

Abstract
In Mycobacterium tuberculosis, mycolic acids and their glycerol, glucose, and trehalose esters (“cord factor”) form the main part of the mycomembrane. Despite their first isolation almost a century ago, full stereochemical evaluation is lacking, as is a scalable synthesis required for accurate immunological, including vaccination, studies. Herein, we report an efficient, convergent, gram‐scale synthesis of four stereo‐isomers of a mycolic acid and its glucose ester. Binding to the antigen presenting protein CD1b and T cell activation studies are used to confirm the antigenicity of the synthetic material. The absolute stereochemistry of the syn‐methoxy methyl moiety in natural material is evaluated by comparing its optical rotation with that of synthetic material.
Keywords: CD1b, cross coupling, mycolic acid, total synthesis, tuberculosis
The total synthesis of four methoxy mycolic acid diastereomers is described. Key steps involve a Suzuki–Fu cross‐coupling, an anti‐selective Abiko–Masamune asymmetric aldol reaction, and an enantioselective Charette cyclopropanation. CD1b‐free mycolic acid tetramer staining experiments clearly favoured one diastereomer over the other three.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, is by far the most lethal member of the family of Mycobacteriaceae, causing around 10 million new infections yearly.1 The difficult diagnosis and treatment of tuberculosis is reflected in high mortality rates (1.5 million in 2018), which makes it the leading cause of death by a single infectious agent worldwide.1 This difficult treatment is in part caused by the presence of the mycomembrane in the cell envelope of Mtb, which consists largely of α‐alkyl, β‐hydroxy long‐chain (C70–C90) fatty acids, known as mycolic acids.2, 3 These highly lipophilic fatty acids are categorized according to the presence of unsaturations or cyclopropyl groups (“α‐mycolic acid”), a methoxy, or a keto function in the main chain. As the number of methylene units between these substituents is variable, mycolic acids are found as an inseparable mixture of homologues. Mycolic acids are found either as the free acids or esterified to the arabinogalactan layer, trehalose, glucose (glucose monomycolate; GMM) or glycerol.
The overall molecular structure of mycolic acids has been elucidated in the late 1960s,7, 8, 9 and several members have been prepared by the group of Baird10, 11, 12, 13 including two methoxy mycolic acid diastereomers (Figure 2, 1 c and 1 d). The complete elucidation of their stereochemistry, however, has so far not been achieved. Moody and co‐workers demonstrated that R,R stereochemistry of the α‐alkyl, β‐hydroxy carboxylic acid is crucial for T cell recognition of glucose monomycolate.14 The stereochemistry of the α‐methyl methoxy moiety in methoxy mycolic acids was inferred by Asselineau et al. to be S,S by comparison of molar optical rotations of natural samples with those of reference compounds.7 For this, the authors applied the principle of optical superposition, originally hypothesized by Van′t Hoff.15 This hypothesis states that stereocenters that are remotely located do not influence each other's optical rotation, which therefore can be added up to provide the overall optical rotation. The analysis was supported by the synthesis work of Baird et al., but it remained uncertain whether this hypothesis was valid for mycolic acids as the optical rotation of the cis‐cyclopropyl ring in model systems was immeasurably small, making the determination of its absolute stereochemistry using this strategy impossible. Due to the lack of other viable analytical techniques, the stereochemistry of the cyclopropyl ring still remains to be determined.
Figure 2.
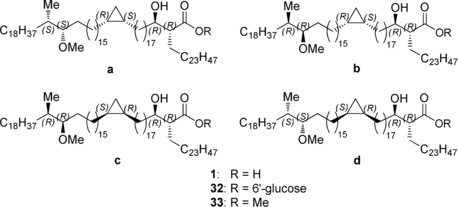
Overview of the four diastereomeric mycolic acids (1 a–d), glucose mycolates (32 a–d), and methyl esters (33 a–d).
Methoxy mycolic acids (Figure 1) appear, with some exceptions, to be present only in pathogenic mycobacteria3 and are antigenic in serological assays4 and T cell assays.5, 6 Upon binding to the antigen‐presenting protein CD1b, mycolic acids and their glucose mycolates activate T cells.16, 17 For glucose monomycolates the type and length of the mycolate does not influence the T cell stimulatory capacity.17 Crystallography of the trimolecular complex of CD1b, glucose monomycolate and T cell receptor showed that the glucose forms the T cell epitope on top of the CD1b protein and stabilizes the complex.18 No crystallographic data of CD1b–mycolic acid are available, but it is known that the type of functional groups in the chain and its length determine the level of T cell activation.5, 6 We hypothesized that the stereochemistry of the cyclopropyl and the α‐methyl methoxy moiety of mycolic acid might influence its capacity to bind CD1b and activate T cells.
Figure 1.
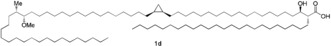
The structure of the major methoxy mycolic acid homologue.
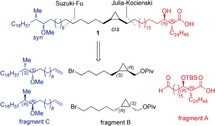
The development of mycolic acids and glucose monomycolates as potential antigens in vaccine development and diagnostics requires synthetic material strictly free from immunologically‐active impurities. This was our incentive to undertake the total synthesis of four stereoisomers of methoxy mycolic acid (Scheme 1, so S,S and R,R‐stereochemistry in the α‐methyl methoxy moiety plus R,S and S,R‐stereochemistry in the cis‐cyclopropyl moiety) and their glucose esters. This endeavour required a drastic decrease in the number of synthetic transformations compared to the previous synthesis by Baird et al.11 Therefore we opted for incorporation of the Suzuki–Fu cross‐coupling on several places in the design. Although the Suzuki‐Fu cross‐coupling19 has hardly been applied in natural product synthesis, we advocate the use of this Pd‐catalysed sp3–sp3 cross‐coupling reaction as it avoids the various functional group transformations and basic reaction conditions required for Wittig‐type and Julia‐type reactions.
Scheme 1.
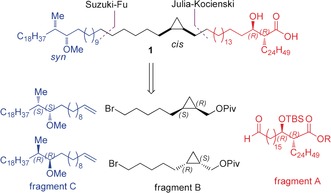
Retrosynthetic analysis.
In order to gain access to the four diastereomers in a convergent manner and on gram scale, we retrosynthetically dissected the molecule in three fragments, each containing a chiral segment (Scheme 1). Both enantiomers of fragments B and C had to be prepared, and combined with the natural enantiomer of fragment A. Whereas fragment B and C were planned to be connected via a Suzuki–Fu cross‐coupling reaction, unification of C–B to A using this reaction could not be achieved since, as model studies showed, with a cyclopropylmethyl bromide the reaction failed. A Julia–Kocienski olefination was projected instead.
Results and Discussion
Synthesis of the Fragments
The α‐branched β‐hydroxy acid moiety in fragment A was installed via an anti‐selective Abiko–Masamune asymmetric aldol reaction20 between aldehyde 6 and chiral ester 9 (Scheme 2 a). The synthesis of 6 started with a DIBAL reduction of commercially available 2 to the corresponding lactol 3, directly followed by a Wittig olefination with 4 21 resulting in 5 in 70 % yield over these two steps. The α,β‐unsaturated thioester 5 was then silylated, saturated, and reduced to the aldehyde in a single step in 82 % yield by a Fukuyama reduction. Esterification of chiral auxiliary 8 with the acyl bromide of 7 gave 9 in nearly quantitative yield. The subsequent Abiko–Masamune aldol reaction of 9 and 6 resulted in a rewarding dr of 9:1, and partial hydrolysis of the TES ether. Therefore, the crude was treated with aqueous HCl in THF and subsequently purified by column chromatography to provide diol 10 in 55 % yield and dr >97:3. Bis‐TBS protection of 10, followed by a Suzuki‐Fu coupling reaction19 with 1‐hexadecene resulted in 11 in good yields, and effectively installed the required α‐chain. Selective deprotection of the primary TBS ether with pyridine⋅HF, followed by DMP oxidation of the primary alcohol to the corresponding aldehyde resulted in fragment A.
Scheme 2.
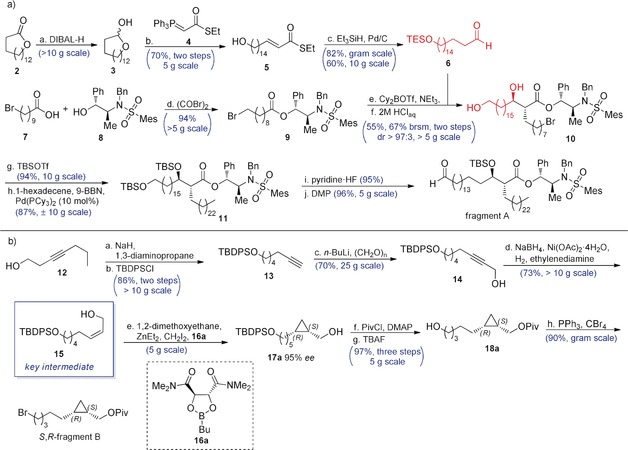
a. The synthesis of fragment A. a) DIBAL, CH2Cl2, −78 °C, 1 h; b) 4, THF/toluene (2:1), 75 °C, 16 h, (70 % over two steps); c) Et3SiH, Pd/C, acetone, 0 °C to rt, 6 h, (82 %); d) (COBr)2, 8, pyridine, CH2Cl2, 0 °C to rt, 3 h (94 %); e) Cy2BOTf, NEt3, THF, −78 °C, then 6, 16 h; f) 2 m HClaq, THF, rt, 2 h (55 % over two steps, 67 % yield brsm); g) TBSOTf, 2,6‐lutidine, CH2Cl2, 0 °C to rt, 3 h (94 %); h) 1‐hexadecene, 9‐BBN, Pd(PCy3)2 (10 mol %), K3PO4⋅H2O, THF, rt, 16 h (87 %); i) pyridine⋅HF, THF, 0 °C to rt, 4 h (95 %); j) DMP, CH2Cl2, rt 1 h (96 %). b. Synthesis of fragment B. a) NaH, 1,3‐diaminopropane, 70 °C, 1.5 h; b) TBDPSCl, CH2Cl2, 0 °C to rt, 16 h (86 % over two steps); c) n‐BuLi, paraformaldehyde, THF, 0 °C to rt, 16 h (70 %); d) NaBH4, Ni(OAc)2⋅4 H2O, H2, ethylenediamine, EtOH, rt, 15 min (73 %); e) 1,2‐dimethoxyethane, diethylzinc, diiodomethane, 16, CH2Cl2, −15 °C to rt, 16 h; f) PivCl, DMAP, pyridine, CH2Cl2, 0 °C to rt, 16 h; g) TBAF, THF, 0 °C to rt, 16 h ( 97 % over three steps); h) CBr4, PPh3, CH2Cl2, 0 °C to rt, 3 h (90 %).22 DIBAL=diisobutylaluminium hydride, THF=tetrahydrofurane, TBSOTf=tert‐Butyldimethylsilyltrifluoromethanesulfonate, 9‐BBN=9‐borabicyclo(3.3.1)nonane, DMP=Dess–Martin periodinane, TBDPSCl=tert‐Butyldiphenylsilyl chloride, PivCl=pivaloyl chloride, TBAF=tetrabutylammonium fluoride.
The synthesis of fragment B started with the alkyne zipper reaction23 applied to commercial 12, followed by protection of the alcohol to provide terminal alkyne 13 in 86 % yield (Scheme 2 b). Deprotonation of 13 with n‐BuLi at 0 °C followed by addition to paraformaldehyde resulted in 14 in 70 % yield. In our hands, Lindlar reduction of 14 24 to cis allylic alcohol 15 resulted in a mixture with the trans isomer, inseparable on silica and Ag‐impregnated silica. Although this problem was avoided by subjecting 14 to a P‐2 nickel reduction,25 this time 1H‐NMR indicated concomitant over‐reduction. Fortunately, separation of 15 from approximately 15 % of the aliphatic alcohol succeeded with a single run on a Ag‐impregnated silica column in 73 % yield. Alkene 15 was cyclopropanated with 95 % ee (see SI), mediated by dioxaborolane 16 a or 16 b according to Charette's procedure yielding the enantiomeric alcohols 17 a and 17 b, respectively.26 The alcohols 17 a and 17 b were subsequently converted into their pivaloyl esters, and desilylated using TBAF to yield both enantiomers of 18 in excellent yield over three steps,22 and bromination resulted in both enantiomers of fragment B. Fragment B decomposed upon extended storage (longer than one month) at 0 °C, and was therefore used within one week after synthesis.
The synthesis of fragment C (Scheme 3 a) started with a diastereoselective conjugate addition of MeLi to 19 a and its enantiomer 19 b.11, 27 Then, LiAlH4 reduction followed by bromination of 21 resulted in bromide 22 in good yields over two steps. Chain extension was achieved by subjecting 22 to a Suzuki‐Fu cross‐coupling19 with 1‐hexadecene. Acid hydrolysis of the acetal, followed by a one‐pot tosylation of the primary alcohol and intramolecular SN2 reaction went smoothly, and yielded the corresponding epoxide 25 in excellent yield over two steps. Epoxide opening with Grignard reagent 26 using catalytic CuCl minimized the formation of halohydrin by‐products28 and afforded the secondary alcohol 27 in good yield. Finally, methylation of the secondary hydroxyl using an excess of MeI and NaH resulted in both enantiomers of fragment C in nearly quantitative yields.
Scheme 3.
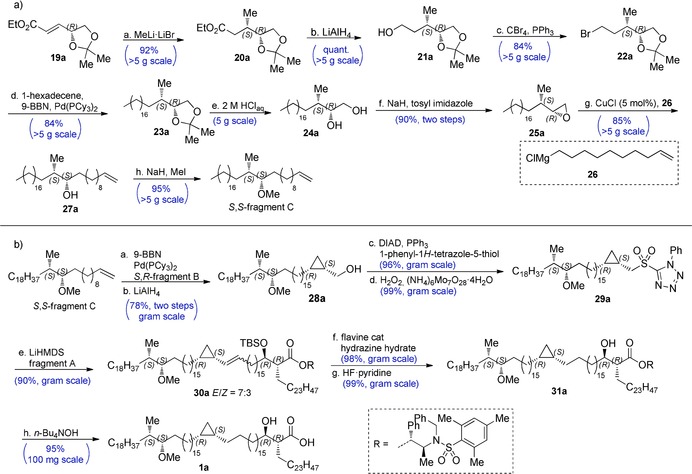
a) The synthesis of fragment C. a: MeLi⋅LiBr, Et2O, −78 °C, 4 h (92 %); b: LiAlH4, THF, 0 °C, 1 h (quant.); c: CBr4, PPh3, CH2Cl2, 0 °C to rt, 4 h (84 %); d: 9‐BBN, 1‐hexadecene, K3PO4, cat. Pd(PCy3)2, THF, 16 h (84 %); e: 2 m HClaq, THF, 95 °C, 16 h; f: NaH, tosylimidazole, THF, rt, 16 h (90 % over two steps); g: 26, CuCl (5 mol %), THF, −10 °C, 2 h (85 %); h: MeI, NaH, THF, rt, 16 h (95 %).22 b) The endgame synthesis. a: 9‐BBN, 1‐hexadecene, fragment B, K3PO4, Pd(PCy3)2 (7 mol %), THF, 16 h; b: LiAlH4, THF, 0 °C, 15 min (78 % over two steps); c: phenyl‐1H‐tetrazole‐5‐thiol, PPh3, DIAD, THF, 0 °C to rt, 1 h (96 %); d: H2O2 (30 %), (NH4)6Mo7O28⋅4 H2O, THF, EtOH, n‐BuOH, rt, 5 d (99 %); e: LiHMDS, fragment A, −35 °C, 1 h (90 %); f: flavine cat.,30 hydrazine hydrate, THF, EtOH, n‐BuOH, rt, 5–7 d (98 %); g: HF⋅pyridine, CHCl3, 0 °C, 2 h (99 %); h: n‐Bu4NOH, (1.5 m in water), THF, rt, 16 h (95 %).22 Tosylimidazole=p‐toluenesulfonylimidazole, DIAD=diisopropylazodicarboxylate, LiHMDS=lithium hexamethyldisilazane.
Endgame of the Synthesis
With all three fragments in hand, we initiated the endgame by yet another Suzuki‐Fu cross‐coupling to connect the fragments B and C (Scheme 3 b). Reductive removal of the pivaloate, and a subsequent Mitsunobu reaction with 1‐phenyl‐1H‐tetrazole‐5‐thiol afforded the thio‐ether in good yields over three steps. Unfortunately, ammonium heptamolybdate/H2O2 oxidation to sulfone 29 proved to be very sluggish due to limited solubility of the starting material in EtOH/THF, whereas oxidation with m‐CPBA resulted in a complex mixture of unidentifiable products. Although oxidation with RuO4 was possible,29 concomitant oxidation of the methoxy function to the ketone was hard to suppress. Fortunately, acceptable rates in the ammonium heptamolybdate/H2O2 oxidation were achieved by addition of n‐BuOH as co‐solvent, resulting in nearly quantitative yields of the sulfone. Subsequently, we applied a Julia–Kocienski olefination for the final coupling of 29 with fragment A. Although initially excellent yields (85–90 %) were achieved in this reaction, at some point we suffered from irreproducible yields, even when we applied starting materials that had provided satisfactory yields earlier. We could circumvent this problem by using an excess of lithiated 29, which was nearly quantitatively recovered. With the complete backbone in place, we subjected 30 to a diimide reduction30 of the olefin, followed by TBS deprotection with pyridine⋅HF in excellent yield. The use of n‐BuOH as cosolvent proved again to be essential for acceptable conversion rates. As we were reluctant to store large quantities of the mycolic acids because of the potentially sensitive hydroxy acid moiety, the final step was carried out portion‐wise at sub‐gram scale. Removal of the chiral auxiliary with LiOH, LiOH/H2O2 or hydrogenolysis were unsuccessful, but hydrolysis with 2 equiv of tetrabutylammonium hydroxide (n‐Bu4NOH) in THF at rt resulted in complete and clean conversion, and yielded the desired mycolic acids in sufficient purity and quantitative yields after a straightforward pentane/acetonitrile extraction. Overall, the total synthesis of mycolic acid 1 a was achieved in a longest linear sequence of just 17 steps, in 15 % yield. The corresponding glucose esters (Figure 2, 32 a–d) were obtained in two steps using a method developed by Prandi31 (see SI for experimental details and yields).
The Stereochemistry of Natural Methoxy Mycolic Acid
With all four diastereomers in hand, it was now possible to prove or disprove the ascription of the absolute stereochemistry (S,S) of the syn‐methoxy methyl moiety by Asselineau et al.7 An aliquot of the carboxylic acids was treated with (trimethylsilyl)diazomethane in toluene/methanol to provide the methyl esters (see SI). The [α]D of both methyl esters with S,S configuration in the α‐methyl methoxy segment (1 a and 1 d) was 0°, which corresponds with the specific rotation of the natural sample reported before.7 Consequently, also the specific molar rotations [Φ]D were 0°. This supports the previous assignment of the stereochemistry by Asselineau, and Baird (for 1 d). The [α]D for the diastereomers with R,R configuration in the α‐methyl methoxy segment was +7.8° for 1 b and +7.9° for 1 c and similar to those reported by Baird and co‐workers for (synthetic) diastereomer 1 c.11 The [Φ]D of 1 b and 1 c was +99° and +100°, respectively, and comparable with the sum of the values of the [Φ]D of the α‐methyl methoxy and the β‐hydroxy acid segments prepared earlier by Asselineau (+90°). Therefore, it is reasonable to conclude that the remotely located stereocenters in methoxy mycolic acid do not influence each other's optical rotation. The principle of optical superposition is therefore applicable here and the assignment of the absolute stereochemistry by Asselineau is correct, although the stereochemistry in the cyclopropyl ring remains to be determined (see below).
Next, we desired to determine whether our synthetic methoxy mycolic acids are biologically active as antigens for human T cells. In the presence of CD1b‐expressing antigen‐presenting cells, all four synthetic stereoisomers of glucose monomycolate activated the LDN5 T cell line in a dose‐dependent manner, as determined by release of the cytokine Interleukine‐2 (IL‐2) (Figure 3 a). The level of activation by the four synthetic diastereomers, the natural long glucose mycolate from Mycobacterium phlei, and the natural short glucose monomycolate from Rodococcus equi, were comparable. This was consistent with previous observations that glucose monomycolate‐specific T cells are insensitive to variations in the distal parts of mycolic acid, but highly sensitive to changes in or near the glucose.17
Figure 3.
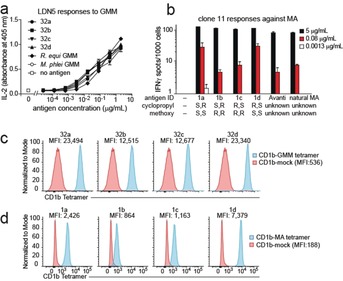
Biological validation of synthetic glucose methoxy mycolates (a,c) and methoxy mycolic acids (b,d). Methods are provided in the Supporting Information.
As for the T cell activation assays with free mycolic acid, it is known from studies with natural mycolic acid isolates that a T cell clone is typically more potently activated by a certain type of mycolic acid (α‐, keto‐, or methoxy‐) than by another. The pattern of antigen potency is different for each T cell clone. In the presence of CD1b‐expressing antigen‐presenting cells, all four synthetic mycolic acids stimulated the T cell clone 11 well at the highest concentration tested, 5 μg mL−1 (Figure 3 b). The two synthetic mycolic acids with the S,S configuration at the α‐methyl methoxy segment, however, were better agonists than those with R,R configuration, and better than the natural mixture of Mtb‐derived mycolic acids when tested at the suboptimal concentration of 0.08 μg mL−1. No significant difference was found for the stereochemistry at the cyclopropyl group.
As an alternative bioassay, mycolic acid and glucose monomycolate can be loaded into fluorescently labelled CD1b tetramers in vitro. Tetramerized, fluorescently labelled CD1b–lipid complexes (“tetramers”) can be used to study glucose mycolate32 and mycolic acid6 specific T cells at single‐cell level, in combination with fluorescently labelled antibodies, in flow cytometry. The fluorescence level of the cells is an indication of the strength of the interaction between T cell receptor and antigenic target, CD1b–mycolate.
All synthetic glucose mycolates, when loaded into CD1b tetramers, stained LDN5 well (Figure 3 c). A minor but noticeable difference in the mean fluorescence intensity (MFI) is observed depending on the stereochemistry of the α‐methyl methoxy unit. The MFI with the GMMs with the S,S configuration is twice as high as in those with the R,R configuration. No noticeable difference is seen between the tetramers loaded with the glucose mycolates that differ in the stereochemistry of their cyclopropyl group.
For the mycolic acids, however, more distinct differences are observed (Figure 3 d). Tetramers loaded with mycolic acids with the S,S‐methoxy methyl unit give much higher MFIs than their R,R counterparts.
In addition to the effects of the configuration of the methoxy methyl group, mycolic acids with an R,S‐cyclopropyl unit have a moderately increased MFI compared to their S,R counterparts (MFI compound 1 d>1 a and compound 1 c>1 b in Figure 3 d). If we assume that the naturally occurring configuration of the cyclopropyl provides the best CD1b loading or the highest affinity for the T cell receptor, this observation suggests the R,S‐stereochemistry of the cyclopropyl group in natural mycolic acids.
Conclusion
In conclusion, the synthetic route presented here provides access to gram amounts of methoxy mycolic acid. The successful repetitive application of the Suzuki–Fu cross‐coupling and the anti‐selective Abiko–Masamune asymmetric aldol reaction in this natural product synthesis are instrumental to this success. The availability of all four diastereomers allowed to confirm the S,S stereochemistry of the methoxy methyl function in natural methoxy mycolic acid, as inferred by Asselineau and Baird. The T cell receptor is sensitive to variations in the stereochemistry of the side chain of free mycolic acid, and not in that of glucose monomycolate. Although the absolute stereochemistry of the cis‐cyclopropyl function could not be established, the CD1b‐free mycolic acid tetramer staining experiments favour R,S stereochemistry. The availability of large amounts of fully synthetic methoxy mycolic acid and glucose mycolate (GMM) allows further study into their application in vaccine development and diagnostics.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank P. van der Meulen, Dr. J. Kemmink, J. L. Sneep and T. D. Tiemersma‐Wegman (University of Groningen) for assistance on analyses. The Dutch Science Foundation NWO and the Bill & Melinda Gates Foundation are acknowledged for funding. Unloaded CD1b monomers were obtained through the NIH Tetramer Core Facility.
N. Tahiri, P. Fodran, D. Jayaraman, J. Buter, M. D. Witte, T. A. Ocampo, D. B. Moody, I. Van Rhijn, A. J. Minnaard, Angew. Chem. Int. Ed. 2020, 59, 7555.
References
- 1.World Health Organization, Global Tuberculosis Report, 2019.
- 2. Marrakchi H., Lanéelle M.-A., Daffé M., Chem. Biol. 2014, 21, 67–85. [DOI] [PubMed] [Google Scholar]
- 3. C. E. Barry III , Lee R. E., Mdluli K., Sampson A. E., Schroeder B. G., Slayden R. A., Yuan Y., Prog. Lipid Res. 1998, 37, 143–179. [DOI] [PubMed] [Google Scholar]
- 4. Beukes M., Lemmer Y., Deysel M., Al Dulayymi J. R., Baird M. S., Koza G., Iglesias M. M., Rowles R. R., Theunissen C., Grooten J., Toschi G., Roberts V. V., Pilcher L., Van Wyngaardt S., Mathebula N., Balogun M., Stoltz A. C., Verschoor J. A., Chem. Phys. Lipids 2010, 163, 800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grant E. P., Beckman E. M., Behar S. M., Degano M., Frederique D., Besra G. S., Wilson I. A., Porcelli S. A., Furlong S. T., Brenner M. B., J. Immunol. 2002, 168, 3933–3940. [DOI] [PubMed] [Google Scholar]
- 6. Van Rhijn I., Iwany S. K., Fodran P., Cheng T.-Y., Gapin L., Minnaard A. J., Moody D. B., Eur. J. Immunol. 2017, 47, 1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asselineau C., Tocanne G., Tocanne J.-F., Bull. Soc. Chim. Fr. 1970, 1455–1459. [PubMed] [Google Scholar]
- 8. Minnikin D. E., Polgar N., Tetrahedron Lett. 1966, 7, 2643–2647. [DOI] [PubMed] [Google Scholar]
- 9. Minnikin D. E., Polgar N., Chem. Commun. 1967, 1172–1174. [Google Scholar]
- 10. Al Dulayymi J. R., Baird M. S., Roberts E., Chem. Commun. 2003, 228–229. [DOI] [PubMed] [Google Scholar]
- 11. Al Dulayymi J. R., Baird M. S., Roberts E., Deysel M., Verschoor J., Tetrahedron 2007, 63, 2571–2592. [Google Scholar]
- 12. Al Dulayymi J. R., Baird M. S., Roberts E., Tetrahedron 2005, 61, 11939–11951. [Google Scholar]
- 13. Koza G., Muzael M., Schubert-Rowles R. R., Theunissen C., Al Dulayymi J. R., Baird M. S., Tetrahedron 2013, 69, 6285–6296. [Google Scholar]
- 14. Moody D. B., Guy M. R., Grant E., Cheng T.-Y., Brenner M. B., Besra G. S., Porcelli S. A., J. Exp. Med. 2000, 192, 965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van′t Hoff J. H., Bull. Soc. Chim. Fr. 1875, 23, 298. [Google Scholar]
- 16. Beckman E. M., Porcelli S. A., Morita C. T., Behar S. M., Furlong S. T., Brenner M. B., Nature 1994, 372, 691–694. [DOI] [PubMed] [Google Scholar]
- 17. Moody D. B., Reinhold B. B., Guy M. R., Beckman E. M., Frederique D. E., Furlong S. T., Ye S., Reinhold V. N., Sieling P. A., Modlin R. L., Besra G. S., Porcelli S. A., Science 1997, 278, 283–286. [DOI] [PubMed] [Google Scholar]
- 18. Gras S., Van Rhijn I., Shahine A., Cheng T.-Y., Bhati M., Tan L. L., Halim H., Tuttle K. D., Gapin L., Le Nours J., Moody D. B., Rossjohn J., Nat. Commun. 2016, 7, 13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Netherton M. R., Dai C., Neuschuetz K., Fu G. C., J. Am. Chem. Soc. 2001, 123, 10099–10100; [DOI] [PubMed] [Google Scholar]; Lou S., Fu G. C., Org. Synth. 2010, 87, 299–309; For a Highlight on sp3–sp3 coupling reactions in the synthesis of natural products and biologically active molecules, see: [DOI] [PubMed] [Google Scholar]; Geist E., Kirschning A., Schmidt T., Nat. Prod. Rep. 2014, 31, 441–448. [DOI] [PubMed] [Google Scholar]
- 20. Abiko A., Liu J. F., Masamune S., J. Am. Chem. Soc. 1997, 119, 2586–2587. [Google Scholar]
- 21. Keck G. E., Boden E. P., Mabury S. A., J. Org. Chem. 1985, 50, 709–710. [Google Scholar]
- 22.See SI for more detailed information regarding the other enantiomers or diastereomers.
- 23. Huang H., Forsyth C. J., J. Org. Chem. 1997, 62, 8595–8599. [DOI] [PubMed] [Google Scholar]
- 24. Nicolaou K. C., Prasad C. V. C., Somers P. K., Hwang C., J. Am. Chem. Soc. 1989, 111, 5335–5340. [Google Scholar]
- 25. Brown C. A., Ahuja V. K., J. Org. Chem. 1973, 38, 2226–2230. [Google Scholar]
- 26. Charette A. B., Juteau H., J. Am. Chem. Soc. 1994, 116, 2651–2652. [Google Scholar]
- 27. Leonard J., Mohialdin S., Reed D., Ryan G., Swain P., Tetrahedron 1995, 51, 12843–12858. [Google Scholar]
- 28. Alam M., Wise C., Baxter C. A., Cleator E., Walkinshaw A., Org. Process Res. Dev. 2012, 16, 435–441. [Google Scholar]
- 29. Su W., Tetrahedron Lett. 1994, 35, 4955–4958. [Google Scholar]
- 30. Smit C., Fraaije M. W., Minnaard A. J., J. Org. Chem. 2008, 73, 9482–9485. [DOI] [PubMed] [Google Scholar]
- 31. Prandi J., Carbohydr. Res. 2012, 347, 151–154. [DOI] [PubMed] [Google Scholar]
- 32. Kasmar A. G., Van Rhijn I., Magalhaes K. G., Young D. C., Cheng T.-Y., Turner M. T., Schiefner A., Kalathur R. C., Wilson I. A., Bhati M., Gras S., Birkinshaw R. W., Tan L. L., Rossjohn J., Shires J., Jakobsen S., Altman J. D., Moody D. B., J. Immunol. 2013, 191, 4499–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary