

Supplemental Digital Content is available in the text.
Keywords: acute respiratory distress syndrome, baby lung, mechanical power, mechanical ventilation, ventilator-induced lung injury
Abstract
Objectives:
To examine the potentially modifiable drivers that injure and heal the “baby lung” of acute respiratory distress syndrome and describe a rational clinical approach to favor benefit.
Data Sources:
Published experimental studies and clinical papers that address varied aspects of ventilator-induced lung injury pathogenesis and its consequences.
Study Selection:
Published information relevant to the novel hypothesis of progressive lung vulnerability and to the biophysical responses of lung injury and repair.
Data Extraction:
None.
Data Synthesis:
In acute respiratory distress syndrome, the reduced size and capacity for gas exchange of the functioning “baby lung” imply loss of ventilatory capability that dwindles in proportion to severity of lung injury. Concentrating the entire ventilation workload and increasing perfusion to these already overtaxed units accentuates their potential for progressive injury. Unlike static airspace pressures, which, in theory, apply universally to aerated structures of all dimensions, the components of tidal inflation that relate to power (which include frequency and flow) progressively intensify their tissue-stressing effects on parenchyma and microvasculature as the ventilated compartment shrinks further, especially during the first phase of the evolving injury. This “ventilator-induced lung injury vortex” of the shrinking baby lung is opposed by reactive, adaptive, and reparative processes. In this context, relatively little attention has been paid to the evolving interactions between lung injury and response and to the timing of interventions that worsen, limit or reverse a potentially accelerating ventilator-induced lung injury process. Although universal and modifiable drivers hold the potential to progressively injure the functional lung units of acute respiratory distress syndrome in a positive feedback cycle, measures can be taken to interrupt that process and encourage growth and healing of the “baby lung” of severe acute respiratory distress syndrome.
The disabled lung units of acute respiratory distress syndrome (ARDS) coexist with functional ones that receive the entirety of each tidal volume (Vt) and conduct all gas exchange with blood. This smaller capacity functioning lung tissue has become known as a “baby lung” (BL) (1, 2). Whatever the number, efficiency, and distribution of units that comprise this operational “BL,” protective strategies for ventilation are directed toward preserving its size and functionality, with the aim of avoiding ventilator-induced lung injury (VILI). Ventilation and perfusion of the BL rise to maintain adequate gas exchange when injury is severe and both may influence the extent of VILI expression. Importantly, the reduced size and capacity for gas exchange imply loss of ventilatory capability, which dwindles in proportion to severity of injury (3). Concentrating the entire ventilation workload on an already overtaxed BL accentuates its potential for progressive injury. In this context, relatively little attention has been paid to the evolving interactions between lung injury and repair and to the timing of interventions that worsen, limit or reverse a potentially accelerating VILI process. The wisdom of applying or withdrawing a given intervention varies with the nature of the underlying lung injury as well as the balance of these competing activities.
CONTRIBUTORS TO VILI
In recent years, better understanding of the biophysical causes of VILI has shifted our traditional focus from optimizing the inflation pattern of the single tidal cycle (e.g., Vt and airway pressures) toward avoiding exposures of the alveolar-capillary barrier to damaging levels of tidal energy and power (4, 5). These newer “ergocentric” variables account not only for static characteristics, as do plateau, positive end-expiratory pressure (PEEP), and driving pressures, but also for the dynamic ones of flow and cycling frequency. With this revised orientation and concern for the energetics of VILI in mind, the relatively neglected vascular side of the alveolar-capillary interface has also begun to receive needed attention (6–8). When lung injury is initiated by a disease that directly inflames the endothelium, exemplified by coronavirus disease 2019 (COVID-19) pneumonia/ARDS, these airspace and microvascular stresses reenforce one another. The energy flowing through the gas exchanging units of the BL exceeds their normal values, both from the airspace and vascular sides of their shared interface. But there is an inherent imbalance in the relative matching of gas flow and blood flow across the BL. During severe ARDS, the ventilated compartment typically comprises only 20–30% of its healthy volume but must perform 100% of ventilation, whereas its nonshunted blood flow generally remains at 60–70% of the cardiac output (2). Here it is worth mentioning that although computed “physiologic dead space” (Vd/Vt), a strong correlate of mortality risk (9, 10), implies ineffective ventilation and has been attributed primarily to narrowed and obstructed microvessels and to systemic recirculation of venous Co2 through shunt units (11), an important contributor to the clinically estimated Vd/Vt ratio is the need for the well-perfused BL to be hyperventilated; its ventilation must exceed its perfusion. Therefore, ARDS severity, BL capacity, and Vd/Vt are understandably intercorrelated.
To clear the same amount of Co2 per minute across the reduced gas exchanging surface area of the BL, its alveolar ventilation (and total ventilation) must increase (Supplemental Fig. 1, Supplemental Digital Content 1, http://links.lww.com/CCM/F521). Of note, this mandatory hyperventilation and VILI-promoting power exposure of the functioning BL tissues also exposes the cells that line its epithelium to locally high pH values that may contribute to impaired surfactant function (12–14). For the same BL size, the level of hyperventilation and its consequences for lung injury and repair vary with the amount of Co2 to be cleared per minute (influenced by oxygen consumption and metabolic acidosis) and with the set equilibration target for Paco2.
Due to the reduced dimensions and low absolute compliance of the BL, the increase in total ventilation required to achieve a physiologic arterial pH is usually obtained by keeping Vt relatively low while increasing the respiratory rate, a combination which, for a given expired volume per minute (Ve), tends to further increase the “anatomic” (series) component of the total “dead space” (15). In severe ARDS, the total ventilation required to maintain a normal Paco2 usually proves unattainable or clinically inadvisable. Consequently, unless aided by a reduction of Co2 production or by extracorporeal support, the alveolar Pco2 of the BL must rise to boost the volume of Co2 discharged per breath.
DETERMINANTS OF VILI RISK AND PROGRESSION
Repeated exposure to tidal cycles that cause excessive, fracturing strain of structural microelements is currently believed the primary mechanical stimulus for tissue injury caused by ventilation (4, 5). Overt VILI results eventually when the rate of such disruptions exceeds the lung’s endogenous capability to prevent or repair them (Fig. 1). Accepting this view, it follows that a sufficient critical strain must first occur during tidal inflation before repeated cycles of that type produce damage (4, 16). The critical airspace pressures required to initiate VILI are almost certain to differ with the cause and stage of ARDS (16, 17), with the velocity of their application (14, 18–20), as well as with disease co-factors, status of pulmonary vascular filling and pressure, and certain therapeutic interventions (e.g., IV fluids, Fio2) (7, 21–23). Consequently, in experimental studies, the critical tidal plateau pressure has been reported to vary from ≈ 11 to 60 cm H2O (21, 22), depending on species type, priming presence and model of injury, supportive measures, and initial state of health or disease. Furthermore, critical airspace pressures and strains are likely to differ site-by-site within the lung parenchyma and with the extent and histologic stage of lung damage.
Figure 1.

Progressive drop-out of stress-bearing matrix elements. A, Ultra-structure of the stress-bearing network. B, Principle of progressive loading and predisposition to sequential failure of parallel stress-bearing elements. Dropout of weak strands increases the strain on those remaining.
Two time-dependent processes compete to determine whether the threshold for mechanical injury rises (resisting mechanical damage and promoting repair) or falls (favoring iatrogenic injury). Successful therapies directed at disease control and innate healing processes (e.g., micro-wound organization and fibrosis) proceed steadily over time, in opposition to the accumulating number of potentially intolerable straining cycles (5, 17) (see What Slows VILI Vortex, following). Regarding the latter, externally measured airway pressures, flows, and volumes do not faithfully indicate the true stresses and strains that influence the locally specific power applied to tissues at the parenchymal level, nor do they accurately reflect the changing mechanical properties of units within the BL compartment (24). Mechanical heterogeneity, which increases with damage extent and severity, focuses stress at junctional interfaces and accentuates effective strain (25, 26). Stress amplifiers include local forces arising from geometrical interdependence, flow-accentuated viscoelastic drag, and the progressive strain experienced as weaker stress-bearing elements of the extracellular fibrillar matrix fracture or drop away, further encumbering those fibrils of the supporting matrix network that remain intact or altering the conformation and alveolar wall tension of the affected units (Fig. 2) (27). This ongoing transfer and amplification of stress promote a positive feedback cycle of intolerable strain, progressive loading and dwindling reserve of the remaining BL units to withstand an unchanging global burden for ventilation.
Figure 2.
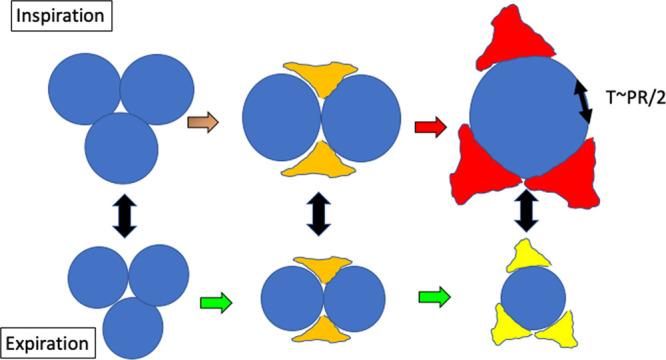
At a constant tidal volume, size, and strain of the individual units of the “baby lung” increase with advancing injury. Deformation of remaining airspaces may accentuate the alveolar tensions that correspond to a given pressure, as suggested by a simplistic analogy to the law of La Place: Tension (T) is proportional to the product of distending pressure (P) and unit radius (R).
Simultaneously, altered mechanical properties of the infiltrated and edematous lung, as well as more slowly maturing processes of organization, fibrosis, and repair proceed along a parallel but opposing trajectory that acts to reduce BL vulnerability (17). Consequently, the specific compliance and time constant of each individual gas-accepting lung unit may rise or fall as ARDS and its management proceed over an extended time. In our present-day practice that emphasizes lung protection, the competition between forces favoring less damage and lung repair and those that favor worsening severity and progressive life-threatening VILI is usually won eventually by the former. Unfortunately, in advanced cases of extreme severity and those that are mismanaged (28), any competitive advantage favoring healing may be overwhelmed.
Progressive Vulnerability of the Shrinking “Baby” Lung to VILI
The limited capacity of the small BL encountered in severe disease predisposes to accelerating injury by mechanical forces, especially when tidal strains above the injury threshold are repeated without letup. Power (Pwr) applied to the respiratory system is the product of cycling frequency and inflation energy per tidal cycle (4, 5, 29). The latter is influenced by the amplitude and profile of inspiratory flow as well as by Vt and airway pressures. In highly simplified form, Pwr = Ve × (FLR + DP/2 + PEEPT), where FL is mean inspiratory flow, R is resistance, DP is driving pressure, and PEEPT (PEEP plus any auto-PEEP) is the starting and ending pressure baseline. Unlike static airspace pressures, which, in theory, apply universally to structures of all dimensions, the components of tidal inflation that relate to power (which include frequency and flow) intensify their tissue-stressing effects as the BL shrinks, especially during the first phase of the evolving injury (30). In other words, assuming that each lung unit that makes up the BL has similar elastance, the same power load that is borne without damaging stress or strain by a lung with large capacity for expansion may injure a smaller one whose cumulative energy exposure per lung unit rises in inverse proportion to reduced BL capacity (2, 5, 31).
To illustrate, even when ventilated with the same tidal package of PEEP, plateau, and driving pressure, the cycling frequency (and pace of energy exposure) must increase as the available lung shrinks in order to compensate for the reduced absolute compliance and smaller Vt of the BL. Conversely, if the shrinking BL were ventilated with the same Vt and frequency as before onset of ARDS injury, its tidal driving pressure would need to rise, increasing the power applied to it (5, 30). Furthermore, assuming that cycling frequency (and minute ventilation) were also kept unchanged, fewer open air channels for ventilation would obligate higher velocity of gas flow through them, as well as a greater rate of parenchymal expansion, boosting both epithelial forces applied to the small airways and viscoelastic amplification of microfibril stress, respectively (27). It follows that vulnerability to damage of the shrinking BL steadily rises as tidal cycling continues, again provided that intolerable tidal strains remain unrelieved. Duration of the noxious ventilating pattern is clearly important; if unsuccessfully opposed by injury-altered lung mechanics and processes of tissue repair, this positive feedback “vortex of VILI” has the potential to progress (Fig. 3).
Figure 3.
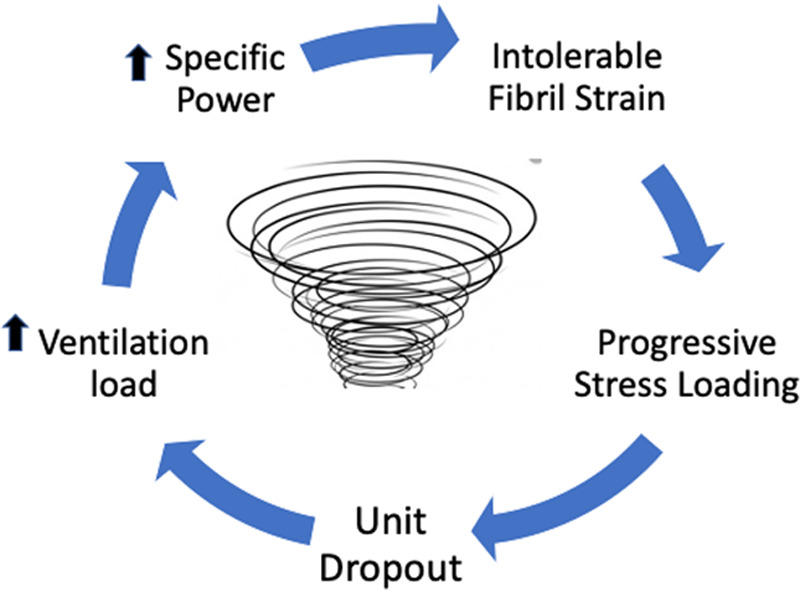
Positive feedback and the “ventilator-induced lung injury vortex” of the shrinking baby lung.
Importance of Hemodynamics.
Unlike the normal lung, whose vasculature can withstand relatively high luminal pressures (≈20–25 mm Hg) before leaking significant fluid into the interstitium and alveolar spaces, the relationship of edema to microvascular luminal pressure becomes a quasi-linear, direct function during acute lung injury that lacks a definable threshold for leakage (32). Precapillary and postcapillary microvessels share this fluid permeability (33, 34). Higher than normal cardiac output, often a component of the systemic stress response, of insufficient sedation of the agitated patient, or of unnecessary inotropes and IV fluid, requires an increased gradient of luminal microvascular pressure to drive it across the increased vascular resistance. Consequently, mean microvascular pressure rises. VILI, therefore, should be considered more than cellular inflammation and disrupted alveoli resulting from high airspace pressures and strain; the vascular side is also integral to injury expression. A hydrodynamic component of edema always accompanies damage to the gas exchanging membrane and that edema is rather easily increased by fluid administration or other interventions that alter pulmonary vascular pressures.
Finally, although seldom considered, the reduced capacity of the BL should also boost the potential for injury to propagate via the air passages that connect functioning alveoli with adjacent ones that are already flooded by inflammatory edema (35). This propensity increases as aerated volume contracts because the smaller BL accommodates and compartmentalizes fluid less well than its larger counterpart.
WHAT CHECKS OR REVERSES THE “VILI VORTEX”?
Disease in Evolution
Reactive and reparative responses to excessive stress tend to interrupt and reverse the vicious cycle of progressive VILI. Unaltered and intrinsically compliant units embedded in zones of inflammatory debris and edema may become “externally” buttressed by what surrounds them and therefore experience lower tidal stretching forces and more tolerable strain as their “functional specific elastance” rises (Fig. 4). Early on, open alveoli in gravitationally dependent zones of the layered BL experience “crowding” from tissues above and below them, raising their opening pressures and proclivity to collapse. Over time, organization of proteinaceous edema and inflammatory debris may transition the make-up of the BL from normally compliant units in any zone to ones with higher specific elastance that opposes potentially damaging strain.
Figure 4.
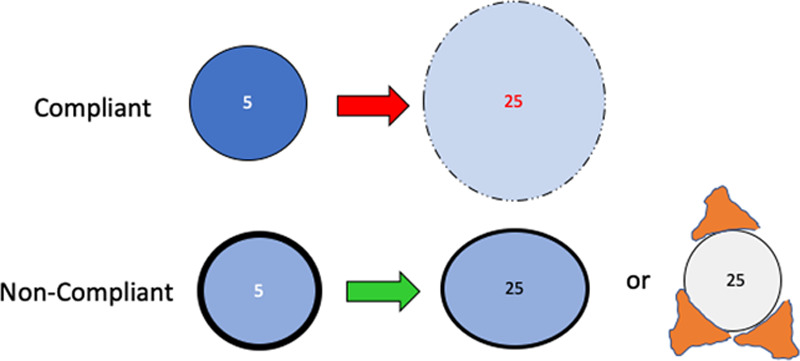
Some baby lung units may be protected (not made more vulnerable) as injury advances by reduced intrinsic compliance or their being embedded among already damaged units that surround them.
Therapeutic Interventions
Apart from recruitment of lung units that, speaking metaphorically, “grow” the BL and help diffuse stress and strain throughout the aerated network, one of the primary benefits from low to moderate PEEP applied in the early phase of ARDS management may be to provide counterpressure that redistributes and trans-locates alveolar edema from alveolus to the interstitial matrix (36). In this process, PEEP also brakes, interrupts or prevents tidal trans-airway propagation of protein and mediator-laden fluid. Potentially adverse consequences of higher and unnecessary levels of PEEP, however, include augmented ventilating power, hemodynamic compromise of systemic vital organs, and increased stress on already open units of the BL, particularly when ventilation occurs in the supine position. In contrast, regional transpulmonary forces are more evenly distributed by prone positioning (37, 38). Proning is a key intervention to improve V/Q matching and to avoid the regional application of tidal strains above the VILI threshold—even when total functional residual capacity (FRC) and overall BL size are not improved by the position change. Moderate PEEP may or may not complement these protective benefits. In theory, the value of prone positioning as a “VILI vortex” countermeasure would be greatest in the initial (most recruitable and perhaps most edematous) stages of ARDS (37, 38).
CLINICAL CONSIDERATIONS
Most experienced intensivists have encountered patients with initially localized infiltrates that proceed within hours to the radiographic “white out” that characterizes catastrophic ARDS. Lacking a clearly obvious explanation for the acceleration, such rapid deterioration is often attributed to the natural progression of ARDS. Undoubtedly, some of these adverse progressions are inescapable, as they result from unusually high vascular permeability, for example, H1N1 (39), the need for copious IV fluids to support systemic blood pressure and cardiac output, or combinations of such factors. Even so, we propose that in other cases, catastrophic decline that occurs despite protocol-guided care may be avoided by earlier intervention to preserve the size and capacity of the BL. Disturbingly, COVID-19 ARDS, a disease that attacks from the vascular (endothelial) side has prompted early-phase protocol-driven choices that encourage using higher PEEP to confront the problems of hypoxemia in what initially is a rather flexible lung predisposed to overstretch. Because vascular dysregulation appears to be the likely cause of hypoxemia during this early phase, that unwise choice to raise mean airway pressure and avoid lung unit collapse usually (but not invariably) improves arterial oxygenation, but itself may accentuate VILI and promote clinical deterioration. Later, when the underlying physiology transitions to resemble more familiar “stiff lung” ARDS, that same familiar approach may prove beneficial.
If our admittedly conceptual hypothesis of “BL-concentrated power→progressive VILI” were correct, it would be clearly desirable to quantify and serially track the size of the “BL” to help gauge risk of VILI and assess the pace of worsening and healing. Arguments continue as to which ventilatory choices (e.g., PEEP, Vt) are safest to apply to the diffusely injured lung (40); some thoughtful experts question whether any safe “lung protective” Vt exists for managing established ARDS and, therefore, strive to reduce Vt to a practicable minimum so as to attenuate further injury and promote recovery (24, 41). At the bedside, the effects of VILI are not separable from those resulting from the underlying process causing ARDS itself. Nonetheless, a progressive decline in BL volume beginning soon after a change in ventilation pattern would strongly suggest additional VILI superimposed by that intervention that at least temporarily overwhelms the opposing reactive or reparative responses outlined above. Conversely, for the same PEEP and body position, stabilization or steady reversal of deterioration over time would signal healing of the natural and/or iatrogenic injuries.
How best might BL size be measured and tracked in daily clinical practice? Although theoretically attractive, currently available imaging methods for determining and monitoring absolute lung volume require passive conditions and are either impractical (e.g., quantitative CT), inexact (e.g., electrical impedance tomography), or both (42). Direct measurements of FRC by gas dilution methodology have been incorporated into at least one commercially available ventilator system (43). Despite obvious appeal, such measurements currently offer limited precision and have not yet been validated as sufficiently accurate and reproducible for this specific purpose. Serial recordings of respiratory system (or preferably lung) compliance are universally available and theoretically justified for bedside monitoring of sequential changes in size of the BL when position, PEEP and (very importantly) specific compliance remain unchanged. Unfortunately, these compliance signals, too, are relatively insensitive, are influenced by PEEP and position, vary over time, and may not decline in linear proportion to the number of BL units, as intimated earlier (see Progressive Vulnerability and Disease in Evolution sections). Accurate tracking of dead space and dead-space fraction (Vd/Vt) is fraught with similar difficulties (44, 45). In addition, Vd/Vt measurements are influenced by spontaneous changes in metabolism and BL perfusion.
CONCLUSIONS
A Pragmatic Approach to Interrupting Progressive BL Shrinkage (the “VILI Vortex”)
In light of the innate vulnerability of the BL to a positive feedback cycle of power concentration and progressive VILI, clinicians must address the underlying cause of ARDS and maintain vigilance to avoid the potentially adverse consequences of their medical management (Table 1). Precise numerical values, mode choices, etc. depend on patient, disease type, and stage of illness, but key bedside “rules of thumb” are presented in Table 2. Important negative factors include inappropriately vigorous spontaneous breathing and inappropriate ventilation patterns (whether or not directed by “standard and approved” protocols), suboptimal supine body positioning, and liberal administration of unneeded fluids. For the moment, perhaps the most logical and practical approach to avoid progressing down the “VILI vortex” of the shrinking BL would appear to be to intervene as early as possible in that cycle and to minimize ventilatory demand and its associated mechanical power. In severe cases, quieting the efforts of the agitated and/or aggressively breathing patient, adopting modest permissive hypercapnia, prone positioning to reduce and balance transpulmonary forces, and extracorporeal Co2 removal are rational and widely available ventilatory support measures. At present, however, these are inconsistently applied until overt and ongoing deterioration becomes evident (28). By that point, however, the BL already may be launched well along its downward VILI spiral.
TABLE 1.
Encouraging and Preventing Shrinkage of the “Baby Lung”
TABLE 2.
Bedside Rules of Thumb to Avoid the Ventilator-Induced Lung Injury Vortex in Acute Respiratory Failure

Supplementary Material
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (http://journals.lww.com/ccmjournal).
The authors have disclosed that they do not have any potential conflicts of interest.
REFERENCES
- 1.Gattinoni L, Pesenti A. The concept of “baby lung.” Intensive Care Med 2005; 31:776–784 [DOI] [PubMed] [Google Scholar]
- 2.Gattinoni L, Marini JJ, Pesenti A, et al. The “baby lung” became an adult. Intensive Care Med 2016; 42:663–673 [DOI] [PubMed] [Google Scholar]
- 3.Dantzker DR, Brook CJ, Dehart P, et al. Ventilation-perfusion distributions in the adult respiratory distress syndrome. Am Rev Respir Dis 1979; 120:1039–1052 [DOI] [PubMed] [Google Scholar]
- 4.Marini JJ, Gattinoni L. Energetics and the root mechanical cause for ventilator-induced lung injury. Anesthesiology 2018; 128:1062–1064 [DOI] [PubMed] [Google Scholar]
- 5.Marini JJ, Rocco PRM, Gattinoni L. Static and dynamic contributors to VILI in clinical practice: Pressure, energy, and power. Am J Respir Crit Care Med 2020; 201:767–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spieth PM, Silva PL, Garcia CS, et al. Modulation of stress versus time product during mechanical ventilation influences inflammation as well as alveolar epithelial and endothelial response in rats. Anesthesiology 2015; 122:106–116 [DOI] [PubMed] [Google Scholar]
- 7.Hotchkiss JR, Jr, Blanch L, Naveira A, et al. Relative roles of vascular and airspace pressures in ventilator-induced lung injury. Crit Care Med 2001; 29:1593–1598 [DOI] [PubMed] [Google Scholar]
- 8.Villar J, Zhang H, Slutsky AS. Lung repair and regeneration in ARDS: Role of PECAM1 and wnt signaling. Chest 2019; 155:587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nuckton TJ, Alonso JA, Kallet RH, et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med 2002; 346:1281–1286 [DOI] [PubMed] [Google Scholar]
- 10.Morales-Quinteros L, Schultz MJ, Bringué J, et al. ; MARS Consortium: Estimated dead space fraction and the ventilatory ratio are associated with mortality in early ARDS. Ann Intensive Care 2019; 9:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robertson HT. Dead space: The physiology of wasted ventilation. Eur Respir J 2015; 45:1704–1716 [DOI] [PubMed] [Google Scholar]
- 12.Albert RK. The role of ventilation-induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am J Respir Crit Care Med 2012; 185:702–708 [DOI] [PubMed] [Google Scholar]
- 13.Haddad IY, Holm BA, Hlavati L, et al. Dependence of surfactant function on extracellular pH: Mechanisms and modifications. J Appl Physiol (1985) 1994; 76:657–662 [DOI] [PubMed] [Google Scholar]
- 14.Bersten AD, Krupa M, Griggs K, et al. Reduced surfactant contributes to increased lung stiffness induced by rapid inspiratory flow. Lung 2020; 198:43–52 [DOI] [PubMed] [Google Scholar]
- 15.Petrini MF, Robertson HT, Hlastala MP. Interaction of series and parallel dead space in the lung. Respir Physiol 1983; 54:121–136 [DOI] [PubMed] [Google Scholar]
- 16.Protti A, Cressoni M, Santini A, et al. Lung stress and strain during mechanical ventilation: Any safe threshold? Am J Respir Crit Care Med 2011; 183:1354–1362 [DOI] [PubMed] [Google Scholar]
- 17.Tomashefski JF., Jr Pulmonary pathology of the adult respiratory distress syndrome. Clin Chest Med 1990; 11:593–619 [PubMed] [Google Scholar]
- 18.Garcia CS, Abreu SC, Soares RM, et al. Pulmonary morphofunctional effects of mechanical ventilation with high inspiratory air flow. Crit Care Med 2008; 36:232–239 [DOI] [PubMed] [Google Scholar]
- 19.Maeda Y, Fujino Y, Uchiyama A, et al. Effects of peak inspiratory flow on development of ventilator-induced lung injury in rabbits. Anesthesiology 2004; 101:722–728 [DOI] [PubMed] [Google Scholar]
- 20.Protti A, Maraffi T, Milesi M, et al. Role of strain rate in the pathogenesis of ventilator-induced lung edema. Crit Care Med 2016; 44:e838–e845 [DOI] [PubMed] [Google Scholar]
- 21.Laffey JG, Kavanagh BP. Fifty years of research in ARDS. Insight into acute respiratory distress dyndrome. From models to patients. Am J Respir Crit Care Med 2017; 196:18–28 [DOI] [PubMed] [Google Scholar]
- 22.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med 2013; 369:2126–2136 [DOI] [PubMed] [Google Scholar]
- 23.Sinclair SE, Altemeier WA, Matute-Bello G, et al. Augmented lung injury due to interaction between hyperoxia and mechanical ventilation. Crit Care Med 2004; 32:2496–2501 [DOI] [PubMed] [Google Scholar]
- 24.Terragni PP, Del Sorbo L, Mascia L, et al. Tidal volume lower than 6 ml/kg enhances lung protection: Role of extracorporeal carbon dioxide removal. Anesthesiology 2009; 111:826–835 [DOI] [PubMed] [Google Scholar]
- 25.Mead J, Takishima T, Leith D. Stress distribution in lungs: A model of pulmonary elasticity. J Appl Physiol 1970; 28:596–608 [DOI] [PubMed] [Google Scholar]
- 26.Cressoni M, Chiurazzi C, Gotti M, et al. Lung inhomogeneities and time course of ventilator-induced mechanical injuries. Anesthesiology 2015; 123:618–627 [DOI] [PubMed] [Google Scholar]
- 27.Faffe DS, Zin WA. Lung parenchymal mechanics in health and disease. Physiol Rev 2009; 89:759–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellani G, Laffey JG, Pham T, et al. ; LUNG SAFE Investigators; ESICM Trials Group: Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016; 315:788–800 [DOI] [PubMed] [Google Scholar]
- 29.Cressoni M, Gotti M, Chiurazzi C, et al. Mechanical power and development of ventilator-induced lung injury. Anesthesiology 2016; 124:1100–1108 [DOI] [PubMed] [Google Scholar]
- 30.Marini JJ, Jaber S. Dynamic predictors of VILI risk: Beyond the driving pressure. Intensive Care Med 2016; 42:1597–1600 [DOI] [PubMed] [Google Scholar]
- 31.Rylander C, Högman M, Perchiazzi G, et al. Functional residual capacity and respiratory mechanics as indicators of aeration and collapse in experimental lung injury. Anesth Analg 2004; 98:782–789, table of contents [DOI] [PubMed] [Google Scholar]
- 32.Sibbald WJ, Anderson RR, Holliday RL. Pathogenesis of pulmonary edema associated with the adult respiratory distress syndrome. Can Med Assoc J 1979; 120:445–450 [PMC free article] [PubMed] [Google Scholar]
- 33.Lamm WJ, Luchtel D, Albert RK. Sites of leakage in three models of acute lung injury. J Appl Physiol (1985) 1988; 64:1079–1083 [DOI] [PubMed] [Google Scholar]
- 34.Albert RK, Lakshminarayan S, Charan NB, et al. Extra-alveolar vessel contribution to hydrostatic pulmonary edema in in situ dog lungs. J Appl Physiol Respir Environ Exerc Physiol 1983; 54:1010–1017 [DOI] [PubMed] [Google Scholar]
- 35.Marini JJ, Gattinoni L. Propagation prevention: A complementary mechanism for “lung protective” ventilation in acute respiratory distress syndrome. Crit Care Med 2008; 36:3252–3258 [DOI] [PubMed] [Google Scholar]
- 36.Paré PD, Warriner B, Baile EM, et al. Redistribution of pulmonary extravascular water with positive end-expiratory pressure in canine pulmonary edema. Am Rev Respir Dis 1983; 127:590–593 [DOI] [PubMed] [Google Scholar]
- 37.Guérin C, Reignier J, Richard JC, et al. ; PROSEVA Study Group: Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013; 368:2159–2168 [DOI] [PubMed] [Google Scholar]
- 38.Gattinoni L, Taccone P, Carlesso E, et al. Prone position in acute respiratory distress syndrome. Rationale, indications, and limits. Am J Respir Crit Care Med 2013; 188:1286–1293 [DOI] [PubMed] [Google Scholar]
- 39.Rohani P, Jude CM, Chan K, et al. Chest radiological findings of patients with severe H1N1 pneumonia requiring intensive care. J Intensive Care Med 2016; 31:51–60 [DOI] [PubMed] [Google Scholar]
- 40.Walkey AJ, Goligher EC, Del Sorbo L, et al. Low tidal volume versus non-volume-limited strategies for patients with acute respiratory distress syndrome. A systematic review and meta-analysis. Ann Am Thorac Soc 2017; 14:S271–S279 [DOI] [PubMed] [Google Scholar]
- 41.Bein T, Weber-Carstens S, Goldmann A, et al. Lower tidal volume strategy (≈3 ml/kg) combined with extracorporeal CO2 removal versus ‘conventional’ protective ventilation (6 ml/kg) in severe ARDS: The prospective randomized Xtravent-study. Intensive Care Med 2013; 39:847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pesenti A, Musch G, Lichtenstein D, et al. Imaging in acute respiratory distress syndrome. Intensive Care Med 2016; 42:686–698 [DOI] [PubMed] [Google Scholar]
- 43.Graf J, Santos A, Dries D, et al. Agreement between functional residual capacity estimated via automated gas dilution versus via computed tomography in a pleural effusion model. Respir Care 2010; 55:1464–1468 [PubMed] [Google Scholar]
- 44.Ferluga M, Lucangelo U, Blanch L. Dead space in acute respiratory distress syndrome. Ann Transl Med 2018; 6:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beydon L, Uttman L, Rawal R, et al. Effects of positive end-expiratory pressure on dead space and its partitions in acute lung injury. Intensive Care Med 2002; 28:1239–1245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.