

Abstract
After realizing mirror‐image genetic replication, transcription, and reverse transcription, the biggest challenge in establishing a mirror‐image version of the central dogma is to build a mirror‐image ribosome‐based translation machine. Here, we chemically synthesized the natural and mirror‐image versions of three ribosomal proteins (L5, L18, and L25) in the large subunit of the Escherichia coli ribosome with post‐translational modifications. We show that the synthetic mirror‐image proteins can fold in vitro despite limited efficiency and assemble with enzymatically transcribed mirror‐image 5S ribosomal RNA into ribonucleoprotein complexes. In addition, the RNA–protein interactions are chiral‐specific in that the mirror‐image ribosomal proteins do not bind with natural 5S ribosomal RNA and vice versa. The synthesis and assembly of mirror‐image 5S ribonucleoprotein complexes are important steps towards building a functional mirror‐image ribosome.
Keywords: chemical protein synthesis, chirality, native chemical ligation, ribonucleoproteins, solid-phase peptide synthesis
The natural and mirror‐image versions of three ribosomal proteins (L5, L18, and L25) were chemically synthesized with post‐translational modifications. The synthetic mirror‐image proteins can fold in vitro despite limited efficiency and assemble with enzymatically transcribed mirror‐image 5S ribosomal RNA into ribonucleoprotein complexes.
Introduction
Towards building functional mirror‐image biology systems, an imperative step is to establish a chirally inverted version of the central dogma of molecular biology.1, 2, 3, 4 We have previously demonstrated mirror‐image genetic replication and transcription by chemically synthesizing the d‐amino acid African swine fever virus polymerase X (ASFV pol X),1 followed by the synthesis of more efficient and thermostable Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) by us and others,5, 6, 7 leading to the realization of mirror‐image polymerase chain reaction (MI‐PCR). More recently, we developed more efficient mirror‐image transcription and reverse transcription systems based on designed Dpo4 mutants,8 and demonstrated the enzymatic transcription of Escherichia coli (E. coli) 5S rRNA gene rrfB into full‐length 120‐nt l‐ 5S ribosomal RNA (rRNA).8 The realization of a functional mirror‐image ribosomal translation apparatus faces considerable challenges in that the ribosome is a highly complex molecular machine assembled from rRNAs and ribosomal proteins (r‐proteins).9, 10, 11 However, synthesizing its protein components is potentially achievable as, taking the example of bacterial ribosomal translation apparatus, it consists of a large number of small r‐proteins, of typically no more than 300 amino acid residues,12 with the exception of the non‐essential S1 protein,13, 14 as well as several key translation factors.15, 16 Here, we set out to synthesize three r‐proteins (L5, L18, and L25) in the large subunit of the E. coli ribosome that are known to bind with 5S rRNA (Figures 1 A, 2 A, 3 A, and 4 A).9, 17, 18 We show that the synthetic mirror‐image r‐proteins with post‐translational modifications can fold in vitro despite limited efficiency, and interact chiral‐specifically with l‐ 5S rRNA to form mirror‐image ribonucleoprotein (RNP) complexes.
Figure 1.
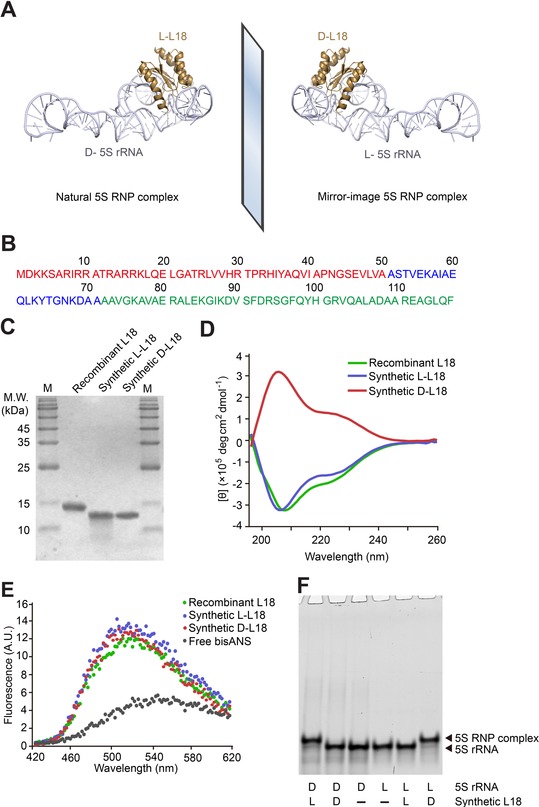
Chemical synthesis, in vitro folding, and binding of L18 with 5S rRNA. A) Schematic assembly of 5S rRNA with L18 into RNP complex. Structural data from PDB 4YBB. B) L18 amino acid sequence, consisting of 117 amino acid residues. The colors of the three peptide segments correspond to the peptide segment colors used in Figure S3. C) Chemically synthesized 12.8 kDa l‐ and d‐L18, as well as recombinant L18 (with His6 tag) expressed and purified from the E. coli strain BL21 (DE3) were analyzed by 15 % SDS‐PAGE, stained by Coomassie Brilliant Blue. M=protein marker. D) Far‐UV CD spectra of synthetic l‐ and d‐L18, as well as recombinant L18 expressed and purified from E. coli. CD curves were averaged from three independent measurements and background signal subtracted. E) bisANS fluorescence when added to l‐ and d‐L18, as well as recombinant L18 expressed and purified from E. coli. F) Binding chiral specificity between different chiral combinations of 5′‐FAM‐labeled 5S rRNA and synthetic, in vitro folded L18. The concentrations of 5S rRNA and L18 were 0.1 μm and 0.8 μm, respectively. All samples were incubated in TMK buffer at 33 °C for 45 min and analyzed by 8 % native PAGE.
Figure 2.
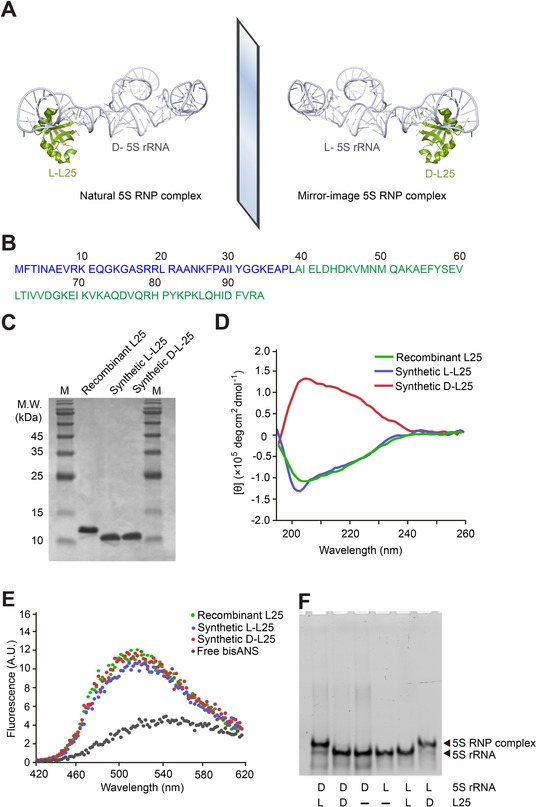
Chemical synthesis, in vitro folding, and binding of L25 with 5S rRNA. A) Schematic assembly of 5S rRNA with L25 into RNP complex. Structural data from PDB 4YBB. B) L25 amino acid sequence, consisting of 94 amino acid residues. The colors of the two peptide segments correspond to the peptide segment colors used in Figure S5. C) Chemically synthesized 10.7 kDa l‐ and d‐L25, as well as recombinant L25 (with His6 tag) expressed and purified from the E. coli strain BL21 (DE3) were analyzed by 15 % SDS‐PAGE, stained by Coomassie Brilliant Blue. M=protein marker. D) Far‐UV CD spectra of the synthetic l‐ and d‐L25, as well as recombinant L25 expressed and purified from E. coli. CD curves were averaged from three independent measurements and background signal subtracted. E) bisANS fluorescence when added to l‐ and d‐L25, as well as recombinant L25 expressed and purified from E. coli. F) Binding chiral specificity between different chiral combinations of 5′‐FAM‐labeled 5S rRNA and synthetic, in vitro folded L25. The concentrations of 5S rRNA and L25 were 0.1 μm and 0.8 μm, respectively. All samples were incubated in TMK buffer at 33 °C for 45 min and analyzed by 8 % native PAGE.
Figure 3.
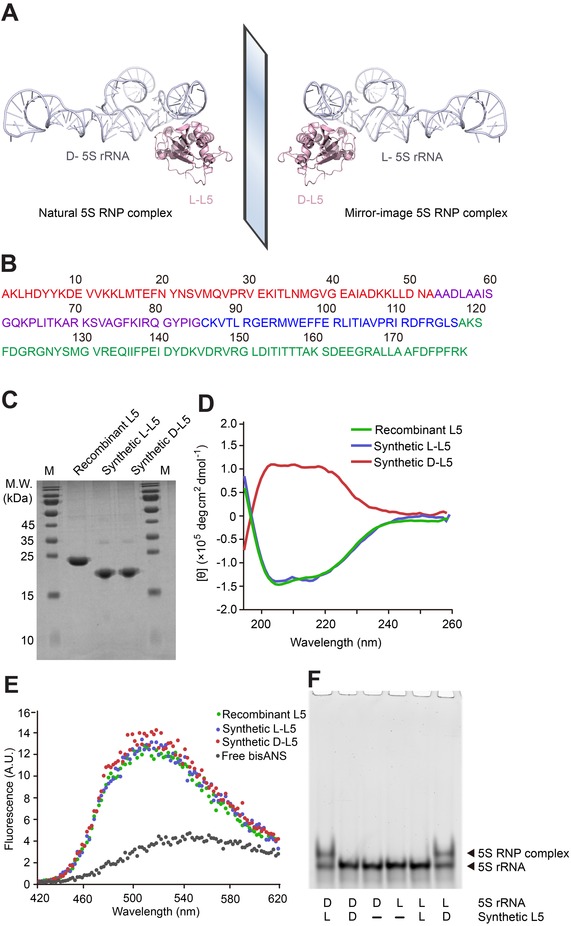
Chemical synthesis, in vitro folding, and binding of L5 with 5S rRNA. A) Schematic assembly of 5S rRNA with L5 into RNP complex. Structural data from PDB 4YBB. B) L5 amino acid sequence, consisting of 178 amino acid residues. The colors of the four peptide segments correspond to the peptide segment colors used in Figure S6. C) Chemically synthesized 20.3 kDa l‐ and d‐L5, as well as recombinant L5 (with His6 tag) expressed and purified from the E. coli strain BL21 (DE3) were analyzed by 15 % SDS‐PAGE, stained by Coomassie Brilliant Blue. M=protein marker. D) Far‐UV CD spectra of the synthetic l‐ and d‐L5, as well as recombinant L5 expressed and purified from E. coli. CD curves were averaged from three independent measurements and background signal subtracted. E) bisANS fluorescence when added to l‐ and d‐L5, as well as recombinant L5 expressed and purified from E. coli. F) Binding chiral specificity between different chiral combinations of 5′‐FAM‐labeled 5S rRNA and synthetic, in vitro folded L5. The concentrations of 5S rRNA and L5 were 0.1 μm and 2.0 μm, respectively. All samples were incubated in TMK buffer at 33 °C for 45 min and analyzed by 8 % native PAGE.
Figure 4.
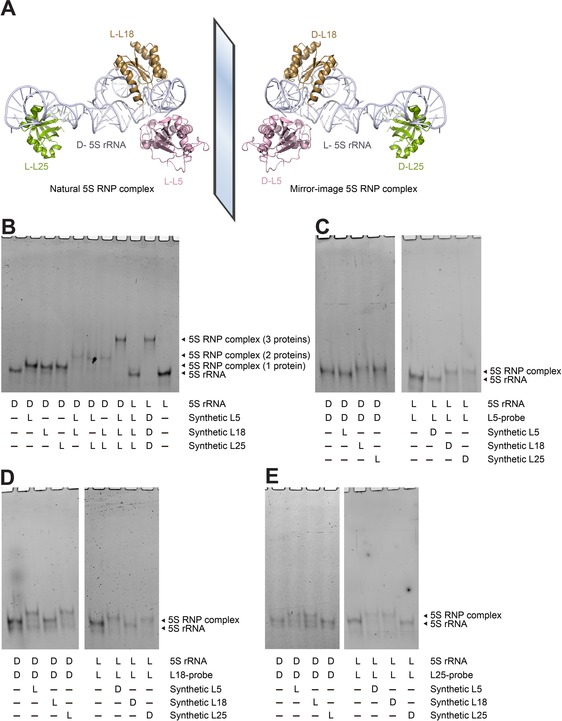
Simultaneous binding of L5, L18, and L25 with 5S rRNA. A) Schematic assembly of 5S rRNA with L5, L18, and L25 into RNP complex simultaneously. Structural data from PDB 4YBB. B) Binding chiral specificity between different chiral combinations of 5′‐FAM‐labeled 5S rRNA with L5, L18, and L25 simultaneously. C) Binding of 5S rRNA with L5, L18, and L25 in the presence of synthetic DNA probe complementary to the binding site of L5 (L5‐probe). D) Binding of 5S rRNA with L5, L18, and L25 in the presence of synthetic DNA probe complementary to the binding site of L18 (L18‐probe). E) Binding of 5S rRNA with L5, L18, and L25 in the presence of synthetic DNA probe complementary to the binding site of L25 (L25‐probe). All samples were incubated in TMK buffer at 33 °C for 45 min and analyzed by 8 % native PAGE.
Results and Discussion
Chemical Synthesis, In Vitro Folding, and Binding of L18 with 5S rRNA
It has been suggested that the binding of L18 with 5S rRNA depends on its phosphorylation modification, with a putative phosphorylation site at Ser57 in the Bacillus stearothermophilus L18 (which corresponds to Ser52 in the E. coli L18).19 More recently, tandem mass spectrometry suggested possible phosphorylation at Ser45 or Ser52 in the E. coli L18 protein,20 although the results were inconclusive. The apparent inconsistency in the existing literature on L18 post‐translational modifications (PTMs) is in part due to the difficulties of expressing recombinant proteins with or without specific PTMs, whereas chemical synthesis can control the presence and absence of PTMs at specific sites to obtain different variants of L18 and help address the question on r‐protein PTM functionality in rRNA binding. Thus, we began with testing the role of L18 PTM in 5S rRNA binding by synthesizing three l‐amino acid protein variants: L18 without modification (L18), L18 with phosphorylated Ser52 (L18S52p), and L18 with phosphorylated Ser45 (L18S45p).
All peptide segments were synthesized by Fmoc‐based solid‐phase peptide synthesis (SPPS), for it is more compatible with acid‐sensitive phosphorylation modification than Boc‐SPPS. We applied native chemical ligation (NCL) using peptide hydrazide as the thioester surrogate to assemble peptide segments1, 5, 6, 21, 22, 23 and desulfurization to convert unprotected cysteine into alanine.24, 25 We divided the L18 protein, which consists of 117 amino acid residues, into three peptide segments and chose Ala51 and Ala72 as ligation sites for synthesizing L18 and L18S52p (Figures 1 B and S3 in the Supporting Information), and Ala37 and Ala72 as ligation sites for L18S45p (Figure S4). After the assembly of peptide segments in the N‐ to C‐terminus direction and HPLC purification, we obtained final products of l‐amino acid L18, L18S52p, and L18S45p at 5 mg, 7.3 mg, and 6 mg, respectively (Figures S7–S15). The purified full‐length proteins were analyzed by reversed‐phase high‐performance liquid chromatography (RP‐HPLC) and electrospray ionization mass spectrometry (ESI‐MS) with an observed molecular weight (M.W.) of 12 770.7 Da (calculated M.W. 12 769.6 Da) for L18 (Figure S9), 12 849.0 Da (calculated M.W. 12 850.6 Da) for L18S52p (Figure S12), and 12 849.3 Da (calculated M.W. 12 850.6 Da) for L18S45p (Figure S15).
The lyophilized synthetic proteins were each dissolved into denaturation buffer containing 6 m guanidine hydrochloride (Gn⋅HCl), and folded by two steps of dialysis against 1 m KCl, followed by another step of dialysis against TMK buffer (30 mm Tris‐HCl, pH 7.5, 350 mm KCl, 20 mm MgCl2, 1 mm DTT) (see the Materials and Methods Section in the Supporting Information).26 The dialyzed synthetic proteins were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE; Figure 1 C), compared to recombinant L18 (with His6 tag) expressed and purified from E. coli (see the Materials and Methods Section in the Supporting Information). The folding of the synthetic proteins was validated by far‐UV circular dichroism (CD) spectroscopy (Figures 1 D and S1 A) and by the bisANS (8‐anilino‐1‐naphthalene sulfonate) assay (Figures 1 E and S1 B).27 The differences between the CD and bisANS spectra of synthetic and recombinant L18 likely result from the low folding efficiency of the synthetic protein in vitro. Next, we performed a gel shift assay to show the binding between chemically synthesized L18 with 5S rRNA in the natural chirality system. We used a 19‐nt 5′‐FAM‐labeled l‐RNA as the primer and a 120‐nt single‐stranded DNA sequence coding for the 5S rRNA gene rrfB as the template to transcribe the full‐length 5S rRNA in the natural chirality system.8 The three synthetic variants of L18 protein (L18, L18S52p, and L18S45p) all bind to 5S rRNA in a similar concentration‐dependent fashion (Figure S2 A), suggesting that in contrary to the previously reported results with recombinant proteins,19 the tested PTMs per se are likely nonessential for E. coli L18 assembly with 5S rRNA into RNP complex in vitro. Nonetheless, the evolutionarily conserved phosphorylation at Ser52 could still make this modification potentially important for ribosome assembly and translation in vivo, and thus we chose the L18S52p variant as a model for the synthesis of mirror‐image L18.
After the synthesis, folding, and binding of L18 with 5S rRNA in the natural chirality system, we performed the synthesis of d‐amino acid version L18 with Ser52 phosphorylation. NCL using peptide hydrazide as the thioester surrogate was applied to assemble the d‐peptide segments, yielding 4.8 mg of d‐amino acid L18S52p (Figures S16–S18). The purified full‐length protein was analyzed by RP‐HPLC and ESI‐MS with an observed M.W. of 12 847.2 Da (calculated M.W. 12 850.6 Da; Figure S18). The mirror‐image L18S52p also folds in vitro as validated by far‐UV CD spectroscopy (Figure 1 D) and by the bisANS assay (Figure 1 E), and assembles with enzymatically transcribed l‐ 5S rRNA into mirror‐image RNP complex (Figure 1 F). However, the mirror‐image L18 does not interact with d‐ 5S rRNA and vice versa, and thus the 5S rRNA–L18 binding displays chiral specificity (Figures 1 F and S2 B).
Chemical Synthesis, In Vitro Folding, and Binding of L25 with 5S rRNA
We next carried out the synthesis of L25 starting from the l‐amino acid version. The L25 protein consists of 94 amino acid residues. We divided L25 into two peptide segments and chose A39 as the ligation site (Figures 2 B and S5). NCL using peptide hydrazide as the thioester surrogate was applied to assemble peptide segments, yielding 6.5 mg of l‐amino acid L25 (Figures S19 and S20), and the purified full‐length protein was analyzed by RP‐HPLC and ESI‐MS with an observed M.W. of 10 693.5 Da (calculated M.W. 10 693.4 Da; Figure S20). The lyophilized synthetic protein was dissolved into denaturation buffer containing 6 m Gn⋅HCl, and folded by dialysis (see the Materials and Methods Section in the Supporting Information). The synthetic protein was analyzed by SDS‐PAGE (Figure 2 C), compared to recombinant L25 (with His6 tag) expressed and purified from E. coli (see the Materials and Methods Section in the Supporting Information). The folding of the synthetic protein was validated by far‐UV CD spectroscopy (Figures 2 D and S1 C) and by the bisANS assay (Figures 2 E and S1 D). The differences in the CD and bisANS spectra between synthetic and recombinant L25 likely result from the low folding efficiency of the synthetic protein in vitro. Additionally, we performed a gel shift assay to show the binding between chemically synthesized L25 with 5S rRNA in the natural chirality system (Figures 2 F and S2 B).
We then performed the synthesis of d‐amino acid version L25. NCL using peptide hydrazide as the thioester surrogate was applied to assemble the d‐peptide segments, yielding 5.4 mg of d‐amino acid L25 (Figures S21 and S22), and the purified full‐length protein was analyzed by RP‐HPLC and ESI‐MS with an observed M.W. of 10 694.0 Da (calculated M.W. 10 693.4 Da; Figure S22). The mirror‐image L25 also folds in vitro as validated by far‐UV CD spectroscopy (Figure 2 D) and by the bisANS assay (Figure 2 E), and assembles with enzymatically transcribed l‐ 5S rRNA into the mirror‐image RNP complex (Figure 2 F). Again, the mirror‐image L25 binding displays chiral specificity as it does not interact with d‐ 5S rRNA and vice versa (Figures 2 F and S2 B).
Chemical Synthesis, In Vitro Folding, and Binding of L5 with 5S rRNA
Next, we carried out the synthesis of L5 starting from the l‐amino acid version. The L5 protein consists of 178 amino acid residues. We divided L5 into four peptide segments and chose A53, C86, and A118 as ligation sites (Figures 3 B and S6). NCL using peptide hydrazide as the thioester surrogate was applied to assemble peptide segments, with the orthogonal cysteine‐protecting group acetamidomethyl (Acm) used to prevent cysteine desulfurization and cyclization,28 which can be removed by palladium chloride (PdCl2) catalysis in the following ligation steps.29 The purified full‐length l‐amino acid L5 was obtained with a yield of 7.5 mg (Figures S23–S28) and analyzed by RP‐HPLC and ESI‐MS with an observed M.W. of 20 167.6 Da (calculated M.W. 20 170.4 Da; Figure S28). The lyophilized synthetic protein was dissolved into denaturation buffer containing 6 m Gn⋅HCl, and folded by dialysis (see the Materials and Methods Section in the Supporting Information). The synthetic protein was analyzed by SDS‐PAGE (Figure 3 C), compared to recombinant L5 (with His6 tag) expressed and purified from E. coli (see the Materials and Methods Section in the Supporting Information). The folding of the synthetic protein was validated by far‐UV CD spectroscopy (Figures 3 D and S1 E) and by the bisANS assay (Figures 3 E and S1 F). The differences in the CD and bisANS spectra between synthetic and recombinant L5 likely result from the low folding efficiency of the synthetic protein in vitro. Additionally, we performed a gel shift assay to show the binding between chemically synthesized L5 with 5S rRNA in the natural chirality system (Figures 3 F and S2 B).
We next performed the synthesis of d‐amino acid version L5 using the same synthetic route, except that Lys2 was acylated, which has been reported as its PTM in the E. coli ribosome.30 NCL using peptide hydrazide as the thioester surrogate was applied to assemble the d‐peptide segments, yielding 8.8 mg of d‐amino acid L5 (Figures S29–S34), and the purified full‐length protein was analyzed by RP‐HPLC and ESI‐MS with an observed M.W. of 20 210.6 Da (calculated M.W. 20 212.3 Da; Figure S34). Mirror‐image L5 also folds in vitro as validated by far‐UV CD spectroscopy (Figure 3 D) and by the bisANS assay (Figure 3 E), and assembles with enzymatically transcribed l‐ 5S rRNA into the mirror‐image RNP complex (Figure 3 F). Again, the mirror‐image L5 binding displays chiral specificity as it does not interact with d‐ 5S rRNA and vice versa (Figures 3 F and S2 B).
Simultaneous Binding of L5, L18, and L25 with 5S rRNA
Finally, we tested the simultaneous binding of L5, L18, and L25 with 5S rRNA in both the natural and mirror‐image systems (Figure 4 A). We performed a gel shift assay to show the chiral‐specific binding between 5′‐FAM‐labeled 5S rRNA and L5, L18, L25 simultaneously (Figure 4 B). In addition, the binding of r‐proteins with 5S rRNA was specific in that the synthetic r‐proteins did not bind transfer RNA (tRNA; Figure S2 C). Furthermore, through binding experiments using synthetic DNA probes complementary to the binding sites of the r‐proteins (Figures 4 C–E), we showed that the binding of synthetic L5, L18, and L25 with 5S rRNA likely fell into the same binding sites identified by previous studies based on nuclease digestion.31
Conclusion
We have chemically synthesized the natural and mirror‐image versions of three r‐proteins in the large subunit of E. coli ribosome with post‐translational modifications, and shown that the mirror‐image r‐proteins can fold in vitro and assemble with enzymatically transcribed l‐ 5S rRNA into mirror‐image RNP complexes with chiral specificity. However, the concentration‐dependent synthetic r‐protein binding with rRNA, with a much higher r‐protein/rRNA molar concentration ratio required (ca. 16:1, 8:1, and 4:1 for L5, L18, and L25, respectively) than those for recombinant r‐proteins (ca. 10:1, 4:1, and 2:1 for L5, L18, and L25, respectively), suggested low folding efficiency of the synthetic proteins in vitro without chaperones and other protein folding machinery. Thus, towards the goal of assembling synthetic r‐proteins with rRNAs into a functional mirror‐image ribosome, the folding of synthetic r‐proteins in vitro remains a major challenge and needs to be addressed, and developing approaches to purify the correctly folded r‐proteins would also be helpful.
Another obstacle towards building a complete mirror‐image ribosome is to transcribe mirror‐image 16S and 23S rRNAs, followed by the in vitro assembly of all other ribosomal components.32, 33 The lengths of 16S and 23S rRNAs (1.5 kb and 2.9 kb, respectively) require efficiencies and fidelities of mirror‐image PCR and transcription that are much higher than those of current polymerase systems.1, 6, 8 Potential strategies include developing better chemical protein synthesis techniques to synthesize larger mirror‐image polymerases, or applying directed evolution34, 35 on small polymerases such as Dpo4 to select for mutants with improved processivity and fidelity, which may enable the enzymatic transcription of long rRNAs and mRNAs.
Furthermore, r‐proteins, rRNAs (except 5S rRNA), and tRNAs are modified in vivo, and although in vitro transcribed rRNAs and tRNAs without modifications have been used for translation, the efficiency could be compromised.32, 36, 37, 38, 39, 40 While it is relatively straightforward to synthesize r‐proteins with PTMs as we have shown in this work, it is more challenging to prepare modified rRNAs and tRNAs through enzymatic transcription,41 and thus new methods for the preparation of modified mirror‐image rRNAs and tRNAs need to be developed. If a minimal mirror‐image ribosomal translation apparatus with all the essential components such as translation factors can be synthesized and assembled,15, 16, 42, 43 it will enable the enzymatic production of all of the molecular components required for building itself. Such a mirror‐image translation machine may result in many applications such as direct translation of mirror‐image enzymatic tools and selection of d‐peptide drugs, and ultimately lead to the creation of mirror‐image life.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported in part by funding from the National Natural Science Foundation of China (21750005, 21925702, 31711530153), the Swedish Research Council (2017‐00738), the Beijing Nova Program (Z171100001117011), the China Postdoctoral Science Foundation (2019M650636), the Tsinghua University‐Peking University Center for Life Sciences (CLS), the Beijing Advanced Innovation Center for Structural Biology, and the Beijing Frontier Research Center for Biological Structure.
J.-J. Ling, C. Fan, H. Qin, M. Wang, J. Chen, P. Wittung-Stafshede, T. F. Zhu, Angew. Chem. Int. Ed. 2020, 59, 3724.
References
- 1. Wang Z., Xu W., Liu L., Zhu T. F., Nat. Chem. 2016, 8, 698–704. [DOI] [PubMed] [Google Scholar]
- 2. Peplow M., Nature 2016, 533, 303–304. [DOI] [PubMed] [Google Scholar]
- 3. Bohannon J., Wired 2010, https://www.wired.com/2010/11/ff_mirrorlife/. [Google Scholar]
- 4. Peplow M., ACS Cent. Sci. 2018, 4, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu W., Jiang W., Wang J., Yu L., Chen J., Liu X., Liu L., Zhu T. F., Cell Discovery 2017, 3, 17008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang W., Zhang B., Fan C., Wang M., Wang J., Deng Q., Liu X., Chen J., Zheng J., Liu L., Zhu T. F., Cell Discovery 2017, 3, 17037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pech A., Achenbach J., Jahnz M., Schulzchen S., Jarosch F., Bordusa F., Klussmann S., Nucleic Acids Res. 2017, 45, 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang M., Jiang W., Liu X., Wang J., Zhang B., Fan C., Liu L., Pena-Alcantara G., Ling J. J., Chen J., Zhu T. F., Chem 2019, 5, 848–857. [Google Scholar]
- 9. Noeske J., Wasserman M. R., Terry D. S., Altman R. B., Blanchard S. C., Cate J. H., Nat. Struct. Mol. Biol. 2015, 22, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Green R., Noller H. F., Annu. Rev. Biochem. 1997, 66, 679–716. [DOI] [PubMed] [Google Scholar]
- 11. Nissen P., Hansen J., Ban N., Moore P. B., Steitz T. A., Science 2000, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 12. Stelzl U., Connell S., Nierhaus K. H., Wittmann-Liebold B., eLS 2001, 10.1038/npg.els.0000687. [DOI] [Google Scholar]
- 13. Tedin K., Resch A., Blasi U., Mol. Microbiol. 1997, 25, 189–199. [DOI] [PubMed] [Google Scholar]
- 14. Isono K., Isono S., Proc. Natl. Acad. Sci. USA 1976, 73, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T., Nat. Biotechnol. 2001, 19, 751–755. [DOI] [PubMed] [Google Scholar]
- 16. Shimizu Y., Kanamori T., Ueda T., Methods 2005, 36, 299–304. [DOI] [PubMed] [Google Scholar]
- 17. Spierer P., Bogdanov A. A., Zimmermann R. A., Biochemistry 1978, 17, 5394–5398. [DOI] [PubMed] [Google Scholar]
- 18. Herold M., Nierhaus K. H., J. Biol. Chem. 1987, 262, 8826–8833. [PubMed] [Google Scholar]
- 19. Bloemink M. J., Moore P. B., Biochemistry 1999, 38, 13385–13390. [DOI] [PubMed] [Google Scholar]
- 20. Soung G. Y., Miller J. L., Koc H., Koc E. C., J. Proteome Res. 2009, 8, 3390–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng J. S., Tang S., Qi Y. K., Wang Z. P., Liu L., Nat. Protoc. 2013, 8, 2483–2495. [DOI] [PubMed] [Google Scholar]
- 22. Fang G. M., Li Y. M., Shen F., Huang Y. C., Li J. B., Lin Y., Cui H. K., Liu L., Angew. Chem. Int. Ed. 2011, 50, 7645–7649; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7787–7791. [Google Scholar]
- 23. Dawson P., Muir T., Clark-Lewis I., Kent S., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 24. Wan Q., Danishefsky S. J., Angew. Chem. Int. Ed. 2007, 46, 9248–9252; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 9408–9412. [Google Scholar]
- 25. Yan L. Z., Dawson P. E., J. Am. Chem. Soc. 2001, 123, 526–533. [DOI] [PubMed] [Google Scholar]
- 26. Moore P. B., Abo S., Freeborn B., Gewirth D. T., Leontis N. B., Sun G., Methods Enzymol. 1988, 164, 158–174. [DOI] [PubMed] [Google Scholar]
- 27. Pastukhov A. V., Ropson I. J., Proteins Struct. Funct. Genet. 2003, 53, 607–615. [DOI] [PubMed] [Google Scholar]
- 28. Raibaut L., Ollivier N., Melnyk O., Chem. Soc. Rev. 2012, 41, 7001–7015. [DOI] [PubMed] [Google Scholar]
- 29. Maity S. K., Jbara M., Laps S., Brik A., Angew. Chem. Int. Ed. 2016, 55, 8108–8112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8240–8244. [Google Scholar]
- 30. Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C. F., Grishin N. V., Zhao Y., Mol. Cell. Proteomics 2009, 8, 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huber P. W., Wool I. G., Proc. Natl. Acad. Sci. USA 1984, 81, 322–326.6364140 [Google Scholar]
- 32. Jewett M. C., Fritz B. R., Timmerman L. E., Church G. M., Mol. Syst. Biol. 2013, 9, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J., Haas W., Jackson K., Kuru E., Jewett M. C., Fan Z. H., Gygi S., Church G. M., ACS Synth. Biol. 2017, 6, 1327–1336. [DOI] [PubMed] [Google Scholar]
- 34. Pinheiro V. B., Taylor A. I., Cozens C., Abramov M., Renders M., Zhang S., Chaput J. C., Wengel J., Peak-Chew S. Y., McLaughlin S. H., Herdewijn P., Holliger P., Science 2012, 336, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Larsen A. C., Dunn M. R., Hatch A., Sau S. P., Youngbull C., Chaput J. C., Nat. Commun. 2016, 7, 11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nesterchuk M. V., Sergiev P. V., Dontsova O. A., Acta Naturae 2011, 3, 22–33. [PMC free article] [PubMed] [Google Scholar]
- 37. Sergiev P. V., Golovina A. Y., Sergeeva O. V., Osterman I. A., Nesterchuk M. V., Bogdanov A. A., Dontsova O. A., Nucleic Acids Res. 2012, 40, 5694–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thiaville P. C., El Yacoubi B., Kohrer C., Thiaville J. J., Deutsch C., Iwata-Reuyl D., Bacusmo J. M., Armengaud J., Bessho Y., Wetzel C., Cao X., Limbach P. A., RajBhandary U. L., de Crecy-Lagard V., Mol. Microbiol. 2015, 98, 1199–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krzyzosiak W., Denman R., Nurse K., Hellmann W., Boublik M., Gehrke C. W., Agris P. F., Ofengand J., Biochemistry 1987, 26, 2353–2364. [DOI] [PubMed] [Google Scholar]
- 40. Murase Y., Nakanishi H., Tsuji G., Sunami T., Ichihashi N., ACS Synth. Biol. 2018, 7, 576–583. [DOI] [PubMed] [Google Scholar]
- 41. Liu Y., Holmstrom E., Zhang J., Yu P., Wang J., Dyba M. A., Chen D., Ying J., Lockett S., Nesbitt D. J., Ferre-D'Amare A. R., Sousa R., Stagno J. R., Wang Y. X., Nature 2015, 522, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mears J. A., Cannone J. J., Stagg S. M., Gutell R. R., Agrawal R. K., Harvey S. C., J. Mol. Biol. 2002, 321, 215–234. [DOI] [PubMed] [Google Scholar]
- 43. Shoji S., Dambacher C. M., Shajani Z., Williamson J. R., Schultz P. G., J. Mol. Biol. 2011, 413, 751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary