

Abstract
Pd‐mediated reactions have emerged as a powerful tool for the site‐selective and bioorthogonal late‐stage diversification of amino acids, peptides and related compounds. Indole moieties of tryptophan derivatives are susceptible to C2H‐activation, whereas halogenated aromatic amino acids such as halophenylalanines or halotryptophans provide a broad spectrum of different functionalisations. The compatibility of transition‐metal‐catalysed cross‐couplings with functional groups in peptides, other biologically active compounds and even proteins has been demonstrated. This Review primarily compiles the application of different cross‐coupling reactions to modify halotryptophans, halotryptophan containing peptides or halogenated, biologically active compounds derived from tryptophan. Modern approaches use regio‐ and stereoselective biocatalytic strategies to generate halotryptophans and derivatives on a preparative scale. The combination of bio‐ and chemocatalysis in cascade reactions is given by the biocompatibility and bioorthogonality of Pd‐mediated reactions.
Keywords: cross-coupling, enzymatic halogenation, late-stage diversification, palladium catalysis, tryptophans
Pd‐mediated reactions have emerged as a powerful tool for the site‐selective and bioorthogonal late‐stage diversification of amino acids, peptides and related compounds. Halotryptophans are accessible by biocatalytic approaches opening the application of a variety of cross‐coupling reactions for late‐stage modification of halotryptophan containing biomolecules.
1. Introduction
Late‐stage diversifications of biomolecules such as amino acids, peptides or pharmaceutically active compounds comprise powerful tools for the generation of functionalised derivatives with modified properties or biological effects. In particular, improvement of the biological efficacy leads to optimised active substances or drugs showing, for instance, enhanced selectivity or stability. Compared to early‐stage modifications, late‐stage approaches have several benefits. Firstly, the preparation of a compound library is simplified because one precursor molecule can result in several modified derivatives. Selective reactions make the use of protecting groups in many cases obsolete; hence, reaction as well as work‐up and purification steps are saved leading to more efficient and sustainable processes. In addition, late‐stage reactions have a great potential for the application as bioorthogonal modification tools for biological macromolecules even in lysates or the presence of living cells. However, these obvious advantages imply several challenges to be overcome. As there are many functional groups present in biomolecules, side reactions might occur or some functional groups may be incompatible with the reaction conditions and for example, might inhibit the reaction. Application in bioorthogonal chemistry is even more challenging. Additionally, biomolecules often contain stereogenic centres, which is why late‐stage diversification reactions need to be performed under mild conditions to avoid epimerisation.
Pd‐mediated reactions have been proven useful for the selective modification of biomolecules, which underscores the versatility of this transition metal.1, 2, 3, 4, 5, 6 The indole moiety of tryptophan constitutes an interesting target to be addressed by such approaches. Tryptophan plays a unique role in peptides and proteins. Considering the low abundance in proteins, it represents at the same time the most abundant amino acid in protein–protein interactions.7 Moreover, the spectrophotometric properties of tryptophan render it an interesting probe to study protein dynamics.8 Hence, modulation of the spectrophotometric properties of tryptophan is of great interest. Apart from that, tryptophan scaffolds are widely present in bioactive natural products featuring both halogenated and non‐halogenated indoles.9 Modification of those indoles may lead to novel pharmaceutically active compounds. This minireview deals with late‐stage diversification of indoles and indole‐based biomolecules beginning with a brief overview on Pd‐mediated C2−H activation strategies. Complementary to C2−H activation, the regioselective, enzymatic synthesis of different halotryptophans at various positions at the indole ring provides a handle for diversification. Accordingly, the main focus will be on addressing haloindoles describing and discussing different Pd‐mediated cross‐coupling strategies.
2. Pd‐Catalysed C2−H Activation of Tryptophans
The C2 position of indoles and tryptophans can be selectively addressed by transition‐metal‐catalysed C(sp2)−H activation.2, 4, 5, 6 Encouraged by protocols enabling Pd‐mediated regioselective functionalisation of indoles,10 Ruiz‐Rodríguez et al. diversified tryptophans and tryptophan containing tri‐ and tetrapeptides with aryl iodides as arylation reagents along with Pd(OAc)2 as catalyst and AgBF4 and 2‐nitrobenzoic acid as additives in DMF or phosphate buffer (Scheme 1 A).11
Scheme 1.
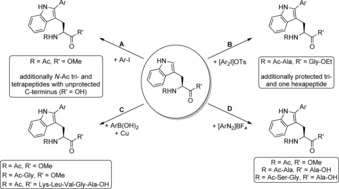
Overview of different, selective Pd‐catalysed C2‐arylation reactions using different aryl sources. A) I) Pd(OAc)2 (5 mol %), 2‐nitrobenzoic acid (1.5 equiv), AgBF4 (1.0 equiv) in DMF, 150 °C, μwave, 5 min;11 II) Pd(OAc)2 (5 mol %), TFA (1.0 equiv), AgBF4 (2.0 equiv) in DMF, 90 °C, μwave, 20 min;13 III) Pd(OAc)2 (5 mol %), 2‐nitrobenzoic acid (1.5 equiv), AgBF4 (1.0 equiv), phosphate buffer, 80 °C, μwave, 10 min.11 B) Pd(OAc)2 (5 mol %), AcOH or H2O, 23 °C, 17–24 h.16 C) Pd(OAc)2 (5–30 mol %), Cu(OAc)2 (10–60 mol %), AcOH, 40 °C, 16 h.17 D) Pd(OAc)2 (5–20 mol %), EtOAc (protected tryptophan) or MeOH (peptides), 25–37 °C, 16 h.18
To expand the substrate scope, tryptophan‐based diketopiperazine brevianamide F was modified resulting in analogues with exceptional cytotoxicities in HeLa cells.12 Additionally, optimisation of the reaction conditions such as replacing 2‐nitrobenzoic acid by trifluoroacetic acid even allowed for conversion of unprotected tryptophan.13 Apart from that, the strategy was used by Dong et al. for the macrocyclisation of peptidic substrates14 and by Mendive‐Tapia et al. for an intramolecular side chain‐to‐side chain cyclisation approach between iodinated phenylalanine or tyrosine derivatives and tryptophan to develop a novel approach towards peptide stapling (Scheme 2).15 Although analysis of the secondary structure by circular dichroism revealed conformationally constrained peptides, the generation of α‐helical peptides was not reported as it would have been expected for stapled peptides. Besides aryl iodides as arylation reagents, further investigations showed the utilisation of different aryl sources under mild conditions. Zhu et al. studied the application of diaryliodonium salts and the reaction proceeded smoothly under very mild conditions in acetic acid or water at 23 °C (Scheme 1 B).16 Moreover, the authors investigated the fluorescence properties of modified tripeptides revealing enhanced fluorescence emission intensities for electron‐rich aryl substituents at the tryptophans. Alternative arylation agents and the performance at mild temperatures were also studied in the group of Fairlamb beginning with aryl boronic acids. In this approach, Cu(OAc)2 is essential as cocatalyst mediating the reaction of N‐ and C‐terminally protected tryptophans (Scheme 1 C).17 Notably, a hexapeptide with free C‐terminus was also accepted as substrate upon six‐fold increase of the Pd and Cu loadings. However, since Cu also caused oxidative side reactions, subsequent studies demonstrated a Cu‐free approach with aryl diazonium salts as diversification reagents (Scheme 1 D).18
Scheme 2.
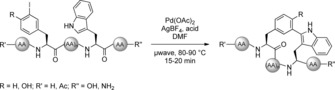
Pd‐catalysed side chain‐to‐side chain macrocyclisation as approach to peptide stapling. Conditions: Pd(OAc)2 (5 mol %), AgBF4 (1.0–2.0 equiv), acid (1.5 equiv 2‐nitrobenzoic acid or 1.0 equiv TFA).15
3. Biocatalytic Preparation of Halotryptophans
Halogen substituents represent advantageous handles to address varying positions of the indole ring and enable a broad range of Pd‐mediated modifications. However, the introduction by chemical procedures is often hampered by a lack of regioselectivity and by harsh reaction conditions like elementary halogens and Lewis acids. Enzymatic halogenations on the contrary are characterised by a unique regioselectivity under mild conditions at ambient temperature in aqueous solution. Flavin‐dependent tryptophan halogenases are one enzyme class catalysing such biocatalytic C−H activations, thus accomplishing halogenation by the use of halide salts, FADH2 and oxygen as oxidant. Mechanistically, hypohalous acid is formed through a flavin‐C4a‐hydroperoxide intermediate in the flavin‐binding site and is shuttled through a 10 Å tunnel to the substrate‐binding site, in which it undergoes an electrophilic aromatic substitution at tryptophan.19 Because FADH2 is consumed for the generation of the hypohalous acid, application in preparative biocatalytic procedures requires a cofactor regeneration system (Scheme 3).20, 21 However, the major bottlenecks of tryptophan halogenases are low stability accompanied by low activity. Hence, reaction scales were hitherto limited and suffered from incomplete conversions.20, 21 In 2015, Frese et al. described an immobilisation approach, in which tryptophan halogenases together with the auxiliary enzymes for cofactor regeneration are cross‐linked by glutaraldehyde forming Schiff bases with surface‐exposed lysine residues. This procedure furnishes a solid biocatalyst called combiCLEAs (cross‐linked enzyme aggregates).22
Scheme 3.
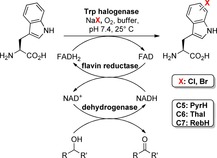
Application of FAD‐dependent tryptophan halogenases (e.g. PyrH, Thal or RebH) leads to regioselective chlorination or bromination in combination with concomitant cofactor regeneration mediated by a flavin reductase (Fre, PrnF or RebF) and a dehydrogenase (ADH or GDH).
Since then, the CLEA methodology has successfully been employed for different types of FAD‐dependent halogenases providing halogenated products on a preparative scale.23, 24, 25 Compared to the use of purified enzymes or lysates, in which the scale had been limited to 100 mg,20 the application of combiCLEAs allows the halogenation on a gram scale.22 As an alternative to an enzymatic cofactor regeneration system, Ismail et al. showed the feasibility of NADH mimics combined with flavin‐dependent tryptophan halogenases.26 The NADH mimics are used in stoichiometric amounts and replace the enzymes flavin reductase and dehydrogenase for cofactor regeneration. Besides chemical or enzymatic regeneration of FADH2, Schröder et al. proved the applicability of a photochemically driven approach.27
Since many amino acids are produced by fermentation on industrial multi‐ton scales, the fermentative production of l‐7‐chlorotryptophan in Corynebacterium glutamicum has very recently been developed by metabolic engineering.28 For this purpose, rebH and rebF genes encoding for a tryptophan 7‐halogenase and a flavin reductase, respectively, were introduced affording a recombinant, l‐tryptophan producing C. glutamicum strain.
Apart from that, enzymatic synthesis of halotryptophans can be performed by tryptophan synthase from serine and (substituted) indoles. This enzyme class depends on the prosthetic group pyridoxal phosphate (PLP), which forms a Schiff base with serine. This intermediate is dehydrated generating a reactive Michael system‐like electrophile, which is attacked by indole as the carbon nucleophile. Preparative scale procedures use E. coli lysates with overexpressed tryptophan synthase as biocatalyst in buffer at 37 °C.29 Besides halogenated tryptophans, other substituted derivatives with electron‐withdrawing and electron‐donating groups can be generated, particularly by engineered variants of the tryptophan synthase β‐subunit (TrpB) at elevated temperatures (Scheme 4).30 However, efficiency of the reaction is influenced by both the indole substituent and the substitution position reflected by the conversion and accordingly the yield of substituted tryptophans.
Scheme 4.
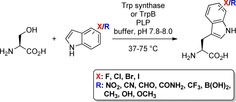
Application of tryptophan synthase. Besides synthesis of halotryptophans, different functional groups at the indole moiety can be introduced.
In addition to the synthesis of l‐halotryptophans, there are recent reports on the preparation of the d‐enantiomers. In contrast to the tryptophan synthase approach, the combiCLEA route also allows the conversion of d‐tryptophan although with reduced efficiency.22 Independently of each other, our group and the group of Chica developed stereoinversion processes to obtain substituted d‐tryptophans from the corresponding l‐isomers.31, 32 Schnepel et al. combined the enzymatic halogenation of l‐tryptophan using halogenase combiCLEAs with a dynamic stereoinversion.32 The l‐selective amino acid oxidase RebO provides the α‐imino acid, which is then unselectively reduced in a one‐pot process. Consequently, the d‐enantiomer accumulates because it is not accepted by RebO as a substrate. Parmeggiani et al. on the other hand designed a completely biocatalytic one‐pot procedure using tryptophan synthase to generate substituted l‐tryptophans followed by a stereoinversion cascade consisting of l‐amino acid deaminase (LAAD) and an engineered variant of aminotransferase DAAT providing desired d enantioselectivity.31
4. Suzuki–Miyaura Cross‐Coupling
The Suzuki–Miyaura reaction (SMC) is a very robust C−C bond forming cross‐coupling reaction between organic halides or pseudohalides and organoboron compounds, which is highly tolerant towards a broad range of functional groups, water‐based solvents, mild reaction temperatures and the presence of oxygen (Scheme 5).33 In particular, aerobic conditions and transition‐metal catalysis seem contradictory due to oxidation‐sensitive catalysts. These limitations may be true for classical approaches using phosphines or N‐heterocyclic carbenes as ligands. Recent developments put forth oxygen‐stable catalysts, whereby ligand‐free, oxygen‐stabilised Pd nanoparticles are even used in oxygen‐promoted SMCs.34 Beyond that, a great variety of organoboron compounds allows arylation and alkenylation as well as alkylation utilising boronic acids, boronic esters or trifluoroborates.35 The reaction mechanism is well studied and includes the cross‐coupling characteristic steps oxidative addition, transmetalation and reductive elimination.36
Scheme 5.

General overview of the SMC. OTf=trifluoromethanesulfonate, OTs=toluenesulfonate, OPiv=pivalate, pin=pinacolato, 9‐BBN=borabicyclo[3.3.1]nonane.33
The following section compiles different (cascade) procedures for the derivatisation of halotryptophans, halotryptophan containing peptides or halogenated, biologically active compounds derived from tryptophan by SMC and shows its biocompatibility and bioorthogonality. Although a variety of functional groups are tolerated, there are moieties such as thiols, amines or carboxylates inhibiting or poisoning the palladium catalyst under biocompatible conditions. These moieties are widely present in biomolecules or biological systems, which is why the reaction conditions need to be carefully optimised for this purpose.3
4.1. SMC as tool for late‐stage diversification of amino acids and peptides
Several methods are reported to utilise protected halotryptophans for derivatisation by SMC although different functional groups are known to coordinate to palladium and inhibit the catalysis.37 The first procedure for the conversion of unprotected halotryptophans to the corresponding aryltryptophans in aqueous medium was depicted by Deb Roy et al. in 2008.38
5‐Bromo‐, 5‐chloro‐, and 7‐bromotryptophan were prepared utilizing tryptophan synthase followed by SMC using a combination of water‐soluble catalyst components comprising Na2PdCl4 and triphenylphosphine‐3,3′,3′′‐trisulfonate (TPPTS) together with potassium carbonate as a base in degassed water at 80 °C for four to six hours. Different arylboronic acids were employed, whereas 5‐bromotryptophan gave the corresponding aryl‐substituted tryptophan in moderate to excellent yields depending on the substituent located at the aryl moiety of the boronic acid. Electron‐donating residues led to a more efficient cross‐coupling than electron‐withdrawing ones. The derivatisation of 5‐chloro‐ and 7‐bromotryptophan with phenylboronic acid is considerably less efficient compared to 5‐bromotryptophan. Upon replacement of TPPTS by the dimethylated analogue tris(4,6‐dimethylphenyl)phosphine‐3,3′,3′′‐trisulfonate (TXPTS) the reaction temperature could be decreased to 40 °C for 5‐bromotryptophan (reaction time 22 h). In addition, the authors were also able to arylate a 5‐bromotryptophan containing dipeptide (Scheme 6 A). Notably, the aryltryptophans showed modified fluorescence properties in methanol and water compared to tryptophan providing an interesting perspective for fluorescence labelling of biomolecules. Since phosphine‐based catalyst systems are prone to oxidation,42 Chalker et al. evaluated the use of a bench‐stable pre‐catalyst, which is pre‐prepared from Pd(OAc)2 and the water‐soluble ligand 2‐amino‐4,6‐dihydroxypyrimidin (ADHP) in aqueous sodium hydroxide.43 The pre‐catalyst Pd‐ADHP enabled SMC applying different halophenylalanines or ‐tyrosines in an aqueous buffer at 37 °C although chlorides were not accepted as substrates. Remarkably, Pd‐ADHP even allowed for SMC of an aryl iodide modified cysteine on the surface of the model protein serine protease SBL. This phosphine‐free pre‐catalyst was then utilised by Willemse et al. in 2015 for the phenyl substitution of unprotected 5‐bromotryptophan and 5‐bromotryptophan containing dipeptides by SMC in pure water requiring a catalyst loading of 5 mol % and potassium carbonate as a base while heating to 80 °C for 2–24 h (Scheme 6 B).39 Likewise, the C6‐ and C7‐brominated regioisomers were also successfully converted with phenylboronic acid. However, for vinylation bromotryptophan had to be replaced by the more reactive iodide combined with PdCl2(dppf) as precatalyst and 50 % (v/v) isopropanol as a cosolvent. In addition, the authors also studied halophenylalanines and halophenylalanine containing peptides in mixtures of isopropanol and water, in which SMC was facilitated by PdCl2(dppf), a ferrocene‐based, bidentate phosphine ligand that tolerates a broad range of functional groups present in amino acid side chains. Notably, histidine and asparagine were found to inhibit the cross‐coupling reaction and PdCl2(dppf) proved to be inefficient for the conversion of halotryptophans. The influence of the cross‐coupling reaction on the enantiopurity was examined by derivatisation with the chiral reagent 1‐fluoro‐2,4‐dinitrophenyl‐5‐l‐alanine amide (FDAA, Marfey's reagent) followed by RP‐HPLC analysis.44 This procedure is called Marfey's method and revealed no epimerisation of the analysed cross‐coupling products.45 A similar system based on Pd‐ADHP was explored by Sharma et al. to perform SMC with bromotryptophan also within peptides at biocompatible temperatures (Scheme 6 C).40 These optimisations of the reaction conditions also paved the way for application of SMC in living cells, which will be discussed in detail in section 4.3.
Scheme 6.
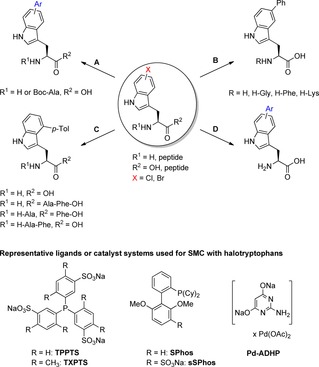
Late‐stage modifications of halotryptophans and halotryptophan containing peptides by SMC. Reaction conditions for SMC: A) Ar‐B(OH)2 (1 equiv), Na2PdCl4 (2.5 mol %), TPPTS or TXPTS (6.25 or 25 mol %), K2CO3 (5 equiv), H2O, 40–80 °C, 4–6 h.38 B) Ph‐B(OH)2 (2.5 equiv), Pd‐ADHP (5 mol %), K2CO3 (5 equiv), H2O, 80 °C, 2–24 h.39 C) p‐Tol‐B(OH)2 (3 equiv), Pd‐ADHP (5 mol %), K2CO3 (6 equiv), H2O/EtOH 4:1, 45 °C, 48 h.40 D) Ar‐B(OH)2 (2 equiv), Na2PdCl4 (5 mol %), sSPhos (15 mol %), K3PO4 (5 equiv), H2O/dioxane 3:1, 95 °C, μwave, 30–60 min.41
The application of catalyst systems including the sterically more demanding and electron‐rich Buchwald phosphine ligand SPhos are known to promote the SMC of challenging substrates such as aryl chlorides or electron‐rich substrates, which is the case for halotryptophans.46 Hence, SPhos‐based catalysts were investigated in our group to mediate the cross‐coupling between unprotected bromotryptophan and different aryl boronic acids. These provided initial studies for the establishments of a high‐throughput (HT) fluorescence assay to determine tryptophan halogenase activity relying on fluorescent aryltryptophans (cf. section 4.3)41 and a chemoenzymatic three‐step one‐pot process combining bio‐ and chemocatalysis (cf. section 4.4).23 As conclusion, a catalyst system composed of water‐soluble Na2PdCl4 as Pd source and the water‐soluble, sulfonated derivative of SPhos (sSPhos) combined with tripotassium phosphate as a base proved suitable for SMC in water or mixtures of water and dioxane at 100 °C even in the presence of air (Scheme 6 D). Similar conditions were also reported to impart the diversification of complex natural products47, 48 or indole‐derived pharmacologically active compounds (cf. section 4.3).49
4.2. SMC as tool for late‐stage cyclisations
Aside from late‐stage diversification of linear halopeptides with different boronic acids, SMC can also be utilised as tool for an intramolecular macrocyclisation affording constrained peptides with potential benefits regarding affinity, stability or even membrane permeability.50 Intramolecular cyclisations between bromo‐ or boronotryptophan and boronated or halogenated amino acid side chain residues have been developed for protected peptides in solution51 and additionally, there are also reports outlining a cross‐coupling between boronated and halogenated phenylalanines, tyrosines or histidines on resin.52, 53 The synthesis of bicyclic peptides containing a biaryl bridge and a cyclic backbone was described by García‐Pindado et al. in 2017, in which the cross‐coupling reaction took place between a boronophenylalanine and a iodophenylalanine to generate the biaryl bridge on resin.54 This approach was extended to bromotryptophan containing peptides in 2018.55 The methodology comprises an on‐resin Miyaura borylation of the first introduced bromotryptophan followed by completion of the linear precursor sequence including the second bromotryptophan moiety. Intramolecular SMC is performed on resin as the first cyclisation step. Then, the peptide is cleaved from the resin and an intramolecular head‐to‐tail cyclisation in solution results in the formation of a peptide bond as the second cyclisation step (Scheme 7).
Scheme 7.
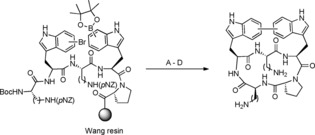
Synthesis of bicyclic peptides by two late‐stage cyclisations beginning with an intramolecular on‐resin SMC followed by cleavage from resin and in‐solution backbone cyclisation. 5‐, 6‐ and 7‐bromotryptophan regioisomers were used. Reaction conditions: A) Pd(PPh3)4 (10 mol %), Cs2CO3 (3 equiv), DMF, 60 °C, 72 h. B) TFA:H2O:TIS 95:2.5:2.5. C) PyBOP, HOAt, DCM with 1 % DMF, RT, 24–48 h. D) NaS2O4, H2O/ACN/EtOH 1:1:1, 48 h.55
The cyclic peptides were analysed in different biological tests. It was hypothesised that the increased rigidity of the bicyclic peptides might be advantageous to cross the challenging blood–brain barrier (BBB) by passive diffusion. As a result, the 7,7‐bicyclic peptide showed the highest transport rate in a parallel artificial membrane permeability assay, which is a model for the BBB. The serum stability of the bicyclic tryptophan peptides was not investigated.55 However, the previous study on a bicyclic peptide with a phenylalanine–phenylalanine biaryl bridge did not show any significant degradation in human serum within 24 h.54
Very recently, our research group reported on the side chain‐to‐tail cyclisation of RGD peptides by SMC.56 The cross‐coupling reaction was performed between l‐ or d‐bromotryptophan and alkyl or aryl boronic acids attached to the N‐terminus of the peptide. Both in solution and on‐resin strategies were evaluated; however, the on‐resin approach was beneficial because no side reactions such as deboronation were observed. Additionally, conducting as many steps as possible on resin avoids purification steps, which usually lead to decreased yields. The on‐resin cross‐coupling step required a catalyst system comprising Pd2(dba)3 and sSPhos combined with potassium fluoride as base in a solvent mixture of dimethoxyethane (DME), ethanol and water.53 Microwave irradiation and 120 °C lead to full conversion after 30 min (Scheme 8).56 Different connectivities involving different regioisomers were generated and compared to the backbone cyclised lead peptide c(RGDfV), phenylalanine and/or valine were substituted against hydrophobic biaryl moieties resulting from SMC cyclisation. The affinity and selectivity of these novel cyclic RGD peptides towards αVβ3 and α5β1 integrins were tested. It could be demonstrated that the connectivity between the indole and the aromatic moiety of the former aryl boronic acid has a major influence on affinity and selectivity. A representative example was chosen, and a good stability was proven in human plasma. Apart from that, NMR and HPLC analyses revealed the presence of multiple isomers. To exclude undesired epimerisation, MD simulations were performed for representative side chain‐to‐tail cyclised peptides exposing stable and distinguishable conformers or atropisomers. The promising results of that study show again the high potential of cross‐coupling reactions in the development of modified bioactive compounds that have not been thoroughly explored until now in context of drug design.
Scheme 8.
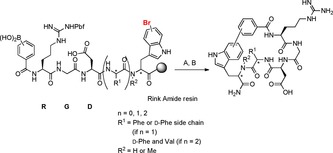
Side chain‐to‐tail cyclisation by on resin SMC for the generation of bioactive RGD peptides. Reaction conditions: A) Pd2(dba)3 (20 mol %), sSPhos (40 mol %), KF (4.0 equiv), DME:EtOH:H2O (9:9:2), μwave, 120 °C, 30 min. B) TFA:H2O:TIS 95:2.5:2.5.56
4.3. SMC as bioorthogonal tool for late‐stage diversification
In 2010, Deb Roy et al. reported on late‐stage SMC modification of a chlorotryptophan containing derivative of the uridyl peptide antibiotic pacidamycin.47 Since the previously described reaction conditions utilising TPPTS as ligand38 did not cause satisfactory conversion of the relative unreactive chloropacidamycin, turning to sSPhos in combination with 20 % (v/v) acetonitrile as cosolvent led to full conversion with phenyl‐ and 4‐methoxyphenylboronic acid during one hour under microwave irradiation at 80 °C. Remarkably, chloropacidamycin was generated in vivo by integration of the gene prnA encoding the tryptophan 7‐halogenase into the producer strain Streptomyces coeruleorubidus presenting a chemogenetic process. The following cross‐coupling proceeded successfully both with purified chloropacidamycin and the crude extract. This proves the biocompatibility of the SMC, because on the one hand this chlorinated natural product is a complex peptidic substrate and on the other hand other biomolecules are also present in crude extracts including different functional groups, which could interfere with the reaction (Scheme 9 A).
Scheme 9.
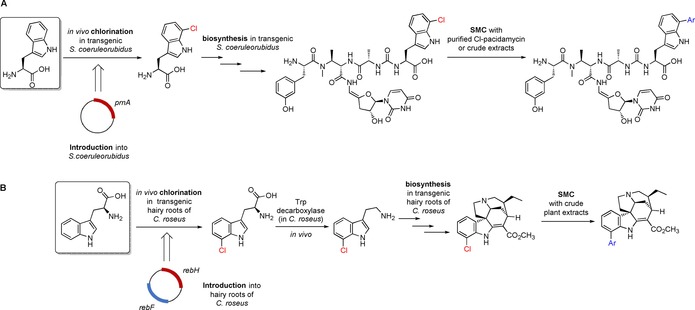
Chemogenetic processes for the synthesis of halogenated derivatives of natural products containing indole scaffolds, which are formed in vivo starting from tryptophan as precursor. The halogenated metabolites can be further diversified by SMC. A) Incorporation of prnA gene encoding for tryptophan 7‐halogenase PrnA in S. coeruleorubidus leading to formation of 7‐chlorotryptophan in vivo, which is used as precursor in the biosynthesis of chloropacidamycin. This halogenated derivative can be further diversified by late‐stage SMC. Reaction conditions for SMC: Na2PdCl4 (5 mol %), sSPhos (12.5 mol %), Ar‐B(OH)2 (1.1 equiv), K2CO3 (5 equiv), H2O:ACN 5:1, 80 °C, μwave, 60 min.47 B) Incorporation of rebH and rebF genes encoding for tryptophan 7‐halogenase RebH and flavin reductase RebF in hairy roots of C. roseus leading to formation of 7‐chlorotryptophan in vivo, which is used as a precursor in the biosynthesis of 12‐chloro‐19,20‐dihydroakuammicine through 7‐chlorotryptamin as intermediate. SMC with the crude plant extracts results in aryl‐diversified analogues. Reaction conditions for SMC: Pd(OAc)2 (5 mol %), SPhos (13 mol %), Ar‐B(OH)2 (3 equiv), K3PO4 (5 equiv), n‐butanol, 90–100 °C, 10–60 min.48
A related approach has been developed by Runguphan et al. in 2013 to modify halogenated monoterpene indole alkaloid derivatives of 19,20‐dihydroakuammicine.48 Halo‐19,20‐dihydroakuammicines were generated by either exogenous addition of halotryptamin or by integration of the tryptophan 7‐halogenase encoding rebH gene into the producer plant yielding transgenic hairy roots of Catharanthus roseus. To ensure sufficient cofactor regeneration to afford significant chlorination of tryptophan in vivo, the rebF gene encoding for a flavin reductase was also recombinantly introduced into C. roseus. Chlorotryptophan was then converted by an endogenous Trp‐decarboxylase to chlorotryptamin,57 which is a precursor in the biosynthesis of chloro‐19,20‐dihydroakuammicine. After one week of cultivation, adequate amounts of the chlorinated alkaloid had accumulated, and the resulting crude extract was used for SMC diversification without any previous purification step. Inspired by the approach of Deb Roy et al., the authors also utilised a catalyst system including the Buchwald ligand SPhos to convert the less reactive aryl chloride. Since the chlorinated alkaloid substrate is much less polar than chloropacidamycin, Pd(OAc)2 was applied as Pd source in n‐butanol as solvent combined with the base K3PO4 at 90–100 °C running reaction times ranging from 10 to 60 min to investigate the reaction with six different arylboronic acids (Scheme 9 B).
To prove bioorthogonality, Yusop et al. performed the SMC within living HeLa cells between exogenously added, reactive aryltriflates and pinacol boronates.58 Moreover, Sharma et al. showed in 2017 the concomitant biosynthesis of brominated metabolites and chemical modification of these less reactive substrates in the presence of living cells.40 To reach this goal, previously reported reaction conditions47 had to be modified to perform SMC at mild temperatures and under aerobic conditions. Catalyst systems based on the water‐soluble Buchwald ligand sSPhos did not lead to satisfactory conversion of halotryptophans due to Pd catalyst inhibition by the free amino group and also, to a lesser extent, by the carboxylic acid. Hence, a phosphine‐free approach exploring Pd‐ADHP was investigated. Optimised reaction conditions caused high conversions (≥90 %) of bromotryptophans at 45 °C in an aqueous, aerobic system with 20 % (v/v) ethanol as cosolvent. A conversion of only 30 % was observed for 7‐chlorotryptophan. Therefore, subsequent experiments employed bromotryptophan. Additionally, 7‐bromotryptophan containing tripeptides were reacted under these conditions and provided good isolated yields (cf. also section 4.1, Scheme 6 C). To perform the SMC in the presence of living, engineered E. coli cells continuously producing 7‐bromotryptophan due to an incorporated prnA gene, reaction conditions had again to be modified with two crucial changes leading to a biocompatible process: 1) a special growth medium named cross‐coupling medium (CCM) was employed since common media contain, for instance, amino acids and glucose, which inhibit SMC at mild temperatures; 2) an even more active catalyst system had to be applied to shorten the reaction time at 37 °C changing the ligand ADHP to its dimethylated analogue DMADHP or tetramethylguanidine (TMG) as more efficient variants (Scheme 10). By these final improvements, enzymatic halogenation of tryptophan using a whole cell catalyst and SMC derivatisation of the formed 7‐bromotryptophan in a sequential process could be realised. The cross‐coupling was performed after dilution of the E. coli culture with phosphate buffer; the cell viability was proven. The enantiopurity of the resulting cross‐coupling product l‐7‐tolyltryptophan was proven by Marfey's method (cf. section 4.1).45 Moreover, this approach was utilised to generate p‐tolyl‐pacidamycin from bromopacidamycin. For this purpose, the producer strain had to be changed from S. coeruleorubidus to S. coelicolor as a modification of the previously reported process to tolerate the bromide salt KBr, which is mandatory to generate the more reactive brominated metabolite.
Scheme 10.
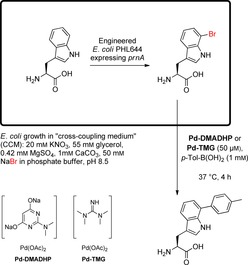
Concomitant bromination of tryptophan by an engineered E. coli whole cell catalyst producing the tryptophan 7‐halogenase PrnA and SMC derivatisation of the formed 7‐bromotryptophan in special growth medium CCM.40
Inspired by the demonstrated biocompatibility and bioorthogonality of the SMC as well as the modulated spectrophotometric properties of aryltryptophans, Schnepel et al. developed a high‐throughput (HT) fluorescence assay to quantify halogenated tryptophan by late‐stage arylation using SMC in lysate to screen for novel halogenase variants, which were engineered by directed evolution.41 The reaction conditions were initially optimised using purified tryptophan (cf. also section 4.1, Scheme 6 D). They were finally adapted for application in E. coli crude lysate employing a catalyst system consisting of water‐soluble Na2PdCl4 as Pd source and the water‐soluble Buchwald ligand sSPhos at a loading of 0.5 equivalents combined with 15 equivalents of tripotassium phosphate and 10 equivalents of 3‐aminophenylboronic acid in air. The reaction can be carried out in a microtiter plate in a drying oven at 95 °C for 120 min providing >98 % conversion. The emission maximum λ em of the product (3‐aminophenyl)‐tryptophan is 430 nm, whereas tryptophan is not fluorescent at that wavelength, which is a mandatory criterion for a selective quantification assay. To prove the applicability of the developed assay, a thermostable variant of the tryptophan 6‐halogenase Thal was engineered by directed evolution consisting of error‐prone PCR, transformation, cultivation, expression, cell lysis and temperature treatment at 49 °C as selection criteria (Scheme 11). Activity determination by HT fluorescence assay yielded in Thal‐GR (mutations: S359G and K374R) as more thermostable enzyme with a 2.5 times increased specific activity.
Scheme 11.
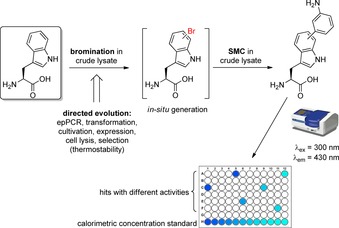
HT fluorescence assay utilising SMC in E. coli crude lysate in order to screen for engineered tryptophan halogenase variants. SMC reaction conditions: Na2PdCl4 (0.5 equiv), sSPhos (1.5 equiv), K3PO4 (15 equiv), 3‐aminophenylboronic acid (10 equiv), 95 °C, 120 min in a microtiter plate.41
4.4. Chemoenzymatic multistep one‐pot processes for application on larger scale
The above‐mentioned approaches by Deb Roy et al., Runguphan et al., Sharma et al. and Schnepel et al. are premier examples for cascade reactions combining bio‐ and chemocatalysis with application potential.40, 41, 47, 48 However, these processes have usually been performed on small (analytical) scales. In this section the perspective of preparative scale multi‐step one‐pot processes without intermediate work‐up and minimum purification steps is described. Pioneering investigations combining Pd and enzyme catalysis have been published by Gröger and co‐workers, in which a SMC in aqueous medium is followed by enzymatic asymmetric reduction as a two‐step one‐pot process. This approach requires only a final work‐up and purification providing improved efficiency and sustainability.59 Motivated by this proof of concept, Frese et al. reported on a three‐step one‐pot process consisting of regioselective, enzymatic halogenation utilising immobilised tryptophan halogenases, SMC and Nα‐Boc (tert‐butoxycarbonyl) protection as final step.23 For immobilisation of an appropriate FAD‐dependent tryptophan halogenase along with the auxiliary enzymes flavin reductase PrnF and alcohol dehydrogenase RR‐ADH for concomitant cofactor regeneration, the combiCLEA methodology was employed (cf. section 3). This provides a solid biocatalyst leading to selective bromination at positions C5 (halogenase PyrH), C6 (halogenase Thal), and C7 (halogenase RebH). Once halogenation has gone to completion, the solid biocatalyst can simply be filtered off. The subsequent cross‐coupling reaction can be performed in the filtrate and requires a suitable Pd catalyst, base and boronic acid. The conditions described by Schnepel et al. are suitable for this reaction.41 Remarkably, a conversion of 85 % can be observed with phenylboronic acid in air and could be rounded out by conducting the reaction under argon.23 Some unprotected SMC products are quite soluble in organic solvents. However, for a simplified isolation by extraction, a Nα‐Boc protection can easily be performed as third step. Purification by RP‐HPLC affords the Boc‐protected aryltryptophans in moderate to excellent isolated yields. Unlike the findings of Deb Roy et al.,38 C7‐substituted derivatives give higher yields than C6‐substituted ones and C5 modifications resulted in only modest yields (Scheme 12). In contrast to the mentioned experimental findings, the most electron‐deficient l‐6‐bromotryptophan was expected to be the most reactive derivative resulting in the highest yields because electron‐poor aryl halides undergo faster oxidative addition which is the rate‐determining step.36 Additionally, there was generally a decreased yield observed for C5‐ and C6‐substituted derivatives when reacted with electron‐poor boronic acids trifluoromethyl‐ and carboxy‐phenylboronic acid.
Scheme 12.
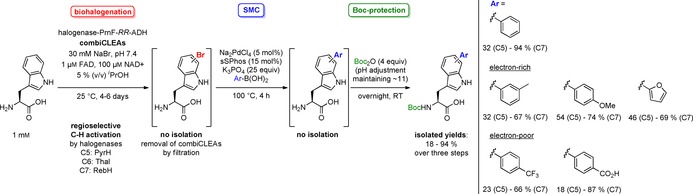
Three‐step one‐pot approach combining enzymatic bromination, SMC and Nα‐Boc protection.23
Besides converting tryptophan, it has been demonstrated that tryptophan halogenases are also able to halogenate tryptophan derivatives either using wild type enzymes or engineered variants opening up the possibility to modify indole‐based biological active compounds.20, 21, 49, 60 The Lewis group explored the substrate scope of the tryptophan 7‐halogenase RebH and engineered variants towards different tryptophan and indole derivatives.20, 61 They also reported on a chemoenzymatic arylation process utilising RebH variant 3‐SS with a higher bromination activity on tryptoline. Enzymatic halogenation was followed by late‐stage SMC performed in the crude extract.49 To further improve the integration of enzymatic halogenation and Pd catalysis, Latham et al. employed tryptophan halogenases for bromination of tryptophol and 3‐indole propionate with subsequent modification by SMC in different one‐pot approaches.24 In this work, the combiCLEA methodology pioneered by Frese et al.22 (cf. section 3) was used for enzymatic bromination of the indole derivatives tryptophol or 3‐indole propionate.
Subsequently, after removal of the heterogenous biocatalyst, anaerobic SMC was performed applying quite similar reaction conditions as described by Deb Roy et al. in 2008.38 For regioselectively addressing the indole substrates, RebH provided halogenation of tryptophol in C7‐position whereas PyrH and SttH lead to bromination of indole‐3 propionate in C5‐ and C6‐position, respectively. To establish an even more efficient and oxygen tolerating, sequential one‐pot process, the authors then investigated a compartmentalisation process (Scheme 13), in which polydimethylsiloxane (PDMS) thimbles containing the reagents necessary for SMC are applied (compartment B).24 These thimbles are placed in a larger reaction vessel (compartment A), in which the enzymatic halogenation has been performed beforehand. PDMS is a hydrophobic polymer functioning as semipermeable membrane through which only the non‐polar substrates can diffuse. At the same time, the exchange of charged compounds, which might vice versa inhibit the coupled process (enzymatic halogenation or SMC), is prevented. Moreover, to achieve an aerobic SMC, the more oxygen‐sensitive phosphine ligand based catalyst system was replaced by Pd‐DMADHP, which has also been later used by Sharma et al. for SMC in the presence of living cells.40 Optimised reaction conditions for the compartmentalisation process utilised RebH‐combiCLEAs combined with enzymatic bromination reagents in compartment A and Pd‐DMADHP along with cross‐coupling reagents in compartment B for regioselective derivatisation of tryptophol in C7‐position (Scheme 13).24 The zwitterionic amino acid tryptophan cannot be modified as the exchange between the compartments is prevented by the PDMS thimble.
Scheme 13.
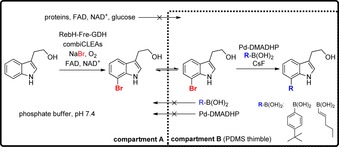
Compartmentalisation one‐pot approach separating reagents for enzymatic halogenation of tryptophol and subsequent SMC, respectively, to suppress inhibitory effects. The mixture is incubated overnight at room temperature followed by heating to 80 °C for 24 h. Reagents for SMC: Pd‐DMADHP (10 mol %), R‐B(OH)2 (5 equiv) and CsF (10 equiv). Fre: flavin reductase, GDH: glucose dehydrogenase.24
5. Cross‐Coupling Reactions beyond SMC
The SMC is a powerful technique for the modification of different haloindole scaffolds and has been proven to be biocompatible and bioorthogonal as described in section 4. Beyond that, there are other promising cross‐coupling reactions for carbon–carbon bond formations such as widely used Sonogashira–Hagihara (SHC) or Mizoroki–Heck reaction (MHC). Beyond that, Pd catalysis can also be applied in the generation of carbon–heteroatom bonds. Until now, several publications outline the importance of SHC to functionalise unprotected amino acids, peptides or proteins bearing aryliodides such as iodophenylalanine or iodotyrosine,62, 63 but only a few of them report on aqueous MHC to modify those substrates.63, 64, 65 However, the possibility to selectively address proteins by MHC has also been proven.63, 65
5.1. C−C bond formations
Recently, an aqueous SHC66 on halotryptophans as well halotryptophan containing tripeptides and the natural product cystargamide was published by Corr et al.67 Initial studies evaluated suitable reaction conditions with 5‐bromoindole as model substrate and were shown to be also compatible with unprotected bromotryptophan utilising a catalyst system consisting of PdCl2(CH3CN)2 as Pd source and the water‐soluble sterically demanding and electron‐rich Buchwald phosphine ligand sulfonated XPhos (sXPhos) in a 1:1 mixture of acetonitrile and water. The use of the base caesium carbonate led to a copper‐free variant of SHC and, moreover, microwave irradiation proved to be more efficient than conventional heating at 100 °C. With optimal reaction conditions in hand, the substrate scope was monitored. Excellent conversions were observed with arylacetylenes irrespective of the bromotryptophan regioisomer used. Noteworthy, iodotryptophan is not more reactive than bromotryptophan, whereas the less reactive chlorotryptophan is not accepted as aryl halide. Besides arylacetylenes, seven aliphatic alkynes possessing different functional groups were tested for derivatisation (Scheme 14 A). Among these, SHC proceeded with 4‐butyn‐1‐ol, 4‐phenyl‐1‐butyne and 1,7‐octadiyne giving good to excellent conversions, but failed with propiolic acid, propargylamine and TMS acetylene. The enantiopurity of the cross‐coupling products were proven by Marfey's method (cf. section 4.1).45 Since both SMC and SHC have the same substrate selectivity and comparable catalyst systems, the biocompatibility of SHC was evaluated.67 The influence of oxygen on the conversion of bromotryptophan was explored at first and found to cause only a moderate decrease in reactivity. Secondly, the cross‐coupling was performed with two 7‐bromotryptophan containing tripeptides as more challenging substrates. 3‐Fluorophenylacetylene was chosen as coupling partner because the highest isolated yield (97 %) was obtained when reacted with 5‐bromotryptophan. Because both brominated peptides were successfully converted, and isolated in 47 and 99 % yield, respectively (Scheme 14 B), an even more challenging cyclic natural product substrate was employed as a benchmark: 6‐bromocystargamide was produced by precursor directed biosynthesis with Kitasatospora cystarginea. Feeding experiments on an analytical scale with chloro‐ and bromotryptophans had been proven effective for the generation of halocystargamides. A scale‐up was conducted with 6‐bromotryptophan and the resulting 6‐bromocystargamide was submitted to SHC as semi‐pure starting material. The brominated natural product was completely converted on an analytical scale and a mixture of SHC‐diversified cyclic cystargamide was formed accompanied by a linear hydrolysis product (Scheme 14 C).
Scheme 14.
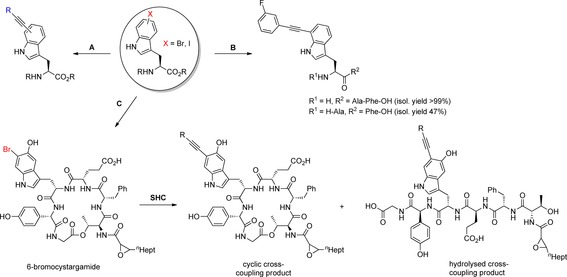
Late‐stage diversification by SHC with halotryptophans as well as halotryptophan containing peptides and natural products. A) Diversification of unprotected bromo‐ or iodotryptophans. Reaction conditions: alkyne (3.0 equiv), PdCl2(CH3CN)2 (5 mol %), sXPhos (15 mol %), Cs2CO3 (2.5 equiv), water/acetonitrile 1:1, 100 °C, μwave, 2 h. R=phenyl, 3‐fluorophenyl, 4‐cyanophenyl, 3‐thiopheneyl, 2‐hydroxyethyl, 2‐phenylethyl, 4‐ethynylbutyl, cyclohexylmethyl. B) Diversification of 7‐bromotryptophan containing tripeptides with 3‐fluorophenylacetylene. Reaction conditions: See A). C) Production of 6‐bromocystargamide by precursor‐directed biosynthesis with K. cystarginea. Subsequent SHC was performed with semi‐pure starting material; partial hydrolysis of cyclic cross‐coupling product to the linear analogue was observed. R=3‐fluorophenyl.67
Very recently, we reported on the utilisation of an aqueous MHC70 to react unprotected 7‐bromotryptophan with different substituted styrenes.68 To find suitable reaction conditions, a catalyst system consisting of Pd(OAc)2 as Pd source and the water‐soluble phosphine ligand TPPTS was tested because it had been successfully used by Yokoyama et al. in the total synthesis of the ergot alkaloid clavicipitic acid.64 Potassium carbonate was used as the base and the reaction with the water‐soluble 4‐carboxystyrene led to full conversion in degassed water at 100 °C in 18 h (Scheme 15 A‐i).68 The addition of the cosolvent dioxane was necessary to guarantee sufficient solubilisation of all components to expand the substrate scope towards water‐insoluble styrenes. Under these conditions six different E‐configured and enantiopure 7‐styryltryptophans were obtained in moderate to good yields. The enantiopurity was confirmed by Marfey's method (cf. section 4.1).45 Apart from this, application of the MHC in a chemoenzymatic three‐step one‐pot process including enzymatic bromination, cross‐coupling and Boc protection was studied as previously developed in our group for the SMC.23 The biocompatibility of the MHC was demonstrated for the water‐soluble 4‐carboxystyrene involving a concentration step after the enzymatic halogenation to favour the Pd‐catalysed alkenylation over dehalogenation (Scheme 15 B). Noteworthy, the generated styryltryptophans showed modulated spectroscopic properties as already mentioned for aryltryptophans. Compared to aryltryptophans, styryltryptophans have bathochromically shifted excitation wavelengths, which is an interesting parameter for the application as fluorophore in living cells to avoid irradiation damage.68 Later, Goss and co‐workers also described the MHC with unprotected halotryptophans and additionally, brominated natural products were explored as substrates.69 The optimised reaction conditions comprise a Na2PdCl4‐TXPTS catalyst system with sodium carbonate as base in a mixture of acetonitrile and water under microwave irradiation at 90 °C (Scheme 15 A‐ii). The use of acrylic acid as activated alkene reagent was also investigated and interestingly, no Michael‐like addition of the free α‐amino group was observed. As reported previously,68 E‐configuration predominates for the 7‐styryltryptophans. However, for 5‐styryltryptophans E/Z mixtures have been obtained.69 To prove the compatibility of MHC with more complex brominated natural products, barettin, a brominated cyclic diketopiperazine‐like peptide and bromopacidamycin were modified with 4‐fluorostyrene leading to full conversion in both cases (Scheme 15 C). However, as for SHC, Goss and co‐workers performed the reactions only on an analytical scale due to low quantities of starting material isolated from biotransformations.
Scheme 15.
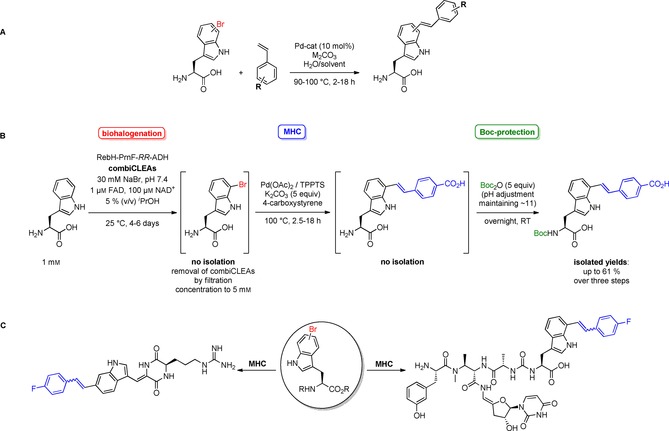
MHC with unprotected bromotryptophans and bromotryptophan containing natural products. A) i) Pd(OAc)2 (10 mol %), TPPTS (30 mol %), K2CO3 (1.5 equiv), styrene (5 equiv), water/(dioxane), 100 °C, 18 h; R=H, 4‐carboxy, 4‐methoxy, 4‐trifluoromethyl, 3‐fluoro, 3‐nitro;68 ii) Na2PdCl4 (10 mol %), TXPTS (23 mol %), Na2CO3 (4 equiv), styrene (2 equiv), water/(acetonitrile), 90 °C, μwave, 2 h; R=H, 4‐methyl, 4‐amino, 4‐fluoro, 3‐fluoro, 4‐pyridyl; additionally, acrylic acid was used as coupling partner.69 B) Chemoenzymatic three‐step one‐pot approach combining enzymatic bromination, MHC and Nα‐Boc protection; Pd loading 10–20 mol %;68 C) MHC diversification of the natural products barettin (left) and 7‐bromopacidamycin (right); reaction conditions: see A) ii).69
Besides SMC, SHC, and MHC as three of the most prominent examples for cross‐coupling reactions, a Pd‐catalysed α‐arylation of ketones using aryl halides as electrophile was reported in 1997.71 Recently, Marelli et al. established a procedure of this C−C bond forming reaction under aqueous conditions to modify ketones with indoles and Nα‐Boc‐protected halotryptophans in α‐position utilising PdCl2(dtbpf), a ferrocene‐based, bidentate phosphine ligand, as catalyst together with sodium hydroxide as base in a mixture of dioxane and water at 60 °C (Scheme 16).72
Scheme 16.
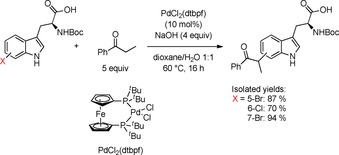
α‐Arylation of Nα‐Boc‐protected halotryptophans in aqueous medium.72
Both bromo‐ and chlorotryptophans were successfully diversified by the procedure and interestingly, no epimerisation was observed even though the reaction was performed under quite basic reaction conditions at elevated temperatures. The enantiopurity was verified using Marfey's method (cf. section 4.1).45
5.2. C−Heteroatom bond formations
Beyond C−C bond formations, Pd‐catalysis opens a versatile toolbox for the formation of C−heteroatom bonds such as C−N,73 C−O74 and even C−S.75 In 2016, Durak et al. published chemoenzymatic reactions combining regioselective halogenation using RebH variants and Pd‐mediated Buchwald‐Hartwig amination as well as alkoxylation with crude extracts.49 Whereas the indole derivative 5‐bromotryptoline was imparted in C−N bond formations with aminopyridines by a catalyst system consisting of Pd(OAc)2 and the dialkylbiaryl phosphine ligand BrettPhos in the presence of sodium tert‐butoxide in dioxane at 100 °C (Scheme 17), attempts to form aryl ethers failed underlining the difficulty to perform alkoxylation reactions with unprotected haloindole scaffolds. The brominated derivative of the drug thenalidine on the other hand was successfully converted with trifluoroethanol albeit only in moderate isolated yield. It remains to be clarified whether the application of enantiopure halotryptophans or halotryptophan containing molecules in Pd‐mediated amination reactions is feasible since strong bases such as sodium tert‐butoxide may be associated with epimerisation.
Scheme 17.
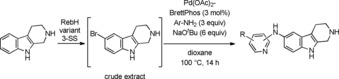
Buchwald–Hartwig amination of 5‐bromotryptoline with aminopyridines. The cross‐coupling reaction was performed with crude extract obtained from enzymatic halogenation.49
6. Summary and Outlook
C2−H activation of indole has been established as important tool for the late‐stage diversification of indole containing biomolecules. However, limitations are obvious as only one position can selectively be addressed. Moreover, in case of substrates containing several indole moieties, no regioselectivity can be achieved.
Regio‐ and stereoselective enzymatic syntheses of halotryptophans allow for introduction of a variety of substituents at the indole ring when combined with diverse Pd‐mediated cross‐coupling reactions in efficient cascade reactions. Apart from multiple applications of the SMC, recent developments also employ other prominent C−C bond forming cross‐coupling reactions. Representative examples demonstrated that the conditions of the Pd‐catalysed cross‐coupling reactions have no impact on the enantiopurity of the resulting products. Aqueous conditions are readily tolerated in all cases and additionally, the biocompatibility including the presence of a wide scope of functional groups and oxygen was demonstrated for many processes. Absolute bioorthogonality as a premier achievement was proven for the SMC. Since there are strategies known to modify proteins containing reactive aryl iodides, incorporation of bromotryptophan in proteins followed by late‐stage diversification by SMC needs to be optimised. Although brominated substrates are even more challenging, current developments suggest the possibility for protein modifications. As substituted tryptophans reveal very interesting spectrophotometric properties, such compounds show promise for fluorescence labelling or to study protein dynamics. Labelling of tryptophan is particularly interesting considering its low abundance in proteins.
Beyond that, only little has been done for C−heteroatom bond formation utilising haloindole substrates and hence, research focus is required on those procedures. Additionally, different transition metal catalysts offer great potential to introduce various functionalities to haloindole containing substrates.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Hendrik Gruß received his B.Sc., M.Sc. and Dr. rer. nat. degrees in biochemistry from Bielefeld University and specialised in chemical biology. From 2016–2019 he was a Ph.D. student in the Organic and Bioorganic Chemistry research group under supervision of Prof. Dr. N. Sewald. He is particularly interested in late‐stage modifications of bromotryptophans and peptides by cross‐coupling reactions as well as peptide stapling.

Biographical Information
Norbert Sewald studied chemistry at the Technical University of Munich. Having obtained his Ph.D. degree in organic chemistry, he worked in the group of Prof. J. E. Baldwin from 1991 to 1992 at the Dyson Perrins Laboratory, Oxford University. In 1998 he finished his habilitation at the University of Leipzig. He was appointed Full Professor of Organic and Bioorganic Chemistry at Bielefeld University in 1999.

Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft (SE 609/16‐1) is gratefully acknowledged. The authors thank Dr. C. Schnepel, S. Dachwitz, and Dr. I. Kemker for helpful discussions. C. Belu is acknowledged for the graphical design.
H. Gruß, N. Sewald, Chem. Eur. J. 2020, 26, 5328.
References
- 1.
- 1a. Ramil C. P., Lin Q., Chem. Commun. 2013, 49, 11007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Chankeshwara S. V., Indrigo E., Bradley M., Curr. Opin. Chem. Biol. 2014, 21, 128; [DOI] [PubMed] [Google Scholar]
- 1c. Zhang C., Vinogradova E. V., Spokoyny A. M., Buchwald S. L., Pentelute B. L., Angew. Chem. Int. Ed. 2019, 58, 4810; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4860; [Google Scholar]
- 1d. Willemse T., Schepens W., Vlijmen H., Maes B., Ballet S., Catalysts 2017, 7, 74. [Google Scholar]
- 2. Noisier A. F. M., Brimble M. A., Chem. Rev. 2014, 114, 8775. [DOI] [PubMed] [Google Scholar]
- 3. Spicer C. D., Davis B. G., Nat. Commun. 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- 4. Sengupta S., Mehta G., Tetrahedron Lett. 2017, 58, 1357. [Google Scholar]
- 5. Wang W., Lorion M. M., Shah J., Kapdi A. R., Ackermann L., Angew. Chem. Int. Ed. 2018, 57, 14700; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14912. [Google Scholar]
- 6. Mondal S., Chowdhury S., Adv. Synth. Catal. 2018, 360, 1884. [Google Scholar]
- 7.
- 7a. Bogan A. A., Thorn K. S., J. Mol. Biol. 1998, 280, 1; [DOI] [PubMed] [Google Scholar]
- 7b. Moreira I. S., Fernandes P. A., Ramos M. J., Proteins Struct. Funct. Bioinf. 2007, 68, 803; [DOI] [PubMed] [Google Scholar]
- 7c. Bullock B. N., Jochim A. L., Arora P. S., J. Am. Chem. Soc. 2011, 133, 14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y., Barkley M. D., Biochemistry 1998, 37, 9976. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Ramani S., Patil N., Nimbalkar S., Jayabaskaran C. in Natual Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes (Eds.: K. G. Ramawat, J. M. Merillon), Springer, Heidelberg, 2013, pp. 575–604; [Google Scholar]
- 9b. Gribble G. W., The Alkaloids: Chemistry and Biology, Vol. 71, Academic Press, Amsterdam, The Netherlands, 2012, pp. 1 ff. [Google Scholar]
- 10.
- 10a. Deprez N. R., Kalyani D., Krause A., Sanford M. S., J. Am. Chem. Soc. 2006, 128, 4972; [DOI] [PubMed] [Google Scholar]
- 10b. Deprez N. R., Sanford M. S., Inorg. Chem. 2007, 46, 1924; [DOI] [PubMed] [Google Scholar]
- 10c. Lebrasseur N., Larrosa I., J. Am. Chem. Soc. 2008, 130, 2926; [DOI] [PubMed] [Google Scholar]
- 10d. Yang S.-D., Sun C.-L., Fang Z., Li B.-J., Li Y.-Z., Shi Z.-J., Angew. Chem. Int. Ed. 2008, 47, 1473; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1495. [Google Scholar]
- 11. Ruiz-Rodríguez J., Albericio F., Lavilla R., Chem. Eur. J. 2010, 16, 1124. [DOI] [PubMed] [Google Scholar]
- 12. Preciado S., Mendive-Tapia L., Torres-García C., Zamudio-Vázquez R., Soto-Cerrato V., Pérez-Tomás R., Albericio F., Nicolás E., Lavilla R., MedChemComm 2013, 4, 1171. [Google Scholar]
- 13. Preciado S., Mendive-Tapia L., Albericio F., Lavilla R., J. Org. Chem. 2013, 78, 8129. [DOI] [PubMed] [Google Scholar]
- 14. Dong H., Limberakis C., Liras S., Price D., James K., Chem. Commun. 2012, 48, 11644. [DOI] [PubMed] [Google Scholar]
- 15. Mendive-Tapia L., Preciado S., García J., Ramón R., Kielland N., Albericio F., Lavilla R., Nat. Commun. 2015, 6, 7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu Y., Bauer M., Ackermann L., Chem. Eur. J. 2015, 21, 9980. [DOI] [PubMed] [Google Scholar]
- 17. Reay A. J., Williams T. J., Fairlamb I. J. S., Org. Biomol. Chem. 2015, 13, 8298. [DOI] [PubMed] [Google Scholar]
- 18. Reay A. J., Hammarback L. A., Bray J. T. W., Sheridan T., Turnbull D., Whitwood A. C., Fairlamb I. J. S., ACS Catal. 2017, 7, 5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Bitto E., Huang Y., Bingman C. A., Singh S., Thorson J. S., Phillips G. N., Proteins Struct. Funct. Bioinf. 2008, 70, 289; [DOI] [PubMed] [Google Scholar]
- 19b. Dong C., Flecks S., Unversucht S., Haupt C., van Pée K.-H., Naismith J. H., Science 2005, 309, 2216; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Flecks S., Patallo E. P., Zhu X., Ernyei A. J., Seifert G., Schneider A., Dong C., Naismith J. H., van Pée K.-H., Angew. Chem. Int. Ed. 2008, 47, 9533; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9676; [Google Scholar]
- 19d. Yeh E., Blasiak L. C., Koglin A., Drennan C. L., Walsh C. T., Biochemistry 2007, 46, 1284; [DOI] [PubMed] [Google Scholar]
- 19e. Yeh E., Cole L. J., Barr E. W., Bollinger J. M., Ballou D. P., Walsh C. T., Biochemistry 2006, 45, 7904; [DOI] [PubMed] [Google Scholar]
- 19f. Zhu X., de Laurentis W., Leang K., Herrmann J., Ihlefeld K., van Pée K.-H., Naismith J. H., J. Mol. Biol. 2009, 391, 74; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19g. Schnepel C., Sewald N., Chem. Eur. J. 2017, 23, 12064; [DOI] [PubMed] [Google Scholar]
- 19h. Latham J., Brandenburger E., Shepherd S. A., Menon B. R. K., Micklefield J., Chem. Rev. 2018, 118, 232. [DOI] [PubMed] [Google Scholar]
- 20. Payne J. T., Andorfer M. C., Lewis J. C., Angew. Chem. Int. Ed. 2013, 52, 5271; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5379. [Google Scholar]
- 21. Frese M., Guzowska P. H., Voß H., Sewald N., ChemCatChem 2014, 6, 1270–1276. [Google Scholar]
- 22. Frese M., Sewald N., Angew. Chem. Int. Ed. 2015, 54, 298; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 302. [Google Scholar]
- 23. Frese M., Schnepel C., Minges H., Voß H., Feiner R., Sewald N., ChemCatChem 2016, 8, 1799. [Google Scholar]
- 24. Latham J., Henry J.-M., Sharif H. H., Menon B. R. K., Shepherd S. A., Greaney M. F., Micklefield J., Nat. Commun. 2016, 7, 11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Neubauer P. R., Widmann C., Wibberg D., Schröder L., Frese M., Kottke T., Kalinowski J., Niemann H., Sewald N., PLoS One 2018, 13, e0196797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Ismail M., Frese M., Patschkowski T., Ortseifen V., Niehaus K., Sewald N., Adv. Synth. Catal. 2019, 361, 2475. [Google Scholar]
- 26. Ismail M., Schroeder L., Frese M., Kottke T., Hollmann F., Paul C. E., Sewald N., ACS Catal. 2019, 9, 1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schroeder L., Frese M., Müller C., Sewald N., Kottke T., ChemCatChem 2018, 10, 3336. [Google Scholar]
- 28. Veldmann K. H., Minges H., Sewald N., Lee J.-H., Wendisch V. F., J. Biotechnol. 2019, 291, 7. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Wilcox M., Anal. Biochem. 1974, 59, 436; [DOI] [PubMed] [Google Scholar]
- 29b. Lee M., Phillips R. S., Bioorg. Med. Chem. Lett. 1992, 2, 1563; [Google Scholar]
- 29c. Phillips R. S., Tetrahedron: Asymmetry 2004, 15, 2787; [Google Scholar]
- 29d. Goss R. J. M., Newill P. L. A., Chem. Commun. 2006, 4924; [DOI] [PubMed] [Google Scholar]
- 29e. Smith D. R. M., Willemse T., Gkotsi D. S., Schepens W., Maes B. U. W., Ballet S., Goss R. J. M., Org. Lett. 2014, 16, 2622. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Murciano-Calles J., Romney D. K., Brinkmann-Chen S., Buller A. R., Arnold F. H., Angew. Chem. Int. Ed. 2016, 55, 11577; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11749; [Google Scholar]
- 30b. Romney D. K., Murciano-Calles J., Wehrmüller J. E., Arnold F. H., J. Am. Chem. Soc. 2017, 139, 10769; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Boville C. E., Romney D. K., Almhjell P. J., Sieben M., Arnold F. H., J. Org. Chem. 2018, 83, 7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parmeggiani F., Rué Casamajo A., Walton C. J. W., Galman J. L., Turner N. J., Chica R. A., ACS Catal. 2019, 9, 3482. [Google Scholar]
- 32. Schnepel C., Kemker I., Sewald N., ACS Catal. 2019, 9, 1149. [Google Scholar]
- 33.
- 33a. Miyaura N., Yamada K., Suzuki A., Tetrahedron Lett. 1979, 20, 3437; [Google Scholar]
- 33b. Miyaura N., Suzuki A., J. Chem. Soc. Chem. Commun. 1979, 866; [Google Scholar]
- 33c. Kürti L., Czakó B., Strategic Applications of Named Reactions in Organic Synthesis: Background and Detailed Mechanisms; 250 Named Reactions, Elsevier, Amsterdam, 2009, pp. 448–449. [Google Scholar]
- 34.
- 34a. Liu C., Li X., Chem. Rec. 2016, 16, 84; [DOI] [PubMed] [Google Scholar]
- 34b. Biffis A., Centomo P., Del Zotto A., Zecca M., Chem. Rev. 2018, 118, 2249. [DOI] [PubMed] [Google Scholar]
- 35. Boronic Acids. Preparation and Applications in Organic Synthesis Medicine and Materials (Ed.: D. G. Hall), Wiley-VCH, Weinheim, 2012, pp. 213 ff. [Google Scholar]
- 36. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457. [Google Scholar]
- 37.
- 37a. Elder A. M., Rich D. H., Org. Lett. 1999, 1, 1443; [DOI] [PubMed] [Google Scholar]
- 37b. Kaiser M., Groll M., Renner C., Huber R., Moroder L., Angew. Chem. Int. Ed. 2002, 41, 780; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 817; [Google Scholar]
- 37c. Basse N., Piguel S., Papapostolou D., Ferrier-Berthelot A., Richy N., Pagano M., Sarthou P., Sobczak-Thépot J., Reboud-Ravaux M., Vidal J., J. Med. Chem. 2007, 50, 2842. [DOI] [PubMed] [Google Scholar]
- 38. Deb Roy A., Goss R. J. M., Wagner G. K., Winn M., Chem. Commun. 2008, 4831. [DOI] [PubMed] [Google Scholar]
- 39. Willemse T., van Imp K., Goss R. J. M., Van Vlijmen H. W. T., Schepens W., Maes B. U. W., Ballet S., ChemCatChem 2015, 7, 2055. [Google Scholar]
- 40. Sharma S. V., Tong X., Pubill-Ulldemolins C., Cartmell C., Bogosyan E. J. A., Rackham E. J., Marelli E., Hamed R. B., Goss R. J. M., Nat. Commun. 2017, 8, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schnepel C., Minges H., Frese M., Sewald N., Angew. Chem. Int. Ed. 2016, 55, 14159; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14365. [Google Scholar]
- 42. Laughlin S. T., Bertozzi C. R., Nat. Protoc. 2007, 2, 2930. [DOI] [PubMed] [Google Scholar]
- 43. Chalker J. M., Wood C. S. C., Davis B. G., J. Am. Chem. Soc. 2009, 131, 16346. [DOI] [PubMed] [Google Scholar]
- 44. Marfey P., Carlsberg Res. Commun. 1984, 49, 591. [Google Scholar]
- 45. Bhushan R., Brückner H., Amino Acids 2004, 27, 231. [DOI] [PubMed] [Google Scholar]
- 46. Barder T. E., Walker S. D., Martinelli J. R., Buchwald S. L., J. Am. Chem. Soc. 2005, 127, 4685. [DOI] [PubMed] [Google Scholar]
- 47. Deb Roy A., Grüschow S., Cairns N., Goss R. J. M., J. Am. Chem. Soc. 2010, 132, 12243. [DOI] [PubMed] [Google Scholar]
- 48. Runguphan W., O'Connor S. E., Org. Lett. 2013, 15, 2850. [DOI] [PubMed] [Google Scholar]
- 49. Durak L. J., Payne J. T., Lewis J. C., ACS Catal. 2016, 6, 1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.
- 50a. Milroy L.-G., Grossmann T. N., Hennig S., Brunsveld L., Ottmann C., Chem. Rev. 2014, 114, 4695; [DOI] [PubMed] [Google Scholar]
- 50b. Valeur E., Guéret S. M., Adihou H., Gopalakrishnan R., Lemurell M., Waldmann H., Grossmann T. N., Plowright A. T., Angew. Chem. Int. Ed. 2017, 56, 10294; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10428. [Google Scholar]
- 51.
- 51a. Jia Y., Bois-Choussy M., Zhu J., Angew. Chem. Int. Ed. 2008, 47, 4167; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4235; [Google Scholar]
- 51b. Kaiser M., Siciliano C., Assfalg-Machleidt I., Groll M., Milbradt A. G., Moroder L., Org. Lett. 2003, 5, 3435. [DOI] [PubMed] [Google Scholar]
- 52. Afonso A., Feliu L., Planas M., Tetrahedron 2011, 67, 2238. [Google Scholar]
- 53. Afonso A., Cussó O., Feliu L., Planas M., Eur. J. Org. Chem. 2012, 6204. [Google Scholar]
- 54. García-Pindado J., Royo S., Teixidó M., Giralt E., J. Pept. Sci. 2017, 23, 294. [DOI] [PubMed] [Google Scholar]
- 55. García-Pindado J., Willemse T., Goss R. J. M., Maes B. U. W., Giralt E., Ballet S., Teixidó M., Biopolymers 2018, 109, e23112. [DOI] [PubMed] [Google Scholar]
- 56. Kemker I., Schnepel C., Schröder D. C. C., Marion A., Sewald N., J. Med. Chem. 2019, 62, 7417. [DOI] [PubMed] [Google Scholar]
- 57. Runguphan W., Qu X., O'Connor S. E., Nature 2010, 468, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yusop R. M., Unciti-Broceta A., Johansson E. M. V., Sánchez-Martín R. M., Bradley M., Nat. Chem. 2011, 3, 239. [DOI] [PubMed] [Google Scholar]
- 59.
- 59a. Burda E., Hummel W., Gröger H., Angew. Chem. Int. Ed. 2008, 47, 9551; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9693; [Google Scholar]
- 59b. Borchert S., Burda E., Schatz J., Hummel W., Gröger H., J. Mol. Catal. B Enzym. 2012, 84, 89; [Google Scholar]
- 59c. Gröger H., Hummel W., Curr. Opin. Chem. Biol. 2014, 19, 171. [DOI] [PubMed] [Google Scholar]
- 60. Glenn W. S., Nims E., O'Connor S. E., J. Am. Chem. Soc. 2011, 133, 19346. [DOI] [PubMed] [Google Scholar]
- 61. Payne J. T., Poor C. B., Lewis J. C., Angew. Chem. Int. Ed. 2015, 54, 4226; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4300. [Google Scholar]
- 62.
- 62a. Dibowski H., Schmidtchen F. P., Angew. Chem. Int. Ed. 1998, 37, 476; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 487; [Google Scholar]
- 62b. Hoffmanns U., Metzler-Nolte N., Bioconjugate Chem. 2006, 17, 204; [DOI] [PubMed] [Google Scholar]
- 62c. Bong D. T., Ghadiri M. R., Org. Lett. 2001, 3, 2509; [DOI] [PubMed] [Google Scholar]
- 62d. Hauke S., Best M., Schmidt T. T., Baalmann M., Krause A., Wombacher R., Bioconjugate Chem. 2014, 25, 1632. [DOI] [PubMed] [Google Scholar]
- 63. Kodama K., Fukuzawa S., Nakayama H., Sakamoto K., Kigawa T., Yabuki T., Matsuda N., Shirouzu M., Takio K., Yokoyama S., Tachibana K., ChemBioChem 2007, 8, 232. [DOI] [PubMed] [Google Scholar]
- 64. Yokoyama Y., Hikawa H., Mitsuhashi M., Uyama A., Hiroki Y., Murakami Y., Eur. J. Org. Chem. 2004, 1244. [Google Scholar]
- 65. Kodama K., Fukuzawa S., Nakayama H., Kigawa T., Sakamoto K., Yabuki T., Matsuda N., Shirouzu M., Takio K., Tachibana K., Yokoyama S., ChemBioChem 2006, 7, 134. [DOI] [PubMed] [Google Scholar]
- 66. Sonogashira K., Tohda Y., Hagihara N., Tetrahedron Lett. 1975, 16, 4467. [Google Scholar]
- 67. Corr M. J., Sharma S. V., Pubill-Ulldemolins C., Bown R. T., Poirot P., Smith D. R. M., Cartmell C., Abou Fayad A., Goss R. J. M., Chem. Sci. 2017, 8, 2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gruß H., Belu C., Bernhard L. M., Merschel A., Sewald N., Chem. Eur. J. 2019, 25, 5880. [DOI] [PubMed] [Google Scholar]
- 69. Pubill-Ulldemolins C., Sharma S. V., Cartmell C., Zhao J., Cárdenas P., Goss R. J. M., Chem. Eur. J. 2019, 25, 10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.
- 70a. Mizoroki T., Mori K., Ozaki A., Bull. Chem. Soc. Jpn. 1971, 44, 581; [Google Scholar]
- 70b. Heck R. F., Nolley J. P., J. Org. Chem. 1972, 37, 2320. [Google Scholar]
- 71.
- 71a. Hamann B. C., Hartwig J. F., J. Am. Chem. Soc. 1997, 119, 12382; [Google Scholar]
- 71b. Palucki M., Buchwald S. L., J. Am. Chem. Soc. 1997, 119, 11108; [Google Scholar]
- 71c. Satoh T., Kawamura Y., Miura M., Nomura M., Angew. Chem. Int. Ed. Engl. 1997, 36, 1740; [Google Scholar]; Angew. Chem. 1997, 109, 1820. [Google Scholar]
- 72. Marelli E., Renault Y., Sharma S. V., Nolan S. P., Goss R. J. M., Chem. Eur. J. 2017, 23, 3832. [DOI] [PubMed] [Google Scholar]
- 73.
- 73a. Louie J., Hartwig J. F., Tetrahedron Lett. 1995, 36, 3609; [Google Scholar]
- 73b. Guram A. S., Rennels R. A., Buchwald S. L., Angew. Chem. Int. Ed. Engl. 1995, 34, 1348; [Google Scholar]; Angew. Chem. 1995, 107, 1456. [Google Scholar]
- 74. Mann G., Hartwig J. F., J. Am. Chem. Soc. 1996, 118, 13109. [Google Scholar]
- 75.
- 75a. Kosugi M., Shimizu T., Migita T., Chem. Lett. 1978, 7, 13; [Google Scholar]
- 75b. Migita T., Shimizu T., Asami Y., Shiobara J.-i., Kato Y., Kosugi M., Bull. Chem. Soc. Jpn. 1980, 53, 1385. [Google Scholar]