

Abstract
Peripheral blood hematopoietic stem and progenitor cells (HSPCs), mobilized by granulocyte colony‐stimulating factor, are widely used as a source for both autologous and allogeneic stem cell transplantation. The use of mobilized HSPCs has several advantages over traditional bone marrow–derived HSPCs, including a less invasive harvesting process for the donor, higher HSPC yields, and faster hematopoietic reconstitution in the recipient. For years, the mechanisms by which cytokines and other agents mobilize HSPCs from the bone marrow were not fully understood. The field of stem cell mobilization research has advanced significantly over the past decade, with major breakthroughs in the elucidation of the complex mechanisms that underlie stem cell mobilization. In this review, we provide an overview of the events that underlie HSPC mobilization and address the relevant cellular and molecular components of the bone marrow niche. Furthermore, current and future mobilizing agents will be discussed.
Keywords: hematopoietic stem cells, stem cell mobilization, stem cell niche, G‐CSF
Hematopoietic stem and progenitor cells (HSPCs) mobilized from the bone marrow to the peripheral blood by granulocyte colony‐stimulating factor (G‐CSF) are widely used for stem cell transplantation and have advantages over traditional bone marrow–derived HSPCs. This review provides an overview of the events that underlie HSPC mobilization and addresses the relevant cellular and molecular components of the bone marrow niche from which the HPSCs are mobilized from.
Introduction
The transplantation of hematopoietic stem and progenitor cells (HSPCs) is a widely used procedure to treat malignant and nonmalignant diseases of the blood and bone marrow (BM). Transplantation of HSPCs in an autologous setting provides hematopoietic rescue after high‐dose cytoreductive therapy; transplantation in an allogeneic setting provides immune tolerance to donor cells, thereby allowing donor T cells to mediate a graft‐versus‐tumor or graft‐versus‐leukemia effect. In addition, HSPCs can be transplanted as a rescue therapy to treat immunodeficiency. HSPCs that have been mobilized by granulocyte colony‐stimulating factor (G‐CSF) from the BM to the peripheral blood have largely replaced BM‐derived HSPCs as a source for autologous stem cell transplantation and are currently used in the majority of allogeneic stem cell transplantations.1, 2
The use of mobilized HSPCs has several advantages over traditional BM‐derived HSPCs, for both donor and patient. The collection of peripheral blood HSPCs through apheresis is a less invasive procedure than harvesting HSPCs from BM and is associated with a decreased occurrence of adverse reactions in the donor. This results in a reduced recovery time for donors of mobilized HSPCs compared with BM donors.3 Patients transplanted with mobilized HSPCs generally receive a higher median number of HSPCs (expressed as CD34+ cell dose) and are more likely to maintain their graft in comparison with patients receiving BM‐derived allografts.4 It has been established that a minimum number of 2.0 × 106 CD34+ cells/kg of body weight is required for autologous transplantation.5 This higher HSPC yield obtained through the mobilization of HSPCs has allowed for the development of novel HSPC transplantation modalities, such as unrelated transplantation, haploidentical transplantation, and nonmyeloablative transplantation. For myeloablative and nonmyeloablative allogeneic transplantation, a minimum threshold of 3.0 × 106 CD34+ cells/kg of body weight is commonly recommended. However, to improve engraftment and overcome rejection in haplotype‐mismatched transplantations, doses exceeding a threshold of 10×106 CD34+ cells/kg of body weight are needed.6 Since higher CD34+ cell doses accelerate hematopoietic recovery, the transplantation of high numbers of CD34+ cells is also important for transplantations in elderly patients, who have an increased risk of transplantation‐related morbidity and mortality.7
Unfortunately, many donors are “poor mobilizers,” as they fail to mobilize in response to G‐CSF. Depending on the study population, this mobilization failure rate can be as high as 40%.5 Several factors are associated with mobilization failure, such as advanced age, a diagnosis of lymphoma, previous radiotherapy or extensive chemotherapy, treatment with immunomodulatory drugs or purine analogs, previous mobilization failure, and low preapheresis circulating peripheral blood CD34+ cells.5 Moreover, diabetes mellitus also correlates with a lower CD34+ yield after cytokine‐induced HSPC mobilization.8 This “mobilopathy” is probably multifactorial; the factors that have been suggested to result in defective HSPC mobilization include microangiopathy, which results in quantitative and qualitative defects in BM microvasculature; sympathetic nervous system (SNS) dysfunction; an increase in BM adipocytes; and an increase in inflammatory macrophages.9
However, it is difficult to predict mobilization failure in an individual donor, because poor mobilization is observed even in patients lacking high‐risk characteristics.5 It is therefore important to gain knowledge about the underlying mechanisms of HSPC mobilization in order to devise efficient strategies to obtain the maximum yield of mobilized HSPCs from stem cell donors.
In this review, we will briefly address the cellular components of the BM niche and provide an overview of the HSPC mobilization mechanisms. Finally, current and future mobilizing agents will be discussed.
Hematopoietic stem cells and their niche
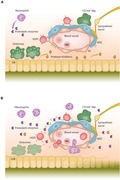
Hematopoietic stem cells (HSCs) reside at the top of the hematopoietic hierarchy and give rise to increasingly committed hematopoietic progenitor cells (HPCs). These HPCs subsequently differentiate into lineage‐restricted progenitors and early differentiated cells that lack proliferative potential. In the BM, HSCs are located in specific BM niches where they are part of a complex microenvironment. HSC niches are composed of different subsets of cells, including osteoprogenitors, osteoblastic cells, vascular endothelial cells (ECs), mesenchymal stromal cells (MSCs), neuronal cells, and hematopoietic cells, such as macrophages and megakaryocytes (MGKs); each of these subsets has specialized functions (Fig. 1A).10, 11, 12 Since the majority of HSCs in the BM are perivascular in location, it is likely that distinct perivascular niches regulate HSC function.11, 13
Figure 1.
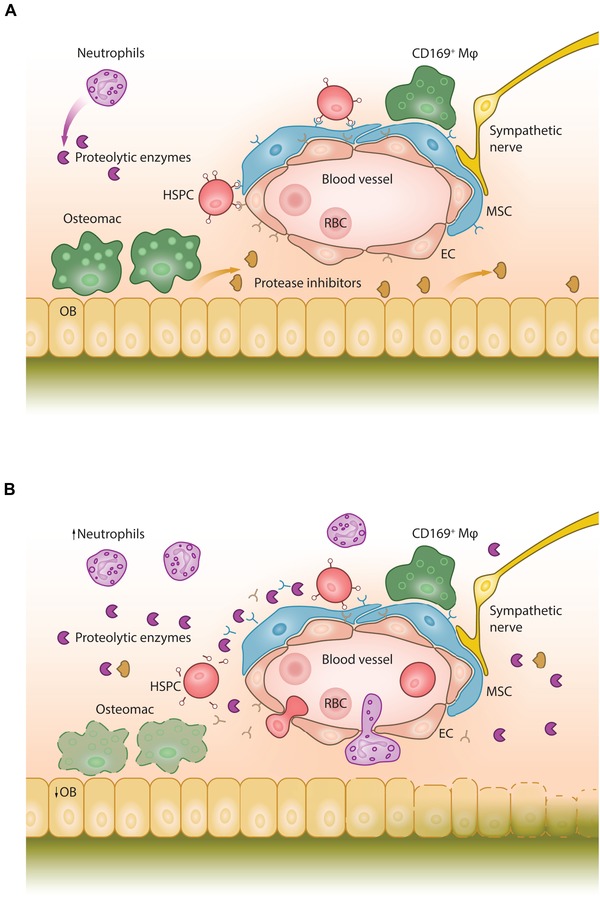
The BM niche in steady state and during G‐CSF–induced HSPC mobilization. (A) Steady state. Mesenchymal stromal cells (MSCs) and endothelial cells (ECs) express chemokine and adhesion molecules that retain hematopoietic stem and progenitor cells (HSPCs) in the BM niche. Osteoblasts (OB) secrete protease inhibitors that inhibit the proteolytic activity of neutrophil‐derived proteases. Osteoblast‐supportive endosteal macrophages (osteomacs) form a canopy over the bone‐lining osteoblasts; CD169+ macrophages (CD169+ Mφ) support the stromal cells in the niche. RBC, red blood cell. (B) G‐CSF–induced mobilization. Following G‐CSF administration, neutrophils in the BM expand, initiating the release of proteolytic enzymes that cleave and inactivate chemokines and adhesion factors, such as CXCL12, SCF, and VCAM‐1. Osteomacs are depleted, coinciding with osteoblast depletion and reduced secretion of protease inhibitors, such as alpha‐1‐antitrypsin. This is associated with decreased expression of CXCL12, SCF, and VCAM‐1, which are required to maintain and retain HSPCs in their BM niches. Increased sympathetic nerve activity leads to the downregulation of CXCL12, SCF, and VCAM‐1 by stromal cells. Together, these processes result in HSPC mobilization to the peripheral blood.
The nonhematopoietic cells in the perivascular niche mainly comprise MSCs, ECs, and osteoprogenitors. Studies in mice that express green fluorescent protein (GFP) under the control of the promoter and the second intronic enhancer of nestin (Nes‐GFP) indicate that HSCs commonly colocalize with Nes‐GFP+ MSCs, mostly around arterioles.14 These Nes‐GFP+ MSCs express the β3‐adrenergic receptor, and also CXCL12 (stromal cell‐derived factor 1, SDF‐1), which is involved in the retention of HSCs in the BM.15 The BM is richly innervated with myelinated and nonmyelinated nerve fibers, with a close association between sympathetic nerve fiber endings and bone‐lining osteoblasts, osteoclasts, and perivascular Nes‐GFP+ MSCs.16 In steady state conditions, circadian noradrenaline secretion by the SNS in the perivascular HSC niche decreases CXCL12 expression by perivascular stromal cells, which results in the circadian release of HSCs from the BM niche and their subsequent mobilization into the bloodstream.15, 17 Sympathetic nerve fibers are sheathed by nonmyelinating Schwann cells that express not only Nes, but also HSC niche factor genes such as Cxcl12 and Scf (Kitl). This further indicates the important role of the SNS in regulating the HSC niche.18 CXCL12 is also expressed by leptin receptor (LEPR)+ perivascular cells.13, 19, 20 Deep confocal imaging studies have indicated that nearly all HSCs colocalize with LEPR+ and CXCL12high cells.21 LEPR+ perivascular cells and also vascular ECs are major sources of stem cell factor (SCF) in the BM; the conditional deletion of Scf in these cells leads to HSC depletion in the BM.22
A direct role for osteoblasts in supporting HSCs has been previously suggested by experiments in which the manipulation of osteoblast numbers, either pharmacologically or genetically, correlated with HSC numbers in the BM.23, 24 Immature, CD166+ osteoblasts promote HSC function through homotypic interactions with CD166 on murine and human HSCs, showing that specific osteoblastic lineage subpopulations play a role in the regulation of HSC–niche interactions.25 However, the current understanding is that mature osteoblasts only have an indirect role in modulating HSC maintenance and differentiation.10
The niche itself is also regulated by hematopoietic cells, such as macrophages and MGKs. Macrophages indirectly support HSCs by influencing the activity of other, nonhematopoietic niche cells.26, 27, 28 Several macrophage populations have been identified in the BM, based on their surface antigen expression, location, and function.28 Osteal tissue macrophages (osteomacs) are Ly6G+F4/80+ cells that regulate osteoblast function by forming a canopy over bone‐lining osteoblasts.29 CD169+ macrophages have been identified as critical stromal niche supportive cells that indirectly regulate both HSC cycling and pool size.27, 30 Depletion of either osteomacs or CD169+ macrophages is associated with increased numbers of circulating HSCs.26, 27 In the BM, MGKs are often closely associated with sinusoidal endothelium because they extend cytoplasmic protrusions into the sinusoids. Several MGK‐derived factors support HSC maintenance, including CXCL4 (or platelet factor 4), transforming growth factor beta‐1 (TGF‐β1), and thrombopoietin.31, 32, 33 Through reduced levels of biologically active TGF‐β1 in the BM, the depletion of MGKs results in increased HSC proliferation and the activation of quiescent HSCs.31, 33
hus, during homeostasis, a complex interaction exists between the hematopoietic and nonhematopoietic compartments in the BM. This interaction results in the retention and support of HSCs in the BM niche, mainly via chemokine and adhesion molecules, such as CXCL12 and SCF, primarily expressed by MSCs and ECs, with a supporting role for the SNS and hematopoietic cells, such as MGKs and macrophages.
Hematopoietic stem and progenitor cell mobilization
Under steady state conditions, the vast majority of HSCs reside in the BM, with only a small minority of HSCs present in the circulation. The mobilization of HSPCs from the BM to the peripheral blood was first described in 1977, when a fourfold increase of HSPCs was found in the peripheral blood of healthy volunteers after the administration of endotoxin.34 Thereafter, many agents, including hematopoietic growth factors, chemokines, and other molecules, have been identified as being capable of inducing HSPC mobilization. The process of HSPC mobilization has been extensively studied in the past decades, mainly through experiments in mice. These experiments, in combination with observations in humans, have led to the current understanding of the complex pathways and cellular components involved in HSPC mobilization.
Hematopoietic cells in HSPC mobilization
The BM contains several types of hematopoietic cells that contribute to HSPC mobilization, such as neutrophils, macrophages, osteoclasts, and erythrocytes.
Neutrophils
Administration of G‐CSF leads to neutrophil expansion. Neutrophils play an essential role in HSPC mobilization induced by the cytokine interleukin‐8 (IL‐8) or by the chemokines GROβ/CXCL2 and GROβT/CXCL2δ4.35, 36 In G‐CSF–induced HSPC mobilization, the role of neutrophils is not as clearly defined. Mice lacking the G‐CSF receptor (G‐CSFR, also known as CSF3R) are neutropenic and do not mobilize after exogenous administration of IL‐8, suggesting that G‐CSFR+ neutrophils are required for mobilization.37 In mice that are chimeric for wild‐type and Csf3r −/– BM cells, treatment with G‐CSF leads to the mobilization of equal proportions of both Csf3r −/– and Csf3r +/+ HSPCs.38 This suggests that G‐CSF–induced mobilization is not dependent on the expression of G‐CSFR on HSPCs, but rather on intermediate cells such as neutrophils. However, this requirement was challenged by a study that used transgenic mice, in which G‐CSFR was only expressed on CD68+ cells of the monocytic lineage.39 In these transgenic mice, G‐CSF–induced HSPC mobilization was not reduced, which suggests that G‐CSFR signaling in monocytic cells is sufficient to induce HSPC mobilization.39
Furthermore, through increased reactive oxygen species production, neutrophil expansion in response to G‐CSF is associated with suppression of osteolineage cell populations in the BM, resulting in MSC and osteoblast apoptosis.40 In turn, this decrease in functional MSCs and osteoblasts leads to reduced expression of HSC retention factors in the BM.40
G‐CSF also indirectly activates mature neutrophils by activating the G‐CSFR on myeloid precursor cells, leading to an increased expression of Fcɣ receptor I (FcɣRI, CD64), CD11b, CD66b, and FcɣRIII (CD16) by neutrophils.41 Upon administration of G‐CSF, neutrophils are activated, which leads to the release of the serine proteases neutrophil elastase (NE) and cathepsin G (CG) from their granules. These proteases accumulate in the BM during HSPC mobilization, which leads to a highly proteolytic environment and allows for the degradation of factors that anchor HSCs in the niche.42 The vascular cell adhesion molecule 1 (VCAM‐1) is cleaved by NE and CG, which leads to a disrupted interaction between VCAM‐1 and very late antigen‐4 (VLA‐4) and an increase in soluble VCAM‐1 levels in the peripheral blood.43 Moreover, NE, CG, and matrix metalloproteinase 9 (MMP9) disrupt the interaction between CXCL12 and its receptor, CXCR4, which is expressed by HSPCs and mature leukocytes. This disruption leads to the egress of HSPCs from the BM toward the peripheral blood.44 Earlier studies by our group already suggested a role for MMP9 in IL‐8–induced HSPC mobilization, as the administration of neutralizing anti–MMP9 antibodies prevented IL‐8–induced mobilization in nonhuman primates.45 MMP9 also plays a role in GROβ/CXCL2‐ and GROβT/CXCL2δ4‐induced HSPC mobilization, which is associated with elevated levels of plasma and BM MMP9.36 Interestingly, the level of HSPC mobilization by GROβ/CXCL2 differs between mouse strains and correlates with polymorphisms in the MMP9 gene.46
The accumulation of NE, CG, and MMP9 in the BM also affects the interaction of SCF with its receptor, c‐Kit (CD117).47 These proteases cleave both human and murine c‐Kit, which reduces c‐Kit expression by human and murine mobilized HSPCs.47 As in mice, the serum levels of MMP9 and NE in healthy stem cell donors increase after 3–5 days of G‐CSF administration, and these levels correlate with the extent of CD34+ mobilization.48
Under physiologic conditions, the accumulation of high levels of proteolytically active proteases is strictly regulated by the presence of protease inhibitors. Protease inhibitors able to block serine proteases are called serpins, of which SERPINA1 (or alpha‐1‐antitrypsin (AAT)) and SERPINA3 (or alpha‐1‐antichymotrypsin (ACT)) inhibit NE and CG, respectively.49 Both AAT and ACT are present in the BM extracellular fluid of mice.50 AAT is locally produced, mainly by osteoblasts, which suggests this protease inhibitor plays a protective role in the HSC niche.51
MMPs are inhibited by the so‐called tissue inhibitors of metalloproteinases (TIMPs).52 Although MMPs play a role in HSPC mobilization, the role of TIMPs in HSPC mobilization is probably less critical.53
Protease inhibitor expression is also tightly regulated in the BM. Upon administration of G‐CSF or cyclophosphamide in mice, the levels of AAT and ACT in the BM significantly decrease, with a concomitant increase in NE and CG activity.50 This increase in protease activity subsequently results in the cleavage of adhesion molecules from the surface of the HSPCs in the BM. As a consequence, interactions between VCAM‐1/VLA‐4 and CXCL12/CXCR4 are impaired.50 Low‐dose total body irradiation (0.5 Gy) of mice resulted in significant inhibition of G‐CSF– and IL‐8–induced HSPC mobilization as a result of increased levels of AAT in the BM.54 Furthermore, the administration of human AAT almost completely blocked IL‐8–induced HSPC mobilization, which demonstrates an important role for protease inhibition in the retention of HSPCs in the BM niche.54 These data indicate that the balance between proteases and their inhibitors is important in the regulation of homeostasis within the BM microenvironment as well as in HSPC mobilization.53, 54, 55
Nevertheless, the notion that proteases are essential for HSPC mobilization has been challenged by experiments that used transgenic mice that are deficient for one or more proteases. The targeted deletion of MMP9 or NE and CG in C57BL/6 mice did not affect the mobilizing capacity of HSPCs.56 This result might be explained by the existence of redundant pathways in these mice, which allows for alternative mechanisms of HSPC mobilization.
In healthy human stem cell donors, AAT serum levels increase during G‐CSF–induced HSPC mobilization. This positively correlates with the increase in peripheral blood CD34+ cells.57 When compared with controls, patients deficient in AAT do not significantly differ with respect to the number of steady state peripheral blood HSPCs.57
These findings suggest that both protease‐dependent and ‐independent pathways play a role in HSPC mobilization. The extent to which each contributes to HSPC mobilization must be further elucidated.
Macrophages
The depletion of osteal macrophages on the endosteal surface of osteoblasts, either through the administration of G‐CSF or upon administration of macrophage‐depleting agents, is associated with the downregulation of Scf, Cxcl12, and Vcam1 expression and subsequent HSPC mobilization.26 Similarly, the depletion of BM‐resident CD169+ macrophages leads to the selective downregulation of HSC retention genes (including Cxcl12, Angpt1, Scf, and Vcam1) in Nes‐GFP+ MSCs, resulting in reduced CXCL12 levels and concomitant HSPC mobilization.27 In steady‐state conditions, the depletion of BM resident macrophages increases both HSC proliferation and the absolute number of quiescent HSCs.30 Furthermore, CD169+ macrophages are essential for supporting erythropoiesis due to the fact that these macrophages are an integral part of erythroblastic islands, where a central macrophage is surrounded by erythroid precursors in varying stages of development.58 The depletion of CD169+ macrophages, as a consequence of the administration in mice of G‐CSF or fms‐like tyrosine kinase 3 ligand (Flt3 ligand, FL), leads to a transient decrease in intramedullary erythropoiesis.59, 60 CD169+ macrophages are also essential for the circadian fluctuations in circulating HSCs. Upon clearance of aged (CD62lo/CXCR4hi) neutrophils by CD169+ macrophages in the BM, the size and function of the hematopoietic niche is reduced and the release of HPCs into the periphery enhanced.61 Macrophages also play a role in HSPC mobilization induced by leukocyte cell‐derived chemotaxin 2 (LECT2), as the LECT2 receptor (CD209a) is mainly expressed on macrophages and osteolineage cells.62 Together, these results unequivocally point to a role for macrophages in HSPC mobilization. However, more research is needed to delineate the nature of the involved macrophage subpopulations.
Osteoclasts
Osteoclasts are large, multinucleated, hematopoietic‐derived cells located adjacent to osteoblasts and osteocytes, where they are responsible for the dissolution and resorption of bone. There is controversy with respect to the role of osteoclasts during steady‐state HSC maintenance and HSPC mobilization.
Osteoclast inhibition, either through administration of the osteoclast inhibitor zoledronate or using transgenic mouse models, enhances G‐CSF–induced HSPC mobilization and decreases Cxcl12, Jag1, and Scf expression.26, 63 Activation of osteoclasts using receptor activator of nuclear factor kappa‐B ligand (RANKL) also decreases CXCL12 levels in the BM and induces HSPC mobilization.64 In contrast, several other studies have reported that osteoclasts are dispensable for HSC maintenance in adult mice.65, 66, 67 Although the data seem to be conflicting, these studies may suggest that HSC numbers and HSPC mobilization are regulated by the level of osteoclast inhibition or activation.
Erythrocytes and the complement system
The complement system contributes to the retention and mobilization of HSPCs. In comparison to wild‐type mice, G‐CSF–induced mobilization is significantly increased in mice deficient in complement factor C3 and the C3a receptor.68 Additionally, mice treated with the C3a receptor antagonist SB 290157 show significantly accelerated and enhanced G‐CSF–induced mobilization.68 Furthermore, mice that are deficient in mannan‐binding lectin (MBL) or its MBL‐associated serine proteases (MASP‐1 and ‐2), which can trigger the classical complement cascade, are poor mobilizers in response to G‐CSF.69 Interestingly, MBL deficiency is seen in around 10% of humans, but it is yet unknown if this results in impaired HSPC mobilization.
The in vivo administration of G‐CSF results in the activation of the complement cascade, with the subsequent formation of the membrane attack complex that lyses peripheral blood erythrocytes. Since erythrocytes are major reservoirs of the bioactive lipid sphingosine‐1‐phosphate (S1P), this hemolysis results in the massive release of S1P in the peripheral blood. As S1P acts as a potent chemoattractant of HSPCs in a dose‐dependent manner, the formation of this counter‐gradient contributes to HSPC mobilization.70 HSPCs express the S1P receptor S1P1; the inhibition of the S1P/S1P1 axis significantly reduces the egress of steady state HSPCs from the BM and diminishes G‐CSF–induced HSPC mobilization, which demonstrates the important role of S1P in HSPC mobilization.71
Integrins and the CXCL12/CXCR4 axis in mobilization
Integrins, such as LFA‐1 (leukocyte function‐associated antigen‐1, αLβ2 integrin, and CD11a/CD18), VLA‐4 (α4β1 integrin), and VLA‐5 (α5β1 integrin), are not only involved in the engraftment of HSCs in mice and humans, but also in HSPC retention and mobilization from the BM to the peripheral blood. Our group showed that IL‐8–induced mobilization of HSPCs in mice is inhibited after a single injection of neutralizing anti‐LFA‐1 antibodies.72 Moreover, injection of neutralizing antibodies to the LFA‐1 ligand intercellular adhesion molecule‐1 significantly inhibited IL‐8–induced HSPC mobilization.72 In contrast, G‐CSF–induced HSPC mobilization is more than twofold enhanced by a blockade of LFA‐1 in comparison to the administration of G‐CSF only, while the administration of anti‐LFA‐1 antibodies alone does not result in HSPC mobilization.73 In LFA‐1 (Itgal) knockout mice, G‐CSF–induced mobilization remains unaffected, which might be explained by the presence of redundant pathways that compensate for the loss of LFA‐1.73
In mice, the conditional deletion of either VLA‐4 or its receptor VCAM‐1, which is constitutively expressed by BM stromal cells, induces a significant migration of HSPCs to the peripheral blood.74, 75 In mice and primates, blocking of the receptor–ligand interaction by neutralizing anti‐VLA‐4 or anti‐VCAM‐1 monoclonal antibodies also results in significant HSPC mobilization.76, 77
The chemokine CXCL12 strongly attracts human and murine HSPCs, which express its receptor CXCR4. In the BM, CXCL12 is constitutively produced at high levels by various BM stromal niche cells and plays an important role in the homing and retention of HSPCs.78, 79 The conditional deletion of CXCR4 or CXCL12 results in dramatically increased HSPC numbers in the peripheral blood and spleen.79 Through the downregulation of CXCR4 on HSPCs and the alteration of the plasma‐to‐marrow CXCL12 gradient, the CXCR4 agonist peptide, CTCE‐0021, rapidly mobilizes neutrophils and HSPCs to the peripheral blood in mice.80 This peptide works synergistically with G‐CSF, which results in a more than fivefold increase in HSPC mobilization.80 Blocking of CXCR4 by the selective chemical antagonist AMD3100 (plerixafor) results in rapid mobilization of HPSCs in mice, nonhuman primates, and humans, with increased in vivo repopulation potential in comparison to G‐CSF–mobilized HSPCs.81, 82 Several intracellular signaling enzymes, such as Rac1 and Rac2, are activated in response to CXCL12‐signaling in HSPCs. The depletion of both Rac1 and Rac2 in a transgenic mouse model leads to a massive egress of HPCs into the blood from the BM.83 Through phosphoproteomic profiling of murine HSPCs, it was recently shown that other proteins, such as the Rac1 activation protein ARHGAP25, are involved in HSPC mobilization via the CXCL12/CXCR4 pathway. Moreover, inhibition of ARHGAP25 activity leads to augmented CXCL12 signaling.84
In vivo, CXCL12 is truncated by the membrane‐bound extracellular peptidase CD26 (dipeptidyl peptidase‐4, DPP4), which is expressed on the surface of many cell types in the BM, including ECs and a subset of HPCs.85, 86 This peptidase is essential for G‐CSF–induced HSPC mobilization, as Dpp4 −/– mice show significantly decreased G‐CSF–induced HSPC mobilization compared with wild‐type mice.87 This is most likely due to altered CD26‐dependent neuropeptide Y (NPY) signaling in sinusoidal ECs, resulting in increased vascular permeability and subsequent HSPC egress.86 Experiments in rats suggest that, in diabetes mellitus, reduced CD26 activity in the BM contributes to impaired HSPC mobilization in response to G‐CSF.88
Role of stromal cells in HSPC mobilization
Osteolineage cells
The BM is encapsulated by highly mineralized bone that is produced by osteolineage cells. These cells go through various differentiation stages, ultimately forming mature osteoblasts and osteocytes. Osteoblasts form a layer of bone‐generating cells, the endosteum, and differentiate into osteocytes when embedded in the mineralized compact bone matrix. Based on recent studies, the role of osteolineage cells in HSPC mobilization is limited.13, 22, 89, 90, 91 Manipulation of osteoblast numbers does not result in HSPC mobilization into the blood.90, 91 Furthermore, animal models have indicated that conditional deletion of Cxcl12 or Scf from mature osteoblasts does not result in HSC mobilization.13, 22, 89 Administration of G‐CSF results in the depletion of osteoblasts at the endosteal surface, probably as a result of osteomac depletion. This coincides with a reduction in the number of pre‐pro‐B, pro‐B, pre‐B, and surface IgM+ mature B cells in the BM, owing to increased B cell apoptosis.92 Medullary B lymphopoiesis is dependent on cell–cell contact between B cell progenitors and osteoblasts, as well as the presence of osteoblast‐derived CXCL12 and IL‐7. This suggests that inhibition of medullary B lymphopoiesis may be the result of G‐CSF–induced osteoblast depletion.92, 93
The role of osteocytes in HSPC mobilization is still unclear; targeted ablation of osteocytes in a transgenic mouse model results in the failure to mobilize HSPCs in response to G‐CSF. However, the validity of this model was questioned, since not only osteolineage cells but also CXCL12‐abundant reticular (CAR) cells were targeted in this model.94, 95
Bone contains a high concentration of calcium ions at the HSC‐enriched endosteal surface. HSCs express the seven‐transmembrane‐spanning calcium‐sensing receptor (CaSR) and thus respond to extracellular ionic calcium concentrations.96 Experiments with CaSR‐deficient mice suggest that the CaSR retains HSCs at the BM endosteal surface and that the absence of CaSR on HSCs impairs stem cell engraftment.96 However, a role of CaSR in HSPC mobilization has not been identified.
Mesenchymal stromal cells
MSCs are an essential part of the HSC niche and play an important role in HSPC mobilization. Several types of BM‐resident MSCs, such as CAR cells, Nes‐GFP+ MSCs, and LEPR+ pericytes, express high levels of HSC‐supporting factors, such as CXCL12 and SCF. Injection of G‐CSF leads to the decreased expression of these HSC retention factors, contributing to HSPC mobilization.16
The administration of MSCs in a mouse model results in the downregulation of niche factors, including Cxcl12, Scf, and Vcam‐1, in endosteal cells. These BM changes are similar to events that occur during G‐CSF–induced HSPC mobilization.97 Interestingly, in this model, the coadministration of MSCs and G‐CSF results in a twofold increase in HSPC mobilization in comparison to G‐CSF alone, while the injection of MSCs by itself did not induce HSPC mobilization. The effects observed can possibly be explained by the secretion of extracellular vesicles (EVs) from the injected MSCs, as MSC‐derived EVs induced similar effects in the BM, inducing a permissive state that primes the BM environment for subsequent G‐CSF–induced HSPC mobilization.
Endothelial cells
The exact role of ECs in the egress of HSPCs from the BM into the circulation is not fully understood. Vascular ECs are the most important source of endogenous G‐CSF, which plays a role in the body's response to physiological stress or bacterial infections.16 ECs also express CXCL12, SCF, and VCAM‐1 on the cell surface, which are crucial HSC retention factors.13, 22 However, when Cxcl12 is conditionally deleted from ECs, HSCs are depleted but not mobilized. This is likely related to the fact that the expression of CXCL12 is approximately 100‐fold lower in ECs in comparison to perivascular MSCs.13, 89 In the BM sinusoids, which are lined with ECs, the transmembrane receptor for the ephrin B2 ligand (EPHB4) is widely expressed. Blockade of the interaction between EPHB4 and ephrin B2 on LSK cells reduces HSPC mobilization. This points toward a critical role for EPHB4/ephrin B2 signaling in the mobilization of HSPCs from the BM.98
Sympathetic nervous system
The role of the SNS in HSPC maintenance under steady‐state conditions is well defined. However, in cytokine‐induced HSPC mobilization, its role is less apparent. The administration of G‐CSF leads to increased sympathetic activity in the BM via impaired removal of noradrenaline from the synaptic cleft.99 Interestingly, sympathetic neurons express both G‐CSF and G‐CSF‐R, where G‐CSF likely plays a role as a protector against neurotoxic agents in an autocrine or paracrine fashion. Damage to the SNS due to neurotoxic chemotherapy, such as vincristine or cisplatin, results in impaired hematopoietic regeneration due to the selective loss of adrenergic innervation.100 However, in mice treated by chemotherapy, adjuvant treatment with neuroprotective agents, such as 4‐methylcatechol or glial cell‐derived neurotrophic factor, can rescue BM engraftment and mobilization.100, 101 Neuro‐adrenergic stimulation can be used to increase HSPC mobilization, as was shown in a trial with multiple myeloma patients who were treated with a combination of G‐CSF and the noradrenaline reuptake inhibitor desipramine.102 Sympathetic nerves also secrete NPY, which is one of the most abundant and widely secreted peptides from the brain and SNS. In addition to its role in EC‐regulated vascular permeability, NPY also induces HSPC mobilization through the Y1 receptor in osteoblasts by activating MMP9.103
Clinical application of mobilizing agents
A wide variety of hematopoietic growth factors, chemokines, chemotherapeutic agents, and other molecules that can induce HSPC mobilization, have been identified since the first mobilization experiments using endotoxin. Several agents have been approved for HSPC mobilization in a clinical setting, such as G‐CSF, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), SCF, and AMD3100. Other agents, such as IL‐8, FL, VCAM‐1/VLA‐4 inhibitors, and S1P agonists, are mainly used in experimental animal studies or have been tested in early phase trials in human patients.1
Granulocyte colony‐stimulating factor
In the first clinical trials of recombinant human G‐CSF in cancer patients, G‐CSF was shown to increase neutrophil counts and reduce the number of days of neutropenia, resulting in fewer infections and more patients receiving planned chemotherapy.104, 105 Furthermore, it was observed that the frequency of hematopoietic colony‐forming cells in the peripheral blood of these patients increased over 100‐fold.106 This result paved the way to use mobilized peripheral blood HSPCs for transplantation in humans, since it had already been shown that transplanted circulating blood cells could restore hematopoietic function in lethally irradiated animals.107 In 1992, Sheridan et al. showed that patients receiving G‐CSF–mobilized peripheral blood progenitors after high‐dose chemotherapy had significantly faster hematopoietic reconstitution.108 Over the past 25 years, the use of G‐CSF–mobilized HSPCs has largely replaced BM as a source of stem cells for both autologous and allogeneic cell transplantation, facilitating the development of novel transplantation modalities.1
However, the multifaceted and interconnected mechanisms by which G‐CSF induces HSPC mobilization have only come to light in the past few years.109 Upon G‐CSF administration, the number of neutrophils in the BM expands, initiating the release of proteolytic enzymes that cleave and inactivate chemokine and adhesion factors, such as CXCL12, SCF, and VCAM‐1 (Fig. 1B).43 Administration of G‐CSF also activates the complement cascade, resulting in the release of C5a. The interaction of C5a with its receptor expressed on granulocytes subsequently activates phospholipase C‐β2 (PLC‐β2). This, in turn, disrupts HSPC membrane lipid rafts containing adhesion molecules, such as VLA‐4 and CXCR4.110
Furthermore, G‐CSF depletes osteoblast‐supportive endosteal macrophages and CD169+ macrophages, inducing osteoblast ablation and blocking bone formation.15, 26, 111, 112 Together, this results in the reduced expression of chemokines and cytokines, such as CXCL12, SCF, and angiopoietin‐1, which are necessary to maintain and retain HSCs in their BM niches.111 Osteoblast ablation might also result in a decrease in intramedullary AAT and ACT, thus promoting a proteolytic environment and amplifying the effects of G‐CSF.50, 54 Administration of exogenous G‐CSF drives BM‐resident HSCs into the cell cycle, leading to increased HSC numbers in the BM, whereas G‐CSF–mobilized peripheral blood HSCs are predominantly in the G0 or G1 phase.113 It was recently shown that G‐CSF does not uniformly mobilize HSCs and HPCs according to their maturation stage, but instead that the most potent HSCs (defined as HSCs able to serially transplant and reconstitute recipients) are mobilized as early as day 2 of G‐CSF treatment.114 This stands in contrast to the kinetics of FL‐induced HSPC mobilization, where only > 5 days of FL administration induced the mobilization of HSCs with long‐term repopulating capacity.115
AMD3100 and other CXCL12/CXCR4 axis antagonists
Following preclinical studies in which the blockade of the CXCL12/CXCR4 axis resulted in robust HSPC mobilization, several drugs have been approved for clinical use in humans or are in early phase trials. AMD3100 is a small‐molecule bicyclam drug that targets the CXCL12/CXCR4 axis, with no inhibitory effect on other chemokine receptors.116 Since CXCR4 serves as a coreceptor for the human immunodeficiency virus (HIV) to enter the cell, AMD3100 was initially developed as an anti‐HIV drug. During phase I clinical trials in human volunteers, where AMD3100 was still being evaluated for its antiviral activity, it was noted that there was a rapid increase in peripheral white blood cells, peaking 6 h after intravenous administration.117 Subsequent studies have shown that a single injection of AMD3100 is able to mobilize CD34+ HSPCs.118 Furthermore, when administered in conjunction with G‐CSF (5 days of G‐CSF at 10 μg/kg plus AMD3100 at 240 μg/kg on day 5), AMD3100 acts synergistically, resulting in a tripling of circulating CD34+ HSPCs in comparison to either agent alone.119 Based on two phase III clinical trials, AMD3100 is currently approved in the United States and Europe for clinical HSPC mobilization in combination with G‐CSF for patients with multiple myeloma or non‐Hodgkin's lymphoma that have failed to mobilize with G‐CSF alone.120, 121 A recent study investigated the combination of AMD3100 and the CXCR2 agonist GROβ in mice, showing rapid HSPC mobilization after a single coinjection of GROβ and AMD3100, which is equivalent to 5 days of G‐CSF administration.46 Combined administration of GROβ and AMD3100 resulted in the mobilization of HSPCs with increased engraftment potential compared with G‐CSF–mobilized HSPCs. Further studies in humans are needed to evaluate the feasibility of this combination in clinical practice.
Flt3 ligand
A type 1 transmembrane protein, FL, exists in both a membrane‐bound and a soluble form. It binds to the tyrosine kinase type III receptor Flt3/Flk2 (CD135) that is primarily expressed by HPCs. The cytokine FL is involved in cell survival, proliferation, and differentiation during early hematopoiesis. In addition, FL is required for lymphocyte development, but not for differentiation into myeloid lineages. Either alone or in combination with other growth factors, FL stimulates the proliferation of highly enriched human and murine HSCs in vitro, and in vivo leads to the expansion and mobilization of HSPCs in animals and humans.122, 123, 124 As exposure to FL increases the total number of CXCR4+ HPCs, FL interacts with the CXCL12/CXCR4 pathway.125 Mice treated with recombinant FL for 3–5 days mainly mobilize HPCs into the peripheral blood, whereas treatment for up to 10 days results in the mobilization of HSCs with a long‐term repopulation capacity, showing that FL is a slow mobilizing agent.115 Administration of FL in combination with G‐CSF, GM‐CSF, or AMD3100 leads to significantly increased HSPC mobilization, with the combination of FL and AMD3100 being the most potent.124, 126 Soluble, recombinant FL (termed CDX‐301) is well tolerated in humans and able to mobilize sufficient HSPCs for transplantation following 10 days of daily injections.127 So far, there is no clinically approved FL product, and more research is needed to warrant the clinical application of FL as monotherapy or in combination with AMD3100 or G‐CSF as a mobilizing agent in humans.
Nonsteroidal anti‐inflammatory drugs
Prostaglandin E2 (PGE2) is an endogenous lipid produced by cyclooxygenase‐2 (COX‐2) that enhances HSC homing, survival, and proliferation.128 Treatment with nonsteroidal anti‐inflammatory drugs (NSAIDs), like the COX‐1 and COX‐2 inhibitor meloxicam, reduces PGE2 production and is associated with significant HSPC egress from the BM.129 PGE2 receptor knockout mice show an increased number of peripheral blood HSPCs, which is caused by reduced E‐prostanoid 4 (EP4) receptor signaling.129 NSAID‐induced HSPC mobilization is independent of the CXCL12/CXCR4‐axis, but is associated with attenuation of osteolineage cells and a significant reduction in osteopontin, which acts as a niche retention factor.129
Based on these preclinical data, multiple myeloma patients have received meloxicam in combination with G‐CSF as a mobilization regimen. Patients receiving G‐CSF and meloxicam showed increased HSPC mobilization compared with administration of G‐CSF alone. This resulted in fewer patients requiring more than 1 day of stem cell collection and a reduced need for plerixafor administration.130 Hematologic engraftment after transplantation and survival rates were similar between the two groups. Furthermore, treatment with meloxicam was well tolerated, making this a promising supportive strategy for HSPC mobilization.130
Integrin antagonists
Treatment of patients with natalizumab, a recombinant humanized monoclonal antibody against the α4 subunit of VLA‐4 that is approved for the treatment of multiple sclerosis and Crohn's disease, results in the mobilization of HSPCs in these patients.131 However, the association of natalizumab with progressive multifocal leukoencephalopathy precluded its further application. Other α4 antagonists, such as the orally bioavailable drug called firategrast, are being developed but are not yet commercially available.132
The development of integrin antagonists for blocking the α9β1 integrin, whose expression is restricted to HSPCs, is promising. The small molecule N‐(benzenesulfonyl)‐l‐prolyl‐l‐O‐(1‐pyrrolidinylcarbonyl)tyrosine (BOP), which is a dual α9β1/α4β1 integrin antagonist, mobilizes multilineage reconstituting HSPCs after a single dose in mice.133 Administration of a single dose of BOP in combination with a single dose of AMD3100 mobilizes similar numbers of HSPCs as is observed after 4 days of G‐CSF. However, in comparison with G‐CSF, the combination of BOP and AMD3100 results in significantly enhanced short‐ and long‐term engraftment in mice, indicating that this combination may be a rapid and effective alternative to G‐CSF.133
Combinations of these integrin and CXCR4 antagonists have the potential to develop into an effective, single‐dose, 1‐day strategy to mobilize HSPCs in different clinical settings.
Summary and future directions
HSPC mobilization involves a multifaceted and complex interaction of HSPCs and stromal and hematopoietic niche cells, as well as an array of cytokines, chemokines, and small molecules. Stem cell mobilization research has advanced immensely over the past decade. Major steps in the elucidation of the complex mechanisms of stem cell mobilization have been made. Understanding the underlying mechanisms of HSPC maintenance and mobilization has led to a plethora of agents with mobilizing capacity. However, with the exception of AMD3100, only a few of these agents have reached the stage of clinical application, and so far, G‐CSF remains the backbone of HSPC mobilization in humans. G‐CSF has its own limitations, such as the necessity for prolonged parenteral administration and suboptimal efficiency in certain patient groups. Furthermore, although the administration of G‐CSF is generally safe and serious adverse events are rare, bone pain and fatigue are experienced by a majority of donors and patients treated with G‐CSF.3 Therefore, there is an unmet need for innovative mobilizing agents or strategies. The identification of agents that are able to collectively influence the many mechanisms that underlie HSPC mobilization may provide substantial improvements to existing HSPC mobilization methods and subsequent transplant outcomes.134 Ideally, these agents are potent HSPC mobilizers that can be titrated to the required peripheral blood HSPC dose, have an excellent safety profile, can be administered as a single dose, and are not expensive.
Despite all efforts to elucidate the mechanisms underlying HSPC mobilization, there are still questions that must be answered before HSPC mobilization can be fully understood and manipulated. These questions include:
-
(1)
Is the continuous exit of HSPCs into the bloodstream in the steady state regulated by the same mechanisms as cytokine‐induced HSPC mobilization?
-
(2)
What is the relative contribution of each cell population (e.g., macrophages and MSCs) and their respective interactions and signals in cytokine‐induced HSPC mobilization?
-
(3)
Can biomarkers be identified that predict the mobilizing capacity in response to mobilizing agents?
These questions, and likely many others, can drive future research and hopefully lead to better, safer, and more efficient mobilization strategies.
Competing interests
The authors declare no competing interests.
Author contributions
E.J.dK., W.E.F., and M.vP. participated in drafting the manuscript and approved the final version of the submitted manuscript.
References
- 1. Hopman, R.K. & DiPersio J.F.. 2014. Advances in stem cell mobilization. Blood Rev. 28: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Passweg, J.R. et al 2018. Is the use of unrelated donor transplantation leveling off in Europe? The 2016 European Society for Blood and Marrow Transplant activity survey report. Bone Marrow Transplant. 53: 1139–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller, J.P. et al 2008. Recovery and safety profiles of marrow and PBSC donors: experience of the National Marrow Donor Program. Biol. Blood Marrow Transplant. 14: 29–36. [DOI] [PubMed] [Google Scholar]
- 4. Anasetti, C. et al 2012. Peripheral‐blood stem cells versus bone marrow from unrelated donors. N. Engl. J. Med. 367: 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giralt, S. et al 2014. Optimizing autologous stem cell mobilization strategies to improve patient outcomes: consensus guidelines and recommendations. Biol. Blood Marrow Transplant. 20: 295–308. [DOI] [PubMed] [Google Scholar]
- 6. Koh, L.P. , Rizzieri D.A. & Chao N.J.. 2007. Allogeneic hematopoietic stem cell transplant using mismatched/haploidentical donors. Biol. Blood Marrow Transplant. 13: 1249–1267. [DOI] [PubMed] [Google Scholar]
- 7. Heimfeld, S. 2003. Bone marrow transplantation: how important is CD34 cell dose in HLA‐identical stem cell transplantation? Leukemia 17: 856–858. [DOI] [PubMed] [Google Scholar]
- 8. Fadini, G.P. et al 2013. Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes Care 36: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fadini, G.P. , Ciciliot S. & Albiero M.. 2017. Concise review: perspectives and clinical implications of bone marrow and circulating stem cell defects in diabetes. Stem Cells 35: 106–116. [DOI] [PubMed] [Google Scholar]
- 10. Morrison, S.J. & Scadden D.T.. 2014. The bone marrow niche for haematopoietic stem cells. Nature 505: 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boulais, P.E. & Frenette P.S.. 2015. Making sense of hematopoietic stem cell niches. Blood 125: 2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Pel, M. , Fibbe W.E. & Schepers K.. 2015. The human and murine hematopoietic stem cell niches: are they comparable? Ann. N.Y. Acad. Sci. 1370: 55–64. [DOI] [PubMed] [Google Scholar]
- 13. Ding, L. & Morrison S.J.. 2013. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495: 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kunisaki, Y. et al 2013. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502: 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mendez‐Ferrer, S. et al 2010. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466: 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tay, J. , Levesque J.P. & Winkler I.G.. 2017. Cellular players of hematopoietic stem cell mobilization in the bone marrow niche. Int. J. Hematol. 105: 129–140. [DOI] [PubMed] [Google Scholar]
- 17. Mendez‐Ferrer, S. et al 2008. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452: 442–447. [DOI] [PubMed] [Google Scholar]
- 18. Yamazaki, S. et al 2011. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147: 1146–1158. [DOI] [PubMed] [Google Scholar]
- 19. Sugiyama, T. et al 2006. Maintenance of the hematopoietic stem cell pool by CXCL12‐CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25: 977–988. [DOI] [PubMed] [Google Scholar]
- 20. Zhou, B.O. et al 2014. Leptin‐receptor‐expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 15: 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Acar, M. et al 2015. Deep imaging of bone marrow shows non‐dividing stem cells are mainly perisinusoidal. Nature 526: 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ding, L. et al 2012. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Calvi, L.M. et al 2003. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425: 841–846. [DOI] [PubMed] [Google Scholar]
- 24. Zhang, J. et al 2003. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425: 836–841. [DOI] [PubMed] [Google Scholar]
- 25. Chitteti, B.R. et al 2014. CD166 regulates human and murine hematopoietic stem cells and the hematopoietic niche. Blood 124: 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winkler, I.G. et al 2010. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116: 4815–4828. [DOI] [PubMed] [Google Scholar]
- 27. Chow, A. et al 2011. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 208: 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCabe, A. & MacNamara K.C.. 2016. Macrophages: key regulators of steady‐state and demand‐adapted hematopoiesis. Exp. Hematol. 44: 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang, M.K. et al 2008. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo . J. Immunol. 181: 1232–1244. [DOI] [PubMed] [Google Scholar]
- 30. McCabe, A. et al 2015. Macrophage‐lineage cells negatively regulate the hematopoietic stem cell pool in response to interferon gamma at steady state and during infection. Stem Cells 33: 2294–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruns, I. et al 2014. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 20: 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakamura‐Ishizu, A. et al 2014. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem. Biophys. Res. Commun. 454: 353–357. [DOI] [PubMed] [Google Scholar]
- 33. Zhao, M. et al 2014. Megakaryocytes maintain homeostatic quiescence and promote post‐injury regeneration of hematopoietic stem cells. Nat. Med. 20: 1321–1326. [DOI] [PubMed] [Google Scholar]
- 34. Cline, M.J. & Golde D.W.. 1977. Mobilization of hematopoietic stem cells (CFU‐C) into the peripheral blood of man by endotoxin. Exp. Hematol. 5: 186–190. [PubMed] [Google Scholar]
- 35. Pruijt, J.F. et al 2002. Neutrophils are indispensable for hematopoietic stem cell mobilization induced by interleukin‐8 in mice. Proc. Natl. Acad. Sci. USA 99: 6228–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pelus, L.M. et al 2004. Neutrophil‐derived MMP‐9 mediates synergistic mobilization of hematopoietic stem and progenitor cells by the combination of G‐CSF and the chemokines GRObeta/CXCL2 and GRObetaT/CXCL2delta4. Blood 103: 110–119. [DOI] [PubMed] [Google Scholar]
- 37. Liu, F. , Poursine‐Laurent J. & Link D.C.. 1997. The granulocyte colony‐stimulating factor receptor is required for the mobilization of murine hematopoietic progenitors into peripheral blood by cyclophosphamide or interleukin‐8 but not flt‐3 ligand. Blood 90: 2522–2528. [PubMed] [Google Scholar]
- 38. Liu, F. , Poursine‐Laurent J. & Link D.C.. 2000. Expression of the G‐CSF receptor on hematopoietic progenitor cells is not required for their mobilization by G‐CSF. Blood 95: 3025–3031. [PubMed] [Google Scholar]
- 39. Christopher, M.J. et al 2011. Expression of the G‐CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G‐CSF in mice. J. Exp. Med. 208: 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singh, P. et al 2012. Expansion of bone marrow neutrophils following G‐CSF administration in mice results in osteolineage cell apoptosis and mobilization of hematopoietic stem and progenitor cells. Leukemia 26: 2375–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Haas, M. et al 1994. Granulocyte colony‐stimulating factor administration to healthy volunteers: analysis of the immediate activating effects on circulating neutrophils. Blood 84: 3885–3894. [PubMed] [Google Scholar]
- 42. Levesque, J.P. et al 2002. Mobilization by either cyclophosphamide or granulocyte colony‐stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp. Hematol. 30: 440–449. [DOI] [PubMed] [Google Scholar]
- 43. Levesque, J.P. et al 2001. Vascular cell adhesion molecule‐1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony‐stimulating factor. Blood 98: 1289–1297. [DOI] [PubMed] [Google Scholar]
- 44. Levesque, J.P. et al 2003. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J. Clin. Invest. 111: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pruijt, J.F. et al 1999. Prevention of interleukin‐8‐induced mobilization of hematopoietic progenitor cells in rhesus monkeys by inhibitory antibodies against the metalloproteinase gelatinase B (MMP‐9). Proc. Natl. Acad. Sci. USA 96: 10863–10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoggatt, J. et al 2018. Rapid mobilization reveals a highly engraftable hematopoietic stem cell. Cell 172: 191–204.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levesque, J.P. et al 2003. Granulocyte colony‐stimulating factor induces the release in the bone marrow of proteases that cleave c‐KIT receptor (CD117) from the surface of hematopoietic progenitor cells. Exp. Hematol. 31: 109–117. [DOI] [PubMed] [Google Scholar]
- 48. van Os, R. et al 2002. Proteolytic enzyme levels are increased during granulocyte colony‐stimulating factor‐induced hematopoietic stem cell mobilization in human donors but do not predict the number of mobilized stem cells. J. Hematother. Stem Cell Res. 11: 513–521. [DOI] [PubMed] [Google Scholar]
- 49. Potempa, J. , Korzus E. & Travis J.. 1994. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J. Biol. Chem. 269: 15957–15960. [PubMed] [Google Scholar]
- 50. Winkler, I.G. et al 2005. Serine protease inhibitors serpina1 and serpina3 are down‐regulated in bone marrow during hematopoietic progenitor mobilization. J. Exp. Med. 201: 1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuiperij, H.B. et al 2009. Serpina1 (alpha1‐AT) is synthesized in the osteoblastic stem cell niche. Exp. Hematol. 37: 641–647. [DOI] [PubMed] [Google Scholar]
- 52. Brew, K. & Nagase H.. 2010. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803: 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Winkler, I.G. & Levesque J.P.. 2006. Mechanisms of hematopoietic stem cell mobilization: when innate immunity assails the cells that make blood and bone. Exp. Hematol. 34: 996–1009. [DOI] [PubMed] [Google Scholar]
- 54. van Pel, M. et al 2006. Serpina1 is a potent inhibitor of IL‐8‐induced hematopoietic stem cell mobilization. Proc. Natl. Acad. Sci. USA 103: 1469–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Velders, G.A. & Fibbe W.E.. 2005. Involvement of proteases in cytokine‐induced hematopoietic stem cell mobilization. Ann. N.Y. Acad. Sci. 1044: 60–69. [DOI] [PubMed] [Google Scholar]
- 56. Levesque, J.P. et al 2004. Characterization of hematopoietic progenitor mobilization in protease‐deficient mice. Blood 104: 65–72. [DOI] [PubMed] [Google Scholar]
- 57. de Kruijf, E.J. et al 2014. Peripheral blood hematopoietic stem and progenitor cell frequency is unchanged in patients with alpha‐1‐antitrypsin deficiency. Int. J. Hematol. 99: 714–720. [DOI] [PubMed] [Google Scholar]
- 58. Chasis, J.A. & Mohandas N.. 2008. Erythroblastic islands: niches for erythropoiesis. Blood 112: 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jacobsen, R.N. et al 2014. Mobilization with granulocyte colony‐stimulating factor blocks medullar erythropoiesis by depleting F4/80(+)VCAM1(+)CD169(+)ER‐HR3(+)Ly6G(+) erythroid island macrophages in the mouse. Exp. Hematol. 42: 547–561. [DOI] [PubMed] [Google Scholar]
- 60. Jacobsen, R.N. et al 2015. Fms‐like tyrosine kinase 3 (Flt3) ligand depletes erythroid island macrophages and blocks medullar erythropoiesis in the mouse. Exp. Hematol. 44: 207–212. [DOI] [PubMed] [Google Scholar]
- 61. Casanova‐Acebes, M. et al 2013. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 153: 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lu, X.J. et al 2016. LECT2 drives haematopoietic stem cell expansion and mobilization via regulating the macrophages and osteolineage cells. Nat. Commun. 7: 12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mansour, A. et al 2012. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J. Exp. Med. 209: 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kollet, O. et al 2006. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 12: 657–664. [DOI] [PubMed] [Google Scholar]
- 65. Flores, C. et al 2013. Osteoclasts are not crucial for hematopoietic stem cell maintenance in adult mice. Haematologica 98: 1848–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Miyamoto, K. et al 2011. Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J. Exp. Med. 208: 2175–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rao, M. et al 2015. Osteoclasts are dispensable for hematopoietic progenitor mobilization by granulocyte colony‐stimulating factor in mice. Exp. Hematol. 43: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ratajczak, J. et al 2004. Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood 103: 2071–2078. [DOI] [PubMed] [Google Scholar]
- 69. Adamiak, M. et al 2017. Novel evidence that the mannan‐binding lectin pathway of complement activation plays a pivotal role in triggering mobilization of hematopoietic stem/progenitor cells by activation of both the complement and coagulation cascades. Leukemia 31: 262–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ratajczak, M.Z. et al 2010. Novel insight into stem cell mobilization‐plasma sphingosine‐1‐phosphate is a major chemoattractant that directs the egress of hematopoietic stem progenitor cells from the bone marrow and its level in peripheral blood increases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia 24: 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Golan, K. et al 2012. S1P promotes murine progenitor cell egress and mobilization via S1P1‐mediated ROS signaling and SDF‐1 release. Blood 119: 2478–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pruijt, J.F. et al 1998. Anti‐LFA‐1 blocking antibodies prevent mobilization of hematopoietic progenitor cells induced by interleukin‐8. Blood 91: 4099–4105. [PubMed] [Google Scholar]
- 73. Velders, G.A. et al 2002. Enhancement of G‐CSF‐induced stem cell mobilization by antibodies against the beta 2 integrins LFA‐1 and Mac‐1. Blood 100: 327–333. [DOI] [PubMed] [Google Scholar]
- 74. Scott, L.M. , Priestley G.V. & Papayannopoulou T.. 2003. Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol. Cell. Biol. 23: 9349–9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ulyanova, T. et al 2005. VCAM‐1 expression in adult hematopoietic and nonhematopoietic cells is controlled by tissue‐inductive signals and reflects their developmental origin. Blood 106: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Craddock, C.F. et al 1997. Antibodies to VLA4 integrin mobilize long‐term repopulating cells and augment cytokine‐induced mobilization in primates and mice. Blood 90: 4779–4788. [PubMed] [Google Scholar]
- 77. Kikuta, T. et al 2000. Mobilization of hematopoietic primitive and committed progenitor cells into blood in mice by anti‐vascular adhesion molecule‐1 antibody alone or in combination with granulocyte colony‐stimulating factor. Exp. Hematol. 28: 311–317. [DOI] [PubMed] [Google Scholar]
- 78. Kollet, O. et al 2001. Rapid and efficient homing of human CD34(+)CD38(–/low)CXCR4(+) stem and progenitor cells to the bone marrow and spleen of NOD/SCID and NOD/SCID/B2m(null) mice. Blood 97: 3283–3291. [DOI] [PubMed] [Google Scholar]
- 79. Tzeng, Y.S. et al 2011. Loss of Cxcl12/Sdf‐1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood 117: 429–439. [DOI] [PubMed] [Google Scholar]
- 80. Pelus, L.M. et al 2005. The CXCR4 agonist peptide, CTCE‐0021, rapidly mobilizes polymorphonuclear neutrophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony‐stimulating factor. Exp. Hematol. 33: 295–307. [DOI] [PubMed] [Google Scholar]
- 81. Broxmeyer, H.E. et al 2005. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 201: 1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hess, D.A. et al 2007. Human progenitor cells rapidly mobilized by AMD3100 repopulate NOD/SCID mice with increased frequency in comparison to cells from the same donor mobilized by granulocyte colony stimulating factor. Biol. Blood Marrow Transplant. 13: 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cancelas, J.A. et al 2005. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat. Med. 11: 886–891. [DOI] [PubMed] [Google Scholar]
- 84. Wang, L.D. et al 2016. Phosphoproteomic profiling of mouse primary HSPCs reveals new regulators of HSPC mobilization. Blood 128: 1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Christopherson, K.W. , Cooper S. & Broxmeyer H.E.. 2003. Cell surface peptidase CD26/DPPIV mediates G‐CSF mobilization of mouse progenitor cells. Blood 101: 4680–4686. [DOI] [PubMed] [Google Scholar]
- 86. Singh, P. et al 2017. Neuropeptide Y regulates a vascular gateway for hematopoietic stem and progenitor cells. J. Clin. Invest. 127: 4527–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Christopherson, K.W. et al 2003. CD26 is essential for normal G‐CSF‐induced progenitor cell mobilization as determined by CD26–/– mice. Exp. Hematol. 31: 1126–1134. [DOI] [PubMed] [Google Scholar]
- 88. Fadini, G.P. et al 2013. Stem cell compartmentalization in diabetes and high cardiovascular risk reveals the role of DPP‐4 in diabetic stem cell mobilopathy. Basic Res. Cardiol. 108: 313. [DOI] [PubMed] [Google Scholar]
- 89. Greenbaum, A. et al 2013. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem‐cell maintenance. Nature 495: 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kiel, M.J. , Radice G.L. & Morrison S.J.. 2007. Lack of evidence that hematopoietic stem cells depend on N‐cadherin‐mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell 1: 204–217. [DOI] [PubMed] [Google Scholar]
- 91. Lymperi, S. et al 2008. Strontium can increase some osteoblasts without increasing hematopoietic stem cells. Blood 111: 1173–1181. [DOI] [PubMed] [Google Scholar]
- 92. Winkler, I.G. et al 2013. B‐lymphopoiesis is stopped by mobilizing doses of G‐CSF and is rescued by overexpression of the anti‐apoptotic protein Bcl2. Haematologica 98: 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhu, J. et al 2007. Osteoblasts support B‐lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 109: 3706–3712. [DOI] [PubMed] [Google Scholar]
- 94. Asada, N. et al 2013. Matrix‐embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell 12: 737–747. [DOI] [PubMed] [Google Scholar]
- 95. Zhang, J. & Link D.C.. 2016. Targeting of mesenchymal stromal cells by Cre‐recombinase transgenes commonly used to target osteoblast lineage cells. J. Bone Miner. Res. 31: 2001–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Adams, G.B. et al 2006. Stem cell engraftment at the endosteal niche is specified by the calcium‐sensing receptor. Nature 439: 599–603. [DOI] [PubMed] [Google Scholar]
- 97. de Kruijf, E.F.M. et al 2018. Mesenchymal stromal cells induce a permissive state in the bone marrow that enhances G‐CSF‐induced hematopoietic stem cell mobilization in mice. Exp. Hematol. 64: 59–70. [DOI] [PubMed] [Google Scholar]
- 98. Kwak, H. et al 2016. Sinusoidal ephrin receptor EPHB4 controls hematopoietic progenitor cell mobilization from bone marrow. J. Clin. Invest. 126: 4554–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lucas, D. et al 2012. Norepinephrine reuptake inhibition promotes mobilization in mice: potential impact to rescue low stem cell yields. Blood 119: 3962–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lucas, D. et al 2013. Chemotherapy‐induced bone marrow nerve injury impairs hematopoietic regeneration. Nat. Med. 19: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Levesque, J.P. & Winkler I.G.. 2013. It takes nerves to recover from chemotherapy. Nat. Med. 19: 669–671. [DOI] [PubMed] [Google Scholar]
- 102. Shastri, A. et al 2017. Stimulation of adrenergic activity by desipramine enhances hematopoietic stem and progenitor cell mobilization along with G‐CSF in multiple myeloma: a pilot study. Am. J. Hematol. 92: 1047–1051. [DOI] [PubMed] [Google Scholar]
- 103. Park, M.H. et al 2016. Neuropeptide Y induces hematopoietic stem/progenitor cell mobilization by regulating matrix metalloproteinase‐9 activity through Y1 receptor in osteoblasts. Stem Cells 34: 2145–2156. [DOI] [PubMed] [Google Scholar]
- 104. Bronchud, M.H. et al 1987. Phase I/II study of recombinant human granulocyte colony‐stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. Br. J. Cancer 56: 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gabrilove, J.L. et al 1988. Effect of granulocyte colony‐stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional‐cell carcinoma of the urothelium. N. Engl. J. Med. 318: 1414–1422. [DOI] [PubMed] [Google Scholar]
- 106. Duhrsen, U. et al 1988. Effects of recombinant human granulocyte colony‐stimulating factor on hematopoietic progenitor cells in cancer patients. Blood 72: 2074–2081. [PubMed] [Google Scholar]
- 107. Perry, V.P. et al 1965. Protection of lethally irradiated guinea pigs with fresh and frozen homologous peripheral blood leukocytes. Cryobiology 1: 233–239. [DOI] [PubMed] [Google Scholar]
- 108. Sheridan, W.P. et al 1992. Effect of peripheral‐blood progenitor cells mobilised by filgrastim (G‐CSF) on platelet recovery after high‐dose chemotherapy. Lancet 339: 640–644. [DOI] [PubMed] [Google Scholar]
- 109. Winkler, I.G. et al 2012. Hematopoietic stem cell mobilizing agents G‐CSF, cyclophosphamide or AMD3100 have distinct mechanisms of action on bone marrow HSC niches and bone formation. Leukemia 26: 1594–1601. [DOI] [PubMed] [Google Scholar]
- 110. Adamiak, M. et al 2016. Evidence that a lipolytic enzyme–hematopoietic‐specific phospholipase C‐β2–promotes mobilization of hematopoietic stem cells by decreasing their lipid raft‐mediated bone marrow retention and increasing the promobilizing effects of granulocytes. Leukemia 30: 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Levesque, J.P. , Helwani F.M. & Winkler I.G.. 2010. The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 24: 1979–1992. [DOI] [PubMed] [Google Scholar]
- 112. Christopher, M.J. & Link D.C.. 2008. Granulocyte colony‐stimulating factor induces osteoblast apoptosis and inhibits osteoblast differentiation. J. Bone Miner. Res. 23: 1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Morrison, S.J. , Wright D.E. & Weissman I.L.. 1997. Cyclophosphamide/granulocyte colony‐stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc. Natl. Acad. Sci. USA 94: 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Winkler, I.G. et al 2016. Mobilization of hematopoietic stem cells with highest self‐renewal by G‐CSF precedes clonogenic cell mobilization peak. Exp. Hematol. 44: 303–314.e1. [DOI] [PubMed] [Google Scholar]
- 115. de Kruijf, E.J. et al 2010. Hematopoietic stem and progenitor cells are differentially mobilized depending on the duration of Flt3‐ligand administration. Haematologica 95: 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. De Clercq, E. 2009. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil). Biochem. Pharmacol. 77: 1655–1664. [DOI] [PubMed] [Google Scholar]
- 117. Hendrix, C.W. et al 2000. Pharmacokinetics and safety of AMD‐3100, a novel antagonist of the CXCR‐4 chemokine receptor, in human volunteers. Antimicrob. Agents Chemother. 44: 1667–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hubel, K. et al 2004. Leukocytosis and mobilization of CD34+ hematopoietic progenitor cells by AMD3100, a CXCR4 antagonist. Support Cancer Ther. 1: 165–172. [DOI] [PubMed] [Google Scholar]
- 119. Liles, W.C. et al 2005. Augmented mobilization and collection of CD34+ hematopoietic cells from normal human volunteers stimulated with granulocyte‐colony‐stimulating factor by single‐dose administration of AMD3100, a CXCR4 antagonist. Transfusion 45: 295–300. [DOI] [PubMed] [Google Scholar]
- 120. DiPersio, J.F. et al 2009. Phase III prospective randomized double‐blind placebo‐controlled trial of plerixafor plus granulocyte colony‐stimulating factor compared with placebo plus granulocyte colony‐stimulating factor for autologous stem‐cell mobilization and transplantation for patients with non‐Hodgkin's lymphoma. J. Clin. Oncol. 27: 4767–4773. [DOI] [PubMed] [Google Scholar]
- 121. DiPersio, J.F. et al 2009. Plerixafor and G‐CSF versus placebo and G‐CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood 113: 5720–5726. [DOI] [PubMed] [Google Scholar]
- 122. Brasel, K. et al 1996. Hematologic effects of flt3 ligand in vivo in mice. Blood 88: 2004–2012. [PubMed] [Google Scholar]
- 123. Jacobsen, S.E. et al 1995. The FLT3 ligand potently and directly stimulates the growth and expansion of primitive murine bone marrow progenitor cells in vitro: synergistic interactions with interleukin (IL) 11, IL‐12, and other hematopoietic growth factors. J. Exp. Med. 181: 1357–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Papayannopoulou, T. et al 1997. In vivo effects of Flt3/Flk2 ligand on mobilization of hematopoietic progenitors in primates and potent synergistic enhancement with granulocyte colony‐stimulating factor. Blood 90: 620–629. [PubMed] [Google Scholar]
- 125. Williams, K.M. et al 2017. FLT3 ligand regulates thymic precursor cells and hematopoietic stem cells through interactions with CXCR4 and the marrow niche. Exp. Hematol. 52: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. He, S. et al 2014. FLT3L and plerixafor combination increases hematopoietic stem cell mobilization and leads to improved transplantation outcome. Biol. Blood Marrow Transplant. 20: 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Anandasabapathy, N. et al 2015. Efficacy and safety of CDX‐301, recombinant human Flt3L, at expanding dendritic cells and hematopoietic stem cells in healthy human volunteers. Bone Marrow Transplant. 50: 924–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hoggatt, J. et al 2009. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood 113: 5444–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hoggatt, J. et al 2013. Differential stem‐ and progenitor‐cell trafficking by prostaglandin E2. Nature 495: 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Jeker, B. et al 2018. NSAID treatment with meloxicam enhances peripheral stem cell mobilization in myeloma. Bone Marrow Transplant. 53: 175–179. [DOI] [PubMed] [Google Scholar]
- 131. Jing, D. et al 2010. CD49d blockade by natalizumab in patients with multiple sclerosis affects steady‐state hematopoiesis and mobilizes progenitors with a distinct phenotype and function. Bone Marrow Transplant. 45: 1489–1496. [DOI] [PubMed] [Google Scholar]
- 132. Domingues, M.J. , Nilsson S.K. & Cao B.. 2017. New agents in HSC mobilization. Int. J. Hematol. 105: 141–152. [DOI] [PubMed] [Google Scholar]
- 133. Cao, B. et al 2016. Therapeutic targeting and rapid mobilization of endosteal HSC using a small molecule integrin antagonist. Nat. Commun. 7: 11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nilsson, S.K. 2017. Factors that influence a mobilized HSC product. Int. J. Hematol. 105: 116–117. [DOI] [PubMed] [Google Scholar]