

Abstract
Oral insulin administration still represents a paramount quest that almost a century of continuous research attempts did not suffice to fulfill. Before pre‐clinical development, oral insulin products have first to be optimized in terms of encapsulation efficiency, protection against proteolysis, and intestinal permeation ability. With the use of dendritic mesoporous silica nanoparticles (DMSNs) as an insulin host and together with a protein‐based excipient, succinylated β‐lactoglobulin (BL), pH‐responsive tablets permitted the shielding of insulin from early release/degradation in the stomach and mediated insulin permeation across the intestinal cellular membrane. Following an original in vitro cellular assay based on insulin starvation, direct cellular fluorescent visualization has evidenced how DMSNs could ensure the intestinal cellular transport of insulin.
Keywords: beta-lactoglobulin tablets, cellular insulin starvation, dendritic mesoporous silica nanoparticles, oral insulin dosage, transcellular epithelial transport
Insulin delivery: A pH‐responsive protein combined with the key attributes of dendrimer‐like mesoporous silica nanoparticles act synergistically as a smart oral drug‐delivery platform to reduce substantially the premature release and degradation of gastro‐sensitive insulin. As evidenced by confocal microscopy, the designed nanocarriers mediated significant cellular permeation of the nanopore‐confined fluorescent‐labeled insulin across human intestinal cells (see figure).
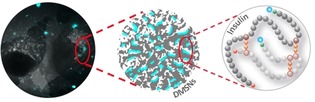
By 2045, the proportion of diabetic patients is expected to increase by 51 % worldwide and yet, the only marketed treatment available is through subcutaneous injection of insulin on a daily basis.1 For both Type 1 and 2 diabetes, the production of insulin by the β‐cells of pancreatic islets is either relatively absent due to autoimmune reaction (Type 1) or inappropriate to cover high blood glucose concentration due to the development of insulin resistance by the body (Type 2). Hence, patients face hyperglycemia, with the symptoms associated to it, and their threatened life relies on insulin delivery from external sources. On top of the discomfort felt during needle injections, complications like skin infections can occur.2 While these major drawbacks would be eliminated by oral administration of insulin, it remains an issue that the pharmaceutical industry is still challenged to tackle. The reason is that the oral bioavailability is far from reaching subcutaneous injection levels, in which 100 % of the injected dose is immediately disponible in the blood. Because of its high molecular weight, insulin is scarcely bioavailable, the susceptibility to gastric and intestinal proteolysis is high and the permeability across the intestinal barrier, low. Hopefully, owing to the recent development of smart drug (nano)carriers, the impact of these parameters on the resulting oral bioavailability can be downgraded. For instance, very recently, a self‐orienting millimeter‐applicator has been developed to inject insulin through the gastric mucosa and plasma concentration comparable to subcutaneous or intragastric injection has been reported for the very first time.3 Even though this represents a major breakthrough, potential chronic side effects such as mucosa perforation or body response to a foreign millimeter‐sized device have to be envisaged. Differently, owing to their nanometer‐scale size, mesoporous silica nanoparticles (MSNs) can simultaneously penetrate the cellular epithelial membrane safely while carrying their protected cargoes.4 More specifically, dendritic (i.e., dendrimer‐like) MSNs (DMSNs) offer customizable pores in the range 6–20 nm, large enough to host insulin, and almost monodispersed particle size distribution in the range 50–100 nm, which is small enough to navigate through the epithelial intestinal barrier.5
Herein, we report the successful nanoconfinement of insulin into the dendritic mesopores of DMSNs as a promising way to significantly reduce the release/degradation in the stomach (pH 1.2) and enhance the transport of insulin towards the intestine (pH 7.4).6 DMSNs of various pore sizes have been designed and a thiol‐functionality was anchored on the free silica surface as a potential permeation and mucoadhesion enhancer.7 Highly reproducible loadings of insulin were achieved through electrostatic attraction between the oppositely charged insulin and DMSNs. The insulin‐confined DMSNs were then tableted with a pH‐responsive whey protein, namely succinylated β‐lactoglobulin (BL). The combination of DMSNs and BL is essential to synergistically refrain the degradation at pH 1.2 and enhance both the release at pH 7.4 and the cellular transport of insulin. Fluorescent images provided evidence to support the hypothesis that DMSNs are nanoparticles of choice to overcome the permeation limitations faced by most peptides and especially insulin. Finally, mechanistic insights into the modulation of the in vitro metabolic activity of human epithelial colon cells (HCEC) through the mediation of DMSNs have been gained using an insulin‐containing/deprived experimental setup.
The formation of DMSNs takes place at the interface of the organic/aqueous phases during the synthesis. DMSNs of various pore sizes have been obtained upon changing the nature of the organic phase. As the organic solvent is suggested to be located in between the alkyl chains of the CTAC surfactant, it can be well understood that from a linear solvent (hexane) to a cyclic one (cyclohexane) and further to an aromatic ring (toluene), the in situ generated steric hindrance differs greatly, which in turn permitted to precisely control the swelling of the pores at the nanometer scale. N2 isotherms supported this hypothesis since from hexane to toluene, the condensation step in the isotherms shifted to higher relative pressure upon increase of the pore size (Figure 1 d–e, Table S1, Supporting Information). This evolution of the pore structure and the increased particle size from DMSN‐Hex to DMSN‐Tol is also well visible in the transmission electron microscopy (TEM) images (Figure 1 a–c). In addition, upon functionalization with thiol groups, the porosity of the resulting DMSN‐SH was reduced as compared to the starting DMSN‐Hex but the particle size did not change (see Figure S1).
Figure 1.
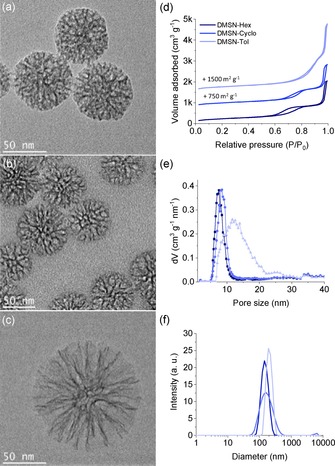
(a–c) TEM images of the DMSNs synthesized with either hexane (DMSN‐Hex), cyclohexane (DMSN‐Cyclo) or toluene (DMSN‐Tol); (d, e) Isotherms and respective pore size distributions obtained from N2 physisorption analyses (−196 °C); (f) Particle size distribution of the DMSNs obtained from dynamic light scattering (DLS) measurements.
Considering the size of insulin and the pore sizes of the DMSNs, both smaller and larger pore size platforms, i.e., DMSN‐Hex and DMSN‐Tol, respectively, were selected (Figure S2a). The optimal pH of the loading was set to 4 as both parts, i.e., insulin and the different DMSNs, have opposite charges at this specific pH (Figure S2b). As a result, the favorable electrostatic attraction between the positively charged insulin and negatively charged DMSNs8 permitted to successfully incorporate about 20 wt % of insulin into DMSN‐Hex, DMSN‐Tol and DMSN‐SH (Figures S3 and S4). Furthermore, owing to the confinement of insulin inside DMSNs, the diffraction peaks originating from the semi‐crystalline structure of insulin disappeared and were replaced by a typical amorphous ‘halo’ (Figure S5).
As depicted in the Scheme 1, either pristine insulin or insulin confined into DMSN‐Hex or DMSN‐SH was further tableted with the succinylated β‐lactoglobulin protein in order to obtain different pH‐responsive formulations which were evaluated for their ability to protect insulin at the acidic stage and releasing it, undegraded, in near neutral conditions. In addition, release profiles obtained with BL‐DMSN‐Tol‐Ins, i.e., insulin confined in larger pores of DMSN‐Tol and mixed homogeneously with BL, and a layer‐by‐layer formulation approach are reported as well and discussed in Figure S6 of the SI. Already at the acidic stage, significant variation in the mean comparison could be observed between BL‐Ins and BL‐DMSN‐Ins, as illustrated in Figure 2 (a different representation of the release profiles is available in SI, Figure S6a). While almost 40 % of insulin is eluted from the BL‐Ins tablets after 2 h, only 5 % was released from BL‐DMSN‐Ins (**** p<0.0001) and less than 10 % from BL‐DMSN‐SH‐Ins. In addition to the pH‐responsiveness of BL, confining insulin into DMSNs has permitted to keep it in the pores, undegraded. Since after the acidic stage, the intestinal tract is normally reached, the same tablets were transferred to pH 7.4. In the case of BL‐Ins, a period of 4 h was required for insulin to dissolve completely. Interestingly, for BL‐DMSN‐Ins, the release profile can be divided into two sequences: a slow sustained release in the first two hours, followed by a faster one for the next two hours until ≈80 % release is observed after 24 h. The slow release observed within the first 2 h at pH 7.4 can therefore offer a window for the DMSNs to transport, within their pores, the loaded insulin through the epithelial barrier. A different release profile was obtained with the thiolated‐DMSNs tablets. Here, thiol groups were anchored through post‐synthesis grafting on the surface of DMSNs, as they are believed to enhance the permeability of the nanocarriers.7 However, in addition to a poor release as compared to BL‐Ins and BL‐DMSN‐Ins (Figure 2 a), the fragmentation of insulin in its chain A, named Frag. 2 with a molecular weight of 2377 Da and chain B, named Frag. 1 with a molecular weight of 3431 Da, was witnessed and is supported by mass spectrometry (MS) data (Figure 2 c–d).9 Once at pH 7.4, the tablets started to erode and thiol groups present at the surface of DMSN‐SH deprotonate, and R‐S− species should predominate under these conditions. Thereafter, the disulfide bond holding the two chains of insulin together is likely to be reduced releasing the two distinct chains. Although, thiol groups have been investigated as permeation enhancer, according to our results, their use would be at the cost of potential protein unfolding which could lead to the loss of the pharmacological activity.10 Thus far, this undesired reductive activity of thiolated‐nanocarriers has not been reported in the literature. Nevertheless, such an issue might be avoided through the replacement of the original disulfide bond with a diselenide bridge which was shown to be resistant against reduction.11
Scheme 1.
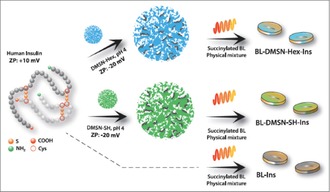
Scheme representing the loading of insulin inside DMSNs and the formulation of the BL tablets for the release tests (ZP stands for Zeta‐potential).
Figure 2.
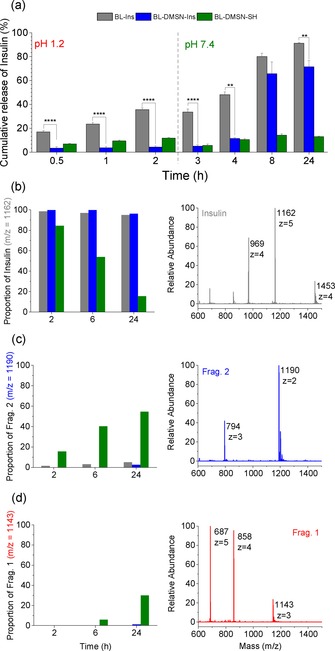
(a) Cumulative release of insulin from the different tablets immersed 2 h at pH 1.2 and subsequently at pH 7.4. Data shown as mean ± SE (n=3), Significant difference express **p<0.01, ****p<0.0001, One‐way ANOVA and Fisher Test; (b–d) MS characterization of the released insulin or the fragments of insulin at different time periods in correlation with their respective proportion.
Once they have reached the intestinal lumen, DMSNs would have to carry insulin across the epithelium. Various pathways can be followed by the nanoparticles to reach the systemic circulation, however the transcellular transport remains the most likely path considering the surface area of the epithelial membrane.12 Apart from the transport mechanism, nanocarriers with smaller particle size (≈100 nm) and negative surface charge are assumed to increase the permeation across the intestinal membrane while reducing their hepatic and splenic accumulation; although, depending on the surface chemistry, some positively charged nanoparticles have also shown enhanced cellular interactions.13 As supported by our characterization, pure DMSNs and DMSN‐SH materials have both a hydrodynamic diameter in water of around 150 nm and a negative surface charge above pH 3 which match well the above‐mentioned criteria. Therefore, the ability of pure DMSNs, DMSN‐SH, and insulin to be efficiently internalized by intestinal cells was tested using HCEC cells through live cell imaging. The resulting confocal microscopy images are presented in Figures 3, 4, S7 and S8. Prior to the experiment, DMSNs and insulin were coupled with Rhodamine (Rhod) or FITC fluorescent probes (see SI for more details). As it can be observed in the Figure S7, Ins‐FITC was insufficiently taken up by the cells until at least 3 h. This supports the interpretation that insulin requires a performant shuttle to be transported through the intestinal epithelium and to help overcome oral bioavailability issues which were previously described in vivo.7b On the other hand, DMSNs penetrated efficiently the cytoplasm, as illustrated in Figure 3. Indeed, the presence of DMSN‐FITC was monitored through time‐dependent kinetic experiments. After already 2 h of incubation, DMSN‐FITC were present in the cytoplasm and the density of internalized DMSNs increased progressively up to 20 h of incubation. In order to prove that the cellular uptake of the DMSNs was not probe‐dependent, a second fluorescent label, i.e., Rhod, was coupled to DMSNs. The ability of DMSN‐Rhod and DMSN‐SH‐Rhod to penetrate the cells was also confirmed, as shown in Figure S8. It makes no doubt that DMSNs are good candidates to enhance protein permeation. Therefore, Ins‐FITC was loaded in non‐labeled DMSN‐Hex and the resulting DMSN‐Ins‐FITC assembly was incubated with the cells for 2 h. The signal intensity from Ins‐FITC confined in DMSNs was compared to Ins‐FITC in its free form, similarly incubated. The resulting images, presented in Figure 4 a, clearly show a higher concentration of Ins‐FITC inside the cells when transported in DMSNs (red arrows). Moreover, from the cross section of the z stack presented in Figure 4 b, we could confirm that within the 2 h period, the DMSN‐Ins‐FITC were reaching the cell membrane of HCEC cells.
Figure 3.
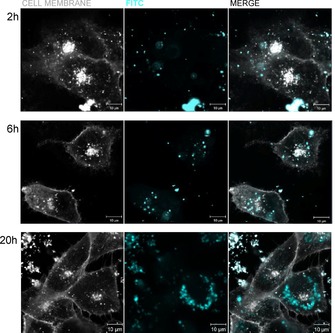
Live cell fluorescence images of the uptake of FITC‐labeled DMSNs by HCEC cells after 2 h, 6 h or 20 h. FITC is represented in light blue and the plasma membrane in white.
Figure 4.
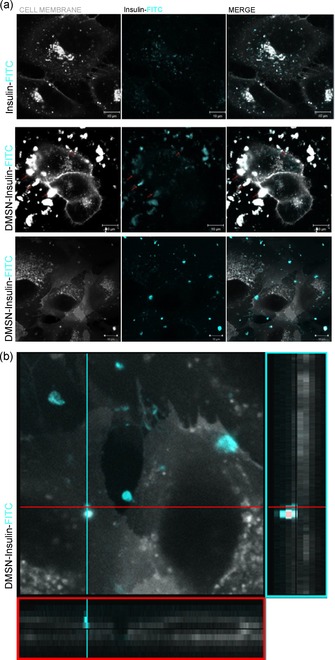
Live cell fluorescence imaging of HCEC cells incubated with Ins‐FITC or DMSN‐Ins‐FITC for 2 h; (a) 2D images and (b) Zoom in the vertical cross‐section of the 3D cut stack. The plasma membrane is represented in white and, the fluorescence coming from Ins‐FITC in light blue.
As insulin confined into DMSNs appeared to interact with the membrane of intestinal cells, we performed additional experiments in order to verify if our observation could be accompanied also by a functional readout. The effects of unconfined insulin and insulin confined in both pure and thiol‐modified DMSNs on the metabolic activity of HCEC cells have thus been compared and the results are presented in Figure 5. To verify the efficacy of the transport, HCEC cells were deprived of insulin supplement (10 μg mL−1) which is originally present in the culture media, thus generating a starvation condition mimicking pathological lack of the protein (see SI for the experimental details). Because of its pivotal role in many metabolic cascade reactions, such as glucose metabolism, lipogenesis, glycogen synthesis, insulin deprivation is supposed to considerably reduce the metabolic activity of the cells.14 In turn, this enabled us to test whether the DMSN‐based systems could maintain viable cell activity under these conditions. In “starvation” condition, i.e., in absence of insulin in the medium, the incubation with DMSN‐Ins and DMSN‐SH‐Ins triggered a significant increase of the metabolism of the Alamar Blue (AB) reagent in comparison to the incubation with the same insulin but in non‐confined form. This difference was reduced by the incubation in complete medium, implying that the metabolic need of the cells enhances the response of the test system. Along that line, longer incubation time (6 h) allowed to observe similar increase in dye metabolism when insulin was applied with the DMSNs both in presence or absence of the supplement, suggesting that insulin starvation might contribute to improve the kinetics of the observed effect.
Figure 5.
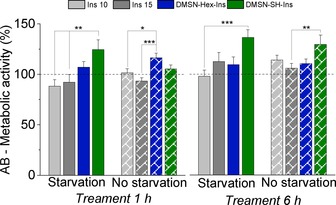
In vitro assay based on the presence (no starvation) or absence (starvation) of insulin in the culture media of HCEC cells prior to the incubation of different treatments: commercial human insulin at the concentration 10 or 15 μg mL−1, insulin confined in pure DMSNs or thiol‐functionalized DMSN‐SH at the concentration equivalent to 15 μg mL−1 of insulin. One‐way ANOVA and Fisher Test expressed significant difference by *p<0.05, **p<0.01, ***p<0.001.
Altogether, our results described a robust and versatile functional solid dosage for the oral delivery of insulin. We have demonstrated that, already in vitro, the acidic release of insulin from BL‐Ins, that is, DMSN‐free tablets, was above the recommended limit and the cellular permeation of unformulated insulin was clearly insufficient. When confined in the pores of DMSNs, the release of insulin in gastric fluid was lowered down to acceptable threshold (less than 10 %) and the enhanced permeation of labeled insulin (Ins‐FITC) was evidenced by confocal fluorescence microscopy. Despite the mucoadhesive properties of thiol groups, proteolysis of insulin was witnessed, and it might therefore not be the most appropriate formulation strategy. In contrast, pure DMSNs provided all the ideal benefits for insulin nanocarriers. Small particle size and tunable large pores along with a negatively charged surface allowed a simultaneous high protein loading and efficient epithelial cell uptake, while incorporation of the nanocarriers in a smart pH‐sensitive protein formulation prevented the premature gastric release and degradation of insulin.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank the University of Vienna (Austria) for the financial support as well as the Mass Spectrometry Centre (MSC) and the Core Facility Multimodal Imaging of the Faculty of Chemistry (members of the VLSI). The authors thank especially Ms. Anna Fabisikova, Ms. Eva Attakpah and Mr. Manuel Felkl for their valuable technical help. The authors are grateful to Prof. Dr. C. Gerner (Department of Analytical Chemistry, Faculty of Chemistry, University of Vienna) for precious suggestions and stimulating discussion.
E. Juère, R. Caillard, D. Marko, G. Del Favero, F. Kleitz, Chem. Eur. J. 2020, 26, 5195.
References
- 1.International Diabetes Atlas, 9th ed., 2019.
- 2. Wong C. Y., Martinez J., Dass C. R., J. Pharm. Pharmacol. 2016, 68, 1093–1108. [DOI] [PubMed] [Google Scholar]
- 3. Abramson A., Caffarel-Salvador E., Khang M., Dellal D., Silverstein D., Gao Y., Frederiksen M. R., Vegge A., Hubálek F., Water J. J., Friderichsen A. V., Fels J., Kirk R. K., Cleveland C., Collins J., Tamang S., Hayward A., Landh T., Buckley S. T., Roxhed N., Rahbek U., Langer R., Traverso G., Science 2019, 363, 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Zhao Y., Trewyn B. G., Slowing I. I., Lin V. S.-Y., J. Am. Chem. Soc. 2009, 131, 8398–8400; [DOI] [PubMed] [Google Scholar]
- 4b. Sun L., Zhang X., Wu Z., Zheng C., Li C., Polym. Chem. 2014, 5, 1999–2009; [Google Scholar]
- 4c. Desai D., Prabhakar N., Mamaeva V., Sen Karaman D., Lähdeniemi I. A. K., Sahlgren C., Rosenholm J. M., Toivola D., Int. J. Nanomed. 2016, 11, 299–313; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Bouchoucha M., Côté M. F., Gaudreault R. C., Fortin M. A., Kleitz F., Chem. Mater. 2016, 28, 4243–4258; [Google Scholar]
- 4e. Nguyen C. T., Webb R. I., Lambert L. K., Strounina E., Lee E. C., Parat M. O., McGuckin M. A., Popat A., Cabot P. J., Ross B. P., ACS Appl. Mater. Interfaces 2017, 9, 9470–9483; [DOI] [PubMed] [Google Scholar]
- 4f. Florek J., Caillard R., Kleitz F., Nanoscale 2017, 9, 15252–15277; [DOI] [PubMed] [Google Scholar]
- 4g. Juère E., Florek J., Bouchoucha M., Jambhrunkar S., Wong K. Y., Popat A., Kleitz F., Mol. Pharm. 2017, 14, 4431–4441. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Wang Y., Nor Y. A., Song H., Yang Y., Xu C., Yu M., Yu C., J. Mater. Chem. B 2016, 4, 2646–2653; [DOI] [PubMed] [Google Scholar]
- 5b. Lei C., Xu C., Nouwens A., Yu C., J. Mater. Chem. B 2016, 4, 4975–4979; [DOI] [PubMed] [Google Scholar]
- 5c. Shen D., Yang J., Li X., Zhou L., Zhang R., Li W., Chen L., Wang R., Zhang F., Zhao D., Nano Lett. 2014, 14, 923–932; [DOI] [PubMed] [Google Scholar]
- 5d. Du X., Xiong L., Dai S., Kleitz F., Qiao S. Z., Adv. Funct. Mater. 2014, 24, 7627–7637; [Google Scholar]
- 5e. Meka A. K., Abbaraju P. L., Song H., Xu C., Zhang J., Zhang H., Yu M., Yu C., Small 2016, 12, 5169–5177; [DOI] [PubMed] [Google Scholar]
- 5f. Möller K., Bein T., Chem. Mater. 2017, 29, 371–388; [Google Scholar]
- 5g. Abeer M. M., Meka A. K., Pujara N., Kumeria T., Strounina E., Nunes R., Costa A., Sarmento B., Hasnain S. Z., Ross B. P., Popat A., Pharmaceutics 2019, 11, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guillet-Nicolas R., Popat A., Bridot J. L., Monteith G., Qiao S. Z., Kleitz F., Angew. Chem. Int. Ed. 2013, 52, 2318–2322; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2374–2378. [Google Scholar]
- 7.
- 7a. Maher S., Mrsny R. J., Brayden D. J., Adv. Drug Delivery Rev. 2016, 106, 277–319; [DOI] [PubMed] [Google Scholar]
- 7b. Shrestha N., Araújo F., Shahbazi M.-A., Mäkilä E., Gomes M. J., Herranz-Blanco B., Lindgren R., Granroth S., Kukk E., Salonen J., Hirvonen J., Sarmento B., Santos H. A., Adv. Funct. Mater. 2016, 26, 3405–3416. [Google Scholar]
- 8. Zhao X., Shan C., Zu Y., Zhang Y., Wang W., Wang K., Sui X., Li R., Int. J. Pharm. 2013, 454, 278–284. [DOI] [PubMed] [Google Scholar]
- 9. Chen Z., Caulfield M. P., McPhaul M. J., Reitz R. E., Taylor S. W., Clarke N. J., Clin. Chem. 2013, 59, 1349–1356. [DOI] [PubMed] [Google Scholar]
- 10. Gori A., Gagni P., Rinaldi S., Chem. Eur. J. 2017, 23, 14987–14995. [DOI] [PubMed] [Google Scholar]
- 11. Arai K., Takei T., Okumara M., Watanabe S., Amagai Y., Asahina Y., Moroder L., Hojo H., Inaba K., Iwaoka M., Angew. Chem. Int. Ed. 2017, 56, 5522–5526; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5614–5618. [Google Scholar]
- 12. Netsomboon K., Bernkop-Schnürch A., Eur. J. Pharm. Biopharm. 2016, 98, 76–89. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Maisel K., Ensign L., Reddy M., Cone R., Hanes J., J. Controlled Release 2015, 197, 48–57; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Dogra P., Adolphi N. L., Wang Z., Lin Y. S., Butler K. S., Durfee P. N., Croissant J. G., Noureddine A., Coker E. N., Bearer E. L., Cristini V., Brinker C. J., Nat. Commun. 2018, 9, 4551–4564; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Shrestha N., Shahbazi M. A., Araújo F., Zhang H., Mäkilä E. M., Kauppila J., Sarmento B., Salonen J. J., Hirvonen J. T., Santos H. A., Biomaterials 2014, 35, 7172–7179; [DOI] [PubMed] [Google Scholar]
- 13d. Andreani T., Miziara L., Lorenzón E. N., de Souza A. L. R., Kiill C. P., Fangueiro J. F., Garcia M. L., Gremião P. D., Silva A. M., Souto E. B., Eur. J. Pharm. Biopharm. 2015, 93, 118–126. [DOI] [PubMed] [Google Scholar]
- 14. Ruegsegger G. N., Creo A. L., Cortes T. M., Dasari S., Sreekumaran Nair K., J. Clin. Invest. 2018, 128, 3671–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary