

Abstract
Background/Objectives
Patients referred for HNF1B testing present very heterogeneous phenotypes. Despite suggestive characteristics, many do not harbor mutations in HNF1B. Our objective was to evaluate the clinical characteristics of probands referred for HNF1B genetic testing through a nationwide monogenic diabetes screening program.
Methods
Probands tested for HNF1B mutations in the 2005‐2018 period (N = 50) were identified in the Polish Monogenic Diabetes Registry, which prospectively recruits primarily pediatric patients and their families on a nationwide scale. Variants that had been reported pathogenic were reassessed using criteria of the American College of Medical Genetics and Genomics (ACMG). A structured medical interview was performed with all available individuals, their parents, and/or their physicians. For each patient, HNF1B score was calculated based on available clinical information.
Results
The study group numbered 36 unrelated probands (28% lost to follow‐up): 14 with pathogenic or likely‐pathogenic variants in HNF1B, one with a variant of uncertain significance, and 21 negative for HNF1B mutations. Presence of cystic kidneys (OR = 9.17, 95% CI:1.87‐44.92), pancreatic abnormalities (OR = 15, 95% CI:1.55‐145.23), elevated liver enzymes (OR = 15, 95% CI:1.55‐145.23) best discriminated HNF1B‐positive cases from the negative ones. Presence of impaired glucose tolerance coupled with kidney disease in the proband and one parent was also highly predictive for HNF1B mutations (OR = 11.11, 95% CI:1.13‐109.36). HNF1B‐score with recommended cutoff distinguished patients with and without HNF1B findings with 100% sensitivity and 47.6% specificity. Addition of four clinical variables to select patients based on HNF1B score improved specificity to 71.4% (95% CI:47.8%‐88.7%) while retaining 100% sensitivity.
Conclusions
Detailed medical interview may enable more accurate patient selection for targeted genetic testing.
Keywords: cystic kidneys, diabetes mellitus, genetic testing, MODY, phenotype
1. INTRODUCTION
Maturity onset diabetes of the young (MODY) is a rare genetic disease with estimated prevalence of 7.52/100000 among children and adolescents,1 and is responsible for up to 4% of all cases of pediatric diabetes.2, 3 Low prevalence prevents genetic testing to be routinely performed in all patients with new onset diabetes. Typically, testing for monogenic diabetes (MD) is carried out in preselected group of patients with clinical features of MD phenotype. This sufficiently limits the population making genetic screening cost‐efficient and easier to interpret through linking the genetic findings with clinical characteristics.
Autosomal dominant mutations in HNF1B cause up to 1%‐2% of MODY cases.3, 4 Pathogenic single nucleotide substitutions, indels, and exon deletions in HNF1B comprise about half of findings in patients with HNF1B‐MODY.5 In the remaining cases the syndrome is caused by heterozygous deletions of the entire gene, frequently as a part of a 17q12 microdeletion that spans 1.5 Mb and 14 other genes.6 Noteworthy, familial history of the disease is often non‐specific or unremarkable, as pathogenic mutations of HNF1B occur spontaneously (de novo) equally often as they are inherited.7
HNF1B‐MODY is a systemic disorder with a very variable clinical presentation which can differ between the carriers of the same mutation, also among affected members from the same family.8 Renal abnormalities and specifically cystic kidney disease (CKD), are the most consistent clinical feature for patients with HNF1B mutations.5, 9 Early‐onset diabetes, present in 34%‐45% of carriers, is the second most common symptom.5, 6, 9 Thus, HNF1B‐MODY is also classified as renal cysts and diabetes syndrome (RCAD). The renal phenotype displays vast heterogeneity in terms of clinical symptoms, ranging from renal cysts, familial hypoplastic glomerulocystic kidney disease, renal malformations (ie, single or horseshoe kidney) to atypical nephropathy.7, 10 Similar variability applies to disorders of glucose metabolism—although most of the patients develop diabetes after 25 years of age,6 the range may span from 15 days to 61 years.7 Other symptoms reported in HNF1B mutation carriers include neurological features, abnormal liver function, pancreatic hypoplasia, genital tract malformation, hypomagnesaemia, hyperuricaemia, early‐onset gout5 or pectus excavatum.11
Preselection of patients toward HNF1B testing is a difficult task due to the low prevalence of the syndrome, variable clinical presentation, and the high rate of de novo mutations (no family history). To aid the process, a clinical HNF1B score was developed by Faguer et al12 based on the most discriminatory features of the RCAD syndrome in literature. To date, the score was validated only in French and British populations.5, 12
Here we present a cross‐sectional study of unrelated RCAD probands recruited from the Polish Monogenic Diabetes Registry.1, 3 We describe the mutations identified in the Polish population re‐evaluated according to the ACMG criteria, summarize phenotypes of mutation carriers, and compare them to patients with negative results searching for the most discriminative features. By retrospectively calculating the HNF1B‐score on this cohort, we assess its performance in the Polish population.
2. METHODS
2.1. Subjects recruitment protocol
The recruitment was based on the patients recorded in the Polish Monogenic Diabetes Registry3 between 2005 and 2018. The Registry tracks the prevalence of monogenic diabetes (MD) in Poland based on a nationwide screening program and is focused on pediatric cases. In Poland diabetes care for underage patients is regionally centralized which eases the patients' referral to genetic centers. However, adult patients or relatives of children with diabetes are also included in the registry and are referred for genetic testing based on their clinical presentation. Thus, when the search for patients with the phenotype in question was performed, the Registry included majority of referred‐as‐children patients (N = 802) and some adult probands (N = 100). Among those we identified the patients tested for mutations in HNF1B. Each referral was based on constellation of impaired glucose tolerance (IGT) and kidney disease, or family history for these conditions, warranting suspicion of the RCAD syndrome. We approached probands who had provided valid consent for contact at the time of genetic testing, as well as their referring physicians. First, we retrieved all available information from the referral time‐point, then the patients underwent a structured medical interview focused on RCAD‐related symptoms. In terms of renal presentation, due to inconsistent reports from ultrasound imaging performed across the whole country by different specialists and at different ages of the evaluated patients, we were able to distinguish only between the presence of singular renal cysts and cystic kidneys. The study protocol was approved by the Bioethics Committee of the Medical University of Lodz (RNN/5/08/KE, RNN/55/06/KE). Parents or guardians of all study participants gave their informed consent.
2.2. Molecular methods
Details on the molecular methods used in the referral period were described earlier.1, 3 Briefly, DNA was isolated from peripheral blood cells using QiaAmp DNA mini kits from Qiagen (Hilden, Germany) and suspended in EDTA buffer. Since 2017, DNA isolation has been performed using automated Maxwell system (Promega, Madison) according to manufacturer's protocol. Point mutations were identified using direct Sanger sequencing of HNF1B exons (years 2005‐2015) or targeted deep‐sequencing on Illumina NextSeq 550 platform (2015‐2018), using either SureSelect (Agilent, Santa Clara) or TruSight One (Illumina, San Diego) assay. Variants detected by deep‐sequencing were subsequently validated by Sanger sequencing. Deep‐sequencing data processing and variant calling was done in Illumina BaseSpace (Illumina) and variant analysis in VariantStudio (Illumina). Copy number alterations were identified by multiplex ligation‐dependent probe amplification (MLPA). A commercially available set MODY‐P241 (MRC‐Holland, the Netherlands) was used according to the manufacturer's protocol. Details of the primer set and protocol are available at the manufacturer's website (http://www.mrcholland.com). Pathogenicity of identified mutations was evaluated using the ACMG criteria.13
2.3. Data analysis
Continuous characteristics were compared between the groups using Mann‐Whitney's U test. For dichotomous variables, odds ratios (ORs) together with corresponding 95% confidence intervals (95% CIs) were calculated. Subsequently, literature‐based HNF1B score12 was calculated by weighing a subset of clinical characteristics. Its utility in the studied group was assessed (for the recommended cutoff of ≥ 8 points) using receiver operating characteristic (ROC) curve to calculate sensitivity and specificity. Furthermore, we attempted to calibrate the HNF1B score for our population. Additional queries to the recommended cutoff of HNF1B score were added, forming a decision tree protocol for inclusion of patients for HNF1B testing. This was done using classification and regression tree (CART) method with relative cost of missing a patient with HNF1B pathogenic mutation set to 300% of a false positive case. We used HNF1B score ≥ 8 and all significant findings from univariate analysis as predictors for the multivariate model. Statistical analysis was carried out in STATISTICA 13.1 (TIBCO Software, Palo Alto, CA). P‐values ≤.05 were considered statistically significant.
3. RESULTS
3.1. Study group and genetic results
In the considered time frame 2254 individuals were tested for monogenic diabetes. Among them, 50 unrelated probands were referred for HNF1B testing (Figure 1). Of those, 14 (28%) were lost to follow‐up due to lack of consent or contact details. Eventually, the studied group included 36 probands of Polish ethnicity.
Figure 1.
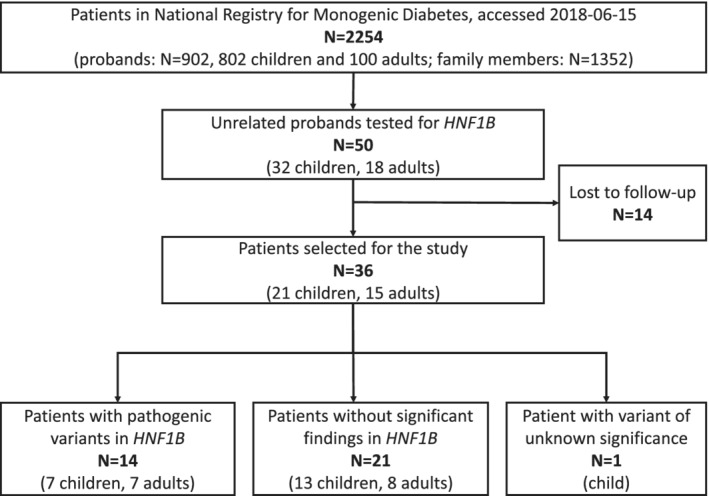
Patient recruitment for the study. Overall, we managed to reach 72% of patients referred for HNF1B testing from the Registry and 87.5% of those with relevant HNF1B findings
The study group included 17 males (48.6%) and was heterogeneous in terms of age (from <1 years old (y.o.) to 58‐y.o. at referral, median age 14.2‐y.o., IQR (interquartile range): 7.8 y.o. to 21.1 y.o.). Detailed characteristics of the patients are provided in Table S1. Fourteen (N = 14) patients harbored pathogenic variants in HNF1B, 21 were negative for pathogenic findings in HNF1B, and 1 carried a variant of unknown significance (VUS). This patient was excluded from the downstream clinical analysis. Two of identified pathogenic mutations have not been previously reported: a missense substitution (c.857 T > G, p.Leu286Arg) and a single nucleotide deletion leading to a frameshift (c.434delT, p.Leu145ArgfsTer16). The Leu286Arg variant occurred de novo, in a homeobox domain of the HNF1B transcription factor, and was not present in 1000 Genomes14 nor gnomAD15 databases. The frameshift variant at Leu145 position introduced a termination codon at position 171, likely causing nonsense‐mediated decay of the gene product. Interestingly, the frameshift started at the amino acid where a pathogenic L145Q substitution was previously reported.16 The list of genetic findings is provided in Table 1 together with pathogenicity evaluation.
Table 1.
Summary of HNF1B mutations
| GRCh37 | Evidence | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Carrier ID | coordinates | Nucleotide change | Protein change | Classification | PVS | PS | PM | PP | |
| 9,15,19,22,28,31,34,36 | ‐ | Whole‐gene deletion | Whole‐gene deletion | pathogenic | 1 | 4 | |||
| 25 | 17:36099540 T/‐ | NM_000458.2 c.434delT | NP_000449.1 p.Leu145Pro_fs_delT | pathogenic | 1 | 2 | 3 | ||
| 10 | 17:36099532 C/T | NM_000458.2 c.443C>T | NP_000449.1 p.Ser148Leu | pathogenic | 1,2 | 2 | 2,3 | 7, 18 | |
| 2 | 17:36093617 C/T | NM_000458.2 c.742C>T | NP_000449.1 p.Gln248Ter | pathogenic | 1 | 1 | 2 | 1 | 8 |
| 16 | 17:36091774 T/G | NM_000458.2 c.857T>G | NP_000449.1 p.Leu286Arg | pathogenic | 2 | 1,2,5 | 2,3 | 22 | |
| 8,20 | 17:36091748 C/T | NM_000458.2 c.883C>T | NP_000449.1:p.Arg295Cys | pathogenic | 1,2 | 1,2,5 | 2,3 | 21, 23 | |
| 30 | 17:36093604 G/A | NM_000458.2 c.755G>A | NP_000449.1 p.Arg252Gln | uncertain | 1 | 2,3,5 | |||
Note: All variants were assessed according to the criteria issued by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Briefly, 14 of 15 discovered variants in HNF1B were classified as pathogenic, with whole‐gene deletions constituting about half of the findings. PVS‐very strong evidence of pathogenicity; PS‐strong evidence; PM‐moderate; PP‐supportive. Numbers indicate specific ACMG criteria.13
Given the national scope of the Registry and its comprehensive coverage of pediatric population, it can be expected that patients diagnosed below 20 y.o. represent virtually all cases in Poland in this age group. Thus, we estimated the lower boundary of prevalence of HNF1B‐MODY in children and young adults (< 20 y.o.) in consecutive years based on available data from Central Statistical Office. Before the year 2010, there were no patients with confirmed HNF1B‐MODY in this age‐group in Poland, and in 2018 the prevalence reached 0.74 / 1 000 000 children (95% CI: 0.30‐1.66) (Figure S1).
3.2. Clinical presentation of selected cases
Phenotypes of patients with novel HNF1B mutations were consistent with HNF1B‐MODY. The proband with missense substitution (p.Leu286Arg, #16) suffered from diabetes, cystic kidneys, and had reported pancreas hyperechogenicity in abdominal ultrasound examination. The carrier of the other novel mutation (p.Leu145Argfs, #25) presented with diabetes, cystic kidneys, and additional genital system malformations: bicornuate uterus, hypoplasia of vaginal part of uteri cervix, hypoplasia of vagina, transverse vaginal septum.
In case of patient #10, a carrier of Ser148Leu substitution which is one of the more commonly reported pathogenic substitutions in HNF1B,9 we note symptoms new to this mutation. The Ser148Leu substitution has been associated with low birth weight, neonatal diabetes, transient neonatal diabetes with recurrence in later life and diabetes diagnosed in adolescence, dysplastic kidneys and associated renal failure, various stages of pancreatic atrophy, and pancreatic exocrine insufficiency (in some cases).7, 17, 18, 19 Our proband partly fits this description (Table S1) with low birth weight, progressive renal failure (at 9 months) and (transient) pancreatic exocrine insufficiency, but expands it with liver fibrosis (4 y.o.), impaired fasting glucose (10 y.o.) and lack of renal or pancreatic dysplasia/hypoplasia. According to our knowledge, the patient currently does not present cirrhosis‐associated abnormalities (no esophageal varices, no signs of portal hypertension, only slightly elevated prothrombin time).
Two unrelated probands (#8, #20) presented with the same aminoacid substitution (Arg295Cys), but divergent clinical presentations demonstrating high variability of HNF1B‐MODY phenotype, even when caused by the same genetic defect. Patient #8 was male, diagnosed with diabetes at the age of 32 (currently he is treated with insulin ~0.9UI kg−1day−1) together with pancreatic atrophy, calcification and exocrine insufficiency, cystic kidneys and elevated liver enzymes; additional reported conditions included non‐specific rectal inflammation and atypical pigmented lesions in the eye. His son developed diabetes at the age of 18, daughter had impaired fasting glucose, father had unspecified IGT, but they were negative for pancreatic, renal, or liver findings. In the pedigree of proband #20, we observed divergent phenotypes of diabetes within the family. The proband was diagnosed with diabetes at the age of 12, currently treated with low‐dose insulin ~0.1UI kg−1day−1, whereas the father diagnosed with diabetes at the age of 29, treated with sulphonylureas. At the same time this family showed a remarkable similarity of extra‐diabetic symptoms—the proband developed cystic kidneys accompanied by a vesicoureteral reflux and chronic pyelonephritis, pancreatic hypoplasia, hepatic steatosis with elevated liver enzymes and abnormalities of the hepatic biliary ducts. The father reported biliary duct pathology and chronic liver insufficiency, two singular cysts in kidneys, history of chronic pyelonephritis in childhood and stage I chronic kidney disease.
Finally, two patients with confirmed HNF1B mutation had detectable autoantibodies (ICA, GAD and ICA, GAD, IAA, respectively) and low C‐peptide concentrations, indicating mixed pathogenesis of diabetes, attributable both to the presence of HNF1B mutation and autoimmunity. However, data for other individuals and precise antibody titers were not available. The first case was a girl (with whole‐gene deletion of HNF1B, #9) who presented with diabetes at 11 y.o., with glycemia ~300 mg/dL, HbA1c concentration of 8%, and low but detectable concentration of C‐peptide (0.2 nmol/L with N: 0.4‐1.2 nmol/L). She was positive for ICA, GAD, and IAA autoantibodies. Currently, she is a non‐obese adult (BMI = 17.6 kg/m2) treated with insulin (daily dose ~1UI kg−1day−1). Second case (L145delT, #25) was a young woman diagnosed with diabetes at the age of 29 years. At diabetes onset, she presented with glycemia ~750 mg/dL, HbA1c = 8.8%, low C‐peptide (0.25 nmol/L) and was positive for ICA and GAD autoantibodies. Currently she is obese (BMI = 32 kg/m2) and treated with medium‐dose of insulin (daily ~0.3‐0.4UI/kg).
3.3. Comparison of HNF1B‐positive and negative probands
Basic clinical comparison of positive and negative cases was presented in Table 2. The group of 14 HNF1B‐mutation carriers consisted of 7 males and 7 females, mainly teenagers and young adults (median age at referral 17.5 y.o. [IQR: 12 to 22.1 y.o.]). Almost all (N = 13, 92.9%) suffered from IGT—diabetes (N = 12, 85.7%) or impaired fasting glucose (IFG; N = 1, 7.1%) at referral. Kidney disease and malformations were equally prevalent (N = 13, 92.9%), most often presenting as cystic kidneys (N = 11, 78.6%) and chronic kidney disease (N = 4, 28.6%). Genitourinary malformations and abnormalities were also noted, however, at lower prevalence (N = 5, 35.7%; ureteral stenosis, vesicoureteral reflux, cysts in epididymis, bicornuate uterus, and others). In some patients two or more abnormalities coexisted, resulting in a complex phenotype.
Table 2.
Comparison of basic clinical features between probands diagnosed with HNF1B‐MODY and those negative for HNF1B mutations
| Feature | HNF1B‐negative | HNF1B‐positive | P‐value |
|---|---|---|---|
| Categorical variables (N [%]) | |||
| Gender (Male) | 10 (47.6%) | 7 (50%) | 1.0000 |
| Cystic kidneys | 6 (28.6%) | 11 (78.6%) | .0059 |
| Diabetes | 17 (81%) | 12 (85.7%) | 1.0000 |
| Continuous variables (median [25%‐75%]) | |||
| Age at referral (years) | 12.7 (4.5‐17.4) | 17.5 (12.0‐22.1) | .1097 |
| Age of diabetes diagnosisa(years) | 12.4 (5.1‐15.9) | 16.3 (13.0‐25.4) | .0661 |
| HbA1c at diabetes diagnosisb (%) | 8.8 (7.3‐10.0) | 7.9 (7.4‐8.7) | .3347 |
| Last known HbA1c (%) | 6.6 (6.1‐7.5) | 6.3 (5.6‐7.1) | .3428 |
| HNF1B score | 8.0 (6.0‐10.0) | 14.0 (10.0‐16.0) | .0006 |
Note: Significant (P < .05) differences were bolded. HNF1B‐mutation carriers presented more often with cystic kidneys, were non‐significantly older at referral and at diabetes diagnosis, and reached higher HNF1B scores.
Only for those with diagnosed diabetes (N = 12 for HNF1B‐positive and N = 17 for HNF1B‐negative cases).
Data available for 13/17 probands in HNF1B‐negative and 6/12 in HNF1B‐positive group.
Data available for 16/17 probands in HNF1B‐negative and 10/12 in HNF1B‐positive group.
Of the 14 HNF1B‐mutation carriers, half were referred for genetic testing as children. The carriers and non‐carriers were of comparable gender structure (50% vs 47.6% males, P = 1.000, Table 2). The age of referral was insignificantly higher in those with confirmed HNF1B‐MODY [median 17.5 y.o. (12.0‐22.1 y.o.) vs 12.7 y.o. (4.5‐17.4 y.o.), P = .1097]. Consistently with this, we observed that the carriers of pathogenic mutations in HNF1B were diagnosed with diabetes slightly later [16.3 y.o. (13.0 y.o. ‐ 25.4 y.o.) vs 12.1 y.o. (2.4‐15.9 y.o.), P = .0661]. None of the HNF1B‐mutation carriers with diabetes (0/12) had diabetic ketoacidosis (DKA) at diabetes diagnosis in comparison to 6/17 (35.3%; P = 0.0280) of diabetic non‐carriers. Furthermore, there were no significant differences in glycated hemoglobin concentrations (HbA1c) at diagnosis [7.9% (7.4% ‐ 8.7%) vs 8.8% (7.3%‐10.0%), P = .3340]. Follow‐up current glycemic control was also similar between carriers and non‐carriers (6.1% [5.6%‐6.9%] vs 6.5% [6.1%‐7.3%], P = .5829). More extensive data from the diabetes diagnosis (such as BMI or initial treatment) was not available for all patients, making it difficult to compare statistically. However, at referral 3/9 HNF1B‐negative and 3/9 HNF1B‐positive cases with relevant data were overweight or obese. Among negative cases, 7/11 patients with IGT were on insulin, 1 was treated with oral hypoglycemic agents, and 3 with diet only. In the group of HNF1B‐positive patients 5/9 used insulin, 1 oral hypoglycemic agents, 2 both insulin and oral hypoglycemic agents, and 1 remained on diet only.
In univariate analysis (Figure 2), three conditions were significantly overrepresented among HNF1B‐mutation carriers: pancreatic abnormalities other than diabetes (including atrophy of pancreas, calcification, and exocrine insufficiency) (OR = 15 [95% CI: 1.55‐145.23]), cystic kidneys (OR = 9.17 [95% CI: 1.87‐44.92]) and elevated liver enzymes (OR = 15, [95% CI: 1.55‐145.23]). IGT and kidney disease in two consecutive generations (ie, proband + parent) distinguished 5 (35.7%) of HNF1B‐mutation carriers from the non‐carriers (OR = 11.11, 95% CI: 1.13‐109.36). No other symptom showed significant discriminatory value.
Figure 2.
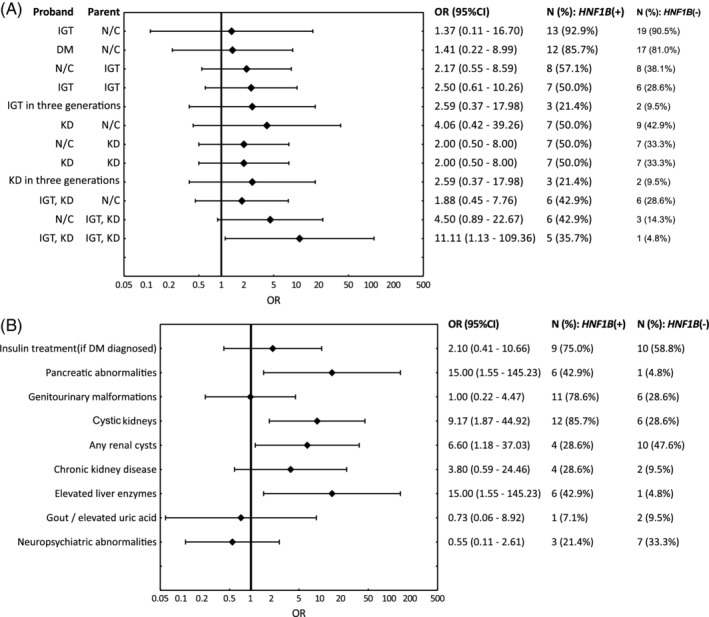
Associations between carrier status and family history (A), and probands' characteristics (B). The forest plot depicts odds ratios (ORs) for each feature (diamonds) together with 95% confidence intervals. IGT, impaired glucose tolerance (impaired fasting glucose or diabetes); DM, diabetes mellitus; KD, kidney disease (includes structural malformations and functional disorders); N/C, not considered for the comparison
For each patient, HNF1B score was calculated. Pathogenic variant carriers presented median score of 14 (25%‐75%: 10‐16) points—a value significantly higher than in the non‐carrier group (median 8 [25‐75%: 6‐10]). Notably, non‐carriers displayed a wide range of HNF1B scores (6 to 14 points). The cut‐off value recommended by the authors (≥8) correctly identified all HNF1B‐mutation carriers yielding 100% sensitivity (95% CI: 76.8% to 100.00%), 47.6% specificity (95% CI: 25.7%‐70.2%), and AUC = 0.847 (95% CI: 0.721‐0.973) (Figure 3). To further improve specificity of the score, a decision tree was constructed. The final model (Figure 4) retained the default HNF1B score cutoff (≥ 8), and included symptoms found discriminatory in our cohort—elevated liver enzymes, diabetes, and kidney disease in proband and parent, presence of pancreatic abnormalities, and cystic kidneys. The refined classification improved specificity to 71.4% (95% CI: 47.8%‐88.7%) retaining 100% sensitivity.
Figure 3.
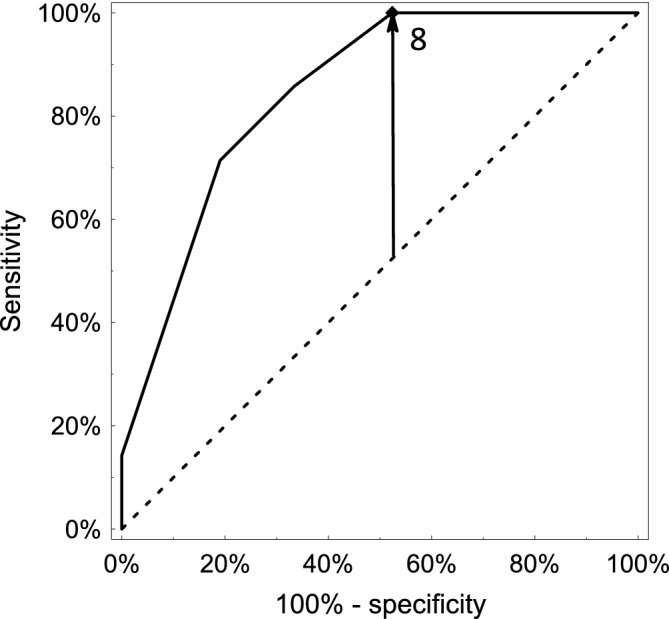
ROC (Receiver operating characteristic) curve for the HNF1B score as a discriminator between HNF1B‐mutation carriers and non‐carriers referred for HNF1B genetic testing based on clinical suspicion. Overall, in the studied group, the HNF1B score demonstrated good performance with area under the curve (AUC) equal to 0.847 (95% CI: 0.721‐0.973)
Figure 4.
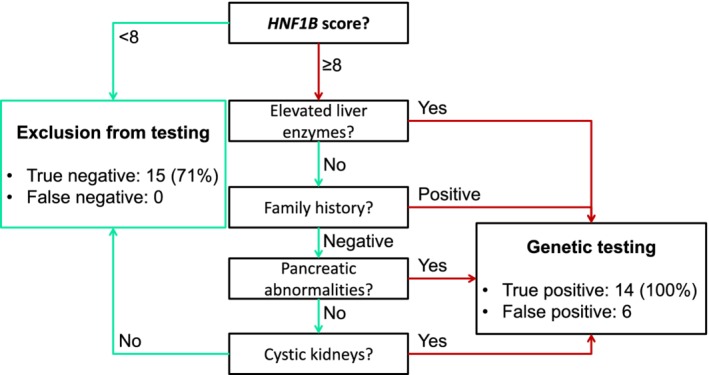
Data‐driven decision tree for streamlining patients for HNF1B genetic testing. Among patients with HNF1B score of at least eight points in our cohort, lack of characteristic features of HNF1B‐MODY could have been used to exclude patients from genetic testing without any loss in sensitivity
4. DISCUSSION
We summarized phenotypes and HNF1B‐related genetic findings in a cohort of patients with HNF1B‐MODY phenotype recruited through monogenic diabetes screening. This is the first such report in the Polish population.
In a long‐term analysis, we observe an increase in the prevalence of HNF1B‐MODY from 0 before establishing the registry (year 2010), to relatively stable 0.75/1 million in 2017 and 2018. By cross‐referencing these data with the prevalence of diabetes among Polish children (data from 2005 to 2011:138/100000 for type 1 and 1.01/100000 for type 2 in ≤18 y.o.),3 we estimate HNF1B mutations to be responsible for at least 0.05% diabetes cases among children, adolescents, and young adults. This estimate matches recently reported 0.05% (35/76836) prevalence of HNF1B‐MODY in multinational diabetes registry Diabetes‐Patienten‐Verlaufsdokumentation,4 suggesting good efficacy of our nationwide screening program, and demonstrating that the study group is representative for the Polish population.
Pathogenic mutations in HNF1B were found in 14 of 36 (38.9%) patients selected for screening because of coexisting diabetes and kidney disease or a positive family history of these disorders. Including patients lost to follow‐up, 14 of the tested 50 (28%) were carriers of HNF1B mutations. This rate is lower than the over 40% diagnostic yield of MODY at our facility1, 3 highlighting the difficulty in patient preselection for this MODY subtype. HNF1B‐carrier detection rates reported for cohorts selected based on renal diseases are typically even lower, with few studies exceeding 25%.5, 20 This is most likely due to less specific inclusion criteria.
In line with previous reports, whole‐gene deletions were identified in approximately half (8/14; 57%) of the carriers of pathogenic HNF1B mutations.5, 12, 21, 22 The remaining causative variants were point mutations, two of which expand the catalog of over 50 known pathogenic HNF1B variants.5 Noteworthy, all mutations previously reported to patients as pathogenic were classified as pathogenic using current ACMG criteria.13
The frequency of individual symptoms in populations of HNF1B mutation carriers is heavily dependent on the selection criteria (phenotype, age).5 Cohorts selected based on renal disease have much lower frequency of diabetes.5, 12, 22, 23 On the other hand, our patients were recruited primarily from diabetology centers, which constrains phenotype‐frequency comparisons to similarly‐recruited cohorts.6 When compared with a much larger French cohort of HNF1B‐MODY patients,6 our group presented similar frequencies of the most commonly observed phenotypes—kidney malformations and disease (92% vs 91% of kidney morphological disorders) and diabetes (85.7% vs 82%). Frequency of genital tract malformations was insignificantly lower among our patients (35% vs 58%). Despite relatively high frequency of morphological abnormalities of the pancreas (62%) and pancreatic exocrine dysfunction (76%) in the French, we noted only 5/14 (36%) of both abnormalities in our cohort. Those discrepancies might be the result of under‐diagnosis of these conditions in Polish population rather than genuine differences, or stem from the adult age of the participants in the French study.
Consistently with a variable phenotypic spectrum of RCAD, we observed a wide range of symptoms in our cohort. However, only the presence of cystic kidneys, pancreatic abnormalities (despite relatively low frequency), elevated liver enzymes, and significant family history (ie, IGT and kidney disease in the proband and the parent) was highly indicative of HNF1B defects. We suggest that an initial screen for such abnormalities in young‐onset diabetes cases might be helpful in preselecting patients for HNF1B testing. Such cases are usually hospitalized at diagnosis and basic liver enzyme tests or abdominal ultrasound are often routinely performed. These data would greatly improve clinical utility of the HNF1B score, a framework aiming at quantifying presence of HNF1B‐associated symptoms.12 Performance of the HNF1B score has been initially tested in a French cohort of patients (total N = 433, HNF1B carriers—56) with reported sensitivity of 98.2%, specificity 41.1%, and negative predictive value over 99%. Later, it was validated in the UK cohort showing 80% sensitivity, 31% PPV, 38% specificity, and 85% NPV. Compared to these studies, our analysis represents a small‐scale re‐evaluation of HNF1B score in a nationwide cohort. Nevertheless, the results are more akin to those reported by Faguer et al suggesting that the score is a very effective tool for preselection of patients for HNF1B testing.
A few carriers of pathogenic HNF1B mutations presented with strong evidence that diabetes might not be entirely attributable to HNF1B‐mutations. High glucose levels at onset, presence of autoantibodies and low C‐peptide levels were more key features of type 1 diabetes, and it is likely that autoimmune diseases coincided with HNF1B defect and affected diabetes phenotype. These cases presented both features of autoimmune disease (type 1 diabetes or latent autoimmune diabetes in adults) and MODY, which makes distinguishing the underlying cause extremely difficult. This highlights the need to assess all the phenotypic features together instead of focusing on single traits, as for instance confident diagnosis of type 1 diabetes might lead to exclusion of a patient from genetic testing.
Finally, 21/36 probands (35/50 including those lost to follow‐up) did not harbor mutations in HNF1B despite suggestive phenotypes. Owing to targeted deep sequencing that has been used in diagnostic testing at our facility since 2015, we were able to identify other mutations related to the RCAD phenotype in four of those HNF1B‐negative individuals. The variants included nonsense substitutions in GCK (p.Lys90Ter) and PKD1 (p.Trp2587Ter), and missense mutations in KCNJ11 and HNF4A (Table S2). Each was present in one individual, explaining either the diabetic (GCK, HNF1A, KCNJ11) or renal phenotype (PKD1), but not both. It is possible that in these cases the other features could be explained by aberrations of other genes, not included on the MODY‐focused sequencing panel, or non‐Mendelian traits (eg, type 1 or type 2 diabetes accompanying autosomal dominant polycystic kidney disease or MODY coinciding with simple renal cysts or other abnormalities). Such co‐existing monogenic phenotypes have been described before,24 also in relation to juvenile diabetes.25 Also, non‐coding changes, typically not interrogated by targeted‐ or exome‐sequencing, could be causative, as it has been shown for pancreatic‐agenesis26 and other disorders.27
By using CART and adding additional follow‐up queries to HNF1B score, we managed to improve the positive predictive value of patient selection. Although the features added to decision tree are already fully or partly represented in the score, extra emphasis on those conditions improved specificity in our setting. This observation must be confirmed on a larger scale and prospective studies, as the small size of our cohort and lack of replication bears the risk of overfitting. In connection with calculating the HNF1B score, we also see major advantages of rigorous phenotyping of RCAD patients prior to genetic testing. An accurate probability score can spare wide‐spectrum tests in the most typical cases. This is a valid point as genome‐wide assays are increasingly used as a first‐tier test.28, 29 Phenotype data at hand are also of great utility when assessing pathogenicity of variants, especially obtained in wider‐scope assays.
We acknowledge the limitations of the study. First, it was a follow‐up of patients included in the registry. All clinical data were gathered retrospectively, which resulted in missing data for some clinical features and underdiagnoses of certain symptoms. Patient recruitment was based primarily on the Monogenic Diabetes Registry, and the presented clinical discrimination between HNF1B‐positive and negative cases could be limited to similarly selected cohorts. Furthermore, patients diagnosed in the studied period followed different genetic testing protocols depending on time of testing and available technology. It is thus likely that the group negative for HNF1B mutations includes individuals with other monogenic defects that have not been discovered by Sanger sequencing of HNF1B. Finally, the size of the study group is modest when compared to other (often international) cohorts, and therefore can be representative only for the Polish population.
5. CONCLUSIONS
The Polish Monogenic Diabetes Registry succeeded in identifying a number of HNF1B‐MODY cases similar to those reported by other registries. The variable phenotype of the disorder and high prevalence of whole‐gene deletions necessitates careful clinical preselection for HNF1B‐testing, which can be improved by detailed medical interviews. Cystic kidneys, significant family history, pancreatic abnormalities and elevated liver enzymes were the best discriminating features, and coupled with a HNF1B score, identified the carriers of pathogenic variants in a sensitive and specific manner. Since the majority of patients clinically diagnosed with RCAD did not present HNF1B mutations, these patients are candidates for whole exome or whole genome screening rather than targeted sequencing.
AUTHOR CONTRIBUTIONS
P.S., W.F., M.B., W.M. designed the study, A.M., P.S., A.Z., A.H. collected the data, P.M, K.A., M.B. performed the experiments, A.M, P.S, B.M analyzed the data, A.M, P.S., B.M., W.F. wrote the article. All authors read and approved the final manuscript.
Supporting information
Supplementary Figure 1 HNF1B‐MODY prevalence in Polish population below 20 years of age ‐ estimated based on the Polish Monogenic Diabetes Registry. Diamonds mark prevalence for a given year and whiskers span over 95% confidence intervals. Owing to the growing awareness of referring physicians, an increasing number of patients is identified and diagnosed every year (r = .95, P < .0001).
Supplementary Table 1 Detailed characteristics of patients included in the study. *lack of more detailed data; DM ‐ diabetes mellitus
Supplementary Table 2 Summary of phenotype‐related non‐HNF1B genetic findings. All variants were assessed according to the criteria issued by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. PVS‐very strong evidence of pathogenicity; PS‐strong evidence; PM‐moderate; PP‐supportive. Numbers indicate specific ACMG criteria.
ACKNOWLEDGEMENTS
We express our gratitude to patients and their families participating in the study for their invaluable contribution. We would like to thank medical centers referring patients for testing at our facility, and physicians helping in collecting clinical data, in particular Joanna Chrzanowska, Małgorzata Stelmach, Dr Eliza Skała‐Zamorowska, Prof Małgorzata Myśliwiec and Prof Anna Materna‐Kiryluk. Project funded by National Science Center in Poland, grants no. 2016/23/P/NZ2/04251, 2014/15/B/NZ5/00144, 2014/15/B/NZ5/01579, 2015/19/B/NZ5/02243, 2018/29/B/NZ5/00330, 2016/21/N/NZ5/01448. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No. 665778.
Sztromwasser P, Michalak A, Małachowska B, et al. A cross‐sectional study of patients referred for HNF1B‐MODY genetic testing due to cystic kidneys and diabetes. Pediatr Diabetes. 2020;21:422–430. 10.1111/pedi.12959
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/pedi.12959.
Funding information Narodowe Centrum Nauki, Grant/Award Numbers: 2014/15/B/NZ5/00144, 2014/15/B/NZ5/01579, 2015/19/B/NZ5/02243, 2016/21/N/NZ5/01448, 2016/23/P/NZ2/04251, 2018/29/B/NZ5/00330; This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie, Grant/Award Number: 665778
Contributor Information
Paweł Sztromwasser, Email: pawel.sztromwasser@umed.lodz.pl.
Wojciech Fendler, Email: wojciech.fendler@umed.lodz.pl.
REFERENCES
- 1. Małachowska B, Borowiec M, Antosik K, et al. Monogenic diabetes prevalence among polish children—summary of 11 years‐long nationwide genetic screening program. Pediatr Diabetes. 2018;19(1):53‐58. 10.1111/pedi.12532. [DOI] [PubMed] [Google Scholar]
- 2. Shepherd M, Shields B, Hammersley S, et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care. 2016;39(11):1879‐1888. 10.2337/dc16-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fendler W, Borowiec M, Baranowska‐Jazwiecka A, et al. Prevalence of monogenic diabetes amongst polish children after a nationwide genetic screening campaign. Diabetologia. 2012;55(10):2631‐2635. 10.1007/s00125-012-2621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Warncke K, Kummer S, Raile K, et al. Frequency and characteristics of MODY 1 (HNF4A mutation) and MODY 5 (HNF1B mutation): analysis from the DPV database. J Clin Endocrinol Metab. 2018;104(3):845‐855. 10.1210/jc.2018-01696. [DOI] [PubMed] [Google Scholar]
- 5. Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B‐associated renal and extra‐renal disease—an expanding clinical spectrum. Nat Rev Nephrol. 2015;11(2):102‐112. 10.1038/nrneph.2014.232. [DOI] [PubMed] [Google Scholar]
- 6. Dubois‐Laforgue D, Cornu E, Saint‐Martin C, Coste J, Bellanné‐Chantelot C, Timsit J. Diabetes, associated clinical spectrum, long‐term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Care. 2017;40(11):1436‐1443. 10.2337/dc16-2462. [DOI] [PubMed] [Google Scholar]
- 7. Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor‐1β and their related phenotypes. J Med Genet. 2006;43(1):84‐90. 10.1136/jmg.2005.032854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hogendorf A, Kosińska‐Urbańska M, Borowiec M, Antosik K, Wyka K, Młynarski W. Atypical phenotypic features among carriers of a novel Q248X nonsense mutation in the HNF1B. Gene. 2014;65(5):15‐21. 10.5603/EP. [DOI] [PubMed] [Google Scholar]
- 9. Chen Y‐Z, Gao Q, Zhao X‐Z, et al. Review article. Chin Med J (Engl). 2010;123(22):3326‐3333. 10.3760/cma.j.issn.0366-6999.2010.22.029. [DOI] [PubMed] [Google Scholar]
- 10. Pearson ER, Badman MK, Lockwood CR, et al. Contrasting diabetes phenotypes associated with hepatocyte nuclear Factor‐1 and ‐1 mutations. Diabetes Care. 2004;27(5):1102‐1107. 10.2337/diacare.27.5.1102. [DOI] [PubMed] [Google Scholar]
- 11. Dubois‐Laforgue D, Bellanne‐ćChantelot C, Subra JF, Timsit J. Pectus excavatum is part of the clinical spectrum of HNF1B MODY5. Diabetes Care. 2014;37(4):72‐74. 10.2337/dc13-2822. [DOI] [PubMed] [Google Scholar]
- 12. Faguer S, Chassaing N, Bandin F, et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int. 2014;86(5):1007‐1015. 10.1038/ki.2014.202. [DOI] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68‐74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lek M, Karczewski K, Minikel E, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016; 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kato T, Tanaka D, Muro S, et al. A novel p.L145Q mutation in the HNF1B gene in a case of maturity‐onset diabetes of the young type 5 (MODY5). Intern Med. 2018;57(14):2035‐2039. 10.2169/internalmedicine.9692-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayer C, Böttcher Y, Kovacs P, Halbritter J, Stumvoll M. Phenotype of a patient with a de novo mutation in the hepatocyte nuclear factor 1β/maturity‐onset diabetes of the young type 5 gene. Metabolism. 2008;57(3):416‐420. 10.1016/j.metabol.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 18. Gonc EN, Ozturk BB, Haldorsen IS, et al. HNF1B mutation in a Turkish child with renal and exocrine pancreas insufficiency, diabetes and liver disease. Pediatr Diabetes. 2012;13(2):e1‐e5. 10.1111/j.1399-5448.2011.00773.x. [DOI] [PubMed] [Google Scholar]
- 19. Edghill EL, Bingham C, Slingerland AS, et al. Hepatocyte nuclear factor‐1 beta mutations cause neonatal diabetes and intrauterine growth retardation: support for a critical role of HNF‐1β in human pancreatic development. Diabet Med. 2006;23(12):1301‐1306. 10.1111/j.1464-5491.2006.01999.x. [DOI] [PubMed] [Google Scholar]
- 20. Groopman EE, Marasa M, Cameron‐Christie S, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2018;380:142‐151. 10.1056/NEJMoa1806891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bellanne‐Chantelot C, Clauin S, Chauveau D, et al. Large genomic rearrangements in the hepatocyte nuclear Factor‐1 (TCF2) gene are the Most frequent cause of maturity‐onset diabetes of the young type 5. Diabetes. 2005;54(11):3126‐3132. 10.2337/diabetes.54.11.3126. [DOI] [PubMed] [Google Scholar]
- 22. Faguer S, Decramer S, Chassaing N, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011;80(7):768‐776. 10.1038/ki.2011.225. [DOI] [PubMed] [Google Scholar]
- 23. Heidet L, Decramer S, Pawtowski A, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol. 2010;5(6):1079‐1090. 10.2215/CJN.06810909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376(1):21‐31. 10.1056/NEJMOA1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lenfant C, Baz P, Degavre A, et al. Juvenile‐onset diabetes and congenital cataract: “double‐gene” mutations mimicking a syndromic diabetes presentation. Genes (Basel). 2017;8(11):309 10.3390/GENES8110309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weedon MN, Cebola I, Patch A‐M, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. 2014;46(1):61‐64. 10.1038/ng.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smedley D, Schubach M, Jacobsen JOB, et al. A whole‐genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am J Hum Genet. 2016;99(3):595‐606. 10.1016/j.ajhg.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole‐exome sequencing as a first‐tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18(11):1090‐1096. 10.1038/gim.2016.1. [DOI] [PubMed] [Google Scholar]
- 29. Doreille A, Raymond L, Mesnard L. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2019;380(21):2079‐2080. 10.1056/NEJMc1903250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 HNF1B‐MODY prevalence in Polish population below 20 years of age ‐ estimated based on the Polish Monogenic Diabetes Registry. Diamonds mark prevalence for a given year and whiskers span over 95% confidence intervals. Owing to the growing awareness of referring physicians, an increasing number of patients is identified and diagnosed every year (r = .95, P < .0001).
Supplementary Table 1 Detailed characteristics of patients included in the study. *lack of more detailed data; DM ‐ diabetes mellitus
Supplementary Table 2 Summary of phenotype‐related non‐HNF1B genetic findings. All variants were assessed according to the criteria issued by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. PVS‐very strong evidence of pathogenicity; PS‐strong evidence; PM‐moderate; PP‐supportive. Numbers indicate specific ACMG criteria.