

Abstract
Lateral meningocele syndrome (LMS) is due to specific pathogenic variants in the last exon of NOTCH3 gene. Besides the lateral meningoceles, this condition presents with dysmorphic features, short stature, congenital heart defects, and feeding difficulties. Here, we report a girl with neurosensorial hearing loss, severe gastroesophageal reflux disease, congenital heart defects, multiple renal cysts, kyphosis and left‐convex scoliosis, dysmorphic features, and mild developmental delay. Exome sequencing detected the previously unreported de novo loss‐of‐function variant in exon 33 of NOTCH3 p.(Lys2137fs). Following the identification of the gene defect, MRI of the brain and spine revealed temporal encephaloceles, inner ears anomalies, multiple spinal lateral meningoceles, and intra‐ and extra‐dural arachnoid spinal cysts. This case illustrates the power of reverse phenotyping to establish clinical diagnosis and expands the spectrum of clinical manifestations related to LMS to include inner ear abnormalities and multi‐cystic kidney disease.
Keywords: encephalocele, lateral meningocele syndrome, NOTCH3
1. INTRODUCTION
Lateral meningocele syndrome (LMS) is an ultra‐rare condition with 22 patients reported to date. Among reported cases, only 8 were shown to carry loss‐of‐function mutations affecting exon 33 of NOTCH3 (Brown, Gupta, & Sayama, 2017; Ejaz et al., 2016; Gripp et al., 2015). Missense variants located in NOTCH3 exons 3–11 are responsible for a distinct disorder, namely cerebral arteriopathy, autosomal dominant, with subcortical infarcts and leukoencephalopathy (Joutel et al., 1996). The core clinical manifestation of LMS is lateral spinal meningoceles, occurring with protrusion of the arachnoid and the dura through the intervertebral foramina causing back pain, paresthesia, paraparesis, and neurologic bladder depending on their size and localization. Chiari type 1 malformation, syringomyelia, and hydrocephalus have also been reported. Other features include a recognizable pattern of dysmorphic facial features (low posterior hair line, coarse hair, arched eyebrows, palpebral ptosis, down‐slanting palpebral fissures, hypertelorism, malar hypoplasia, micrognatia, long or smooth philtrum, thin vermillion, high and narrow palate, and cleft palate), low weight, short stature, hypotonia, congenital heart defects, feeding difficulties (dysphagia and gastroesophageal reflux disease), joint laxity, kyphoscoliosis, and vertebral anomalies (Ejaz et al., 2016; Gripp et al., 2015). Cognitive impairment is reported in only one case (Gripp et al., 2015). Mixed or conductive hearing loss was found in three individuals, while ocular anomalies had been rarely reported (Ejaz et al., 2016; Gripp et al., 2015).
We describe a further case with LMS due to a novel NOTCH3 truncating variant in exon 33 presenting with inner ear abnormalities and cystic kidneys disease that have not been previously reported in LMS.
2. CLINICAL REPORT
We report a 7‐year‐and‐8‐month‐old girl who was the second child of healthy and non‐consanguineous parents. Prenatal ultrasound showed bilateral hydronephrosis and oligohydramnios. She was born at term by vaginal delivery complicated by fracture of a clavicle. Her birth weight was 3,660 g. At birth, she developed acute respiratory distress requiring endotracheal intubation. From the first months of life, she had feeding difficulties with frequent regurgitations, failure to thrive, and recurrent episodes of aspiration pneumonia requiring gastrostomy tube. At 2 years, she was diagnosed with severe gastroesophageal reflux disease treated with Nissen fundoplication. Transient hypothyroidism in the first months of life was treated with hormone replacement therapy up to 2 years of age. Bilateral severe neurosensorial hearing loss (80 dB HL) was diagnosed at 6 months of life, and she was treated with hearing aids from the age of 1 year. Postnatal echocardiogram showed patent ductus arteriosus that spontaneously closed, and an atrial septal defect; the ascending aorta was not dilated. She had severe kyphosis and left‐convex scoliosis requiring brace. Multiple vertebral anomalies were detected by X‐ray, including an odontoid process retroflexion with a cleft of anterior arch of C1, cuneal deformation, and posterior scalloping of multiple vertebral bodies. Abdomen ultrasound showed hypoplasia of the left kidney and multiple bilateral renal cysts the largest measuring 3 cm in diameter. She also had microlithiasis and was treated with potassium citrate.
She sat independently at 6 months, walked unassisted at 24 months, and spoke her first words at 12 months. She attended regular school without learning difficulties.
At the age of 5 years, her weight was 19 kg (fourth centile), height 115 cm (fourth centile), and occipitofrontal circumference was 51.5 cm (14th centile), BMI was at the 18th centile. Dysmorphic features included prominence of the occipital bone, low posterior hairline, arched eyebrows, synophria, bilateral ptosis, down‐slanting of the palpebral fissures, low‐set ears, anteverted nares, protruding columella, smooth philtrum, thin lips, micrognatia, and high and narrow palate (Figure 1). She also had joint hypermobility. Array comparative genomic hybridization was normal and MLL2 and OFD1 sequencing performed for the suspicion of Kabuki and orofaciodigital syndromes, respectively, did not reveal any pathogenic variants.
Figure 1.
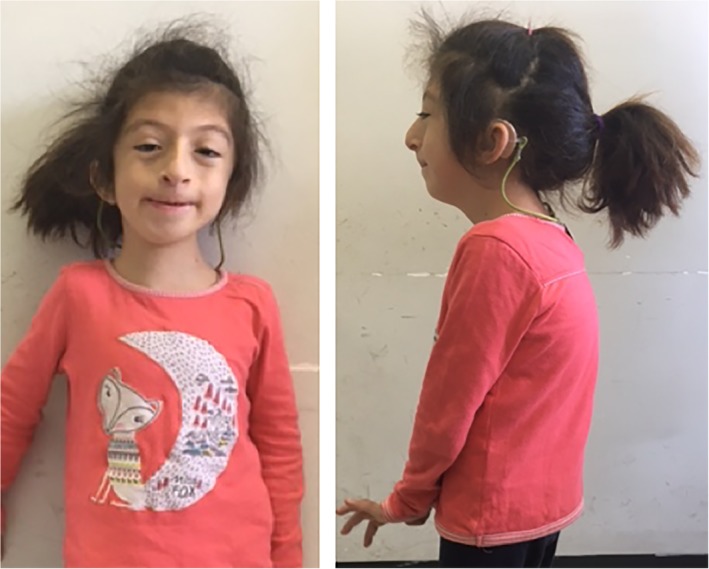
Dysmorphic features of the presented girl including low posterior hair line, arched eyebrows, synophria, bilateral ptosis and down‐slanting palpebral fissures, low‐set ears, anteverted nares, protruding columella, smooth philtrum, thin lips, and micrognatia. Kyphosis is also evident [Color figure can be viewed at http://wileyonlinelibrary.com]
Following informed consent, genomic DNA from the patient and her parents underwent exome sequencing (ES), enriched using the SureSelect Clinical Research Exome (Agilent, Technologies, Santa Clara, CA) and sequences in the NextSeq 500 sequencing system (Illumina, San Diego, CA). A custom pipeline based on Burrows‐Wheeler Alignment tool, Genome Analysis Toolkit, and ANNOVAR (Wang et al., 2010) was used to call, annotate, filter, and prioritize variants. TrioES revealed a novel heterozygous de novo two‐nucleotide deletion in exon 33 of NOTCH3 gene (NM_000435: c.6409_6410delTC; p.Lys2137fs).
Brain and spine MRI performed after the identification of NOTCH3 mutation revealed bilateral meningoencephaloceles of the inferior temporal lobes, multiple spinal extra‐ and intra‐dural arachnoid cysts with tethering of the filum terminale at L4‐L5 and multiple bilateral spinal meningoceles extending from C7 to S3 (Figure 2a–c). MRI of the inner ear showed hypoplasia of the apical turn of the cochlea and of the modiolus, and dysmorphic vestibule (Figure 2d,e).
Figure 2.
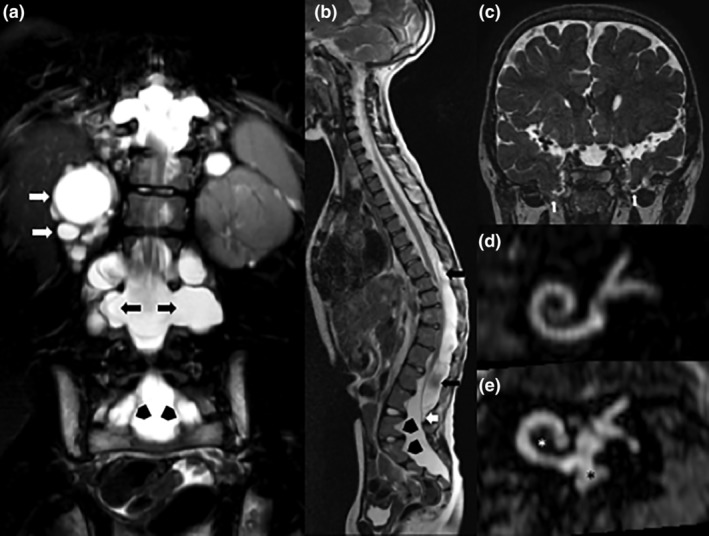
(a) Turbo Spin Echo (TSE) T2‐weighted sequence on coronal plane shows multiple bilateral meningoceles (black arrows), renal cysts (white arrows), and foraminal extra‐dural arachnoid cysts (black arrowheads). (b) TSE T2‐weighted sequence on sagittal plane shows some confluent intra‐dural arachnoid cysts, located posteriorly respect to the spinal cord between T8 and L4‐L5 levels (black arrows). Tethering of the filum terminale at L4‐L5 (white arrow) and posterior scalloping of the lumbar and sacral vertebral bodies (black arrowheads) can also be noted. (c) Driven equilibrium radiofrequency reset pulse (DRIVE) 3D TSE T2‐weighted sequence on coronal plane shows bilateral inferior temporal meningoencephaloceles (white arrows). (d) Oblique coronal multi‐planar reconstructions (MPR) of DRIVE 3D TSE T2‐weighted sequence highlight the differences between a normal internal ear (d) and the inner ear of the subject herein described (e) with hypoplasia of the apical turn of the cochlea (white asterisk), and dysmorphic vestibule (black asterisk)
3. DISCUSSION
A total of eight cases with LMS carrying loss‐of‐function variants affecting exon 33 of NOTCH3 gene have been reported and we herein describe a further LMS individual harboring a novel nonsense variant of NOTCH3 exon 33. Notably, the presented case had inner ear malformation and cystic kidney disease that have not been previously reported. Interestingly, Notch3 is expressed in the mouse inner ear (Eddison, Le Roux, & Lewis, 2000) where it supports the differentiation of auditory and vestibular hair cell progenitors (Ma, Rubel, & Raible, 2008). Moreover, Notch signaling has been involved in regeneration of hair cells (Ma et al., 2008; Shu et al., 2019).
The child herein described showed multiple bilateral kidney cysts without liver cysts or fibrosis that have also not been previously reported in LMS. Genes causing dominant and recessive forms of polycystic kidney diseases (PKD1, GANAB, FCYT, PKD2, DNAJB11, and DZIP1L) had a good coverage in the ES and no pathogenic variants were identified. Disruption of NOTCH pathway during development resulted in proximal tubular cysts and, and its overexpression correlated to rapidly growing cysts in kidneys of autosomal recessive and autosomal dominant polycystic kidney disease mouse models (Idowu et al., 2018). Nevertheless, the description of further individuals with LMS is required to conclude whether inner ear malformation and cystic kidney are indeed features of LMS. In conclusion, we describe a further individual with LMS carrying a novel pathogenic variant in NOTCH3 who presented with inner ear abnormalities and cystic kidney disease that expand the spectrum of phenotypic abnormalities.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
G.C., D.A., and N.B.‐P. contributed to conceptualization. M.A., A.T., M.P., and V.N. contributed to methodology. M.P., A.T., V.N., and B.F. contributed to formal analysis. B.C., E.D.G., and A.D. contributed to investigation. G.C. and M.P. contributed to data curation. G.C., D.A., and N.B.‐P. contributed to writing—original draft preparation. G.C. and N.B.‐P contributed to writing—review and editing. N.B.‐P. contributed to supervision.
ACKNOWLEDGMENTS
The authors would like to thank the patient and her family for their participation. This work was supported by Fondazione Telethon, Telethon Undiagnosed Diseases Program (TUDP, GSP15001), TIGEM NGS Facility and the newco Next Generation Diagnostic SRL: S. Banfi, N. Brunetti‐Pierri, A. Bruselles, G. Cappuccio, V. Caputo, R. Castello, G. Chillemi, A. Ciolfi, M. D'Antonio, M. Dentici, M. Dionisi, G. L. Di Filippo, Esposito, S. Fecarotta, G. Mancano, S. Maitz, L. Monaco, F. Musacchia, M. Mutarelli, V. Nigro, G. Oliva, G. Parenti, M. Pinelli, S. Pizzi, M. Pizzo, F. Radio, S. Riccardo, E. Rizzi, A. Selicorni, G. Sgroi, A. Torella, and R. Turra.
Cappuccio G, Apuzzo D, Alagia M, et al. Expansion of the phenotype of lateral meningocele syndrome. Am J Med Genet Part A. 2020;182A:1259–1262. 10.1002/ajmg.a.61536
Gerarda Cappuccio and Diletta Apuzzo contributed equally to this study.
Funding information Fondazione Telethon, Grant/Award Number: GSP15001
REFERENCES
- Brown, E. C. , Gupta, K. , & Sayama, C. (2017). Neurosurgical management in lateral meningocele syndrome: Case report. Journal of Neurosurgery: Pediatrics, 19(2), 232–238. [DOI] [PubMed] [Google Scholar]
- Eddison, M. , Le Roux, I. , & Lewis, J. (2000). Notch signaling in the development of the inner ear: Lessons from drosophila. Proceedings of the National Academy of Sciences of the United States of America, 97(22), 11692–11699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejaz, R. , Qin, W. , Huang, L. , Blaser, S. , Tetreault, M. , Hartley, T. , … Care4Rare Canada C . (2016). Lateral meningocele (Lehman) syndrome: A child with a novel NOTCH3 mutation. American Journal of Medical Genetics. Part A, 170A(4), 1070–1075. [DOI] [PubMed] [Google Scholar]
- Gripp, K. W. , Robbins, K. M. , Sobreira, N. L. , Witmer, P. D. , Bird, L. M. , Avela, K. , … Sol‐Church, K. (2015). Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome. American Journal of Medical Genetics. Part A, 167A(2), 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idowu, J. , Home, T. , Patel, N. , Magenheimer, B. , Tran, P. V. , Maser, R. L. , … Sharma, M. (2018). Aberrant regulation of Notch3 signaling pathway in polycystic kidney disease. Scientific Reports, 8(1), 3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joutel, A. , Corpechot, C. , Ducros, A. , Vahedi, K. , Chabriat, H. , Mouton, P. , … Tournier‐Lasserve, E. (1996). Notch3 mutations in CADASIL, a hereditary adult‐onset condition causing stroke and dementia. Nature, 383(6602), 707–710. [DOI] [PubMed] [Google Scholar]
- Ma, E. Y. , Rubel, E. W. , & Raible, D. W. (2008). Notch signaling regulates the extent of hair cell regeneration in the zebrafish lateral line. The Journal of Neuroscience, 28(9), 2261–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, Y. , Li, W. , Huang, M. , Quan, Y. Z. , Scheffer, D. , Tian, C. , … Chen, Z. Y. (2019). Renewed proliferation in adult mouse cochlea and regeneration of hair cells. Nature Communications, 10(1), 5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M ., & Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research: 38(16), e164. [DOI] [PMC free article] [PubMed] [Google Scholar]