

Abstract
Chronic obstructive pulmonary disease (COPD) is a devastating lung disease with a high personal and societal burden. Exposure to toxic particles and gases, including cigarette smoke, is the main risk factor for COPD. Together with smoking cessation, current treatment strategies of COPD aim to improve symptoms and prevent exacerbations, but there is no disease‐modifying treatment. The biggest drawback of today's COPD treatment regimen is the ‘one size fits all’ pharmacological intervention, mainly based on disease severity and symptoms and not the individual's disease pathology. To halt the worrying increase in the burden of COPD, disease management needs to be advanced with a focus on personalized treatment. The main pathological feature of COPD includes a chronic and abnormal inflammatory response within the lungs, which results in airway and alveolar changes in the lung as reflected by (small) airways disease and emphysema. Here we discuss recent developments related to the abnormal inflammatory response, ECM and age‐related changes, structural changes in the small airways and the role of sex‐related differences, which are all relevant to explain the individual differences in the disease pathology of COPD and improve disease endotyping. Furthermore, we will discuss the most recent developments of new treatment strategies using biologicals to target specific pathological features or disease endotypes of COPD. © 2020 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: COPD, inflammation, aging, ECM, genome‐wide association study, CELSR1, sex differences, small airways, precision imaging, personalized treatment, biologics
Introduction
Chronic obstructive pulmonary disease (COPD) is an often severely disabling chronic lung disease with a high prevalence of over 250 million cases worldwide. It is currently the fourth leading cause of death globally and is predicted to be the third by 2020 1. In the most recent definition of the Global Initiative for Chronic Obstructive Lung Disease (GOLD), COPD is defined as ‘a common, preventable and treatable disease that is characterized by persistent respiratory systems and airflow limitation that is due to airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases’ 2. This exposure can be quite variable, with smoking being the main risk factor in high‐income countries and indoor cooking and occupational exposures representing important risk factors in low‐income countries. The definition and staging by GOLD is rather uniform, as defined by lung function, symptoms and exacerbation history, despite COPD being a heterogeneous disease in its pathological manifestations in patients. It has long been recognized that inflammation is a central hallmark of COPD, playing a role in the pathological changes in all different lung compartments 3. Next to toxic exposures, genetic predisposition is an important risk factor for COPD and COPD represents a complex disease in which genetic abnormalities in combination with the type and duration of exposures determine the clinical phenotype 4. The development of novel 2D and 3D in vitro and ex vivo models, as well as animal models that accurately recapitulate the main features of COPD pathology, has been very important for the study of disease mechanisms in COPD and recent developments in this area have been amply reviewed elsewhere 5, 6, 7, 8, 9, 10.
In the present review we focus on recent developments related to the abnormal inflammatory response, ECM and age‐related changes, structural changes in the small airways and the role of sex‐related differences, which are relevant to explain the underlying individual differences in the disease pathology of COPD and are important to improve disease endotyping. Where possible, we will underpin the observed pathogenetic changes by their potential genetic drivers.
Finally, we will discuss the most recent developments of new treatment strategies using biologicals to target specific pathological features or disease endotypes (a specific group of patients who share a distinct pathobiological mechanism) of COPD.
Abnormal inflammatory responses in COPD
It has long been known that the innate immune system plays a main role in COPD, as reviewed previously 3. Although it can be envisaged that noxious gases will evoke such an immune response, the peculiarity within COPD is that it is more extensive and damaging and sustained for a longer time than, for example, in smokers without COPD. Neutrophilic inflammation, as observed in the innate response, is strongly dependent on IL‐1‐alpha, which is reported to be increased in COPD patients 11, 12 and also more readily induced in COPD airway epithelial cells 13.
In the adaptive immune response in COPD, the predominant cell is the CD8 cytotoxic T cell. The presence of this cell type in the airways as well as parenchyma remains sustained over a long period of time, even up to 3 years after smoking cessation 14, 15. The finding of lymphoid aggregates and follicles in COPD 16 and, in particular, the confirmation of oligoclonality in these follicles 17, fitted very well with the concept of autoimmunity. In severe COPD, IL‐18, associated with lung lymphoid aggregates, has been shown to drive IFN‐gamma production, contributing to a Th1 response 18. Nevertheless, clonal B cell responses could be a consequence of antigenic exposure due to the disease (matrix components, infectious agents, immune components) and does not necessarily prove that this would also contribute to disease 19. More recently, the role of innate lymphoid cells (ILC) in inflammatory disease has received more attention 20. Although this role in COPD as yet is far from clear, group 3 ILC (ILC3) appear to be the main subtype in COPD 20, suggested to be involved in the initiation of the ectopic lymphoid aggregates 21. In addition, ILC1 were found to be associated with lymphoid cell infiltration and have been postulated to play a role in emphysematous destruction in COPD 22. In COPD exacerbations it was shown that ILC2 can switch to ILC1 and thus also contribute to IFN‐gamma‐driven inflammation 23.
Type 2 inflammation, normally associated with asthma, has also been described in COPD patients without a history of asthma and was suggested to represent an endotype of COPD 24. Obviously, this has also been discussed as a representative of the ‘asthma COPD overlap syndrome’ 25. Furthermore, a key role has been described for Th17 cells and their principal cytokine, IL‐17, at least in a subset of COPD 26, 27, 28. In particular, human lung dendritic cells from emphysema patients stimulated Th1 and Th17 responses 29 and emphysema patients had higher Th17 cell levels, which were responsive to elastin stimulation in vitro 29. Furthermore, the response to elastin stimulation was associated with the percentage of emphysema on CT scans, supporting a possible (T cell‐mediated) autoimmune phenomenon. A very recent study showed an airway epithelial IL‐17A response signature identifying a steroid‐unresponsive COPD patient subgroup 28. The authors suggested that such a gene signature of IL‐17 airway epithelial response distinguishes a biologically, radiographically and clinically distinct COPD subgroup that may benefit from personalized therapy 28.
ECM changes in COPD
The lung ECM forms the main building blocks of the lung. Disturbance of the ECM can have important consequences leading to lung tissue remodeling, which can affect all lung compartments (i.e. airway wall fibrosis and emphysema) in COPD. The main ECM compartments in the lung are the basement membrane, lamina propria of the airways and the alveolar interstitium, where the ECM connects the alveoli and blood vessels, forming the parenchyma. The main protein components of the basement membrane are collagen IV and laminin; in the lamina propria and interstitium they are fibrillar collagens, elastin, fibronectin, glycosaminoglycans and proteoglycans 30, 31.
The main producers of ECM proteins in the lung are fibroblasts and activated myofibroblasts, followed by airway smooth muscle and airway epithelial cells 32, 33. Degradation of the ECM is regulated by endogenous proteases, of which MMPs are the main type in the lung; their activity is regulated by tissue inhibitors of metalloproteinases (TIMPs). The balance between these MMPs and TIMPs is essential for ECM homeostasis 30, 34, 35.
A potential contributor of emphysematous lung tissue destruction in COPD is the inflammation‐related increase in MMPs (i.e. MMP‐2, MMP‐9 and MMP‐12), leading to an imbalance between MMPs and TIMPs and a shift toward increased ECM degradation, especially elastin 36, 37, 38. Several studies have shown a reduction in elastic fibers in COPD, as well as disrupted fibers and disturbed elastogenesis 39, 40, 41. Additionally, increased elastin gene expression was demonstrated in severe COPD 37, as well as an upregulation of several elastogenesis‐related genes, including elastin and fibulin‐5, in COPD lung tissue 42. Of interest, fibulin‐5 appeared to be a key modulator of elastic fiber formation, with fibulin‐5 knockout mice suffering from severe elastinopathy, resulting in loose skin, vascular abnormalities and severe emphysema 43, 44. Of interest, fibulin‐5 protein was shown to be cleaved by serine proteases in vitro, resulting in disturbed elastogenesis 45. Thus, the increase in elastin and fibulin‐5 on the gene expression level and the decreased presence of elastic fibers and elastin fiber abnormalities in COPD suggest a defect in the formation and repair of elastic fibers in COPD, which is possibly related to increased levels of cleaved non‐functional fibulin‐5 protein. In addition to the concept that increased fibulin‐5 gene expression is a response of hampered tissue repair in COPD, it has also been proposed that the increased fibulin‐5 expression in COPD is involved in small airway fibrosis and is thus detrimental 46.
Other important ECM changes in COPD include increased collagen deposition in the (small) airway walls and structural changes in collagen fibrils, with more disorganized collagen fibrils in COPD 47, 48. An important contributing factor to this disorganization of collagen is the lack of decorin deposition in the adventitia of the small airways 49. Decorin is a proteoglycan that binds to collagen fibrils, providing structural support. Decorin also binds many growth factors and their receptors, including TGF‐beta, the main inducer of tissue repair, thereby inhibiting growth factor activity 50, 51, 52.
Together, the collagen changes in the airways and elastic fiber‐related changes with loss of alveolar attachments in peripheral lungs leads to loose airway walls that lack their alveolar support, resulting in increased airway collapsibility and airflow limitation during expiration in COPD. An example of a diseased airway with lack of alveolar support is shown in Figure 1.
Figure 1.
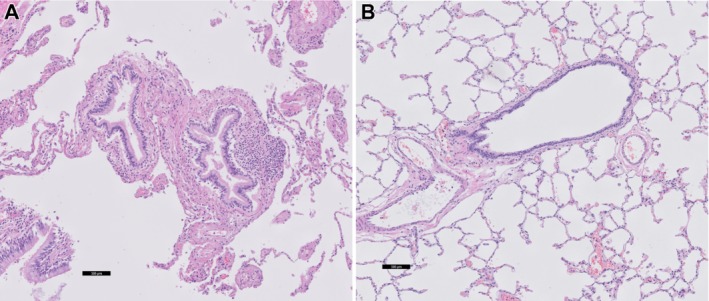
(A) COPD lung tissue with severe emphysema showing a small airway with extensive loss of alveolar attachments. (B) Comparable image of normal lung tissue with a small airway with normal parenchymal surroundings and attachments.
Genetic regulation of ECM changes in COPD
The list of genetic variants associated with COPD or reduced lung function is increasing rapidly 53, 54, 55, 56. Whether and which of these genetic variants have functional consequences for COPD pathology and are underlying ECM changes in COPD is a million dollar question. Of interest, several recent genome‐wide association studies (GWAS) showed an enrichment of, partly the same, ECM‐related genes among their lists of genetic variants for COPD and/or pulmonary function (Table 1). Gharib et al 57 performed GWAS analysis of pulmonary function measures (forced expiratory volume in the first second [FEV1] and FEV1/forced vital capacity [FVC]) in two large consortia (CHARGE and SpiroMeta) and used gene set enrichment analysis to identify pathways linked to pulmonary function. Interestingly, their pathway analysis for airflow obstruction (decreased FEV1/FVC) resulted in a clear enrichment of gene sets involved in ECM remodeling, including the integrin pathway, proteinaceous ECM, collagen and ECM. Subsequent network analysis identified MMP‐10 as a candidate gene and they demonstrated a pathophysiological role for MMP‐10 in a mouse model of smoke‐induced emphysema 57.
Table 1.
Enrichment of genes involved in ECM remodeling processes among genetic variants associated with COPD and lung function
| Study | Phenotype | Gene set* | P value enrichment† | Key enriched genes‡ |
|---|---|---|---|---|
| Gharib et al 57 | Airflow obstruction (FEV1/FVC) | Integrin pathway | 0 | § ACAN, COL1A2, COL3A1, COL4A2, COL4A4, COL5A1, COL5A3, COL18A1, COL15A1, FBN1, FBN2, FBLN1, FBLN2, HAPLN1, LAMA4, LAMC1, LUM, MFAP5, MMP10, NID2, THBS4 |
| Proteinaceous ECM | 1.45E–04 | |||
| ECM part | 1.75E–04 | |||
| ECM | 2.50E–04 | |||
| Collagen | 2.52E–04 | |||
| ECM structural constituent | 9.48E–04 | |||
| Wain et al 54 | Lung function (FEV1, FVC, FEV1/FVC) | Molecules associated with elastic fibers | 0.006 | EFEMP1, TGFB2, LTBP4, MFAP2, FBLN5 |
| Elastic fiber formation | 0.008 | EFEMP1, TGFB2, LTBP4, MFAP2, FBLN5 | ||
| ECM organization | 0.029 | HSD17B12, MMP15, TGFB2, CTSK, ADAMTSL4, EFEMP1, ITGA1, THSD4, NTN4, NPNT, LTBP4,MFAP2, CTSS, LEMD3, FBLN5 | ||
| Extracellular structure organization | 0.019 | HSD17B12, MMP15, TGFB2, CTSK, ADAMTSL4, EFEMP1, ITGA1, THSD4, NTN4, NPNT, LTBP4,MFAP2, CTSS, LEMD3, FBLN5 | ||
| Fibronectin binding | 0.014 | HSD17B12, CTSS, CTSK, MFAP2 | ||
| Sakornsakolpat et al 58 | Severe COPD | Collagen binding involved in cell matrix adhesion | 2.70E–03 | ITGA10, ITGA2, ITGA1 |
| Sakornsakolpat et al 56 | COPD | Basement membrane | <0.01 | COL15A1, COL18A1, PRSS23, ITGA1, GPX8, DSP, TIMP3, SERPING1, GJA1, DST |
| ECM part | <0.01 | COL15A1, SERPING1,GPX8, COL18A1, B3GNT9, PRSS23, GJA1, TIMP3, SCARF2, EMID1 | ||
| Laminin complex | <0.01 | COL15A1, ELMO3, COL18A1, ITGA2, DST, GPX8, DSP, FGD6, ENSG00000228536, ESRP2 | ||
| ECM | <0.05 | COL15A1, GPX8, SERPING1, COL18A1, LRP1, B3GNT9, SCARF2, GJA1, PRSS23, TIMP3 | ||
| Collagen binding | <0.05 | COL15A1, LRP1, TIMP3, GPX8, PRSS23, GJA1, ADAMTSL3, SERPING1, DLC1, SCARF2 | ||
| Proteinaceous ECM | <0.05 | COL15A1, GPX8, COL18A1, SERPING1, B3GNT9, LRP1, GJA1, SCARF2, ADAMTSL3, PRSS23 | ||
| ECM binding | <0.05 | TIMP3, COL15A1, COL18A1, EMP1, PRSS23, ITGAV, ARHGEF17, GPX8, SCARF2, GJA1 | ||
| Cell matrix adhesion | <0.05 | ASAP2, SH3PXD2A, ENSG00000173517, KIAA0754, ADAMTSL1, ITGA1, PARVA, ENSG00000251175, ENSG00000223561, ARHGEF17 | ||
| Shrine et al 55 | Lung function (FEV1, FVC, FEV1/FVC) | Molecules associated with elastic fibers | 9.33E–05 | ITGAV, TGFB2, LTBP4, MFAP2, GDF5 |
| Elastic fiber formation | 0.000104 | ITGAV, TGFB2, LTBP4, MFAP2, GDF5 | ||
| ECM organization | 0.00241 | MMP15, TGFB2, LTBP4, DST, ITGAV, P4HA2, MFAP2, GDF5, ADAM19 | ||
| Alpha6Beta4Integrin | 0.018468 | MET, DST, DSP, SMAD3 | ||
| TGF‐Core | 0.036822 | TGFB2, GDF5, SMAD3 | ||
| TGF‐beta2 production | 0.034539 | TGFB2, SMAD3 | ||
| Extracellular structure organization | 0.045883 | MMP15, TGFB2, THSD4, ITGAV, SMAD3, NPNT, MFAP2 | ||
| TGF‐beta receptor binding | 0.012484 | TGFB2, GDF5, SMAD3 | ||
| TGF‐beta binding | 0.026674 | LTBP4, ITGAV |
Only gene sets involved in ECM remodeling processes were selected from the referred publications.
Gene sets enriched in more than one study are highlighted in bold.
P values are corrected for multiple testing using different methods.
Genes enriched in more than one study are underlined.
§Twenty‐one focus genes derived from network analysis based on enriched gene sets.
Sakornsakolpat et al 58 used a new approach integrating GWAS and existing gene expression data to identify new genes for severe COPD. Using pathway enrichment analysis, they demonstrated enrichment of cell matrix adhesion of collagen binding, which included three ECM‐binding integrin genes (ITGA10, ITGA2, ITGA1).
In 2017, Wain et al 54 published a large GWAS study for lung function and COPD using the UK Biobank. Integration of the genetic variants with existing expression Quantitative Trait Loci (eQTL) datasets resulted in the identification of 234 genes with potentially causal effects on lung function that were enriched for several ECM‐related genes, including molecules related to elastic fibers, elastic fiber formation, ECM organization and extracellular structure organization. These gene sets included fibulin‐5 as a potential causal gene for impaired lung function. More recently, two even larger GWAS studies for lung function and COPD were published by Shrine et al 55 and Sakornsakolpat et al 56, identifying 139 new genetic loci for lung function and 35 new loci for COPD, respectively. Again, pathway analysis of the lung function‐associated genes showed that most significantly enriched pathways were related to elastic fibers and ECM organization and this also included a new signal for an ECM‐binding integrin, integrin alpha‐V. A significant enrichment of ECM‐related pathways was also found among the COPD‐associated genes, including laminin binding, integrin binding, mesenchyme development, cell matrix adhesion and actin filament bundles. Together with the enrichment of lung development pathways among the COPD‐associated genes, these findings strongly support a role for disturbed ECM development and homeostasis in the development of COPD. Follow‐up functional studies are now warranted to disentangle the exact disease‐driving mechanism and to identify new potential targets for intervention.
Aging and lung ECM changes in COPD
Aging is the progressive decline of homeostasis, resulting in increased risk of disease or death 59. The aging lung is characterized by several physiological and structural changes that are also present in COPD, including a decline in lung function, decreased mucociliary clearance, decreased antioxidant levels, senile emphysema and altered ECM proteins 60. Furthermore, mutations in genes responsible for telomere shortening, a hallmark of aging, have been associated with severe emphysema 61, 62. Therefore, it has been suggested that accelerated lung aging plays an important role in the pathogenesis of COPD 59, 63.
Although ECM dysregulation was proposed as an additional hallmark of the aging lung 64, it is not yet completely clear how lung ECM homeostasis is affected by aging and how this may contribute to COPD. The occurrence of airspace enlargement or senile emphysema 65 and the decrease in elastic fibers and increased collagen deposition in the aging lung 66, 67 are, however, indicative of lung remodeling.
It has been proposed that the aged lung is characterized by subtle changes in the ECM and lung architecture, which does not directly cause disease or changes in lung function, so‐called transitional remodeling 68. When the lung is then exposed to an additional insult, like in COPD, these alterations may trigger an aberrant repair response and cause permanent tissue damage. This theory is in line with the findings of a recent lung tissue gene expression study where it was shown that genes involved in ECM–receptor interactions, including three collagen genes, decreased more with age in COPD patients compared with controls, suggesting a defective repair response in COPD with aging 69. In addition, Calhoun et al 70 demonstrated an increase in ECM proteins, including collagen, with increasing age in mice and human lungs, together with an increase in senescent cells. In a more recent study 71, the link between growth differentiation factor 11 (GDF11), a member of the TGF family, and senescence in COPD was investigated. GDF11 was decreased in plasma of COPD patients and the protein was localized in mesenchymal cells in the airway wall and the airway epithelium. Treatment with GDF11 attenuated emphysema and cellular senescence in an elastase‐induced emphysema model, indicating a role for GDF11 in cellular senescence in COPD.
The true role of age‐related ECM changes in COPD pathology is still to be determined, but it certainly represents an interesting area for research. More so because targets for pharmaceutical intervention can potentially be applicable to other age‐related diseases as well. In this respect, it is of interest that novel anti‐aging treatment strategies are emerging that target cellular senescence with beneficial effects on lifespan, lung aging and lung fibrosis in animal models 72, 73, 74 and very recently also with beneficial effects in patients with idiopathic pulmonary fibrosis 75. This first‐in‐human, open‐label pilot study, where 14 idiopathic pulmonary fibrosis patients were treated with the combination of dasatinib (a tyrosine kinase inhibitor that targets anti‐apoptosis pathways; used for the treatment of leukemia) and quercetin (a natural product, flavonoid, targeting anti‐apoptosis pathways) showed significant and clinically meaningful physical improvement in these patients. Further evaluation of these senolytic drugs in larger randomized controlled trials is warranted.
Precision imaging for new insights into COPD pathology
The major site of airflow obstruction in COPD has been shown, by direct measurements of resistance, to occur in the small conducting airways that are <2 mm in internal diameter 76, 77, 78. Since then, there has been much debate over the most important contributor to airflow obstruction: structural changes within small airways or loss of elastic recoil due to emphysematous destruction. To date, clinical thoracic CT scans have been validated as a non‐invasive imaging technique to correlate with regional lung function 79 and to quantify emphysema (<−950 Hounsfield Units) 80, 81. However, the spatial resolution of clinical CT scans, 800–1000 μm, does not permit the analysis of the smallest conducting airways, or parenchymal structures 82, 83, 84. The recent application of micro‐CT, with a spatial resolution of up to 1 μm, has provided the ability to resolve alveolar structures and reliably identify terminal bronchioles, with an average lumen diameter of 424 μm 85. McDonough and colleagues 85 used micro‐CT to image tissue samples from explanted lungs from patients with very severe (GOLD4) COPD, and provided the first evidence that terminal bronchioles are destroyed in end‐stage COPD, even in regions of lung with no emphysema measured using mean linear intercept (a measure for airspace enlargement). These data therefore suggested that loss of terminal bronchioles may occur prior to the development of emphysema. To test this hypothesis, Koo and colleagues 86 used a cross‐sectional analysis of smokers with normal lung function and patients with mild, moderate and very severe COPD, and demonstrated that terminal bronchioles are significantly destroyed by 41% in mild (GOLD1), 43% in moderate (GOLD2) and 69% in end‐stage (GOLD4) COPD patients. Using the robust measure of alveolar surface area, which translates to the functional tissue involved in gas exchange, the study also reported that terminal and transitional (first generation of respiratory airways) bronchioles are lost in lung tissue in which no emphysematous destruction is present, indicating that small airways disease is an early pathological feature of mild and moderate COPD.
Although micro‐CT has the resolution to assess small airways, only CT and MRI modalities hold promise for the early detection of obstructive lung diseases and the characterization of disease pathology over time. The recent application of parametric response mapping (PRM) 87, which uses image registration to match inspiratory and expiratory CT scans to examine local changes in lung density, is now able to detect emphysema (PRMemph) from non‐emphysematous gas trapping, termed functional small airways disease (PRMfSAD). In a subsequent study using excised lungs and micro‐CT 88, it was validated that PRMfSAD is associated with a lower number of terminal bronchioles and a reduced terminal bronchiole lumen area, whereas changes in alveolar surface area only correlated with PRMemph. This is important, as Bhatt and colleagues 89, using the COPDGene cohort of 1508 current and former smokers, have shown that PRMfSAD is associated with early disease, before the development of airflow limitation, and contributed to a faster FEV1 decline than PRMemph. Furthermore, Labaki and colleagues 90, in a cohort of 725 smokers with and without COPD, reported that PRMfSAD is associated with regions of normal tissue over a range of COPD severities and that subjects with the highest PRMfSAD at baseline had the greatest increase in emphysema. PRM has also been shown to correlate with GOLD stage and several clinical parameters used to assess COPD morbidity, such as BMI, 6 min walking distance and quality of life as determined by the St‐George Respiratory Questionnaire (SGRQ) 91. In addition, PRMfSAD has been shown to be sensitive enough for short‐term (1 year) follow‐up of COPD patients, with PRMfSAD decreasing as PRMemph and tissue destruction increase 92.
These studies therefore support the notion that small airways disease is a precursor to the development of emphysema. As a CT biomarker, PRMfSAD has the potential to improve patient care through improved screening, disease subtyping and monitoring the response to treatment as an outcome measure. Future GWAS to associate genes with PRM disease subtypes will probably be informative compared with the traditional lung function measures to identify potential therapeutic targets and early disease markers.
Role of sex differences in COPD pathology
Although COPD has long been considered a disease of males, its prevalence and mortality among females have risen rapidly during the last decades and are now equal to those of males 93. Several studies have shown that sex differences exist with respect to susceptibility to smoking and clinical presentation of COPD. Females report more symptoms of dyspnea and cough and have a faster annual decline in FEV1, even with similar pack‐years of smoking 94, 95, 96. In a population‐based cohort study (SAPALDIA) of 1792 current smokers, Downs et al 96 investigated the effects of the number of cigarettes smoked per day on the course of lung function in males and females measured over a period of 11 years (Figure 2). In both sexes, the number of packs of cigarettes smoked per day was related to lung function decline, the mean annual FEV1 decline being −10.4 ml per pack per day in males and −13.8 ml in females. Interestingly, smoking‐related FEV1 decline was accelerated in females with airflow obstruction (FEV1/FVC <0.7) compared with those without (FEV1/FVC >0.7), the decline being −39.4 versus −12.2 ml/year per pack per day, respectively (p < 0.002), whereas this was not the case in males (FEV1 decline of −12.9 versus −8.8 ml/year per pack, p = 0.07). The difference between men and women with a reduced FEV1/FVC was significant (p = 0.05), indicating that women with airway obstruction experience a greater smoking‐related lung function decline than men (Figure 2). Another argument in support of the notion that females are more susceptible to the adverse effects of smoking is the finding that females are over‐represented among patients with severe early onset COPD, defined as the occurrence of COPD before the age of 53 years with an FEV1 below 40% predicted. In a study by Silverman et al 97, a group of 84 probands with severe early onset COPD (in the absence of alpha1‐antitrypsin deficiency) and 348 of their first‐degree relatives was assembled. Probands with severe early onset COPD were more often female (71%). Among the entire group of first‐degree relatives, males and females demonstrated similar lung function. However, among current and ex‐smokers, female first‐degree relatives had significantly lower FEV1/FVC values compared with males (82 ± 17% in females versus 87 ± 13% in males, p = 0.009) 98, 99. In addition, they more often had severe lung function impairment (FEV1 < 40% predicted) compared with male first‐degree relatives, suggesting that females are more susceptible to developing severe COPD.
Figure 2.
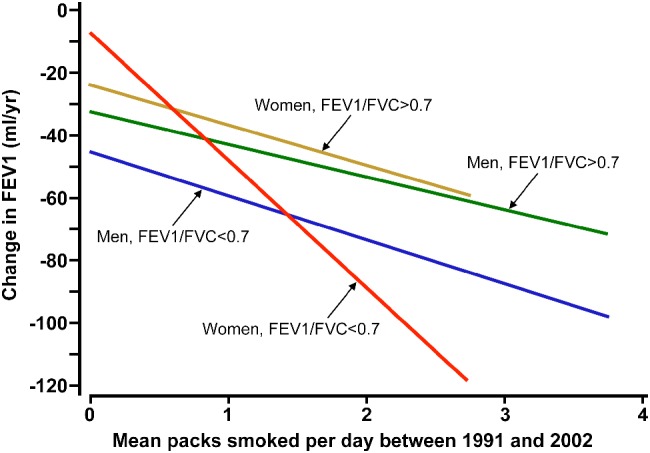
Relationship between mean packs per day and annual change in FEV1 in men and women: greater smoking‐related lung function decline in women compared with men with airflow obstruction. Reproduced from Downs et al 96 © 2005 Downs et al; Licensed under Creative Commons Attribution 4.0 International Public License (https://creativecommons.org/licenses/by/4.0/legalcode).
It has been reported that females develop a different type of COPD than males. They demonstrate less severe emphysematous destruction for the same degree of lung function impairment 95, 98, 99. However, they have more airway wall remodeling, with thicker small airway walls relative to the lumen 95. A recent analysis of COPDGene subjects confirmed earlier findings that males display more emphysema than females overall for the same degree of airflow obstruction 100. However, this was not the case in females with severe early onset COPD, severe emphysema (percentage of lung with low attenuation areas > 25%) and GOLD4 COPD. These three phenotypic subgroups with advanced disease had comparable radiographic emphysema despite fewer pack‐years of smoking. This finding that females with severe COPD demonstrate a similar degree of emphysema with less smoke exposure is in line with the notion that a subgroup is particularly susceptible to developing COPD with smoking. The difference in susceptibility to smoking already starts at an early age, as the growth in lung volumes is less in girls compared with boys when they start smoking as adolescents 101.
Several factors might contribute to the increased susceptibility to smoking. A possible explanation is that females have smaller lungs and therefore one pack of cigarettes may represent a higher dose in females than males. Another possible explanation would be different brands of cigarettes smoked or differences in inhalation technique. Finally, hormonal and genetic factors may drive the sex differences observed in COPD. Tam et al 102 showed increased airway wall thickness in female compared with male smokers at risk for or with GOLD1 COPD. They recapitulated these findings in a smoke‐induced mouse model of COPD, where female mice developed greater airway remodeling, greater small airway resistance, increased TGF‐beta activity and a reduced antioxidant response upon smoke exposure. These effects were prevented by ovariectomy and in part also by tamoxifen treatment, indicating a role for sex hormones. The CYP450 pathway, which is important in drug metabolism and detoxification, has been suggested to play a role, as it is known to be affected by estrogen and several enzymes of this family are increased in women (reviewed in 103). CYP1A1 and CYP1B1 expression increases in cultured bronchial epithelial cells after estrogen stimulation 104 and higher CYP450A1 and CYP450B1 expression has been observed in primary epithelial cells of patients with COPD compared with those without COPD 105. CYP450 enzymes convert the cigarette smoke constituent naphthalene to the far more toxic intermediate metabolite naphthalene oxide, leading to a rapid death of club cells. Another study reporting different underlying mechanisms of COPD in males and females was carried out by Kohler et al 106. They investigated the bronchoalveolar lavage fluid proteome in smokers with and without COPD and found a total of 19 proteins to be significantly differentially expressed 106. Interestingly, COPD‐associated differential protein expressions were completely driven by the female patients and included cathepsin B (downregulated), ATP synthase (upregulated) and chaperonin (upregulated). Pathway analysis revealed that the lysosome activity pathway was significantly downregulated in females with COPD, which was primarily driven by the decrease in cathepsin B. Downregulation of lysosomal function has been linked to autophagy, a conserved homeostatic pathway for protein degradation and regeneration. There is growing evidence in the literature that autophagy plays an important role in the pathogenesis of COPD 107.
Finally, genetic factors may play a role. In a recent study by Hardin et al 108, sex‐related dimorphic genetic risk factors were examined in a GWAS meta‐analysis using three large COPD cohorts (COPDGene, ECLIPSE, GenKOLS). A genome‐wide SNP*sex interaction analysis for COPD was performed. Although no genome‐wide significant hits were identified, several variants were identified that approached the prespecified significance threshold of 5*10−8, including four SNPs located in the CELSR1 gene, a member of the cadherin superfamily. The top‐hit was SNP rs9615358, located in CELSR1. CELSR1 is a known early lung development gene and was found to show higher expression in female compared with male fetal lung tissue, suggesting that in females CELSR1 may play a role in the susceptibility to COPD with a developmental origin.
Emerging treatment strategies in COPD; potential use of biologics
The chronic airway inflammation in COPD has been traditionally characterized by neutrophilic inflammation. However, a subgroup of patients with COPD has an eosinophilic phenotype 109, based on increased peripheral blood eosinophil counts or increased percentages of eosinophils in induced sputum. Increased blood eosinophil counts in COPD patients have been associated with an increased risk of future exacerbations 110, 111; chronic treatment with inhaled corticosteroids decreases this risk 112, 113, 114. This and other observations have led to the concept of treatable traits in chronic airway diseases 115, as in both asthma and COPD the presence of eosinophilic airway inflammation (as evidence by increased blood eosinophil levels) has been shown to be a theragnostic biomarker, predicting the therapeutic response to inhaled corticosteroids.
The next step is to investigate whether a precision medicine approach using specific biologics targeting eosinophilic inflammation would be as successful in patients with COPD as in asthmatics. Indeed, in patients with severe eosinophilic asthma, three monoclonal antibodies targeting the IL‐5/IL‐5 receptor axis have been shown to significantly reduce exacerbation rates 116, 117, 118. Mepolizumab and reslizumab inhibit the ligand IL‐5, whereas benralizumab binds to the IL‐5 receptor expressed on eosinophils and basophils, inducing apoptosis and thus depleting these cells. In two phase III double‐blind, randomized, controlled trials (METREX and METREO), monthly subcutaneous injections of mepolizumab were associated with a lower rate of exacerbations than placebo in patients with COPD and an eosinophilic phenotype 119. However, whereas mepolizumab reduced exacerbation rates in severe eosinophilic asthma by 50%, the effect of mepolizumab in eosinophilic COPD was very modest (approximately 15% reduction), although the same cut‐off values for blood eosinophil counts had been applied as inclusion criteria in these trials for both diseases. Moreover, whereas mepolizumab treatment was associated with significant improvements in quality of life in patients with severe eosinophilic asthma, the effects on patient‐reported outcomes, such as SGRQ and the COPD Assessment Test, did not reach the minimal clinically important difference.
Several mechanisms might underlie the very modest effects of mepolizumab in patients with eosinophilic COPD: first, the role of eosinophils in eliciting acute exacerbations in COPD might be less prominent as compared with severe eosinophilic asthma; second, the eosinophilic inflammation might be driven by other mediators than IL‐5, such as IL‐3, GM‐CSF, eotaxins and leukotrienes. As the cytolytic anti‐IL5 receptor monoclonal antibody benralizumab depletes eosinophils (completely in blood and significantly in sputum and mucosal tissues), clinical studies with benralizumab can help to disentangle the mechanisms of eosinophilic airway inflammation in COPD, as well as their contribution to acute exacerbations. Interestingly, in two large trials, the GALATHEA and TERRANOVA studies, benralizumab – at different doses (10, 30 or 100 mg s.c.) – did not affect the annualized exacerbation rate as compared with placebo in patients with moderate to very severe COPD, a blood eosinophil count of 220/μl or greater and a history of frequent exacerbations 120. Therefore, in contrast to the clear clinical benefits of benralizumab treatment in patients with severe eosinophilic asthma, eosinophil depletion in COPD patients did not translate to a reduction in exacerbations. This important discrepancy highlights that the ‘treatable traits’ concept should not replace but should complement the clinical diagnoses of asthma and COPD, which appear to be dominant in predicting therapeutic responses to targeted biologics in chronic airway diseases.
Take home message
To halt the worrying increase in the burden of COPD, improved and personalized treatment strategies are needed. Genetic risk factors, accelerated aging and sex differences are important contributors to key features of the individual's disease pathology in COPD, i.e. Th2, Th17 and/or autoimmune‐type inflammatory responses, ECM changes and disturbed cell–matrix interactions, and small airways disease in COPD (Figure 3). Improved insight into these pathological features and more accurate diagnosis using precision imaging will enable the discovery of new endotypes in COPD that can be targeted by precision medicine in the future.
Figure 3.
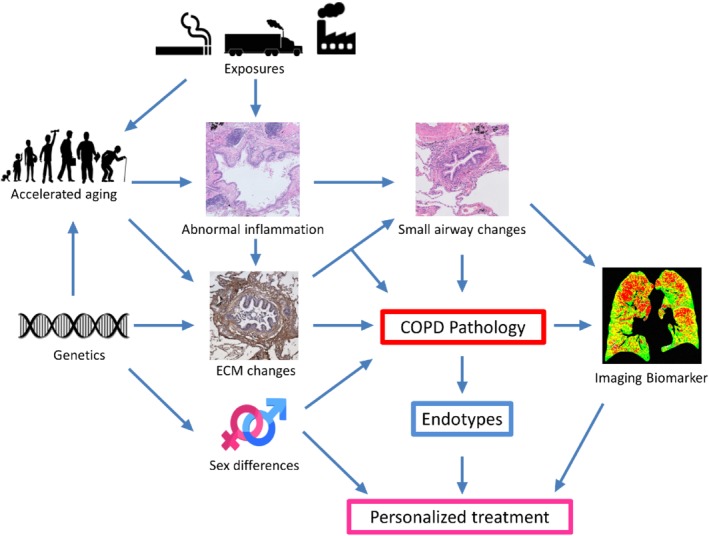
From key contributors to the individual's disease pathology to disease endotyping and personalized treatment in COPD.
Author contributions statement
CAB and WT drafted the initial design. All authors (CAB, MvdB, T‐LH, GB, WT) contributed equally to the contents, contributed to adaptations of the concept and design, and approved the final version of the manuscript.
Abbreviations
COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in the first second; FVC, forced vital capacity; GDF11, growth differentiation factor 11; GOLD, Global Initiative for Chronic Obstructive Lung Disease; GWAS, genome‐wide association study; ILC3, group 3 innate lymphoid cells; PRM, parametric response mapping; SGRQ, St‐George Respiratory Questionnaire; TIMP, tissue inhibitor of metalloproteinase.
Acknowledgements
Thanks are due to Dr Dragos M Vasilescu (UBC Centre for Heart Lung Innovation, St Paul's Hospital, Vancouver, BC, Canada) for providing the PRM mask image (Imaging Biomarker) included in Figure 3.
Conflict of interest statement: Maarten van den Berge reports research grants paid to his institution from GlaxoSmithKline, Chiesi, Astra Zeneca and TEVA Pharma. Guy G. Brusselle has received honoraria for lectures from AstraZeneca, Boehringer‐Ingelheim, Chiesi, GlaxoSmithKline, Novartis and Teva; he is a member of advisory boards for AstraZeneca, Boehringer‐Ingelheim, GlaxoSmithKline, Novartis, Sanofi/Regeneron and Teva. Wim Timens reports fees (all to UMCG) from Pfizer, GSK, Roche Diagnostics/Ventana, Merck Sharp Dohme, Novartis, Lilly Oncology, Boehringer Ingelheim, Astra‐Zeneca, Bristol‐Myers‐Squibb and AbbVie, for lectures and memberships of incidental advisory boards. Corry‐Anke Brandsma and Tillie‐Louise Hackett do not have conflicts of interest to disclose.
References
- 1. World Health Organization . 2019. [Accessed 19 April 2019]. Available from: http://www.who.int/respiratory/copd/en/
- 2.Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD). 2019. [Accessed 19 April 2019]. Available from: http://goldcopd.org/
- 3. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011; 378: 1015–1026. [DOI] [PubMed] [Google Scholar]
- 4. Silverman EK, Spira A, Pare PD. Genetics and genomics of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009; 6: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hiemstra PS, Grootaers G, van der Does AM, et al Human lung epithelial cell cultures for analysis of inhaled toxicants: lessons learned and future directions. Toxicol In Vitro 2018; 47: 137–146. [DOI] [PubMed] [Google Scholar]
- 6. Barkauskas CE, Chung MI, Fioret B, et al Lung organoids: current uses and future promise. Development 2017; 144: 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gkatzis K, Taghizadeh S, Huh D, et al Use of three‐dimensional organoids and lung‐on‐a‐chip methods to study lung development, regeneration and disease. Eur Respir J 2018; 52: 11800876. [DOI] [PubMed] [Google Scholar]
- 8. Gilpin SE, Wagner DE. Acellular human lung scaffolds to model lung disease and tissue regeneration. Eur Respir Rev 2018; 27: 180021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ross JT, Nesseler N, Lee JW, et al The ex vivo human lung: research value for translational science. JCI Insight 2019; 4: 128833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones B, Donovan C, Liu G, et al Animal models of COPD: what do they tell us? Respirology 2017; 22: 21–32. [DOI] [PubMed] [Google Scholar]
- 11. Pauwels NS, Bracke KR, Dupont LL, et al Role of IL‐1α and the Nlrp3/caspase‐1/IL‐1β axis in cigarette smoke‐induced pulmonary inflammation and COPD. Eur Respir J 2011; 38: 1019–1028. [DOI] [PubMed] [Google Scholar]
- 12. Botelho FM, Bauer CM, Finch D, et al IL‐1alpha/IL‐1R1 expression in chronic obstructive pulmonary disease and mechanistic relevance to smoke‐induced neutrophilia in mice. PLoS One 2011; 6: e28457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Osei ET, Noordhoek JA, Hackett TL, et al Interleukin‐1α drives the dysfunctional cross‐talk of the airway epithelium and lung fibroblasts in COPD. Eur Respir J 2016; 48: 359–369. [DOI] [PubMed] [Google Scholar]
- 14. Willemse BW, ten Hacken NH, Rutgers B, et al Effect of 1‐year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J 2005; 26: 835–845. [DOI] [PubMed] [Google Scholar]
- 15. Lapperre TS, Postma DS, Gosman MM, et al Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax 2006; 61: 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hogg JC, Chu F, Utokaparch S, et al The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645–2653. [DOI] [PubMed] [Google Scholar]
- 17. van der Strate BW, Postma DS, Brandsma CA, et al Cigarette smoke‐induced emphysema: a role for the B cell? Am J Respir Crit Care Med 2006; 173: 751–758. [DOI] [PubMed] [Google Scholar]
- 18. Briend E, Ferguson GJ, Mori M, et al IL‐18 associated with lung lymphoid aggregates drives IFNγ production in severe COPD. Respir Res 2017; 18: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brusselle GG, Demoor T, Bracke KR, et al Lymphoid follicles in (very) severe COPD: beneficial or harmful? Eur Respir J 2009; 34: 219–230. [DOI] [PubMed] [Google Scholar]
- 20. Mjosberg J, Spits H. Human innate lymphoid cells. J Allergy Clin Immunol 2016; 138: 1265–1276. [DOI] [PubMed] [Google Scholar]
- 21. Shikhagaie MM, Bjorklund AK, Mjosberg J, et al Neuropilin‐1 is expressed on lymphoid tissue residing LTi‐like group 3 innate lymphoid cells and associated with ectopic lymphoid aggregates. Cell Rep 2017; 18: 1761–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki M, Sze MA, Campbell JD, et al The cellular and molecular determinants of emphysematous destruction in COPD. Sci Rep 2017; 7: 9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silver JS, Kearley J, Copenhaver AM, et al Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 2016; 17: 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garudadri S, Woodruff PG. Targeting chronic obstructive pulmonary disease phenotypes, endotypes, and biomarkers. Ann Am Thorac Soc 2018; 15: S234–S238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Christenson SA, Steiling K, van den Berge M, et al Asthma‐COPD overlap. Clinical relevance of genomic signatures of type 2 inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 191: 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Stefano A, Caramori G, Gnemmi I, et al T helper type 17‐related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol 2009; 157: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roos AB, Sanden C, Mori M, et al IL‐17A is elevated in end‐stage chronic obstructive pulmonary disease and contributes to cigarette smoke‐induced lymphoid neogenesis. Am J Respir Crit Care Med 2015; 191: 1232–1241. [DOI] [PubMed] [Google Scholar]
- 28. Christenson SA, van den Berge M, Faiz A, et al An airway epithelial IL‐17A response signature identifies a steroid‐unresponsive COPD patient subgroup. J Clin Invest 2019; 129: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shan M, Cheng HF, Song LZ, et al Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med 2009; 1: 4ra10. [DOI] [PubMed] [Google Scholar]
- 30. Dunsmore SE, Rannels DE. Extracellular matrix biology in the lung. Am J Physiol 1996; 270: L3–L27. [DOI] [PubMed] [Google Scholar]
- 31. Burgstaller G, Oehrle B, Gerckens M, et al The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J 2017; 50: 1601805. [DOI] [PubMed] [Google Scholar]
- 32. White ES. Lung extracellular matrix and fibroblast function. Ann Am Thorac Soc 2015; 12: S30–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burgess JK, Mauad T, Tjin G, et al The extracellular matrix – the under‐recognized element in lung disease? J Pathol 2016; 240: 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol 2004; 16: 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pelosi P, Rocco PR, Negrini D, et al The extracellular matrix of the lung and its role in edema formation. An Acad Bras Cienc 2007; 79: 285–297. [DOI] [PubMed] [Google Scholar]
- 36. Merrilees MJ, Ching PS, Beaumont B, et al Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease. Respir Res 2008; 9: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deslee G, Woods JC, Moore CM, et al Elastin expression in very severe human COPD. Eur Respir J 2009; 34: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease–antiprotease imbalance in emphysema. Int J Tuberc Lung Dis 2008; 12: 361–367. [PubMed] [Google Scholar]
- 39. Black PN, Ching PS, Beaumont B, et al Changes in elastic fibres in the small airways and alveoli in COPD. Eur Respir J 2008; 31: 998–1004. [DOI] [PubMed] [Google Scholar]
- 40. Annoni R, Lancas T, Yukimatsu Tanigawa R, et al Extracellular matrix composition in COPD. Eur Respir J 2012; 40: 1362–1373. [DOI] [PubMed] [Google Scholar]
- 41. Fukuda Y, Masuda Y, Ishizaki M, et al Morphogenesis of abnormal elastic fibers in lungs of patients with panacinar and centriacinar emphysema. Hum Pathol 1989; 20: 652–659. [DOI] [PubMed] [Google Scholar]
- 42. Brandsma CA, van den Berge M, Postma DS, et al A large lung gene expression study identifying fibulin‐5 as a novel player in tissue repair in COPD. Thorax 2015; 70: 21–32. [DOI] [PubMed] [Google Scholar]
- 43. Yanagisawa H, Davis EC, Starcher BC, et al Fibulin‐5 is an elastin‐binding protein essential for elastic fibre development in vivo. Nature 2002; 415: 168–171. [DOI] [PubMed] [Google Scholar]
- 44. Nakamura T, Lozano PR, Ikeda Y, et al Fibulin‐5/DANCE is essential for elastogenesis in vivo. Nature 2002; 415: 171–175. [DOI] [PubMed] [Google Scholar]
- 45. Hirai M, Ohbayashi T, Horiguchi M, et al Fibulin‐5/DANCE has an elastogenic organizer activity that is abrogated by proteolytic cleavage in vivo. J Cell Biol 2007; 176: 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brusselle G. Dysregulated fibulin‐5 expression and elastogenesis in COPD lungs: pyromaniac or fire fighter? Thorax 2015; 70: 1–2. [DOI] [PubMed] [Google Scholar]
- 47. Abraham T, Hogg J. Extracellular matrix remodeling of lung alveolar walls in three dimensional space identified using second harmonic generation and multiphoton excitation fluorescence. J Struct Biol 2010; 171: 189–196. [DOI] [PubMed] [Google Scholar]
- 48. Tjin G, Xu P, Kable SH, et al Quantification of collagen I in airway tissues using second harmonic generation. J Biomed Opt 2014; 19: 36005. [DOI] [PubMed] [Google Scholar]
- 49. van Straaten JF, Coers W, Noordhoek JA, et al Proteoglycan changes in the extracellular matrix of lung tissue from patients with pulmonary emphysema. Mod Pathol 1999; 12: 697–705. [PubMed] [Google Scholar]
- 50. Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor‐beta by the proteoglycan decorin. Nature 1990; 346: 281–284. [DOI] [PubMed] [Google Scholar]
- 51. Danielson KG, Baribault H, Holmes DF, et al Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol 1997; 136: 729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Orgel JP, Eid A, Antipova O, et al Decorin core protein (decoron) shape complements collagen fibril surface structure and mediates its binding. PLoS One 2009; 4: e7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hobbs BD, de Jong K, Lamontagne M, et al Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet 2017; 49: 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wain LV, Shrine N, Artigas MS, et al Genome‐wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 2017; 49: 416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shrine N, Guyatt AL, Erzurumluoglu AM, et al New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 2019; 51: 481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sakornsakolpat P, Prokopenko D, Lamontagne M, et al Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell‐type and phenotype associations. Nat Genet 2019; 51: 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gharib SA, Loth DW, Soler Artigas M, et al Integrative pathway genomics of lung function and airflow obstruction. Hum Mol Genet 2015; 24: 6836–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sakornsakolpat P, Morrow JD, Castaldi PJ, et al Integrative genomics identifies new genes associated with severe COPD and emphysema. Respir Res 2018; 19: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest 2009; 135: 173–180. [DOI] [PubMed] [Google Scholar]
- 60. Pinkerton KE, Wang L, Smiley‐Jewell SM, et al Developmental and physiological aging of the lung In Molecular Aspects of Aging: Understanding Lung Aging, Rojas M, Meiners S, Jourdan Le Saux C. (eds). Wiley Blackwell: Oxford, 2014; 99–115. [Google Scholar]
- 61. Stanley SE, Merck SJ, Armanios M. Telomerase and the genetics of emphysema susceptibility. Implications for pathogenesis paradigms and patient care. Ann Am Thorac Soc 2016; 13: S447–S451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stanley SE, Chen JJ, Podlevsky JD, et al Telomerase mutations in smokers with severe emphysema. J Clin Invest 2015; 125: 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chilosi M, Poletti V, Rossi A. The pathogenesis of COPD and IPF: distinct horns of the same devil? Respir Res 2012; 13: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meiners S, Eickelberg O, Konigshoff M. Hallmarks of the ageing lung. Eur Respir J 2015; 45: 807–827. [DOI] [PubMed] [Google Scholar]
- 65. Gillooly M, Lamb D. Airspace size in lungs of lifelong non‐smokers: effect of age and sex. Thorax 1993; 48: 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. D'Errico A, Scarani P, Colosimo E, et al Changes in the alveolar connective tissue of the ageing lung. An immunohistochemical study. Virchows Arch A 1989; 415: 137–144. [DOI] [PubMed] [Google Scholar]
- 67. Frette C, Jacob MP, Wei SM, et al Relationship of serum elastin peptide level to single breath transfer factor for carbon monoxide in French coal miners. Thorax 1997; 52: 1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Roman J. Remodeling of the extracellular matrix in the aging lung In Molecular Aspects of Aging: Understanding Lung Aging, Rojas M, Meiners S, Jourdan Le Saux C. (eds). Wiley Blackwell: Oxford, 2014; 145–157. [Google Scholar]
- 69. de Vries M, Fiaz A, Postma DS, et al Accelerated lung aging in COPD: a lung tissue gene expression signature. Am J Respir Crit Care Med 2016; 193: A2627. [Google Scholar]
- 70. Calhoun C, Shivshankar P, Saker M, et al Senescent cells contribute to the physiological remodeling of aged lungs. J Gerontol A 2016; 71: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Onodera K, Sugiura H, Yamada M, et al Decrease in an anti‐ageing factor, growth differentiation factor 11, in chronic obstructive pulmonary disease. Thorax 2017; 72: 893–904. [DOI] [PubMed] [Google Scholar]
- 72. Xu M, Pirtskhalava T, Farr JN, et al Senolytics improve physical function and increase lifespan in old age. Nat Med 2018; 24: 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schafer MJ, White TA, Iijima K, et al Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 2017; 8: 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hashimoto M, Asai A, Kawagishi H, et al Elimination of p19ARF‐expressing cells enhances pulmonary function in mice. JCI Insight 2016; 1: e87732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Justice JN, Nambiar AM, Tchkonia T, et al Senolytics in idiopathic pulmonary fibrosis: results from a first‐in‐human, open‐label, pilot study. EBioMedicine 2019; 40: 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med 1968; 278: 1355–1360. [DOI] [PubMed] [Google Scholar]
- 77. Yanai M, Sekizawa K, Ohrui T, et al Site of airway obstruction in pulmonary disease: direct measurement of intrabronchial pressure. J Appl Physiol 1992; 72: 1016–1023. [DOI] [PubMed] [Google Scholar]
- 78. Van Brabandt H, Cauberghs M, Verbeken E, et al Partitioning of pulmonary impedance in excised human and canine lungs. J Appl Physiol Respir Environ Exerc Physiol 1983; 55: 1733–1742. [DOI] [PubMed] [Google Scholar]
- 79. Hoffman EA, Simon BA, McLennan G. State of the art. A structural and functional assessment of the lung via multidetector‐row computed tomography: phenotyping chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006; 3: 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Coxson HO, Mayo JR, Behzad H, et al Measurement of lung expansion with computed tomography and comparison with quantitative histology. J Appl Physiol 1995; 79: 1525–1530. [DOI] [PubMed] [Google Scholar]
- 81. Gevenois PA, De Vuyst P, de Maertelaer V, et al Comparison of computed density and microscopic morphometry in pulmonary emphysema. Am J Respir Crit Care Med 1996; 154: 187–192. [DOI] [PubMed] [Google Scholar]
- 82. Kirby M, Tanabe N, Tan WC, et al Total airway count on computed tomography and the risk of chronic obstructive pulmonary disease progression. Findings from a population‐based study. Am J Respir Crit Care Med 2018; 197: 56–65. [DOI] [PubMed] [Google Scholar]
- 83. Hasegawa M, Nasuhara Y, Onodera Y, et al Airflow limitation and airway dimensions in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 173: 1309–1315. [DOI] [PubMed] [Google Scholar]
- 84. Kurashima K, Hoshi T, Takaku Y, et al Changes in the airway lumen and surrounding parenchyma in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2013; 8: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McDonough JE, Yuan R, Suzuki M, et al Small‐airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 2011; 365: 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Koo HK, Vasilescu DM, Booth S, et al Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross‐sectional study. Lancet Respir Med 2018; 6: 591–602. [DOI] [PubMed] [Google Scholar]
- 87. Galban CJ, Han MK, Boes JL, et al Computed tomography‐based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progression. Nat Med 2012; 18: 1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vasilescu DM, Martinez FJ, Marchetti N, et al Non‐invasive imaging biomarker identifies small airway damage in severe COPD. Am J Respir Crit Care Med 2019; 200: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bhatt SP, Soler X, Wang X, et al Association between functional small airway disease and FEV1 decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2016; 194: 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Labaki WW, Gu T, Murray S, et al Voxel‐wise longitudinal parametric response mapping analysis of chest computed tomography in smokers. Acad Radiol 2019; 26: 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pompe E, Galban CJ, Ross BD, et al Parametric response mapping on chest computed tomography associates with clinical and functional parameters in chronic obstructive pulmonary disease. Respir Med 2017; 123: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Boes JL, Hoff BA, Bule M, et al Parametric response mapping monitors temporal changes on lung CT scans in the subpopulations and intermediate outcome measures in COPD Study (SPIROMICS). Acad Radiol 2015; 22: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ford ES, Mannino DM, Wheaton AG, et al Trends in the prevalence of obstructive and restrictive lung function among adults in the United States: findings from the National Health and Nutrition Examination surveys from 1988–1994 to 2007–2010. Chest 2013; 143: 1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Watson L, Schouten JP, Lofdahl CG, et al Predictors of COPD symptoms: does the sex of the patient matter? Eur Respir J 2006; 28: 311–318. [DOI] [PubMed] [Google Scholar]
- 95. Martinez FJ, Curtis JL, Sciurba F, et al Sex differences in severe pulmonary emphysema. Am J Respir Crit Care Med 2007; 176: 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Downs SH, Brandli O, Zellweger JP, et al Accelerated decline in lung function in smoking women with airway obstruction: SAPALDIA 2 cohort study. Respir Res 2005; 6: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Silverman EK, Weiss ST, Drazen JM, et al Gender‐related differences in severe, early‐onset chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 162: 2152–2158. [DOI] [PubMed] [Google Scholar]
- 98. Camp PG, Coxson HO, Levy RD, et al Sex differences in emphysema and airway disease in smokers. Chest 2009; 136: 1480–1488. [DOI] [PubMed] [Google Scholar]
- 99. Dransfield MT, Davis JJ, Gerald LB, et al Racial and gender differences in susceptibility to tobacco smoke among patients with chronic obstructive pulmonary disease. Respir Med 2006; 100: 1110–1116. [DOI] [PubMed] [Google Scholar]
- 100. Hardin M, Foreman M, Dransfield MT, et al Sex‐specific features of emphysema among current and former smokers with COPD. Eur Respir J 2016; 47: 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Xuan W, Peat JK, Toelle BG, et al Lung function growth and its relation to airway hyperresponsiveness and recent wheeze. Results from a longitudinal population study. Am J Respir Crit Care Med 2000; 161: 1820–1824. [DOI] [PubMed] [Google Scholar]
- 102. Tam A, Churg A, Wright JL, et al Sex differences in airway remodeling in a mouse model of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2016; 193: 825–834. [DOI] [PubMed] [Google Scholar]
- 103. Ben‐Zaken Cohen S, Pare PD, Man SF, et al The growing burden of chronic obstructive pulmonary disease and lung cancer in women: examining sex differences in cigarette smoke metabolism. Am J Respir Crit Care Med 2007; 176: 113–120. [DOI] [PubMed] [Google Scholar]
- 104. Han W, Pentecost BT, Pietropaolo RL, et al Estrogen receptor alpha increases basal and cigarette smoke extract‐induced expression of CYP1A1 and CYP1B1, but not GSTP1, in normal human bronchial epithelial cells. Mol Carcinog 2005; 44: 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pierrou S, Broberg P, O'Donnell RA, et al Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 175: 577–586. [DOI] [PubMed] [Google Scholar]
- 106. Kohler M, Sandberg A, Kjellqvist S, et al Gender differences in the bronchoalveolar lavage cell proteome of patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 2013; 131: 743–751. [DOI] [PubMed] [Google Scholar]
- 107. Haspel JA, Choi AM. Autophagy: a core cellular process with emerging links to pulmonary disease. Am J Respir Crit Care Med 2011; 184: 1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hardin M, Cho MH, Sharma S, et al Sex‐based genetic association study identifies CELSR1 as a possible chronic obstructive pulmonary disease risk locus among women. Am J Respir Cell Mol Biol 2017; 56: 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Singh D, Kolsum U, Brightling CE, et al Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J 2014; 44: 1697–1700. [DOI] [PubMed] [Google Scholar]
- 110. Bafadhel M, McKenna S, Terry S, et al Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184: 662–671. [DOI] [PubMed] [Google Scholar]
- 111. Brusselle G, Pavord ID, Landis S, et al Blood eosinophil levels as a biomarker in COPD. Respir Med 2018; 138: 21–31. [DOI] [PubMed] [Google Scholar]
- 112. Pascoe S, Locantore N, Dransfield MT, et al Blood eosinophil counts, exacerbations, and response to the addition of inhaled fluticasone furoate to vilanterol in patients with chronic obstructive pulmonary disease: a secondary analysis of data from two parallel randomised controlled trials. Lancet Respir Med 2015; 3: 435–442. [DOI] [PubMed] [Google Scholar]
- 113. Pascoe S, Barnes N, Brusselle G, et al Blood eosinophils and treatment response with triple and dual combination therapy in chronic obstructive pulmonary disease: analysis of the IMPACT trial. Lancet Respir Med 2019; 7: 745–756. [DOI] [PubMed] [Google Scholar]
- 114. van den Berge M, Kerstjens HA. Blood eosinophils as a continuous variable in the treatment of COPD: impact on the guidelines. Lancet Respir Med 2019; 7: 722–723. [DOI] [PubMed] [Google Scholar]
- 115. Agusti A, Bel E, Thomas M, et al Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J 2016; 47: 410–419. [DOI] [PubMed] [Google Scholar]
- 116. Ortega HG, Liu MC, Pavord ID, et al Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med 2014; 371: 1198–1207. [DOI] [PubMed] [Google Scholar]
- 117. Castro M, Zangrilli J, Wechsler ME, et al Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double‐blind, randomised, placebo‐controlled, phase 3 trials. Lancet Respir Med 2015; 3: 355–366. [DOI] [PubMed] [Google Scholar]
- 118. Bleecker ER, FitzGerald JM, Chanez P, et al Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high‐dosage inhaled corticosteroids and long‐acting beta2‐agonists (SIROCCO): a randomised, multicentre, placebo‐controlled phase 3 trial. Lancet 2016; 388: 2115–2127. [DOI] [PubMed] [Google Scholar]
- 119. Pavord ID, Chanez P, Criner GJ, et al Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N Engl J Med 2017; 377: 1613–1629. [DOI] [PubMed] [Google Scholar]
- 120. Criner GJ, Celli BR, Brightling CE, et al Benralizumab for the prevention of COPD exacerbations. N Engl J Med 2019; 381: 1023–1034. [DOI] [PubMed] [Google Scholar]