybridization in plants: old ideas, new techniques. Plant Physiology 173: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichoux E, Garnier‐Géré P, Lagache L, Lang T, Boury C, Petit RJ. 2013. Outlier loci highlight the direction of introgression in oaks. Molecular Ecology 22: 450–462. [DOI] [PubMed] [Google Scholar]
- Harrison RG. 1990. Hybrid zones: windows on evolutionary process. Oxford Surveys in Evolutionary Biology 7: 69–128. [Google Scholar]
- Hedrick PW. 2013. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology 22: 4606–4618. [DOI] [PubMed] [Google Scholar]
- Hipp AL, Manos PS, González‐Rodríguez A, Hahn M, Kaproth M, McVay JD, Avalos SV, Cavender‐Bares J. 2018. Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. New Phytologist 217: 439–452. [DOI] [PubMed] [Google Scholar]
- Hubert F, Grimm GW, Jousselin E, Berry V, Franc A, Kremer A. 2014. Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus . Systematics and Biodiversity 12: 405–423. [Google Scholar]
- Ishida TA, Hattori K, Sato H, Kimura MT. 2003. Differentiation and hybridization between Quercus crispula and Q. dentata (Fagaceae): insights from morphological traits, amplified fragment length polymorphism markers, and leafminer composition. American Journal of Botany 90: 769–776. [DOI] [PubMed] [Google Scholar]
- Ito M. 2009. Variation in leaf morphology of Quercus crispula and Quercus dentata assemblages among contact zones: a method for detection of probable hybridization. Journal of Forest Research 14: 240–244. [Google Scholar]
- Khodwekar S, Gailing O. 2017. Evidence for environment‐dependent introgression of adaptive genes between two red oak species with different drought adaptations. American Journal of Botany 104: 1088–1098. [DOI] [PubMed] [Google Scholar]
- Kim BY, Wei X, Fitz‐Gibbon S, Lohmueller KE, Ortego J, Gugger PF, Sork VL. 2018. RADseq data reveal ancient, but not pervasive, introgression between Californian tree and scrub oak species (Quercus sect. Quercus: Fagaceae). Molecular Ecology 27: 4556–4571. [DOI] [PubMed] [Google Scholar]
- Leroy T, Roux C, Villate L, Bodénès C, Romiguier J, Paiva JAP, Dossat C, Aury J‐M, Plomion C, Kremer A. 2017. Extensive recent secondary contacts between four European white oak species. New Phytologist 214: 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexer C, Kremer A, Petit RJ. 2006. Shared alleles in sympatric oaks: recurrent gene flow is a more parsimonious explanation than ancestral polymorphism. Molecular Ecology 15: 2007–2012. [DOI] [PubMed] [Google Scholar]
- Lind‐Riehl JF, Sullivan AR, Gailing O. 2014. Evidence for selection on a CONSTANS‐like gene between two red oak species. Annals of Botany 113: 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Davey JW, Jiggins CD. 2015. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Molecular Biology and Evolution 32: 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Kawahara T, Kanazashi A, Yoshimaru H, Takahashi M, Tsumura Y. 2009. Differentiation of three closely related Japanese oak species and detection of interspecific hybrids using AFLP markers. Botany–Botanique 87: 145–153. [Google Scholar]
- McVay JD, Hipp AL, Manos PS. 2017. A genetic legacy of introgression confounds phylogeny and biogeography in oaks. Proceedings of the Royal Society of London. Series B: Biological Sciences 284: e20170300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir G, Schlötterer C. 2004. Evidence for shared ancestral polymorphism rather than recurrent gene flow at microsatellite loci differentiating two hybridizing oaks (Quercus spp.). Molecular Ecology 14: 549–561. [DOI] [PubMed] [Google Scholar]
- Nagamitsu T, Shimizu H, Aizawa M, Nakanishi A. 2019. An admixture of Quercus dentata in the coastal ecotype of Q. mongolica var. crispula in northern Hokkaido and genetic and environmental effects on their traits. Journal of Plant Research 132: 211–222. [DOI] [PubMed] [Google Scholar]
- Ohba H. 2006. Fagaceae In: Iwatsuki K, Boufford DE, Ohba H, eds. Flora of Japan, vol. IIa Tokyo, Japan: Kodansha, 42–60. [Google Scholar]
- Ortego J, Gugger PF, Sork VL. 2018. Genomic data reveal cryptic lineage diversification and introgression in Californian golden cup oaks (section Protobalanus). New Phytologist 218: 804–818. [DOI] [PubMed] [Google Scholar]
- Pease JB, Hahn MW. 2015. Detection and polarization of introgression in a five‐taxon phylogeny. Systematic Biology 64: 651–662. [DOI] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. 2012. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non‐model species. PLoS ONE 7: e37135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit RJ, Bodénès C, Ducousso A, Roussel G, Kremer A. 2004. Hybridization as a mechanism of invasion in oaks. New Phytologist 161: 151–164. [Google Scholar]
- Plomion C, Aury J‐M, Amselem J, Leroy T, Murat F, Duplessis S, Faye S, Francillonne N, Labadie K, Le Provost G et al 2018. Oak genome reveals facets of long lifespan. Nature Plants 4: 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorter H, Niinemets Ü, Poorter L, Wright IJ, Villar R. 2009. Causes and consequences of variation in leaf mass per area (LMA): a meta‐analysis. New Phytologist 182: 565–588. [DOI] [PubMed] [Google Scholar]
- Puritz JB, Hollenbeck CM, Gold JR. 2014. ddocent: a RADseq, variant‐calling pipeline designed for population genomics of non‐model organisms. PeerJ 2: e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2016. R: a language and environment for statistical computing, v. 3.1.3. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ramirez‐Valiente JA, Sanchez‐Gomez D, Aranda I, Valladares F. 2010. Phenotypic plasticity and local adaptation in leaf ecophysiological traits of 13 contrasting cork oak populations under different water availabilities. Tree Physiology 30: 618–627. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Kim S‐C, Randell RA, Whitney KD, Gross BL, Lexer C, Clay K. 2007. Hybridization and the colonization of novel habitats by annual sunflowers. Genetica 129: 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sork VL, Riordan E, Gugger PF, Fitz‐Gibbon S, Wei X, Ortego J. 2016. Phylogeny and introgression of California scrub white oaks (Quercus section Quercus). International Oaks 27: 61–74. [Google Scholar]
- Suarez‐Gonzalez A, Hefer CA, Christe C, Corea O, Lexer C, Cronk QCB, Douglas CJ. 2016. Genomic and functional approaches reveal a case of adaptive introgression from Populus balsamifera (balsam poplar) in P. trichocarpa (black cottonwood). Molecular Ecology 25: 2427–2442. [DOI] [PubMed] [Google Scholar]
- Suarez‐Gonzalez A, Hefer CA, Lexer C, Douglas CJ, Cronk QCB. 2018a. Introgression from Populus balsamifera underlies adaptively significant variation and range boundaries in P. trichocarpa . New Phytologist 217: 416–427. [DOI] [PubMed] [Google Scholar]
- Suarez‐Gonzalez A, Lexer C, Cronk QCB. 2018b. Adaptive introgression: a plant perspective. Biology Letters 14: e20170688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubukata M, Itahana N, Kohno K. 1999. Cross‐compatibility between Quercus mongolica var. grosseserrata and Quercus dentata and both the reproductive ability and flowering time of their interspecific hybrids. Journal of Japanese Forest Society 81: 286–290. [Google Scholar]
- Whitney KD, Broman KW, Kane NC, Hovick SM, Randell RA, Rieseberg LH. 2015. Quantitative trait locus mapping identifies candidate alleles involved in adaptive introgression and range expansion in a wild sunflower. Molecular Ecology 24: 2194–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. 2010. Adaptive introgression of abiotic tolerance traits in the sunflower Helianthus annuus . New Phytologist 187: 230–239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Morphological measurements of leaf and shoot traits and coastal stress.
Fig. S2 Distributions of selected ddRAD sites.
Fig. S3 Principal component analysis of genotypes.
Fig. S4 Principal component analysis of phenotypes.
Fig. S5 Relationships between q values of the false discovery rate (FDR) and the Weir and Cockerham's F ST between populations.
Fig. S6 Weir and Cockerham's F ST among populations at ddRAD sites in individual chromosomes.
Notes S1 Morphological measurements.
Notes S2 Preparation of ddRAD library.
Notes S3 Variant calling and site filtering.
Notes S4 Patterson's D with ambiguous derived alleles.