

Summary
Age is a non-modifiable risk factor for the inflammation that underlies age-associated diseases; thus, anti-inflammaging drugs hold promise for increasing health span. Cytokine profiling and bioinformatic analyses showed that Th17 cytokine production differentiates CD4+ T cells from lean, normoglycemic older and younger subjects, and mimics a diabetes-associated Th17 profile. T cells from older compared to younger subjects also had defects in autophagy and mitochondrial bioenergetics that associate with redox imbalance. Metformin ameliorated the Th17 inflammaging profile by increasing autophagy and improving mitochondrial bioenergetics. By contrast, autophagy-targeting siRNA disrupted redox balance in T cells from young subjects and activated the Th17 profile by activating the Th17 master regulator, STAT3, which in turn bound IL-17A and F promoters. Mitophagy-targeting siRNA failed to activate the Th17 profile. We conclude that metformin improves autophagy and mitochondrial function largely in parallel to ameliorate a newly defined inflammaging profile that echoes inflammation in diabetes.
Keywords: inflammaging, T cells, metformin, autophagy, mitochondria
Graphical Abstract
Highlights
-
•
CD4+ T cells from healthy older people preferentially produce a Th17 profile
-
•
Autophagy, but not mitophagy, knockdown activates a Th17 profile in “young” cells
-
•
Mitochondrial ROS is needed, but not sufficient, for a Th17 profile in “young” cells
-
•
Metformin improves autophagy and mitochondria in parallel to decrease inflammaging
We uncovered a dominant Th17 inflammaging profile made by CD4+ T cells. Knockdown of autophagy in T cells from young subjects activates this profile. In vitro metformin improves autophagy and mitochondrial function in parallel to ameliorate Th17 inflammaging. Oral metformin intervention improves T cell autophagy in people, indicating potential use for age-associated inflammation.
Context and Significance
Inflammation increases naturally with age and contributes to many diseases that limit the amount of one’s life spent in good health, including type 2 diabetes, dementias, and many cancers. Researchers at the University of Kentucky and their colleagues identified a source of age-related inflammation that, if targeted by appropriate medications, holds great promise for promoting healthy aging. They manipulated immune cells from 30-year-old people to mimic inflammation in cells from 60-year-old people. This method identified specific breakdowns in the cellular machinery that cause age-related inflammation. The type 2 diabetes drug metformin repaired the broken machinery in cells treated outside the body to drastically lower inflammation, paving the way for clinical trials to test whether metformin lowers age-related inflammation to promote healthy aging.
Introduction
Aging-associated inflammation, or inflammaging, plays roles in increased risk of insulin resistance, type 2 diabetes (T2D), and cardiovascular diseases with age. Inflammaging thereby limits the length of one’s lifespan spent in good health (Mueller and Rose, 1996, Franceschi and Campisi, 2014, Franceschi et al., 2017, Bharath et al., 2017b). Aging-associated inflammation has been defined based on cytokines, such as TNFα and IL-6 (Bruunsgaard et al., 2000, Roubenoff et al., 1998, Ferrucci et al., 2005, Piber et al., 2019), without consideration of the relative importance of the many sources of what is collectively labeled “inflammation.” T cells are a major source of inflammatory cytokines in settings characterized by chronic low-level inflammation, and multiple lines of evidence showed that one particular T cell subset, Th17s, characterizes and mathematically predicts T2D (Ip et al., 2016). Th17s also promote arguably the most prevelant inflammatory disease in the world, periodontal disease, which fuels cardiovascular and other more deadly diseases (Abusleme and Moutsopoulos, 2017). Similarly defining age-associated T cell inflammation will significantly enhance our current appreciation of inflammaging toward the goal of improving health span.
Multiple mediators of cell maintenance are known to decline in aging, raising the possibility that age-associated changes in processes, such as macroautophagy, herein “autophagy,” and mitochondrial bioenergetics (Sun et al., 2016) both parallel and promote inflammaging. Autophagy has multiple immunomodulatory effects, including broad coordination of general immune cell responses, as evidenced by the importance of autophagy in development and function of anti-inflammatory regulatory T cells (Tregs) (Wei et al., 2016, Le Texier et al., 2016). Autophagy controls immune cell function in part by regulating mitochondrial bioenergetics, as shown by demonstrations that CD4+ T cell autophagy negatively regulates glucose metabolism in Tregs (Kabat et al., 2016). Given that ATP generation through mitochondrial oxidative phosphorylation (OXPHOS) versus non-mitochondrial glycolysis (deemed “glycolysis” herein) can determine T cell function (Kominsky et al., 2010, Priyadharshini et al., 2018), these findings suggest that autophagy enhancement may alter the mitochondrial response of T cells to stimulation, and thereby ameliorate inflammaging to prolong health span. Arguably, the top candidate drug for activating autophagy over the immediate term is metformin, a well-tolerated T2D drug that improves glycemic control and, in some studies, chronic inflammation (Cameron et al., 2016, Malínská et al., 2016). Putative effects of metformin on age-associated T cell inflammation justify targeted pre-clinical work to identify metformin-sensitive mechanisms that ameliorate a more sophisticated profile of inflammaging.
Data herein show that a combinatorial Th17 cytokine profile differentiated CD4+ T cell inflammation in healthy sexagenarians compared to ∼30-year-old subjects. Physiologically achievable concentrations of metformin lowered overall T cell cytokine production ex vivo in samples from all subjects, but the Th17 profile was disproportionately susceptible in samples from older (O) subjects. By contrast, metformin reduced a Th2 profile in cells from younger (Y) subjects. Metformin increased autophagy in CD4+ T cells from older subjects and shifted measures of mitochondrial bioenergetics and T cell inflammation to values indistinguishable from young subjects’ cells. siRNA-mediated impairment of autophagy, but not mitophagy, in cells from younger subjects compromised mitochondrial function and activated a Th17 profile indistinguishable from T cell profiles produced by cells from older subjects. We conclude metformin-sensitive defects in immune cell autophagy (1) accompany natural aging in people, (2) shift mitochondrial bioenergetics, and (3) fuel a previously unappreciated Th17 inflammaging profile. Our findings highlight cause-and-effect relationships among defects in non-mitochondrial autophagy, mitochondrial function, and inflammaging to justify clinical trials to extend health span with metformin.
Results
A Th17 Profile Dominates CD4+ T Cell Function from Older Subjects through a Metformin-Sensitive Mechanism
To define an age-associated T cell cytokine profile, we quantified cytokines produced by αCD3/αCD28-stimulated CD4+ T cells from O and Y subjects (Table S1) by bioplex. O cells produced higher amounts of most classically defined Th17-associated/supportive cytokines (IL-6, IL-17A, IL-17F, IL-21, and IL-23) but similar amounts of cytokines typically produced by other CD4+ T cell subsets (Figures 1A and S1A–S1D). Age-associated shifts in CD4+ T cell subset distribution in our cohort was as previously published (Figures 1B and S1E), including CD57+ terminal effectors that were almost unique to samples from O subjects and fewer central memory T cells in Y samples. These results were consistent with recent work showing that Th17 frequency does not increase with age (Alpert et al., 2019). Age-associated changes in CD8+ T cell subsets were also as expected (Figure S1F). Partial least squares discriminant analysis (PLSDA) models, which combine all cytokines from one sample into a compendium multi-dimensional value for “inflammation,” showed that cytokine production differentiates O and Y samples (Figure 1C). Variable importance projection (VIP) calculations, which rank cytokines based on their overall importance for separating cytokine data clouds, showed almost all Th17 cytokines were disproportionately important for identifying higher overall inflammation produced by O-derived CD4+ T cells (VIP > 1.0, bracket in Figure 1D, red bars highlight classical Th17 cytokines). We conclude that a comprehensive Th17 profile defines and mathematically predicts age-related T cell inflammation.
Figure 1.

Metformin Ameliorates an Age-Related Th17 Cytokine Profile
Cytokine production was assessed in T cells from BMI-matched normoglycemic Y and O subjects following 40 h αCD3/αCD28 stimulation ± 100 μm metformin (MET).
(A) Concentrations of IL-17A, IL-17F, IL-21, and IL-6 as indicated. Data are mean ± SEM. n = 10–14. For all panels, each n (i.e., each dot) represents T cells isolated from one subject. ∗p < 0.05 versus Y, #p < 0.05 versus O by ANOVA.
(B) Left: tSNE grouping of CD4+ T cell subsets based on markers shown in Figure S1E identified 5 subsets. Right: two representative analyses from subjects in age groups as indicated. Table shows frequencies (average and SD) of CD4+ T cell subsets in samples from Y or O subjects. ∗p < 0.05 by two-tailed t test.
(C, E, and G) PLSDA shows compendium measures of “inflammation” generated by combining all cytokines measured by (C) Y (blue) or O (green) CD4+ T cells, (E) CD4+ cells from O subjects stimulated in the presence (orange) or absence (green) of metformin (100 μM), or (G) CD4+ cells from Y subjects stimulated in the presence (purple) or absence (blue) of metformin (100 μM).
(D, F, and H) Bar graphs show VIP scores, which rank cytokines as most (leftmost) or least (rightmost) important for differentiating overall cytokine profiles between the groups indicated in key. A VIP score >1 (bracket) is considered important for differentiating inflammatory profiles between groups. All VIP cytokines indicated also differed in post hoc analyses (p < 0.05). n = 10–14.
See also Figure S1.
The glycemic control drug metformin variably impacts inflammation and inflammatory comorbidities like T2D in part through undefined age-associated mechanisms (Chakraborty et al., 2011, Smith et al., 2010, Fidan et al., 2011). We tested the effect of physiologically achievable metformin (100 μM) (Madiraju et al., 2019) added coincidence with T cell-targeted stimuli on the newly defined age-related inflammation profile. Metformin specifically decreased production of Th17 cytokines but failed to decrease most Th2 cytokines (IL-4, IL-5, and IL-10) by O cells (O + met), as indicated by single cytokine (Figures 1A and S1A–S1D) or PLSDA (Figures 1E and 1F) analysis. In contrast, single cytokine analyses showed metformin did not change cytokine production by Y-derived T cells (Y + met; Figures 1A and S1A–S1D), while PLSDA showed metformin ameliorated a Th2/type 2 immune profile produced by Y cells (IL-13, IL-33, IL-31, IL-10, and IL-5; Figures 1G and 1H). Cytokine profiles from O + met cultures were indistinguishable from profile produced in Y cultures as indicated by statistically similar profiles (p > 0.05), no VIP cytokines with values >1.0, and a non-predictive value in “leave-one-out” analysis of p = 0.15 (data not shown). We conclude that metformin restores age-related T cell inflammation to profiles generated by Y cells.
Mitochondria Dysfunction in CD4+ T Cells Is Regulated by an Age-Related, Metformin-Sensitive Mechanism
To identify metformin-sensitive mechanisms that control Th17 inflammaging, we quantified indicators of mitochondrial function that promote pro-inflammatory T cells (De Rosa et al., 2015, Hong et al., 2013, Bharath et al., 2017a) using a mito stress test in extracellular flux (XF, Seahorse). αCD3/αCD28-stimulated CD4+ T cells from O subjects had higher OXPHOS (OXPHOS/oxygen consumption rate [OCR]; baseline and maximal), extracellular acidification rate ratio (OCR:ECAR), and proton leak. Spare respiratory capacity was similar between O and Y cells. CD4+ T cells from O compared Y subjects produced less lactate and had lower ECAR (Figures 2A–2D and S2A–S2D). Mitochondrial membrane potential (MMP) was lower in O cells, as measured by tetramethylrhodamine, ethyl ester (TMRE) (Figure 2E), perhaps in part due to intrinsically lower membrane potential differences in O compared with Y cells (Figure 2F). Addition of metformin (100 μM) concomitant with stimulation decreased basal and maximal OCR, OCR:ECAR ratio, and proton leak of CD4+ T cells from O subjects (Figures 2A, 2B, 2D, S2A, and S2B). Metformin increased lactate production and ECAR (Figures 2C and S2C) and supported a trend toward increase in MMP in CD4+ T cells from O subjects (Figure 2E; p = 0.055). Metformin action on mitochondrial bioenergetics was independent of AMPK, as indicated by similar outcomes from cells treated with AMPK-specific or scrambled siRNA prior to stimulation and extracellular flux (XF) analysis (Figures S2E and S2F). Metformin had no effect on the mitochondrial function of T cells from Y subjects (Figures 2A–2E and S2A–S2D). We conclude that higher OXPHOS corresponds with lower glycolysis and Th17 inflammation in T cells from O compared with Y subjects, and that metformin shifts O cells to recapitulate characteristics of Y cells.
Figure 2.

Metformin Ameliorates OXPHOS and Promotes Non-mitochondrial Glycolysis in CD4+ T Cells from O Subjects
(A) OCR in a mito stress test assayed by XF of CD4+ T cells following 40 h αCD3/αCD28 stimulation ± 100 μm MET as indicated.
(B) OCR:ECAR ratio calculated by profiles in (A) and Figure S2C.
(C) Relative lactate production after 40-h stimulation per (A).
(D) Proton leak calculated from (A) data.
(E) MMP measured with TMRE after stimulation per (A). #p = 0.055 versus O.
(F) MMP measured following addition of the mitochondrial uncoupler fluoro-carbonyl cyanide phenylhydrazone (FCCP) to unstimulated CD4+ T cells from Y or O subjects.
n = 8–10 (A–E) and 12–13 (F).
(G) LDH quantification on western blots. Top: representative blot and bottom averages n = 4–6 of group indicated beneath. ∗p < 0.05 versus Y, #p < 0.05 versus O. Data shown are mean ± SEM.
(H) VIP scores, which rank cytokines as most (leftmost) or least (rightmost) important for differentiating overall cytokine profiles between CD4+ T cells from young subjects stimulated ± the LDH inhibitor OA in an orthagonalized model. A VIP score >1 (bracket) is considered important for differentiating inflammatory profiles between groups. All VIP cytokines indicated also differed in post hoc analyses (p < 0.05). Fold change is compared to Y or Y + FCCP.
See also Figure S2.
Although inflammation is traditionally fueled by glycolysis, our data showing association between mitochondrial respiration and inflammation in CD4+ T cells from O subjects raise the possibility that O cells ineffectively shift to glycolysis to fuel inflammation. To begin testing this possibility, we quantified protein levels of glycolytic pathway enzymes. CD4+ T cells from O subjects had lower expression of lactate dehydrogenase A (LDH), which catalyzes pyruvate ↔ lactate (Figure 2G), providing a mechanistic explanation for low lactate in O cells (Figure 2C). Expression of enzymes that regulate pyruvate production from glucose, including hexokinase and pyruvate kinase M2 (PKM2), was higher or equivalent, respectively, in CD4+ T cells from O compared with Y subjects (Figures S2G and S2H), suggesting age did not change pyruvate production to limit lactate. In contrast to lower glycolysis, pyruvate hydrolysis through the citric acid cycle was not likely compromised by age-dependent changes in citric acid cycle enzymes, as suggested by protein levels of isocitrate dehydrogenase (IDH2) and oxoglutarate dehydrogenase (OGDH) (Figures S2I and S2J). NADH:ubiquinone oxidoreductase (mitochondrial respiratory complex 1) was quantitatively equal in CD4+ T cells from O and Y subjects, although western blots suggested an age-related post-translational modification (Figure S2K). Metformin increased protein levels of LDH (Figure 2G) and OGDH (Figure S2J) in CD4+ T cells from O subjects but had no effect on the other enzymes measured. We conclude age-related decreases in LDH, which are sensitive to metformin, mechanistically explain lower glycolysis and may thereby promote compensatory OXPHOS in CD4+ T cells from O subjects.
To test causal relationships between low LDH expression in O cells and the Th17 inflammaging profile, we pharmacologically inhibited LDH activity in CD4+ T cells from Y subjects with 20 mM oxamic acid (OA; Figure S2L). Some Th17-associated cytokines, including IL-21 and the Th17 supporters IL-6 and IL-23, were activated by OA (Figure S2M). However, PLSDA showed that the majority of Th17 signature cytokines (IL-17A, IL-17F IL-6, and IL-21; Figure 1F) were not important for distinguishing compendium cytokine profiles from LDH inhibitor-treated Y cells. Exceptions were IL-22 and IL-23, highlighted as important by this method (Figure 2H). We conclude that changes in glycolytic machinery do not play critical roles in Th17 inflammaging.
Mitochondrial Dysfunction Disrupts Redox Balance to Support Th17 Cytokine Production by CD4+ T Cells from O Subjects and Is Corrected by Metformin
Higher OXPHOS in the absence of parallel increases in anti-oxidants can generate excess oxidative stress, as measured by reactive oxygen species (ROS), which in turn can support Th17 number and function (Zhi et al., 2012, Ungvari et al., 2009, Murphy, 2009, Liu et al., 2002). CD4+ T cells from O subjects had more ROS than Y counterparts, as measured by DCFDA (Figure 3 A), and consistent with higher ATP-linked respiration (Figure 3B). Lower glutathione (GSH) and more nicotinamide nucleotide transhydrogenase (NNT) in O compared with Y cells indicated that less antioxidant, perhaps in response to “reverse” NNT function (Nickel et al., 2015), contributed to higher oxidative stress (Figures 3C and 3D). Lower expression of mitochondrial manganese superoxide dismutase (MnSOD/SOD2) in T cells from O subjects (Figure 3E) was also consistent with higher ROS, despite age-independent expression of the anti-oxidants SOD1 and PRDX2 (Figures S3A and S3B), all of which associated with lower MMP (Figure 3F). Metformin increased the expression of GSH and SOD1 (Figures 3C and S3A), decreased ROS and NNT (Figures 3A and 3D), and showed a trend toward increased MnSOD (Figure 3E; p = 0.062) in cells from O subjects.
Figure 3.

ROS Amelioration Prevents Th17 Profile Production by CD4+ T Cells from O Subjects
(A–E) ROS production (A), ATP-linked respiration (B), GSH (C), NNT (D), or MnSOD expression (E) by CD4+ cells stimulated for 40 h with αCD3/αCD28 ± 100 μm MET. #p = 0.062 versus O.
(F and G) Outcomes following stimulation the ROS scavenger Tempol ± 100 μM MET as indicated.
(H) Production of Th17 cytokines by CD4+ cells stimulated for 40 h with αCD3/αCD28 ± tempol.
n = 7–10 (A–D, F, and G), 4–10 (H), and 4–7 (E); ∗p < 0.05 versus Y, #p < 0.05 versus O. Data are represented as mean ± SEM. Fold change is compared with Y. See also Figure S3.
To explore the possibility that metformin regulates the Th17 inflammaging profile through effects on ROS, we tested the ability of the ROS-specific scavenger Tempol to recapitulate metformin effects on CD4+ T cells from O subjects. Tempol reduced ROS and trended toward increased MMP (TMRE, p = 0.058) in CD4+ T cells from O subjects (Figures 3F and 3G), but perhaps more importantly, Tempol uniformly decreased Th17 profile cytokines in O cells with no effect on Y cells, though effects on other cytokines somewhat differed from effects of metformin (Figures 3H and S3C–S3F). Tempol and metformin together increased TMRE signal more than either alone in O cells (Figure 3F), further indicating overlapping but non-identical effects of ROS scavenging and metformin, but consistent with previous demonstrations that ROS dissipation is a mechanism of metformin action (Madiraju et al., 2019). We conclude mitochondrial OXPHOS in O cells coincides with lower antioxidant to cause oxidative stress and a ROS-downstream Th17 profile. Metformin-sensitive pathways that are partially redundant with mitochondrial ROS-scavenging re-establish redox balance to ameliorate T cell inflammaging.
Autophagy Defects in CD4+ T Cells from O Subjects Are Corrected by Metformin
Accumulation of defective mitochondria stemming from general age-related declines in autophagy may, in part, explain excessive OXPHOS and thus redox imbalance in CD4+ T cells from O subjects. We quantified mitochondrial accumulation in CD4+ T cells with Mitotracker green fluorescence and flow cytometry. Cells from O subjects had more mitochondrial mass and mitochondrial matrix proteins, such as m-aconitase, consistent with mitochondrial accumulation. Metformin decreased mitochondrial accumulation in O, but not Y cells (Figures 4A–4D). Metformin action was redundant with Tempol-mediated decrease in mitochondrial mass (Figure 4D), raising the possibility that metformin corrects age-related changes in autophagy that impact redox balance that in turn drives T cell inflammaging.
Figure 4.

Metformin Promotes Mitochondrial Turnover and Mitophagy in CD4+ T Cells from O Subjects
(A and B) MitoTracker green fluorescence in CD4+ T cells from O and Y subjects assessed by flow cytometry. n = 6.
(C) Mitochondrial matrix protein m-aconitase in CD4+ T cells from O and Y subjects as measured on western blots. n = 7–10.
(D) Mitochondrial mass assessed via Mitotracker green fluorescence in the presence of Tempol (TEMP) ± MET as indicated. n = 7–8.
(E and F) Expression of the autophagy proteins LC3II (E) or p62 (F) in cells, measured on western blots. n = 8–10.
(G and H) Autophagosome formation as indicated by puncta and quantitated by confocal microscopy. 3-MA is an inhibitor or autophagy thus serves as a negative control. n = 3.
(I) LC3II expression in CD4+ T cells as indicated, following treatment with metformin + BAF A1 as a positive control for autophagy. n = 5.
(J and K) Localization in representative CD4+ cells (J), and quantitation of co-localization of the mitochondrial protein TOM20 and the lysosomal protein (LAMP1) ± metformin as indicated (K). n = 4 with multiple dots from some N’s shown.
For both confocal analyses (G, H, J, and K), 3 cells/field and 3 fields/slide were imaged using 63× oil immersion in Zeiss microscope. The average fluorescence/field is reported.
(L) Expression of mitochondrial fission protein Drp 1 on western blots. n = 5.
(M–O) Indicators of autophagy (M) LC3II, (N) m-aconitase, or (O) GRP78 quantified in CD4+ T cells from pre-diabetes subjects sampled before or after 3 months’ administration of metformin (1,000 mg/day; n = 4). ∗p < 0.05 versus Y or pre-met, #p < 0.05 versus O.
Data are represented as mean ± SEM. Fold change is compared to either Y, Y + MET, or PRE-MET.
To more broadly test the possibility that age-related autophagy defects impact CD4+ T cells and thereby age-related inflammation, we quantified autophagy indicators in CD4+ T cells from O subjects. O cells had less robust autophagy than Y counterparts, as shown by lower LC3II, p62 accumulation, and fewer LC3II-labeled puncta. Metformin improved all measures of autophagy in O cells (Figures 4E–4H). More LC3II in cells treated with metformin + bafilomycin A1 (BAF A1, autophagy inhibitor) indicated that metformin enhanced autophagosome flux rather than stalled cargo degradation (Figure 4I). In addition, O cells had less co-localization of the mitochondrial protein translocase of outer membrane 20 protein (TOM20) and the lysosomal protein lysosome associated membrane protein 1 (LAMP1) than Y cells had, definitively confirming defective mitochondrial turnover was restored by metformin (Figures 4J and 4K). Metformin similarly increased expression of the mitochondrial fission protein dynamin related protein 1 (Drp1), a mitochondrial indicator of improved health span in Drosophila (Rana et al., 2017) in cells from O subjects (Figure 4L). Metformin did not affect general indicators of autophagy, nor specific mitophagy indicators in Y cells (Figures 4E–4H and 4J–4L), and also failed to impact age-related decreases in humanin and prohibitin (Figures S4A and S4B), two mitokines that regulate inflammation in some circumstances (Conte et al., 2019, Zapała et al., 2010, Kathiria et al., 2012). We conclude that mitochondrial turnover is defective in CD4+ T cells from older people, and that this defect, but not age-related declines in mitokines, is neutralized by metformin.
To test the clinical significance of our in vitro demonstration that metformin improves CD4+ T cell autophagy, we collected cells from obese, pre-diabetes subjects (Table S2) before and 3 months after clinically indicated metformin (1,000 mg/day). Metformin activated autophagy in purified CD4+ T cells from these subjects, after cells were stimulated (without additional metformin) ex vivo, as indicated by more LC3II and multiple indicators of organelle clearance (less m-aconitase and GRP78, indicating mitochondrial and endoplasmic reticulum [ER] clearance; Figures 4M–4O). These data indicate that metformin intervention in older subjects is likely to improve autophagy and autophagy-downstream effects of aging on CD4+ T cells.
To begin testing the possibility that metformin-sensitive defects in autophagy, alone or in combination with fundamental changes in mitochondria, fuel Th17 inflammaging, we activated CD4+ T cells from Y subjects in the presence of the autophagy activator trehalose, or the fatty acid oxidation inhibitor trimetazidine (to induce partial mitochondrial dysfunction), alone or in combination. Trimetazidine increased production of the Th17 cytokines IL-17A/F, IL-21, and IL-23 as previously reported in peripheral blood mononuclear cells (PBMCs) (Nicholas et al., 2019), although, in contrast to PBMCs, trimetazidine also activated Th1 cytokine production by CD4+ T cells (Figures 5A and S4C–S4E). Trehalose did not affect cytokine production, as expected from the inability of metformin or other manipulations to alter Y cells. However, trehalose partially blocked trimetazidine-activated cytokine production (Figures 5A and S4C–S4E), supporting the conclusion that autophagy improvement cannot entirely prevent inflammatory cytokine production by cells with compromised mitochondrial function.
Figure 5.

Genetic Inhibition of Autophagy Recapitulates Respiratory Profiles of Cells from O Subjects
(A) Th17-associated cytokine production by CD4+ T cells from Y subjects, with cells stimulated 40 h αCD3/αCD28 in the presence of trimetazidine, a fatty acid oxidation inhibitor; alone; or in combination with trehalose, an autophagy activator. n = 4. ∗p < 0.05 versus Y by one-way ANOVA.
(B) Mito stress test XF profiles from 40 h αCD3/αCD28-stimulated CD4+ T cells from Y or O subjects as indicated. Autophagy dysfunction was induced in cells from Y subjects using siRNA-mediated ATG3 knockdown. n = 8–12.
(C and D) OCR:ECAR ratio (C) and ROS generation (D) measured by DCFDA in CD4+ T cells manipulated as indicated. n = 8–12.
∗p < 0.05 versus Y by SHORE (Nicholas et al., 2017) (B) or one-way ANOVA (C and D). ∗p < 0.05 versus Y (basal), $p < 0.05 versus Y (max) (C).
(E and G) PLSDA analysis differentiated combinatorial “inflammation” of CD4+ cells from Y subjects (blue), Y with siRNA-induced autophagy dysfunction (purple), or autophagy dysfunction and metformin (met; orange). n = 8–9.
(F and H) VIP scores rank cytokines important for differentiating data clouds in (E) and (G). Comparison of Figure 5F with 1D highlights profiles that differentiate Y from either O or Y + ATG3 siRNA conditions. n = 8–9.
(I) Mitochondrial ROS generation measured by MitoSOX in CD4+ T cells manipulated as indicated; 3 cells/field and 4 fields/slide were imaged using 40× in Zeiss microscope. The average fluorescence/field is reported. n = 4–5.
(J) Cytokine production by CD4+ T cells from young subjects, with cells stimulated 40 h αCD3/αCD28 after autophagy inhibition and in the presence of mitoTEMPO, a mitochondrial ROS-specific scavenger (1 μM, added 3 h post αCD3/αCD28 stimulation). n = 5–6.
(A, C, D, I, and J) Data show mean ± SEM. Fold change is compared with Y.
See also Figures S4 and S5.
Genetic Disruption of Autophagy in T Cells from Y Subjects Recapitulates Traits of T Cells from O Subjects and Prevents Metformin-Mediated Repair
To more definitively test the possibility that metformin-mediated autophagy ameliorates age-related Th17 inflammation, we knocked down autophagy in CD4+ T cells from Y subjects using siRNA specific for autophagy related protein 3 (Atg3; Figure S5A). Atg3 siRNA, but not scrambled siRNA, decreased LC3II and increased m-aconitase (Figures S5B and S5C), indicating compromised autophagy. Atg3 knockdown in CD4+ T cells from Y subjects increased basal OCR, maximal OCR, OCR:ECAR ratio, and ROS to values indistinguishable from those of CD4+ T cells from O subjects (Figures 5B–5D). Furthermore, Atg3 knockdown in CD4+ T cells from Y subjects resulted in higher Th17 cytokine production (IL-17A, IL-17F, and IL-21) that was indistinguishable from Th17 cytokines produced by O cells (Figure S5D). CCL-20 and GMCSF, two cytokines produced by both Th17 and Th1 cells, increased with Atg3 knockdown (Figure S5D). Other cytokines generally produced by other CD4+ T cell subsets variably changed with knockdown (Figure S5E). PLSDA analysis showed that Atg3 siRNA treatment of CD4+ T cells from Y subjects induced VIP cytokines that were very similar to the Th17 profile produced by cells from O subjects, and that the Th17 profile induced by siAtg3 was minimally changed by metformin (Figures 5E–5H; compare Figure 5F with Figure 1D).
Autophagy blockade with Atg3-knockdown increased mitochondrial ROS in CD4+ cells from Y subjects (Figure 5I). Given that ROS mitigation is a major action of metformin, we tested the impact of MitoTempo, a mitochondrial ROS-specific scavenger, on mitochondrial ROS in Atg3-knockdown cells. MitoTempo added 3 h after αCD3/αCD28 stimulation decreased mitochondrial ROS of Atg3-knockdown cells to baseline levels (Figure 5I) and had no effect on activation of Th17-associated cytokines in the absence of autophagy (Figure 5J). Addition of MitoTempo concurrent with stimulation resulted in cell death; thus, we cannot eliminate the possibility that an initial burst of mitochondrial ROS initiates measurable production of Th17 cytokines 40 h later. We conclude that mitochondrial ROS does not drive the Th17 signature in cells with compromised autophagy.
Taken together, our data suggest that age-related autophagy decline may activate Th17 inflammaging through a metformin-sensitive mechanism. Corroborating evidence for this model includes metformin-sensitive under-expression of PINK-1, a key regulator of mitochondrial autophagy (mitophagy; Geisler et al., 2010), in CD4+ T cells from O compared with Y subjects (Figure S6A). We used PINK-1 knockdown in CD4+ T cell from Y subjects (Figure S6B) to determine whether compromised mitophagy, rather than general macroautophagy, was a critical driver of the Th17 inflammaging profile. PINK-1 knockdown compromised Y function, as indicated by a metformin-sensitive increase in mitochondrial ROS, quantified by MitoSox fluorescence (Figure S6C), and accumulation of m-aconitase and Mitotracker green (Figures S6D and S6E), a surrogate for mitochondrial accumulation. TOM20/LAMP1 co-location definitely confirmed PINK-1 knockdown in Y cell decrease mitophagy (Figures S6F and S6G), although PINK-1 knockdown failed to more broadly change macroautophagy was indicated by lack of effect on LC3II expression (Figure S6H). PINK-1 knockdown insignificantly affected all cytokines, including those in the Th17 profile (Figure S6I). Our data indicate that Atg3-mediated autophagy activates Th17 cytokines independent of PINK-1-dependent mitophagy to support the conclusion that defective autophagy combines with suboptimal mitochondrial function, largely independent of mitophagy, to support the newly defined Th17 inflammaging profile. Our data support a model in which metformin increases non-mitochondrial autophagy in parallel with mitochondrial improvements to ameliorate one source of inflammaging.
Autophagy Defects Activate the Th17 Master Regulator STAT3 to Promote IL-17F Production in CD4+ T Cells from Y Subjects
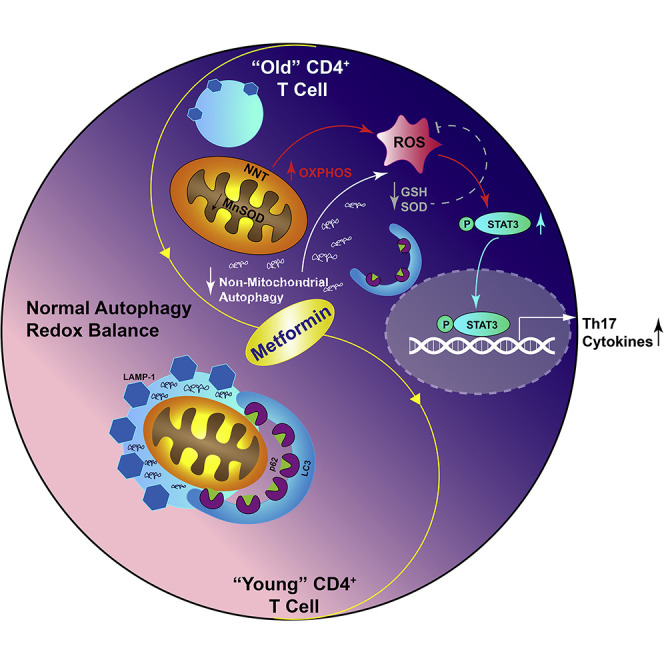
To define molecular mechanisms that link autophagy and mitochondrial defects to Th17 inflammaging, we queried the effect of metformin on STAT3, a regulator of the mitochondrial respiratory chain (Gough et al., 2009) and master regulator of Th17s (Wei et al., 2007, Wei et al., 2008), in CD4+ cells from O and Y subjects. Phospho-STAT3 serine 727 (p-S727 Stat3), modified for mitochondrial localization (Poli and Camporeale, 2015) co-localized more frequently with the mitochondrial complex 1 subunit NDUFA13/GRIMM19 in cells from O subjects (Figures 6A and 6B), suggesting STAT3 may mediate aging-associated changes in mitochondrial function. Metformin significantly lowered p-S727 STAT3 interaction with NDUFA13 in T cells from Y and O subjects (Figures 6A and 6B). Similarly, phospho-STAT3 with the nuclear localization/DNA-binding Y705 modification (Guadagnin et al., 2015) was twice as abundant in CD4+ T cells from O subjects compared with Y subjects (Figure 6C), further suggesting age-related changes in STAT3 function. Metformin, but not the general ROS scavenger Tempol, decreased p-T705 STAT3 in CD4+ T cells from O, but (for metformin) not Y subjects (Figures 6C and 6D), indicating that metformin is not solely acting on STAT modification through ROS mitigation. Conversely, Atg3-specific siRNA increased p-T705 STAT3 in CD4+ T cells from Y subjects in a metformin-independent mechanism (Figure 6C). Binding of p-Y705 STAT3 to the IL-17A and IL-17F promoters was higher in CD4+ T cells from O compared to Y subjects and was metformin-sensitive (Figure 6E). Together these data support the model that less non-mitochondrial autophagy in T cells from O subjects converges with changes in mitochondrial function and thereby redox balance to promote a Th17 inflammaging profile (Figure 6F). Metformin-mediated repair of these defects, either alone or in combination, ameliorates age-related inflammation.
Figure 6.

Aging Promotes STAT3 Activation, Mitochondrial Localization, and IL17A/F Promoter Binding
(A and B) Quantification of STAT3 ser727 and the mitochondrial complex 1 subunit NDUFA13 localization in CD4+ T cells stimulated for 40 h with αCD3/αCD28 ± metformin as indicated. n = 4 with multiple field readings shown per N.
(C) Phospho (p)-STAT3 T705 expression assayed on western blots as indicated on x axis. n = 6–8.
(D) p-STAT3 T705 expression relative to total STAT3 in O cells in the presence of TEMP or MET. n = 4–5. ∗p < 0.05 versus Y or control siRNA or #p < 0.05 versus O. Fold change is compared with Y or O.
(A and D) 3 cells/field and 3–5 fields/slide were imaged using 63× oil immersion in Zeiss microscope. Average fluorescence/field is reported.
(E) Chromatin immunoprecipitation assay showing fold-enrichment of p-STAT3 705 on (left) IL-17A or (right) IL-17F promoters. n = 4. All bar graphs show mean ± SEM.
(F) Model for parallel metformin-sensitive pathways that drive T cell inflammaging. Activated T cells from O subjects displayed blunted autophagy, which was largely independent of changes in mitochondrial bioenergetics/excess ROS that promote STAT3-mediated activation of a CD4+ T cell inflammaging profile.
Discussion
Our data combined with mathematical modeling approaches identified a Th17 profile produced by CD4+ T cells from lean O compared with Y subjects to show that Th17s are an unexpected source of inflammaging. In vitro treatment of cells with metformin in the presence or absence of neutralizers specific to various downstream pathways converged on a model that showed metformin prevented the production of Th17 profile cytokines by correcting age-related changes in autophagy and mitochondrial bioenergetics. Together these changes culminated in phosphorylation of STAT3, a master transcriptional activator of Th17s. STAT3 may or may not be a convergence point of the two demonstrated pathways. In contrast, Th17 cytokines do not dominate the response of CD4+ T cells from Y subjects to stimuli and are not affected by metformin. These data significantly extend demonstrations that metformin alleviates a limited definition of “inflammation” in pre-clinical and clinical studies (Saisho, 2015, Cameron et al., 2016, Lee et al., 2013) by identifying that metformin can act in either of two pathways to exert its anti-inflammatory effect. This work therefore connects previously independent demonstrations: (1) autophagy declines with age (Martinez-Lopez et al., 2015), (2) very recent work specifically showing CD4+ T cell autophagy and mitochondrial respiration declines with age (Bektas et al., 2019), (3) defective mitophagy compromises mitochondrial function (Melser et al., 2015), (4) bioenergetic shifts regulate T cell function (Almeida et al., 2016), and (5) STAT3 activates Th17 cytokines (Wei et al., 2007). The ability of metformin to trigger this anti-inflammation cascade justifies phase IV clinical trials on the potential of metformin to alleviate the generally negative consequences of age-related inflammation.
Numerous studies show that increased ATP demand following αCD3/αCD28 T cell receptor engagement is met through glycolysis, as indicated by a 20- to 50-fold increase in lactate production and higher ECAR (Wang et al., 2011, Greiner et al., 1994, Menk et al., 2018). This shift to glycolysis is important for pro-inflammatory T cell function outside the context of aging (Palmer et al., 2015). Higher OXPHOS in stimulated CD4+T cells from O compared to Y subjects, perhaps due in part to increased mitochondrial mass to compensate for a higher incidence of dysfunctional mitochondria, was entirely unanticipated. Given that OXPHOS naturally generates mitochondrial ROS, and that glycolysis is critical for generation of anti-oxidant regulators, such as GSH (Yarosz and Chang, 2018, Mak et al., 2017), our data indicate that some unknown brake on age-related changes in cell metabolism converge to shift what has been classically considered anti-inflammatory OXPHOS metabolism to a pro-inflammatory outcome. Demonstrations that ROS activates the JAK/STAT pathway (Simon et al., 1998) indicate that metabolically triggered redox imbalance in cells from O subjects would be predicted to converge with defective non-mitochondrial autophagy (Figure 6F) to promote Th17s through STAT3 action as a transcription factor. ROS may also activate autophagy and the removal of unwanted or dysfunctional organelles (beyond mitochondria) in a negative feedback loop (Zorov et al., 2014). As shown by our data, the inability of cells from O subjects to upregulate autophagy in response to stimulation may exaggerate ROS build up from overexuberant mitochondrial OXPHOS. The data also indicate that the initial events are adequate to generate a Th17 profile but that sustained mitochondrial ROS might not be required to maintain the Th17 profile. The importance of STAT3/mitochondrial co-localization, which has seemingly opposite effects in different systems (Rincon and Pereira, 2018), remains unexplored as does the role played by feed forward loops between STAT3 and autophagy (You et al., 2015). Moreover, prior studies have shown that STAT3 activation in autoimmune conditions chronically activated Th1 and Th17 cells that are inadequately suppressed by Tregs (Goodman et al., 2011).
The previously unappreciated Th17 cytokine profile that defined T cell inflammation in lean, healthy sexagenarians is strikingly similar to the Th17 profile that dominates inflammation in middle-aged people with obesity-associated T2D (Ip et al., 2016) and in periodontal disease (Abusleme and Moutsopoulos, 2017). Other age-related changes, like autophagy dysfunction and redox imbalance, are also characteristic of cells from metabolically compromised people (McCormick et al., 2018, Gautam et al., 2010). These cell-intrinsic changes, alone or in association with secreted Th17 cytokines, raise the possibility that mechanisms amenable to drug intervention drive the increased risk of T2D with age (Defronzo, 1979, Chen et al., 1985, Davidson, 1979) in addition to age-related (currently non-druggable) changes in adipose tissue distribution. Outcomes from pre/post-metformin treatment in pre-diabetes subjects further indicated that metformin treatment of O subjects may have impact on age-related pre-diabetes, raising the justification for a full-scale clinical trial with Th17 inflammaging as a primary outcome. Relationships between inflammaging and other age-related diseases with inflammatory underpinnings like cancer, cardiovascular disease and neurodegeneration could be better defined and thus understood/treated by approaches described herein, toward the long-term goal of improving health span.
Limitations of Study
The participants in both cohorts, and in particular the O group, were lean and normoglycemic and therefore may not represent age-related inflammation in more typical Americans who generally have higher BMIs, are metabolically less healthy, take any number of clinically indicated medications, do not meet exercise recommendations, and do not have advanced academic degrees. Our exclusive focus on CD4+ T cells does not address the likely contribution of other important sources of inflammaging, like myeloid cells (monocytes/macrophages). Finally, our ability to rigorously address the mitochondrial phenotype of PINK-1-knockdown cells with an appropriate N was significantly compromised by the 2020 COVID-19/SARS-CoV-2 pandemic, which halted human subject recruitment across the globe for (at the time of revision) an indeterminant amount of time.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Atg3 used at 1:500 | Sigma Aldrich | Cat #A3231; RRID: AB_1078235 |
| anti-AMPK used at 1:500 | Cell signaling technology | Cat #2793; RRID: AB_915794 |
| anti-pAMPK used at 1:500 | Cell signaling technology | Cat# 2535; RRID: AB_331250 |
| anti-Β-Actin used at 1:10,000 | Cell signaling technology | Cat# 3700; RRID: AB_2242334 |
| anti-Complex-1 used at 1:500 | Sigma Aldrich | Cat# ABN302 |
| anti-DRP1 used at 1:500 | Cell signaling technology | Cat# 14647; RRID: AB_2798554 |
| anti-Hexokinase used at 1:500 | Cell signaling technology | Cat# 2867; RRID: AB_2232946 |
| anti-IDH2 used at 1:500 | Santa Cruz Biotechnology | Cat# sc-374476; RRID: AB_10986415 |
| anti-LC3 used at 1:500 for WB, 1:50 for confocal microscopy | Sigma Aldrich | Cat# L7543; RRID: AB_796155 |
| Anti-LDH used at 1:500 | Cell signaling technology | Cat# 3582; RRID: AB_2066887 |
| anti-m-aconitase used at 1:500 | Abcam | Cat# ab110321; RRID: AB_10863392 |
| anti-MnSOD/SOD2 used at 1:500 | Santa Cruz Biotechnology | Cat# sc-137254; RRID: AB_2191808 |
| anti-NDUFA-13 used at 1:50 for confocal microscopy | Abcam | Cat# ab110240; RRID: AB_10863178 |
| anti-NNT used at 1:500 | Abcam | Cat# ab110352; RRID: AB_10887748 |
| anti-OGDH used at 1:500 | Cell signaling technology | Cat# 26865; RRID: AB_2737585 |
| anti-P62 used at 1:500 | Cell signaling technology | Cat# 5114; RRID: AB_10624872 |
| anti-Peroxiredoxin 2 used at 1:500 | Cell signaling technology | Cat# 46855; RRID: AB_2799310 |
| anti-PINK1 used at 1:50 for confocal microscopy | Biolegend | Cat# 846201; RRID: AB_2783414 |
| anti-PKM2 used at 1:500 | Cell signaling technology | Cat# 4053; RRID: AB_1904096 |
| anti-TOM20 used at 1:500 for WB, 1:50 for confocal microscopy | Abcam | Cat# ab56783; RRID: AB_945896 |
| anti-LAMP1 used at 1:500 for WB, 1:50 for confocal microscopy | Abcam | Cat# ab24170; RRID: AB_775978 |
| anti-OPA1 used at 1:500 | Cell signaling technology | Cat# 67589; RRID: AB_2799728 |
| anti-SOD1 used at 1:500 | Cell signaling technology | Cat# 4266; RRID: AB_2193898 |
| anti-STAT3 used at 1:500 | Cell signaling technology | Cat# 4904; RRID: AB_331269 |
| anti-p-STAT3 Tyr 705 used at 1:500 | Cell signaling technology | Cat# 9145; RRID: AB_2491009 |
| anti-p-STAT3 Ser 727 used at 1:500 for WB, 1:50 for confocal microscopy | Cell signaling technology | Cat# 9134; RRID: AB_331589 |
| Anti-mouse Alexa 488 used at 1:500 | Rockland antibodies | Cat# 610-741-124; RRID: AB_1057558 |
| Anti-rabbit Alexa 647 used at 1:500 | Thermo fisher scientific | Cat# A-21244; RRID: AB_2535812 |
| Anti-mouse IgG, HRP-linked used at 1:5000 | Cell signaling technology | Cat# 7076; RRID: AB_330924 |
| Anti-rabbit IgG, HRP-linked used at 1:5000 | Cell signaling technology | Cat# 7074; RRID: AB_2099233 |
| Anti CD-27 BUV395 used at 1:100 | BD Biosciences | Cat# 563815; RRID: AB_2744349 |
| Anti CD8 BUV 805 used at 1:400 | BD Biosciences | Cat# 564912; RRID: AB_2744465 |
| Anti-CD3 BV 510 | Biolegend | Cat# 317331; RRID: AB_2561376 |
| Anti-CD45RO BV605 used at 1:100 | Biolegend | Cat# 304237; RRID: AB_2562143 |
| Anti-CD28 BV650 used at 1:50 | Biolegend | Cat# 302945; RRID: AB_2616854 |
| Anti-CCR7 BV785 used at 1:25 | Biolegend | Cat# 353229; RRID: AB_2561371 |
| anti-CD45RA Alexa 488 used at 1:100 | Biolegend | Cat# 304105; RRID: AB_314409 |
| anti- CD57 PE/Dazzle used at 1:50 | Biolegend | Cat# 359619; RRID: AB_2564062 |
| anti- KLRG1 (MAFA) PE-Cy7-A used at 1:100 | Biolegend | Cat# 138415; RRID: AB_2561735 |
| anti- CD4 antibody Alexa Fluor 700 used at 1:400 | Biolegend | Cat# 100429; RRID: AB_493698 |
| anti- CD14 APC-Cy7-A used at 1:50 | Biolegend | Cat# 325620; RRID: AB_830693 |
| anti- CD19 APC-Cy7-A used at 1:50 | Biolegend | Cat# 363029; RRID: AB_2572092 |
| anti-CD28 used at 1:500 | Thermo fisher scientific | Cat# 16-0289-81; RRID: AB_468926 |
| anti-CD3 used at 1:500 | Thermo fisher scientific | Cat# 16-0037-85; RRID: AB_468855 |
| Biological Samples | ||
| Healthy lean adults CD4+ T cells; Table S1 | This paper | N/A |
| Prediabetic obese adults CD4+ T cells; Table S2 | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Metformin | Invivogen | Tlrl-metf |
| Tempol | Sigma Aldrich | SML0737 |
| Bafilomycin A1 | Sigma Aldrich | B1793 |
| 3-methyladenine | Sigma Aldrich | M9281 |
| 2’,7’ –dichlorofluorescin diacetate (DCFDA) | Sigma- Aldrich | D6883 |
| tert-butyl hydrogen peroxide | Sigma Aldrich | 416665 |
| Mitotracker green | Cell Signaling technology | 9074S |
| Tetramethylrhodamine, ethyl ester (TMRE) | Abcam | ab113852 |
| Critical Commercial Assays | ||
| Lactate Colorimetric/Fluorometric Assay Kit | BioVision | Cat# K607 |
| Lactate dehydrogenase activity assay kit | BioVision | Cat# K726 |
| Milliplex human Th17 25-plex kit | Millipore Sigma | Cat# HT17MG-14K-PX25 |
| Oligonucleotides | ||
| Accell smartpool AMPK siRNA 10nm | Dharmacon | Cat# E-005027-00-0010 |
| Accell Non-target control siRNA 10nm | Dharmacon | Cat# D-001810-01-05 |
| Accell smartpool PINK1 siRNA 10nm | Dharmacon | Cat# E-004030-00-0010 |
| Atg3 siRNA | Bharath et al., 2014, Bharath et al., 2017a | N/A |
| Non-target control siRNA | Bharath et al., 2014, Bharath et al., 2017a | N/A |
| Primers CHIP Assay; Table S3 | This paper | N/A |
| Software and Algorithms | ||
| Seahorse Explorer (SHORE) Analysis program | Nicholas et al. (2017) | https://github.com/elizabethproctor/Seahorse-Analysis |
| FlowJo v.10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| Cytobank | Cytobank | http://premium.cytobank.org |
| t-SNE embeddings were created using opt-SNE Python script | Belkina et al, 2019 | N/A |
| PLSDA was performed using R “ropls” package and “mixOmics” package | Thévenot et al., 2015, Rohart et al., 2017 | N/A |
| SAS 9.4 | SAS Institute Inc | https://support.sas.com |
| GraphPad Prism version 7 for Windows | GraphPad software | https://www.graphpad.com/ |
| Other | ||
| Seahorse XFe96 FluxPak | Agilent Technologies | Cat# 102416 |
| Dynabeads Human T-Activator CD3⁄CD28 for T Cell Activation | GIBCO life technologies | Cat# 11131D |
| 100 5X siRNA Buffer, 100 mL | Dharmacon | Cat# B-002000-UB-100 |
| Accell siRNA Delivery Media, 500 mL | Dharmacon | Cat# B-005000-500 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barbara Nikolajczyk (barb.nik@uky.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets generated and analyzed during the study are included with the published manuscript (and Supplemental Information). All other data are available from the corresponding author upon request.
Experimental Model and Subject Details
Human Subjects Sample Collection
Informed consent for all human samples was obtained following Institutional Review Board-approved protocols in accordance with the Declaration of Helsinki at University of Kentucky, The Forsyth Institute, and Boston University. Study design was cross-sectional. Young lean subjects (“Y” avg. 31.2 yrs,) and BMI-matched older subjects (“O” avg. 62 yrs) have additional characteristics shown in Table S1. All subjects were normoglycemic as indicated by %HbA1c. Prediabetic subjects pre and post metformin (avg. 48 yrs), avg. A1c% 5.84, have additional characteristics shown in Table S2. Exclusion criteria were smoking, long-term or recent use of antibiotics or anti-inflammatory medications (i.e. NSAIDs; low-dose aspirin was not an exclusion), auto-immune disease, allergy medications and pregnancy.
Method Details
Cellular Analyses
Fifty milliliters of peripheral blood were collected into acid/citrate/dextrose containing tubes by venous puncture. PBMCs were purified by histopaque 1077 followed by negative selection with CD4+ cell-excluding magnetic beads (Miltenyi, Auburn, CA) for experiments on purified T cells as we published 4. CD4+ T cells were >93% pure as assessed via flow cytometry. All cells were frozen at -80°C in a Mr. Frosty apparatus (Nalgene). For multi-week storage, cells were moved to -170°C following 1-7 days at -80°C. T cells from BMI-matched young (Y) and old (O) subjects were stimulated in vitro for 40 h with αCD3/αCD28 Dynabeads (Thermo Fisher Scientific, 11132D) at 2μL/100k cells ± 100μM metformin (met) in RPMI media with 5mM glucose (euglycemic). The supernatant was stored at -80°C until analyzed in technical duplicate on a BioRad 3D instrument using a Th17 multiplex kit (Miltenyi). Cells were assayed as outlined below. Metabolic characterization of CD4+ T cells was performed using the Seahorse XF96 Analyzer (Nicholas et al., 2017, van der Windt et al., 2016).
Extracellular Flux Analysis (Mitostress Test)
After thawing rapidly in a 37°C water bath, cells were activated, then adhered onto wells of a poly-D-lysine coated XF96 plate in extracellular flux assay media (non-buffered DMEM containing 10 mM glucose, 4 mM L-glutamine, and 2 mM sodium pyruvate. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the mitochondrial stress test procedure for basal OCR followed by sequential addition of 3.5 mM oligomycin (Calbiochem), 1 mM or 2 mM (for resting or activated cells, respectively) fluoro-carbonyl cyanide phenylhydrazone (FCCP) (Enzo) and 14 mM rotenone + 14 mM antimycin A (Enzo) with the XF96 Extracellular Flux Analyzer (Seahorse Bioscience) as previously described (Nicholas et al., 2017, van der Windt et al., 2016).
Autophagy and Mitochondrial Turnover
Autophagy was assessed in CD4+ T cells as follows. First, we quantified the ratio of the lipidated form of microtubule associated protein light chain (LC3-II) and the adaptor protein p62 (A170/SQSTM1) relative to β-actin on Western blots (Glick et al., 2010, Kabeya et al., 2004) as we published (Bharath et al., 2015, Bharath et al., 2017a). The Key Resources Table lists the antibodies used in this study. Second, we quantified the formation of LC3 puncta after intracellular labeling using fluorescent antibodies to LC3 II protein (Mizushima et al., 2010). Third, we measured mitochondrial turnover (Kubli and Gustafsson, 2012) by Western blotting for the mitochondrial matrix protein m-aconitase. Fourth, we visualized mitophagy through colocalization of TOM20 with LAMP1 using immunofluorescence as detailed below. Finally, to differentiate between trafficking of the autophagosome to the lysosome and lysosomal degradation of the autophagosome, cells were treated with bafilomycin A1 (1 μM; BAF A1, Sigma Aldrich, B1793) to impair autophagosome-lysosome fusion (Kawai et al., 2007, Yoshimori et al., 1991, Glick et al., 2010). Cells treated with 3-methyladenine (5 mM; 3-MA; Sigma Aldrich, M9281) (Wang et al., 2013) were a negative control.
Small Interference RNA
Small interference RNA (siRNA)-mediated knockdown of Atg3 AMPK or PINK1 was performed using published siRNA sequences or Accell siRNA (Dharmacon, Lafayette, CO) following manufacturer’s guidelines. Scrambled sequences were used as controls. One μM siRNA was diluted in Accell siRNA delivery medium and added to CD4+ T cells (Y and O ± met) in 5 mM RPMI medium with 10% FBS and 300 IU of IL-2. Cells were incubated 40 h prior to assessing knockdown efficiency via immunoblotting.
ROS Generation
ROS or mitochondrial ROS was assessed by 2’,7’ –dichlorofluorescin diacetate (DCFDA) fluorescence (Sigma- Aldrich, D6883), or MitoTEMPO (Santa Cruz Bitoechnology, sc-221945) normalized to cell number (Bharath et al., 2014, Bharath et al., 2015). Treatment with 100 mM glucose (Sigma Aldrich, D8270) and 50 μM tert-butyl hydrogen peroxide (TBHP) (Sigma Aldrich, 416665) was the positive control. The free radical scavenger Tempol (10 μM; Sigma Aldrich SML0737) was used to manipulate ROS generation (Bharath et al., 2017a, Dikalova et al., 2010).
Immunohistochemistry/Immunofluorescence
Activated CD4+ T cells (Y and O ± met) were plated on coverslips coated with poly-D-lsine in 6-well plates. After 40 h, the cells were briefly centrifuged, washed 3 times with 1X PBS and incubated in 4% paraformaldehyde for 30 min on ice. The coverslips were washed 3X with PBS and 0.1% triton X-100 (PBST), and were blocked for 30 min in 5% BSA/PBST. Antibodies to TOM20 (Abcam, Cambridge, MA) and LAMP1 (Abcam, Cambridge, MA) were added at 1:50 dilution with incubation overnight at 4°C. The coverslips were rinsed 3X with PBST and incubated with fluorophore-tagged secondary antibodies (TOM20 with anti-mouse Alexa 488 and LAMP1 with anti- rabbit Alexa 680) (Rockland Immunochemicals, Limerick,PA) for 2h at RT. The coverslips were washed 3X with PBST and mounted on glass slides using Fluromount G (Southern Biotech, Birmingham, AL). Cell imaging under a 63X oil immersion lens was performed on a Zeiss confocal microscope. Three cells/field and 3 fields/slide were imaged on N=4 slides (∼20 values per each N) or as detailed in the figure legend, and data were analyzed using Image J (Kirber et al., 2007, Vereb et al., 2000).
Mitochondrial Mass
Mitochondrial mass was assessed in CD4+ T cells by flow cytometry using Mitotracker green fluorescence. CD4+ T cells from Y and O subjects were stimulated with αCD3/αCD28 ± 100μM metformin or 50 μM Tempol, washed 2X with PBS and incubated with 40 nM Mitotracker green (9074S,Cell Signaling Technology, Danvers, MA) in 5 mM RPMI for 30 min. Cells were co-stained with human anti-CD4+APC (Biolegend, San Diego, CA) and Zombie NIR live-dead stain (Biolegend, San Diego, CA) and assessed on a BD LSRII cell analyzer (BD Biosciences) or a fluorescence spectrophotometer (Biotek). Flow cytometry data was analyzed using FlowJo v.10 (FlowJo).
Biochemical Assays
Mitochondrial membrane potential was assessed using tetramethylrhodamine, ethyl ester (TMRE) (Abcam, Cambridge, MA), a cell-permeable positively-charged dye that accumulates in mitochondria with high membrane potential. Activated CD4+ T cells (Y and O ± met ± tempol) were incubated with 400 nm TMRE for 30 min. The cells were assayed using a microplate spectrophotometer (Biotek). Fifty μM FCCP was the positive control. GSH and lactate were measured using fluorometric assays (BioVision, Milpitas, CA) according to the manufacturer’s protocols.
Lactate Dehydrogenase Assay
The activity of the enzyme lactate dehydrogenase was assessed in activated CD4+ T cells (Y ± OA), using colorimetric assay (BioVision, Milpitas, CA) according to manufacturer’s protocols.
Chromatin Immunoprecipitation (ChIP) Assay
The procedures for ChIP assay were conducted as published (Xie et al., 2015, Jiang et al., 2013). Following treatment, cells (1x107 CD4+ cells/sample) were washed with phosphate buffered saline (PBS) and cross-linked with 1% formaldehyde for 15 minutes at room temperature to preserve the protein-DNA interactions. After quenched by glycine, cells were lysed in cold cell lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40 and protease and phosphatase inhibitor cocktail). After cells were lysed, nuclei were collected by centrifugation and re-suspended in 0.5 mL nucleus lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 0.2% SDS and protease and phosphatase inhibitor cocktail) for ultra-sonication (Branson Digital Sonifier 450; micro tip; 7 times for 10 seconds with interval of 50 seconds and 30% power), followed by centrifugation to remove insoluble material. Supernatants were precleared by incubating for 2 h at 4°C with 2 μg sheared salmon sperm DNA (Invitrogen) and 20 μL of a 50% slurry protein A agarose resin (Santa Cruz Biotech) and then subjected to immunoprecipitation with the rabbit anti phospho-Stat3 (Tyr705) (D3A7) XP® (4 μg; Cell signaling) or nonspecific rabbit IgG (4 μg; Vector Laboratory) overnight at 4°C. After washing and centrifugation, immune complexes were eluted and heated at 65°C for 2 hours to reverse protein-DNA cross-links. Purified DNA was used as template to amplify the target region of the IL-17 promoter. The ChIP-PCR primers are described in Table S3. All primer sets for each target gave similar results.
Flow Cytometry
All reagents were from Biolegend unless otherwise stated. For phenotyping, cryopreserved PBMCs were thawed into R10 medium. The cells were then stained with Zombie NIR fixable dye for 15 min at room temperature, then washed with PBS/0.5%BSA/2mM EDTA. Cell pellets were resuspended in PBS/0.5%BSA/2mM EDTA supplemented with anti-human FcR-blocking reagent and incubated for 10 min. A mixture of fluorescent antibodies (listed in Key Resources Table) were resuspended in Brilliant Buffer (BD Biosciences) and supplemented with Monocyte blocker reagent (Biolegend). Single stain controls were prepared with UltraComp capture beads (Thermo Fisher) stained with fluorescent antibodies, or killed PBMCs stained with Live-dead Zombie NIR dye. Samples were analyzed on a BD FACSARIA II SORP and data were captured with BD FACSDIVA 8.0. Compensation matrix was created in BD FACSDIVA using single-stain controls and recorded within each sample data file according to FCS 3.0 standards. All data were preprocessed in FlowJo v10 (FlowJo) to remove debris and dead cells and gate on specific subsets of interest. Equal numbers per sample of live, singlet CD4 or CD8 T cells (defined as CD3+CD4+CD19-CD14- or CD3+CD8+CD19-CD14-) were concatenated and lineage marker fluorescence measurements were mapped into t-SNE space using opt-SNE package (Belkina et al, 2019). Data plots and heatmaps were generated in Cytobank cloud platform (Chen and Kotecha, 2014) Cytobank: providing an analytics platform for community cytometry data analysis and collaboration.
Multiplex Measurement of Cytokine Concentrations
Supernatant samples were centrifuged for 30 s to remove debris before being applied to a 384-well plate for analysis by multiplexing bead-based ELISA using the Milliplex human Th17 25-plex kit (Millipore). Antibodies and magnetic beads were diluted 1:1 with assay buffer and utilized at half-volume to adjust the manufacturer’s protocol to our 384-well plate format. Each sample and standard was assayed in technical triplicate, with the average value of these used for analyses. Outcomes from wells with < 35 beads read for each analyte were excluded from analysis. Plates were washed in between incubations using a BioTek 406 Touch plate washer (BioTek) and read using the Luminex FlexMap 3D system (Luminex). We assayed samples at dilutions of 1:1 and 1:7 (sample:assay buffer) to allow abundant cytokines to be measured in the linear range of the instrument.
Quantification and Statistical Analysis
Data are presented as mean ± standard error of the mean. Non parametric Kruskal-Wallis and Dunn’s post hoc tests were performed using Graph-Pad Prism 7.03 (GraphPad Software) Since the quantitative cytokine expression variables were right-skewed, we first log-transformed each response variable. For each cytokine, a repeated-measures full factorial two-way ANOVA was performed, analyzing differences in cytokine expression between T-cells from young and old individuals and in the presence or absence of metformin treatment. For cytokines whose interaction p-value was significant, we performed the corresponding post hoc t-tests. Across all analyses, a p-value of less than 0.05 was considered significant. All analyses were completed in SAS 9.4 (SAS Institute Inc.). Statistical details of experiments can be found in figure legends in the manuscript and in the Supplemental Information.
Partial Least Squares Discriminant Analysis (PLSDA) was performed to test whether a set of cytokines can distinguish between the groups. This analysis was carried out using R “ropls” package (Thévenot, 2016) and “mixOmics” package (Rohart et al., 2017). We first preprocessed the data by converting the unit of ng/ml to pg/ml and further normalized the data through Z-scoring or dividing each subject’s cytokine profile by the sum of corresponding cytokine profile of all subjects. Then, we performed PLSDA to find the latent variables (LVs) i.e. the cytokines that can differentiate the groups. PLSDA consists of a classical partial least square regression (PLSR) where the dependent variable is a categorical one that represents the groups. PLS components are then built to increase the explained variance between groups so that the separation between them is improved.
We used variable importance in projection (VIP) score to determine which cytokine variables contribute the most to the variance explanation of the dependent variable. VIP score for each cytokine variable was calculated as the weighted sum of squares of the PLS weights with explained variance of the dependent variable. VIP scores that are greater than 1 were considered as important cytokines. Assuming there are cytokine variables and PLS-DA components, then the VIP score for the cytokine variable can be calculated as (Farres et al., 2015):
| (Equation 1) |
where is the weight value for cytokine variable and component, is the sum of squares of explained variance of the dependent variable for the component, and is the sum of squares of explained variance of the dependent variable for all the components.
To validate the model, we used and for assessing the statistical significance, where is the percentage of the variation of dependent variables explained for each LV, and is the percentage of the variation of dependent variables predicted for each LV by cross validation. The higher the values of and , the better the performance of the model. Additional LVs were then added to the model if their corresponding R2Y > 0.01 and Q2Y > 0.05. and were calculated as (Szymańska et al., 2012):
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
where is residual sum of squares, is the total sum of squares, is the predicted residual error sum of squares, is the predicted dependent variable in training sets, is the predicted dependent variable in validation sets, and is the mean of the dependent variable.
The statistical significance of R2Y and Q2Y was estimated using permutation test (100 random permutations) at . We compared the R2Y and Q2Y values of the model built with unpermuted (original) data and with the R2Y_perm and Q2Y_perm values of the model with random permutated data. The p value was then calculated as (Szymańska et al., 2012):
| (Equation 6) |
where ) is the number of elements in the null distribution which are larger than or equal to the original data set, and N is the number of permutations. All VIP outcomes exceeded the p < 0.05 cutoff unless otherwise indicated.
Acknowledgments
We thank Dr. Sara Santa Cruz Calvo for critical reading of the manuscript. This work was supported by grants R01DK108056 and U01DE025383 (B.S.N.), UL1TR001998 from NCATS (P.A.K.), University of Kentucky College of Medicine (B.S.N.), University of Kentucky Markey Cancer Center and Flow Cytometry Core P30CA177558 (B.S.N.), Boston University Flow Cytometry Core Facility T32AI089673, and Boston University Medical Center, Metabolism, Endocrinology and Obesity training grant T32DK007201. This work was also supported by the College of Health Sciences, Faculty Development grant (FDG) and Sakowich Center for Undergraduate Research and Creative Activities grant (SCURCA), Merrimack College (L.P.B.).
Author Contributions
Conceptualization, B.S.N. and L.P.B.; Methodology, L.P.B., J.L., L.C., and A.C.B.; Investigation, L.P.B., M.A., G.M., D.N.A., Z.G., K.J., R.M.F.P., R.L., and A.C.B.; Formal Analysis, J.L., L.C., R.M.F., E.A.P., A.C.B., and K.T.; Supervision and Fund Acquisition, B.S.N.; Writing – Review & Editing, L.P.B. and B.S.N.; Resources, H.H., J.D., C.M.A., J.S.C., and P.A.K.
Declaration of Interests
The authors declare no competing interests.
Published: May 12, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.04.015.
Supplemental Information
References
- Abusleme L., Moutsopoulos N.M. IL-17: overview and role in oral immunity and microbiome. Oral Dis. 2017;23:854–865. doi: 10.1111/odi.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida L., Lochner M., Berod L., Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 2016;28:514–524. doi: 10.1016/j.smim.2016.10.009. [DOI] [PubMed] [Google Scholar]
- Alpert A., Pickman Y., Leipold M., Rosenberg-Hasson Y., Ji X., Gaujoux R., Rabani H., Starosvetsky E., Kveler K., Schaffert S. A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat. Med. 2019;25:487–495. doi: 10.1038/s41591-019-0381-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektas A., Schurman S.H., Gonzalez-Freire M., Dunn C.A., Singh A.K., Macian F., Cuervo A.M., Sen R., Ferrucci L. Age-associated changes in human CD4+ T cells point to mitochondrial dysfunction consequent to impaired autophagy. Aging (Albany NY) 2019;11:9234–9263. doi: 10.18632/aging.102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina A.C., Ciccolella C.O., Anno R., Halpert R., Spidlen J., Snyder-Cappione J.E. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat. Commun. 2019;10:5415. doi: 10.1038/s41467-019-13055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharath L.P., Mueller R., Li Y., Ruan T., Kunz D., Goodrich R., Mills T., Deeter L., Sargsyan A., Anandh Babu P.V. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can. J. Physiol. Pharmacol. 2014;92:605–612. doi: 10.1139/cjpp-2014-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharath L.P., Ruan T., Li Y., Ravindran A., Wan X., Nhan J.K., Walker M.L., Deeter L., Goodrich R., Johnson E. Ceramide-initiated protein phosphatase 2A activation contributes to arterial dysfunction in vivo. Diabetes. 2015;64:3914–3926. doi: 10.2337/db15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharath L.P., Cho J.M., Park S.K., Ruan T., Li Y., Mueller R., Bean T., Reese V., Richardson R.S., Cai J. Endothelial cell autophagy maintains shear stress-induced nitric oxide generation via glycolysis-dependent purinergic signaling to endothelial nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2017;37:1646–1656. doi: 10.1161/ATVBAHA.117.309510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharath L.P., Ip B.C., Nikolajczyk B.S. Adaptive immunity and metabolic health: harmony becomes dissonant in obesity and aging. Compr. Physiol. 2017;7:1307–1337. doi: 10.1002/cphy.c160042. [DOI] [PubMed] [Google Scholar]
- Bruunsgaard H., Pedersen A.N., Schroll M., Skinhøj P., Pedersen B.K. TNF-alpha, leptin, and lymphocyte function in human aging. Life Sci. 2000;67:2721–2731. doi: 10.1016/s0024-3205(00)00851-1. [DOI] [PubMed] [Google Scholar]
- Cameron A.R., Morrison V.L., Levin D., Mohan M., Forteath C., Beall C., McNeilly A.D., Balfour D.J., Savinko T., Wong A.K. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 2016;119:652–665. doi: 10.1161/CIRCRESAHA.116.308445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A., Chowdhury S., Bhattacharyya M. Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res. Clin. Pract. 2011;93:56–62. doi: 10.1016/j.diabres.2010.11.030. [DOI] [PubMed] [Google Scholar]
- Chen T.J., Kotecha N. Cytobank: providing an analytics platform for community cytometry data analysis and collaboration. Curr. Top. Microbiol. Immunol. 2014;377:127–157. doi: 10.1007/82_2014_364. [DOI] [PubMed] [Google Scholar]
- Chen M., Bergman R.N., Pacini G., Porte D., Jr. Pathogenesis of age-related glucose intolerance in man: insulin resistance and decreased beta-cell function. J. Clin. Endocrinol. Metab. 1985;60:13–20. doi: 10.1210/jcem-60-1-13. [DOI] [PubMed] [Google Scholar]
- Conte M., Ostan R., Fabbri C., Santoro A., Guidarelli G., Vitale G., Mari D., Sevini F., Capri M., Sandri M. Human aging and longevity are characterized by high levels of Mitokines. J. Gerontol. A Biol. Sci. Med. Sci. 2019;74:600–607. doi: 10.1093/gerona/gly153. [DOI] [PubMed] [Google Scholar]
- Davidson M.B. The effect of aging on carbohydrate metabolism: a review of the English literature and a practical approach to the diagnosis of diabetes mellitus in the elderly. Metabolism. 1979;28:688–705. doi: 10.1016/0026-0495(79)90024-6. [DOI] [PubMed] [Google Scholar]
- De Rosa V., Galgani M., Porcellini A., Colamatteo A., Santopaolo M., Zuchegna C., Romano A., De Simone S., Procaccini C., La Rocca C. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015;16:1174–1184. doi: 10.1038/ni.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defronzo R.A. Glucose intolerance and aging: evidence for tissue insensitivity to insulin. Diabetes. 1979;28:1095–1101. doi: 10.2337/diab.28.12.1095. [DOI] [PubMed] [Google Scholar]
- Dikalova A.E., Bikineyeva A.T., Budzyn K., Nazarewicz R.R., McCann L., Lewis W., Harrison D.G., Dikalov S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farres P.S., Tsakovski S., Tauler R. Comparison of the variable importance in projection (VIP) and of the selectivity ratio(SR) methods for variable selection and interpretation. J. Chemometrics. 2015;29:528–536. [Google Scholar]
- Ferrucci L., Corsi A., Lauretani F., Bandinelli S., Bartali B., Taub D.D., Guralnik J.M., Longo D.L. The origins of age-related proinflammatory state. Blood. 2005;105:2294–2299. doi: 10.1182/blood-2004-07-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidan E., Onder Ersoz H., Yilmaz M., Yilmaz H., Kocak M., Karahan C., Erem C. The effects of rosiglitazone and metformin on inflammation and endothelial dysfunction in patients with type 2 diabetes mellitus. Acta Diabetol. 2011;48:297–302. doi: 10.1007/s00592-011-0276-y. [DOI] [PubMed] [Google Scholar]
- Franceschi C., Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Franceschi C., Garagnani P., Vitale G., Capri M., Salvioli S. Inflammaging and 'garb-aging. Trends Endocrinol. Metab. 2017;28:199–212. doi: 10.1016/j.tem.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Gautam N., Das S., Kar Mahapatra S., Chakraborty S.P., Kundu P.K., Roy S. Age associated oxidative damage in lymphocytes. Oxid. Med. Cell. Longev. 2010;3:275–282. doi: 10.4161/oxim.3.4.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S., Holmström K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Glick D., Barth S., Macleod K.F. Autophagy: cellular and molecular mechanisms. J. Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman W.A., Young A.B., McCormick T.S., Cooper K.D., Levine A.D. Stat3 phosphorylation mediates resistance of primary human T cells to regulatory T cell suppression. J. Immunol. 2011;186:3336–3345. doi: 10.4049/jimmunol.1001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough D.J., Corlett A., Schlessinger K., Wegrzyn J., Larner A.C., Levy D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner E.F., Guppy M., Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J. Biol. Chem. 1994;269:31484–31490. [PubMed] [Google Scholar]
- Guadagnin E., Narola J., Bönnemann C.G., Chen Y.W. Tyrosine 705 phosphorylation of STAT3 is associated with phenotype severity in TGFbeta1 transgenic mice. BioMed Res. Int. 2015;2015:843743. doi: 10.1155/2015/843743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z., Kutty S., Toth P.T., Marsboom G., Hammel J.M., Chamberlain C., Ryan J.J., Zhang H.J., Sharp W.W., Morrow E. Role of dynamin-related protein 1 (Drp1)-mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circ. Res. 2013;112:802–815. doi: 10.1161/CIRCRESAHA.111.300285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip B., Cilfone N.A., Belkina A.C., DeFuria J., Jagannathan-Bogdan M., Zhu M., Kuchibhatla R., McDonnell M.E., Xiao Q., Kepler T.B. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity (Silver Spring) 2016;24:102–112. doi: 10.1002/oby.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K., Yang Z., Cheng L., Wang S., Ning K., Zhou L., Lin J., Zhong H., Wang L., Li Y. Mediator of ERBB2-driven cell motility (MEMO) promotes extranuclear estrogen receptor signaling involving the growth factor receptors IGF1R and ERBB2. J. Biol. Chem. 2013;288:24590–24599. doi: 10.1074/jbc.M113.467837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat A.M., Harrison O.J., Riffelmacher T., Moghaddam A.E., Pearson C.F., Laing A., Abeler-Dörner L., Forman S.P., Grencis R.K., Sattentau Q. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. eLife. 2016;5:e12444. doi: 10.7554/eLife.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Kathiria A.S., Butcher L.D., Feagins L.A., Souza R.F., Boland C.R., Theiss A.L. Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS One. 2012;7:e31231. doi: 10.1371/journal.pone.0031231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai A., Uchiyama H., Takano S., Nakamura N., Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy. 2007;3:154–157. doi: 10.4161/auto.3634. [DOI] [PubMed] [Google Scholar]
- Kirber M.T., Chen K., Keaney J.F., Jr. YFP photoconversion revisited: confirmation of the CFP-like species. Nat. Methods. 2007;4:767–768. doi: 10.1038/nmeth1007-767. [DOI] [PubMed] [Google Scholar]
- Kominsky D.J., Campbell E.L., Colgan S.P. Metabolic shifts in immunity and inflammation. J. Immunol. 2010;184:4062–4068. doi: 10.4049/jimmunol.0903002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli D.A., Gustafsson Å.B. Mitochondria and mitophagy: the Yin and Yang of cell death control. Circ. Res. 2012;111:1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Texier L., Lineburg K.E., Cao B., McDonald-Hyman C., Leveque-El Mouttie L., Nicholls J., Melino M., Nalkurthi B.C., Alexander K.A., Teal B. Autophagy-dependent regulatory T cells are critical for the control of graft-versus-host disease. JCI Insight. 2016;1:e86850. doi: 10.1172/jci.insight.86850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.M., Kim J.J., Kim H.J., Shong M., Ku B.J., Jo E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Fiskum G., Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Madiraju A.K., Qiu Y., Perry R.J., Rahimi Y., Zhang X.M., Zhang D., Camporez J.G., Cline G.W., Butrico G.M., Kemp B.E. Author correction: metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2019;25:526–528. doi: 10.1038/s41591-018-0220-6. [DOI] [PubMed] [Google Scholar]
- Mak T.W., Grusdat M., Duncan G.S., Dostert C., Nonnenmacher Y., Cox M., Binsfeld C., Hao Z., Brüstle A., Itsumi M. Glutathione primes T cell metabolism for inflammation. Immunity. 2017;46:1089–1090. doi: 10.1016/j.immuni.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Malínská H., Oliyarnyk O., Škop V., Šilhavý J., Landa V., Zídek V., Mlejnek P., Šimáková M., Strnad H., Kazdová L., Pravenec M. Effects of metformin on tissue oxidative and dicarbonyl stress in transgenic spontaneously hypertensive rats expressing human C-reactive protein. PLoS One. 2016;11:e0150924. doi: 10.1371/journal.pone.0150924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez N., Athonvarangkul D., Singh R. Autophagy and aging. Adv. Exp. Med. Biol. 2015;847:73–87. doi: 10.1007/978-1-4939-2404-2_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick J.J., VanDusseldorp T.A., Ulrich C.G., Lanphere R.L., Dokladny K., Mosely P.L., Mermier C.M. The effect of aging on the autophagic and heat shock response in human peripheral blood mononuclear cells. Physiol. Int. 2018;105:247–256. doi: 10.1556/2060.105.2018.3.20. [DOI] [PubMed] [Google Scholar]
- Melser S., Lavie J., Bénard G. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta. 2015;1853:2812–2821. doi: 10.1016/j.bbamcr.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Menk A.V., Scharping N.E., Moreci R.S., Zeng X., Guy C., Salvatore S., Bae H., Xie J., Young H.A., Wendell S.G., Delgoffe G.M. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 2018;22:1509–1521. doi: 10.1016/j.celrep.2018.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller L.D., Rose M.R. Evolutionary theory predicts late-life mortality plateaus. Proc. Natl. Acad. Sci. USA. 1996;93:15249–15253. doi: 10.1073/pnas.93.26.15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas D., Proctor E.A., Raval F.M., Ip B.C., Habib C., Ritou E., Grammatopoulos T.N., Steenkamp D., Dooms H., Apovian C.M. Advances in the quantification of mitochondrial function in primary human immune cells through extracellular flux analysis. PLoS One. 2017;12:e0170975. doi: 10.1371/journal.pone.0170975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas D.A., Proctor E.A., Agrawal M., Belkina A.C., Van Nostrand S.C., Panneerseelan-Bharath L., Jones A.R., Raval F., Ip B.C., Zhu M. Fatty acid Metabolites combine with reduced beta oxidation to activate Th17 inflammation in human type 2 diabetes. Cell Metab. 2019;30:447–461.e5. doi: 10.1016/j.cmet.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel A.G., von Hardenberg A., Hohl M., Löffler J.R., Kohlhaas M., Becker J., Reil J.C., Kazakov A., Bonnekoh J., Stadelmaier M. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–484. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Palmer C.S., Ostrowski M., Balderson B., Christian N., Crowe S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015;6:1. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piber D., Olmstead R., Cho J.H., Witarama T., Perez C., Dietz N., Seeman T.E., Breen E.C., Cole S.W., Irwin M.R. Inflammaging: age and systemic, cellular, and nuclear inflammatory biology in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2019;74:1716–1724. doi: 10.1093/gerona/glz130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli V., Camporeale A. STAT3-mediated metabolic reprograming in cellular transformation and implications for drug resistance. Front. Oncol. 2015;5:121. doi: 10.3389/fonc.2015.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyadharshini B., Loschi M., Newton R.H., Zhang J.W., Finn K.K., Gerriets V.A., Huynh A., Rathmell J.C., Blazar B.R., Turka L.A. Cutting edge: TGF-beta and phosphatidylinositol 3-kinase signals modulate distinct metabolism of regulatory T cell subsets. J. Immunol. 2018;201:2215–2219. doi: 10.4049/jimmunol.1800311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A., Oliveira M.P., Khamoui A.V., Aparicio R., Rera M., Rossiter H.B., Walker D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017;8:448. doi: 10.1038/s41467-017-00525-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon M., Pereira F.V. A new perspective: mitochondrial Stat3 as a regulator for lymphocyte function. Int. J. Mol. Sci. 2018;19:E1656. doi: 10.3390/ijms19061656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohart F., Gautier B., Singh A., Lê Cao K.A. mixOmics: an R package for 'omics feature selection and multiple data integration. PLoS Comput. Biol. 2017;13:e1005752. doi: 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubenoff R., Harris T.B., Abad L.W., Wilson P.W., Dallal G.E., Dinarello C.A. Monocyte cytokine production in an elderly population: effect of age and inflammation. J. Gerontol. A Biol. Sci. Med. Sci. 1998;53:M20–M26. doi: 10.1093/gerona/53a.1.m20. [DOI] [PubMed] [Google Scholar]
- Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr. Metab. Immune Disord. Drug Targets. 2015;15:196–205. doi: 10.2174/1871530315666150316124019. [DOI] [PubMed] [Google Scholar]
- Simon A.R., Rai U., Fanburg B.L., Cochran B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998;275:C1640–C1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- Smith L.M., Yao-Borengasser A., Starks T., Tripputi M., Kern P.A., Rasouli N. Insulin resistance in African-American and Caucasian women: differences in lipotoxicity, adipokines, and gene expression in adipose tissue and muscle. J. Clin. Endocrinol. Metab. 2010;95:4441–4448. doi: 10.1210/jc.2010-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Youle R.J., Finkel T. The mitochondrial basis of aging. Mol. Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymańska E., Saccenti E., Smilde A.K., Westerhuis J.A. Double-check: validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics. 2012;8:3–16. doi: 10.1007/s11306-011-0330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenot E.A., Roux A., Xu Y., Ezan E., Junot C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome. Res. 2015;14:3322–3325. doi: 10.1021/acs.jproteome.5b00354. [DOI] [PubMed] [Google Scholar]
- Ungvari Z., Labinskyy N., Mukhopadhyay P., Pinto J.T., Bagi Z., Ballabh P., Zhang C., Pacher P., Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1876–H1881. doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt G.J.W., Chang C.H., Pearce E.L. Measuring bioenergetics in T cells using a Seahorse extracellular flux analyzer. Curr. Protoc. Immunol. 2016;113:3.16B.1–3.16B.14. doi: 10.1002/0471142735.im0316bs113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereb G., Matkó J., Vámosi G., Ibrahim S.M., Magyar E., Varga S., Szöllosi J., Jenei A., Gáspár R., Jr., Waldmann T.A., Damjanovich S. Cholesterol-dependent clustering of IL-2Ralpha and its colocalization with HLA and CD48 on T lymphoma cells suggest their functional association with lipid rafts. Proc. Natl. Acad. Sci. USA. 2000;97:6013–6018. doi: 10.1073/pnas.97.11.6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Dillon C.P., Shi L.Z., Milasta S., Carter R., Finkelstein D., McCormick L.L., Fitzgerald P., Chi H., Munger J., Green D.R. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Feng X., Zeng Y., Fan J., Wu J., Li Z., Liu X., Huang R., Huang F., Yu X., Yang X. Lipopolysaccharide (LPS)-induced autophagy is involved in the restriction of Escherichia coli in peritoneal mesothelial cells. BMC Microbiol. 2013;13:255. doi: 10.1186/1471-2180-13-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Laurence A., Elias K.M., O'Shea J.J. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J. Biol. Chem. 2007;282:34605–34610. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Laurence A., O'Shea J.J. New insights into the roles of Stat5a/b and Stat3 in T cell development and differentiation. Semin. Cell Dev. Biol. 2008;19:394–400. doi: 10.1016/j.semcdb.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Long L., Yang K., Guy C., Shrestha S., Chen Z., Wu C., Vogel P., Neale G., Green D.R., Chi H. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016;17:277–285. doi: 10.1038/ni.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Su W., Liu S., Zhao G., Esser K., Schroder E.A., Lefta M., Stauss H.M., Guo Z., Gong M.C. Smooth-muscle BMAL1 participates in blood pressure circadian rhythm regulation. J. Clin. Invest. 2015;125:324–336. doi: 10.1172/JCI76881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosz E.L., Chang C.H. The role of reactive oxygen species in regulating T cell-mediated immunity and disease. Immune Netw. 2018;18:e14. doi: 10.4110/in.2018.18.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- You L., Wang Z., Li H., Shou J., Jing Z., Xie J., Sui X., Pan H., Han W. The role of STAT3 in autophagy. Autophagy. 2015;11:729–739. doi: 10.1080/15548627.2015.1017192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapała B., Kaczyński Ł., Kieć-Wilk B., Staszel T., Knapp A., Thoresen G.H., Wybrańska I., Dembińska-Kieć A. Humanins, the neuroprotective and cytoprotective peptides with antiapoptotic and anti-inflammatory properties. Pharmacol. Rep. 2010;62:767–777. doi: 10.1016/s1734-1140(10)70337-6. [DOI] [PubMed] [Google Scholar]
- Zhi L., Ustyugova I.V., Chen X., Zhang Q., Wu M.X. Enhanced Th17 differentiation and aggravated arthritis in IEX-1-deficient mice by mitochondrial reactive oxygen species-mediated signaling. J. Immunol. 2012;189:1639–1647. doi: 10.4049/jimmunol.1200528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the study are included with the published manuscript (and Supplemental Information). All other data are available from the corresponding author upon request.