

Abstract
Aim
Matrix metalloproteinases (MMPs) and their endogenous tissue inhibitors (TIMPs) control proteolysis within the extracellular matrix (ECM) of the brain. Dysfunction of this enzymatic system due to brain inflammation can disrupt the blood‐brain barrier (BBB) and has been implicated in the pathogenesis of epilepsy. However, this has not been extensively studied in the epileptogenic human brain.
Methods
We investigated the expression and cellular localization of major MMPs (MMP2, MMP3, MMP9 and MMP14) and TIMPs (TIMP1, TIMP2, TIMP3 and TIMP4) using quantitative real‐time polymerase chain reaction (RT‐PCR) and immunohistochemistry in resected epileptogenic brain tissue from patients with tuberous sclerosis complex (TSC), a severe neurodevelopmental disorder characterized by intractable epilepsy and prominent neuroinflammation. Furthermore, we determined whether anti‐inflammatory microRNAs, miR146a and miR147b, which can regulate gene expression at the transcriptional level, could attenuate dysregulated MMP and TIMP expression in TSC tuber‐derived astroglial cultures.
Results
We demonstrated higher mRNA and protein expression of MMPs and TIMPs in TSC tubers compared to control and perituberal brain tissue, particularly in dysmorphic neurons and giant cells, as well as in reactive astrocytes, which was associated with BBB dysfunction. More importantly, IL‐1β‐induced dysregulation of MMP3, TIMP2, TIMP3 and TIMP4 could be rescued by miR146a and miR147b in tuber‐derived TSC cultures.
Conclusions
This study provides evidence of dysregulation of the MMP/TIMP proteolytic system in TSC, which is associated with BBB dysfunction. As dysregulated MMP and TIMP expression can be ameliorated in vitro by miR146a and miR147b, these miRNAs deserve further investigation as a novel therapeutic approach.
Keywords: blood‐brain barrier, extracellular matrix, matrix metalloproteinases, microRNA, tuberous sclerosis complex
Introduction
Matrix metalloproteinases (MMPs) are extracellular zinc‐dependent proteases characterized by a conserved methionine residue at the active site 1. Membrane‐bound or secreted latent forms of MMPs require the removal of the propeptide domain for optimal activity. This proteolytic removal can be mediated by other, already active, MMPs or by other proteases, in particular, members of the plasminogen‐plasmin system 2, 3. The activity of MMPs can be regulated by their endogenous tissue inhibitors (TIMPs) of which four forms are present in the brain 4. All TIMPs are capable of inhibiting all MMPs, though with varying efficacy 5. MMPs are able to use as substrates, all components of the extracellular matrix (ECM) 6, as well as several intracellular components 7. Under normal physiological conditions, neuronal activity can stimulate the release of MMPs from neurons 8, 9 and MMP activity is involved in dendritic spine enlargement during hippocampal long‐term potentiation and the increase in glutamatergic neurotransmission 10. They also play an important role in neurite outgrowth, cell migration and neuroinflammation 11, 12, 13, 14, 15. Under pathological conditions, MMPs can induce blood‐brain barrier (BBB) dysfunction via degradation of the basal lamina of cerebral blood vessels and disruption of tight junctions between endothelial cells 16. Due to the accumulation of serum proteins that enter the brain, causing a pro‐inflammatory response and increased neuronal excitability, BBB dysfunction can contribute to epileptogenesis 17, 18, 19, 20.
Epilepsy is one of the major clinical symptoms in patients with tuberous sclerosis complex (TSC) 21. TSC is caused by a mutation in either the TSC1 or TSC2 gene, resulting in a constitutive activation of the mammalian target of rapamycin (mTOR) pathway 22, 23. This multisystem disorder affects a large range of organs including the brain 24. Brain pathology in TSC patients is associated with a complex clinical phenotype including epilepsy (often intractable to medical treatment), autism and intellectual disability 23, 25, 26. Focal malformations in cortical cytoarchitecture, also known as cortical tubers, are a pathological hallmark of TSC. These tubers are characterized by the presence of abnormal cells called dysmorphic neurons and giant cells and their localization is often associated with the presence of an epileptogenic focus in TSC patients 22, 27. Furthermore, prominent brain inflammation and BBB dysfunction are observed 23, 28, 29, 30, 31.
As MMPs may contribute to TSC pathophysiology and epileptogenesis, understanding their role could have implications for the treatment of neurological symptoms. We therefore investigated the expression and localization of MMPs and TIMPs in resected brain tissue of patients with TSC in relation to BBB dysfunction. Furthermore, we determined whether anti‐inflammatory microRNAs (miRs), miR146a and miR147b, could attenuate dysregulated MMP and TIMP expression in TSC tuber‐derived astroglial cultures. miRs, which are short non‐coding RNAs, approximately 18–23 nucleotides in length, have therapeutic potential as they are capable of regulating target gene expression at the post‐transcriptional level 32. Recently, we showed that inhibition of the pro‐inflammatory miR155 could attenuate interleukin (IL)‐1β‐induced overexpression of MMP3 in cultured human astrocytes 33. Moreover, we showed in a previous study that both miR146a and miR147b can act as negative‐feedback regulators of inflammatory responses 34, which may also affect MMP and TIMP expression. Although miR146a and miR147b do not directly target MMPs, we hypothesized that by interfering with brain inflammation using these miRNAs (which directly target pro‐inflammatory genes), we can indirectly reduce MMP expression.
Materials and methods
Subjects
The cases included in this study were obtained from the archives of the departments of Neuropathology of the Amsterdam UMC (University of Amsterdam, The Netherlands) and the University Medical Center Utrecht (Utrecht, The Netherlands). For immunohistochemical analyses and real‐time polymerase chain reaction (RT‐PCR), cortical tubers from, respectively, 6 and 16 TSC patients were examined from whom neocortical tuber tissue was surgically removed. Presurgical evaluation, including long‐term video‐EEG monitoring, high‐resolution MRI and neuropsychological testing was performed in order to characterize the epileptogenic zone. A group of 20 autopsy gender‐ and age‐matched control cases without a history of seizures or other neurologic diseases were also included. Clinical findings of both TSC patients and control autopsy patients can be found in Table S1. None of the TSC patients received mTOR inhibitors. The age and gender did not differ between TSC patients and autopsy controls (P > 0.05, Mann–Whitney U‐Test). Tissue was obtained and used in accordance with the Declaration of Helsinki and the Amsterdam UMC Research Code provided by the Medical Ethics Committee and the local ethical committees of the participating centres gave permission to undertake the study.
Immunohistochemistry
Human brain tissue was fixed in 10% buffered formalin and embedded in paraffin. Paraffin‐embedded tissue was sectioned at 5 μm, mounted on precoated glass slides (Star Frost, Waldemar Knittel, Braunschweig, Germany) and processed for immunohistochemical staining. Sections were deparaffinated in xylene, rinsed in ethanol (100%, 96%, 70%) and incubated for 20 min in 0.3% hydrogen peroxide diluted in methanol. Antigen retrieval was performed using a pressure cooker in 10 mM sodium citrate, pH 6.0 at 121°C for 10 min. Slides were washed with phosphate‐buffered saline (PBS, pH 7.4) and incubated overnight with primary antibody (1:200 rabbit polyclonal IgG anti‐MMP2, Abcam, Cambridge, UK; 1:100 rabbit monoclonal IgG anti‐MMP3, Abcam; 1:100 mouse monoclonal IgG2a anti‐MMP9, Millipore, Amsterdam, The Netherlands; 1:500 mouse monoclonal IgG3/κ anti‐MMP14, Millipore; 1:200 mouse monoclonal clone VT7 anti‐TIMP1, Agilent, Santa Clara, CA, USA; 1:100 mouse monoclonal IgG2a/κ anti‐TIMP2, Millipore; 1:300 mouse monoclonal IgG1/κ anti‐TIMP3, Millipore; 1:600 rabbit polyclonal IgG anti‐TIMP4, Abcam; 1:80 000 rabbit anti‐human albumin, DAKO/Agilent Technologies Netherlands, Amstelveen, The Netherlands; or 1:300 mouse anti‐rat CD163, AbD Serotec, Dusseldorf, Germany) in PBS at 4°C. For single labelling, staining was performed using 3,3′‐diaminobenzidine substrate solution (1:10 in 0.05 M Tris–HCl, pH 7.6; ImmunoLogic, Duiven, The Netherlands) with 0.015% H2O2. The reaction was stopped by washing with distilled water after which sections were dehydrated in alcohol and xylene and coverslipped. Negative controls were performed without primary antibody (the sections were blank).
Cell‐specific double‐labelling of MMPs and TIMPs was performed with glial fibrillary acidic protein (GFAP; 1:4000 polyclonal rabbit, DAKO, Glostrup, Denmark; or 1:4000 monoclonal mouse, Sigma, St. Louis, MO, USA), ionized calcium binding adapter molecule‐1 (IBA‐1; 1:2000 polyclonal rabbit, WAKO, Richmond, VA, USA) or CD11b/c (1:100 OX‐42; DAKO/Agilent Technologies Netherlands, Amstelveen, The Netherlands). After overnight incubation at 4°C and rinsing, sections were incubated with secondary antibodies Alexa Fluor 488 donkey anti‐mouse IgG (H+L) and Alexa Fluor 568 goat anti‐rabbit IgG (H+L) (1:200; Invitrogen, Eugene, OR, USA) for 2 h at room temperature, washed in PBS and coverslipped. Fluorescent microscopy was performed using a confocal microscope (SP8‐X, Leica, Son, The Netherlands).
For double‐labelling of MMPs and TIMPs, specific MMP and TIMP combinations were chosen based on their interaction as described in literature 35. The combinations were: MMP2/TIMP2, MMP3/TIMP1, MMP3/TIMP3, MMP9/TIMP1, MMP14/TIMP2, MMP14/TIMP4. Double‐labelling was performed using BrightVision poly‐alkaline phosphatase (AP)‐anti‐rabbit or anti‐mouse (ImmunoLogic) incubated for 30 min at room temperature and washed with PBS. AP activity was visualized with the AP substrate kit III Vector Blue (SK‐5300; Vector Laboratories Inc., Burlingame, CA, USA). For double‐labelling with the same species antibodies, sections were incubated in 10 mM sodium citrate, pH 6.0 at 121°C for 10 min in a pressure cooker after visualizing the first protein of interest to denature the first primary antibody and prevent non‐specific binding of the second primary antibody. After washing in PBS, sections were stained with a polymer‐based peroxidase immunocytochemistry detection kit (Power‐Vision Peroxidase system; ImmunoVision, Brisbane, CA, USA). Signal was detected using chromogen 3‐amino‐9‐ethylcarbazole (Sigma‐Aldrich) in 0.05‐M acetate buffer (pH 8.2). Sections were dried and coverslipped with Vectashield (Vector Laboratories Inc.).
Evaluation of immunohistochemistry
Immunoreactivity of MMPs and TIMPs was quantified by measuring the optical density (OD) of (dysmorphic) neurons, giant cells and glia within cortical tubers of patients using ImageJ as well as of neurons and glia within the neocortex of controls and perituberal cortex of TSC patients. For each case, photographs of representative brain areas were taken at 20× and the OD of six to 12 individual cells was measured after which the background signal of the respective picture was subtracted. Afterwards, the mean OD was calculated per subject. Additionally, the relative number of positive cells (0: no; 1: single to 10%; 2: 11‐50%; 3: >50%) was evaluated as described previously 36, 37.
TSC cell cultures
Primary TSC cell cultures were derived from surgical brain specimens obtained from two patients (age at surgery: 2.5 and 2 years; gender: male; mutation: TSC2) undergoing epilepsy surgery at the Wilhelmina Children's Hospital of the University Medical Center Utrecht (Utrecht, The Netherlands). Cell isolation was performed as described elsewhere 38, 39. Briefly, after removal of blood vessels, tissue was mechanically minced into smaller fragments and enzymatically digested by incubating at 37°C for 30 min with 2.5% trypsin (Sigma‐Aldrich). Tissue was washed with incubation medium containing Dulbecco's modified Eagle's medium (DMEM)/HAM F10 (1:1) medium (Gibco, Life Technologies, Grand Island, NY, USA), supplemented with 1% penicillin/streptomycin and 10% foetal calf serum (Gibco, Life Technologies) and triturated by passing through a 70‐μm mesh filter. The cell suspension was incubated at 37°C, 5% CO2 for 48 h to let cells adhere the culture flask before it was washed with PBS to remove excess of myelin and cell debris. Cultures were subsequently refreshed twice a week.
Transfection and stimulation of cell cultures
Cells plated in poly‐L‐lysin‐coated plates were transfected with mimic pre‐miRNA for miR146a or miR147b (Applied Biosystems, Carlsbad, CA, USA). Oligonucleotides were delivered to the cells using Lipofectamine® 2000 transfection reagent (Life Technologies) in a final concentration of 50 nM for a total of 24 h before the start of stimulation. Astrocyte cultures were stimulated with human recombinant (r)IL‐1β (10 ng/ml; Peprotech, Rocky Hill, NJ, USA) for 24 h. The viability of human cell cultures was not influenced by the treatment with IL‐1β (as shown before; 40). The cells were harvested after 24 h of stimulation.
RNA isolation and RT‐PCR
For RNA isolation, cell culture or fresh brain material was homogenized in Qiazol Lysis Reagent (Qiagen Benelux, Venlo, The Netherlands). Total RNA was isolated using the miRNeasy Mini kit (Qiagen Benelux) according to the manufacturer's instructions. The concentration and purity of RNA were determined at 260/280 nm using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). To evaluate mRNA expression, 200–500 ng of cell culture‐derived total RNA and 2500 ng of fresh brain‐derived total RNA were reverse‐transcribed into cDNA using oligo dT primers. Expression of MMPs and TIMPs was evaluated as described previously 40 using the following primer sequences: MMP2 (forward primer ataacctggatgccgtcgt, reverse primer aggcacccttgaagaagtagc); MMP3 (forward primer ctccaaccgtgaggaaaatc, reverse primer catggaatttctcttctcatcaaa); MMP9 (forward primer gaaccaatctcaccgacagg, reverse primer gccacccgagtgtaaccata); MMP14 (forward primer gccttggactgtcaggaatg, reverse primer aggggtcactggaatgctc); TIMP1 (forward primer gggcttcaccaagacctaca, reverse primer tgcaggggatggataaacag); TIMP2 (forward primer tgcagatgtagtgatcagggc, reverse primer tctcaggccctttgaacatc); TIMP3 (forward primer gctggaggtcaacaagtacca, reverse primer cacagccccgtgtacatct) and TIMP4 (forward primer ttggtgcagagggaaagtct, reverse primer ggtactgtgtagcaggtggtga). Quantification of data was performed using the LinRegPCR program in which linear regression on the Log (fluorescence) per cycle number data is applied to determine the amplification efficiency per sample 41. The starting concentration of each specific product was divided by the geometric mean of the starting concentration of the reference genes EF1α and C1orf43 and this ratio was compared between groups.
Statistical analysis
Statistical analysis of immunohistochemistry was performed using IBM SPSS Statistics 22 (IBM Nederland, Amsterdam, The Netherlands) using the Mann–Whitney U‐Test. For mRNA data from cell culture and human tissue, statistical analyses were performed with GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA) using the Mann–Whitney U‐Test. A P < 0.05 was assumed to indicate a statistical difference.
Results
Matrix metalloproteinase expression in control cortex and cortical tubers
There was higher gene expression in cortical tubers of patients with TSC vs. neocortical control tissue for MMP9 (P < 0.01; Figure 1 A) and MMP14 (P < 0.001; Figure 1 A). No significant changes in gene expression were detected for MMP2 and MMP3 (Figure 1 A).
Figure 1.
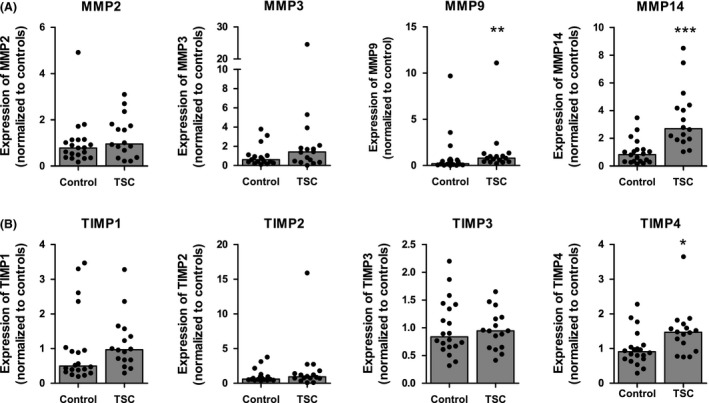
mRNA expression of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinase (TIMPs) in tuberous sclerosis complex (TSC) tubers. Quantitative real‐time PCR shows higher mRNA expression of MMP9, MMP14 (A) and TIMP4 (B) in cortical tubers from TSC patients compared to control tissue. mRNA expression of MMP2, MMP3, TIMP1, TIMP2, TIMP3 and TIMP4 did not change. *P < 0.05, **P < 0.01, ***P < 0.001; Mann–Whitney U‐Test.
Resected neocortical tissue from six patients with TSC was used for immunohistochemical analysis of protein expression of MMPs and TIMPs and compared to neocortical tissue from six controls. Strong MMP2 protein expression was observed in neurons, while no to weak staining was seen in glial cells of control cortex (Figure 2 A,B). In TSC tubers, strong MMP2 expression was observed in dysmorphic neurons and giant cells (Figure 2 C). The OD and the relative number of MMP2‐positive cells were higher in glial cells of cortical tubers compared to controls (P < 0.01; Table 1). Furthermore, the relative number of MMP2‐positive glial cells was higher in cortical tubers compared to perituberal cortex.
Figure 2.
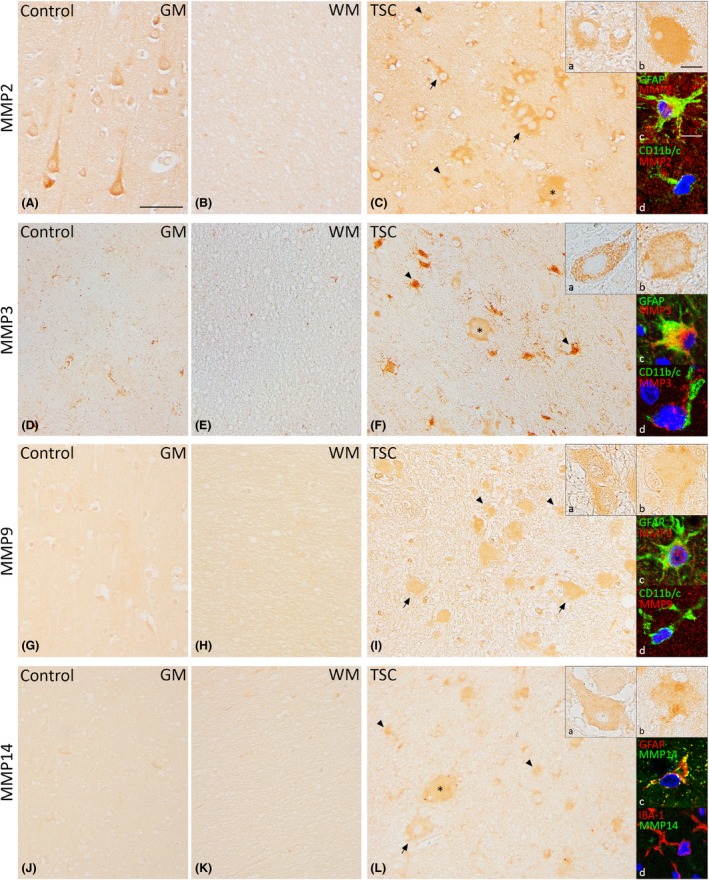
Expression of matrix metalloproteinases 2 (MMP2), MMP3, MMP9 and MMP14 in TSC tubers. Strong MMP2 expression in neurons and weak MMP2 expression in glial cells were seen in control cortex (A,B). In TSC tubers, strong MMP2 expression was observed in dysmorphic neurons, giant cells and glial cells. (C). MMP3 expression was not observed in neurons and weakly in glial cells of controls (D,E). Moderate expression was observed in dysmorphic neurons and giant cells, while expression was strong in glial cells (F). Moderate MMP9 expression was observed in neurons and no to weak expression was seen in glial cells of control cortex (G,H). In TSC tubers, dysmorphic neurons, giant cells and astrocytes showed moderate MMP9 expression (I). MMP14 expression was weak in neurons and not present in glial cells in controls (J,K). Moderate expression of MMP14 was observed in dysmorphic neurons, giant cells and astrocytes of TSC tubers (L). Insets a show magnification of dysmorphic cells, insets b show magnification of giant cells. Insets c and d in C,F,I and L depict double‐labelling of MMPs with glial fibrillary acidic protein (GFAP)‐positive cells (astrocytes) and IBA‐1 or CD11b/c‐positive cells (microglia), counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI, blue). GM, grey matter; WM, white matter; TSC, tuberous sclerosis complex. Arrows depict dysmorphic neurons, arrowheads depict glial cells and asterisks depict giant cells. Scale bar A–I: 100 μm; insets a, b: 25 μm; insets c, d: 12.5 μm.
Table 1.
Immunoreactivity analysis of matrix metalloproteinases 2 (MMP2), MMP3, MMP9 and MMP14
| (Dysmorphic) Neurons | Glia | Giant cells | ||||
|---|---|---|---|---|---|---|
| OD | Rel. no. | OD | Rel. No. | OD | Rel. no. | |
| MMP2 | ||||||
| Control | 25 ± 4 | 3 | 13 ± 3 | 1 (1–2) | – | – |
| Tuber | 30 ± 2 | 3 | 32 ± 3** | 3*** , ^^ | 26 ± 3 | 3 |
| Perituber | 23 ± 8 | 3 (2–3) | 23 ± 6 | 2 (2–3)** | – | – |
| MMP3 | ||||||
| Control | −1 ± 1 | 3 (1–3) | 54 ± 6 | 1 (1–2) | – | – |
| Tuber | 16 ± 5 | 2.5 (1–3) | 58 ± 7 | 2.5 (1–3)* | 10 ± 4 | 2.5 (1–3) |
| Perituber | 2 ± 2 | 3 | 40 ± 7 | 2 (1–3) | – | – |
| MMP9 | ||||||
| Control | 9 ± 2 | 3 (2–3) | 4 ± 3 | 2 (1–3) | – | – |
| Tuber | 13 ± 1 | 3 | 13 ± 2* | 2 (2–3) | 12 ± 2 | 3 |
| Perituber | 8 ± 2 | 3 | 8 ± 5 | 3 (1–3) | – | – |
| MMP14 | ||||||
| Control | 9 ± 2 | 2.5 (1–3) | −1 ± 0 | 1 | – | – |
| Tuber | 15 ± 2 | 3 (2–3) | 17 ± 2** , ^ | 2.5 (2–3)** | 17 ± 5 | 3 (2–3) |
| Perituber | 5 ± 3 | 3 | 5 ± 2 | 3** | – | – |
The background‐corrected optical density (OD) is given as mean ± SE of the mean. The relative number (Rel. no.) of positive cells is given as median (minimum–maximum) and is defined as follows: 0: no; 1: single to 10%; 2: 11–50%; 3: >50%. Different compared to control, *P < 0.05, **P < 0.01, ***P < 0.001. Different compared to tuberous sclerosis complex perituber, ^ P < 0.05, ^^ P < 0.01.
In control tissue, MMP3 protein expression was not observed in neurons and moderately to strongly expressed in the present glial cells (Figure 2 D,E). In TSC tubers, however, the relative number of MMP3‐positive glia was higher compared to controls (P < 0.05; Table 1). Additionally, moderate expression of MMP3 was observed in dysmorphic neurons and giant cells (Figure 2 F).
Moderate MMP9 protein expression was observed in neurons while no to weak expression was observed in glial cells of control tissue (Figure 2 G,H). TSC tubers had moderate expression of MMP9 in dysmorphic neurons and giant cells (Figure 2 I). The OD of MMP9‐positive cells was higher in glia cells in TSC tubers compared to controls (Table 1).
MMP14 protein expression was weak in neurons and not present in glial cells of control tissue (Figure 2 J,K). MMP14 expression in TSC tubers was moderate in dysmorphic neurons and giant cells (Figure 2 L). The OD in TSC tubers was higher compared to both controls (P < 0.01) and perituberal cortex (P < 0.05; Table 1). The relative number of MMP14‐positive glia was higher in both tuber and perituberal cortex compared to control tissue (P < 0.01; Table 1).
Double‐labelling experiments showed that all MMPs co‐localized with the astrocytic marker GFAP (Figure 2 insets c) and that MMP2 and MMP3 also co‐localized with the microglial marker CD11b/c (Figure 2 insets d).
Tissue inhibitor of metalloproteinase expression in control cortex and in cortical tubers
There was higher gene expression in cortical tuber of patients with TSC vs. neocortical control tissue for TIMP4 (P < 0.05; Figure 1 B), while no significant changes in gene expression were detected for TIMP1, TIMP2 and TIMP3 (Figure 1 B).
In control tissue, weak TIMP1 protein expression in neurons and moderate TIMP1 expression was observed in glial cells (Figure 3 A,B). In cortical tubers, moderate expression of TIMP1 was observed in dysmorphic neurons, giant cells and astrocytes (Figure 3 C). The OD of TIMP1‐positive glia in tubers was higher compared to perituberal cortex (P < 0.01; Table 2). The relative number of TIMP1‐positive glia was higher in TSC tubers compared to controls (P < 0.05, Table 2).
Figure 3.
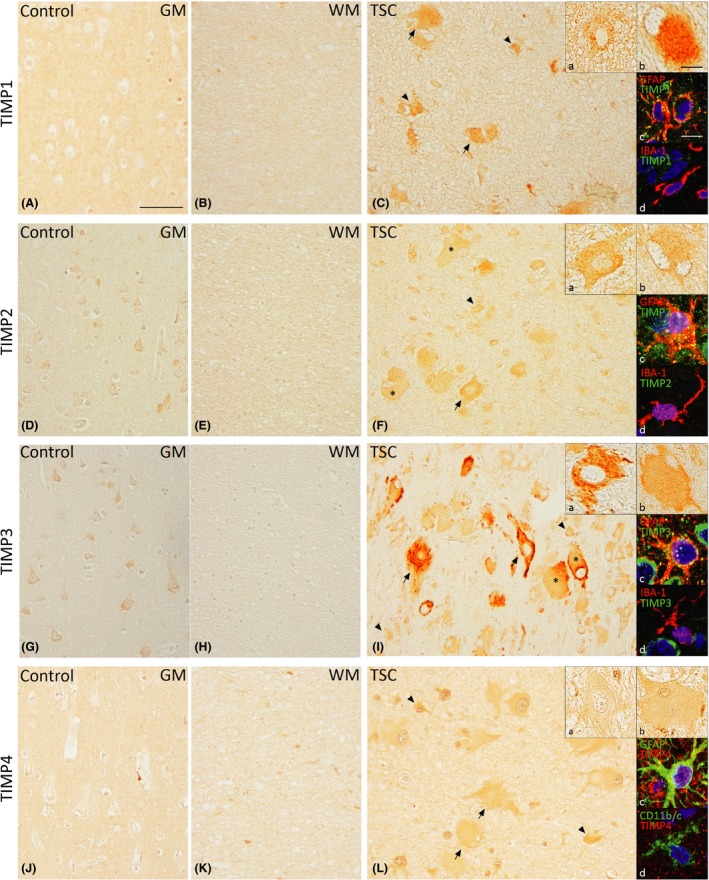
Expression of tissue inhibitor of metalloproteinase 1 (TIMP1), TIMP2, TIMP3 and TIMP4 in TSC tubers. Weak TIMP1 expression in neurons and moderate expression in glial cells were observed in control cortex (A,B). In TSC, TIMP1 was moderately expressed in dysmorphic neurons, giant cells and astrocytes (C). Moderate expression of TIMP2 in neurons and weak expression in glial cells were seen in control tissue (D,E). In TSC, dysmorphic neurons and giant cells showed strong TIMP2 expression while astrocytes showed moderate TIMP2 expression (F). TIMP3 was moderately expressed in neurons only in controls (G,H). Strong TIMP3 expression was seen in dysmorphic neurons, giant cells and astrocytes in TSC tubers (I). In control cortex, TIMP4 expression was moderate in glial cells only (J,K). Strong TIMP4 expression was seen in dysmorphic neurons, giant cells and astrocytes of TSC tubers (L). Insets a show magnification of dysmorphic cells, insets b show magnification of giant cells. Insets c and d in C,F,I and L depict double‐labelling of matrix metalloproteinases (MMPs) with glial fibrillary acidic protein (GFAP)‐positive cells (astrocytes) and IBA‐1 or CD11b/c‐positive cells (microglia), counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI, blue). GM, grey matter; WM, white matter; TSC, tuberous sclerosis complex. Arrows depict dysmorphic neurons, arrowheads depict glial cells and asterisks depict giant cells. Scale bar A–I: 100 μm; insets a, b: 25 μm; insets c, d: 12.5 μm.
Table 2.
Immunoreactivity analysis of tissue inhibitor of metalloproteinase 1 (TIMP1), TIMP2, TIMP3 and TIMP4
| (Dysmorphic) Neurons | Glia | Giant cells | ||||
|---|---|---|---|---|---|---|
| OD | Rel. no. | OD | Rel. no. | OD | Rel. no. | |
| TIMP1 | ||||||
| Control | 4 ± 6 | 2 (1–3) | 27 ± 6 | 1.5 (1–2) | – | – |
| Tuber | 25 ± 5 | 2.5 (1–3) | 36 ± 5^^ | 2.5 (2–3)* | 14 ± 3 | 2.5 (1–3) |
| Perituber | 0 ± 4 | 3 (2–3) | 3 ± 5* | 1 (1–3) | – | – |
| TIMP2 | ||||||
| Control | 4 ± 6 | 3 (2–3) | 27 ± 6 | 1 | – | – |
| Tuber | 33 ± 4 | 3 | 39 ± 5 | 3*** , ^^ | 29 ± 4 | 3 |
| Perituber | 21 ± 2 | 3 | 24 ± 4 | 2** | – | – |
| TIMP3 | ||||||
| Control | 24 ± 2 | 2.5 (2–3) | 8 ± 1 | 1 | – | – |
| Tuber | 93 ± 11 | 3 | 58 ± 12** | 2.5 (2–3)** | 48 ± 11 | 3 |
| Perituber | 44 ± 6 | 3 | 32 ± 9* | 2.5 (2–3)** | – | – |
| TIMP4 | ||||||
| Control | 1 ± 4 | 3 (2–3) | 64 ± 7 | 1 (1–2) | – | – |
| Tuber | 50 ± 13 | 3 (2–3) | 64 ± 6 | 3 (2–3)** , ^ | 38 ± 8 | 3 (2–3) |
| Perituber | 10 ± 5 | 3 | 53 ± 8 | 2 (2–3)** | – | – |
The background‐corrected optical density (OD) is given as mean ± SE of the mean. The relative number (Rel. no.) of positive cells is given as median (minimum–maximum) and is defined as follows: 0: no; 1: single to 10%; 2: 11–50%; 3: >50%. Different compared to control, *P < 0.05, **P < 0.01, ***P < 0.001. Different compared to tuberous sclerosis complex perituber, ^ P < 0.05, ^^ P < 0.01.
Moderate protein expression of TIMP2 was observed in neurons, while weak expression was seen in glial cells of control brain tissue (Figure 3 D,E). In cortical tubers, dysmorphic neurons and giant cells showed strong expression of TIMP2 (Figure 3 F). The relative number of TIMP2‐positive glia was higher in both tubers (P < 0.001) and perituber (P < 0.01) compared to controls, with TIMP2 having the highest relative number in tubers (P < 0.05; Table 2).
TIMP3 showed a moderate protein expression in neurons in control tissue, while glial cells showed no expression of TIMP3 (Figure 3 G,H). Strong TIMP3 expression was observed in dysmorphic neurons and moderate expression in giant cells of cortical tubers (Figure 3 I). Both the OD and relative number of positive cells are higher in tubers (P < 0.01) and perituberal tissue (P < 0.05, P < 0.01 respectively) compared to controls (Table 2).
Protein expression of TIMP4 was not present in neurons of control cortex; however, moderate expression was seen in glial cells (Figure 3 J,K). Strong TIMP4 expression was observed in dysmorphic neurons and giant cells of cortical tubers (Figure 3 L). The relative number of TIMP4‐positive glia was higher within tubers and perituberal cortex as compared to controls (P < 0.01) with glia in tubers displaying the highest relative number (P < 0.05; Table 2).
Fluorescent double‐labelling showed co‐localization of all TIMPs with GFAP (Figure 3 insets c), while co‐localization with microglia markers was not evident.
To study whether specific MMPs and TIMPs co‐localize, double‐labelling was performed. Co‐localization of MMP2 and TIMP2 was observed in giant cells and dysmorphic neurons while many glial cells were only immunopositive for MMP2 (Figure S1 A). MMP3 co‐localized with TIMP1, while co‐localization with TIMP3 was only seen in some giant cells, but not in glia (Figure S1 B,C). Co‐localization of MMP9 and TIMP1, as well as MMP14 with TIMP2 and TIMP4 was observed in the majority of cells (Figure S1 D–F).
Increased albumin and CD163 immunoreactivity in cortical tubers
To assess BBB dysfunction, extravasation of albumin and the presence of CD163‐positive perivascular macrophages were investigated in tubers and the adjacent tissue of TSC patients using immunohistochemistry. In perituberal cortex, albumin extravasation was not observed (Figure 4 A). In tubers, however, strong albumin immunoreactivity was seen in the parenchyma. Additionally, astrocytes, dysmorphic neurons and giant cells were albumin‐positive (Figure 4 B). CD163‐positive cells were sparsely present around vessels in the perituberal cortex (Figure 4 C), while considerably more CD163‐positive cells were observed in tubers. Moreover, CD163‐positive cells in tubers had the morphology of activated macrophages with large cell bodies and coarse processes (Figure 4 D inset).
Figure 4.
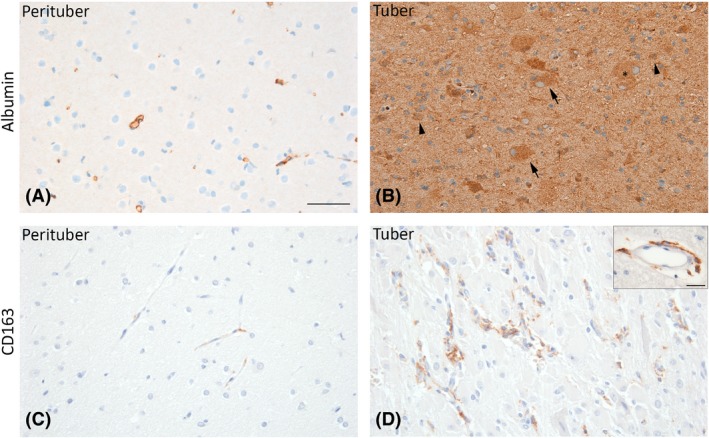
Albumin and CD163 immunoreactivity in tuberous sclerosis complex (TSC) tubers. Albumin extravasation was not observed in perituberal brain tissue of TSC patients (A). In tubers, the parenchyma was strongly positive for albumin. Albumin staining was also seen in glial cells, dysmorphic neurons and giant cells within the tuber (B). CD163‐positive perivascular macrophages were sparsely present around vessels in perituberal brain tissue (C). In tubers, CD163‐positive cells were more frequently observed around blood vessels (D) and had an activated morphology with large cell bodies and coarse processes (inset). Arrows depict dysmorphic neurons, arrowheads depict glial cells and asterisks depict giant cells. Scale bar A–D: 200 μm, scale bar inset: 25 μm.
Modulation of MMPs and TIMPs by miR146a and miR147b in tuber‐derived cultures
To investigate whether the expression of MMPs and TIMPs could be modulated, TSC tuber‐derived cell cultures were transfected with either miR146a or miR147b mimic molecules and subsequently stimulated with IL‐1β to mimic an inflammatory response. After transfection, Taqman PCRs confirmed the overexpression of miR146a and miR147b 34 indicating successful transfection. MMP3 expression was higher after IL‐1β stimulation compared to control (P = 0.002; Figure 5 B), while MMP2, MMP9 and MMP14 expression did not change (Figure 5 A,C,D). IL‐1β‐induced upregulation of MMP3 could be attenuated by approximately 50% after transfection with miR146a or miR147b (P = 0.002). Following IL‐1β stimulation, expression of TIMP2, TIMP3 and TIMP4 was lower compared to control (P = 0.002, P = 0.002, P = 0.026 respectively; Figure 6 B–D). Decreased TIMP2 and TIMP3 expression could be attenuated by both miR146a (P = 0.002) and miR147b (P = 0.009) transfection.
Figure 5.

Effects of interleukin (IL)‐1β stimulation and microRNA transfection on matrix metalloproteinases (MMP) expression. Quantitative real‐time PCR of MMP2, MMP3, MMP9 and MMP14 in tuberous sclerosis complex (TSC) tuber‐derived cell cultures. (B) Stimulation of cells with IL‐1β led to higher MMP3 expression, which was attenuated by transfection with either miR146a or miR147b. (A, C, D) The expression of MMP2, MMP9 and MMP14 did not change after stimulation and/or transfection with microRNAs. **P < 0.01; Mann–Whitney U‐Test.
Figure 6.
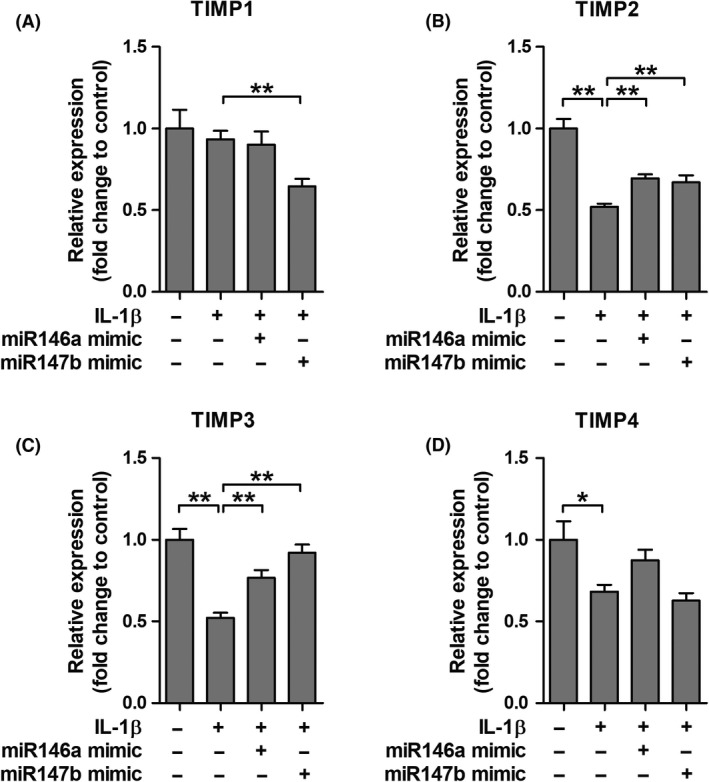
Effects of interleukin (IL)‐1β stimulation and microRNA transfection on tissue inhibitor of metalloproteinase (TIMPs) expression. Quantitative real‐time PCR of TIMP1, TIMP2, TIMP3 and TIMP4 in tuberous sclerosis complex (TSC) tuber‐derived cell cultures. (B,C,D) Stimulation of cells with IL‐1β led to higher TIMP2, TIMP3 and TIMP4 expression. Decreased TIMP2 and TIMP3 mRNA expression could be attenuated by transfection with both miR146a and miR147b. *P < 0.05, **P < 0.01; Mann–Whitney U‐Test.
Discussion
This study provides insight into the dysregulated MMP/TIMP proteolytic system in TSC brain. We demonstrated higher expression of several MMPs and TIMPs in TSC tubers as compared to normal neocortical brain tissue and perituberal cortex, particularly in dysmorphic neurons and giant cells, as well as in glial cells, which was associated with BBB dysfunction. Furthermore, IL‐1β‐induced upregulation of MMP3 and downregulation of TIMP2 and TIMP3 in tuber‐derived cultures could be attenuated by miR146a or miR147b transfection. These findings will be discussed in further detail in the following paragraphs.
Dysregulation of MMP and TIMP in cortical tubers
We observed that besides higher mRNA expression of MMP9, MMP14 and TIMP4, protein expression of MMP2, 3, 9 and 14 and TIMP1, 2, 3 and 4 is abundantly present in cortical tubers of patients with TSC and that particularly glial cells, dysmorphic neurons and giant cells were strongly immunopositive for MMPs. To the best of our knowledge, this is the first study providing a complete picture of the expression and cellular distribution of major brain MMPs and TIMPs in resected tubers of TSC patients. The discrepancy between mRNA and protein expression of MMPs/TIMPs may be attributed to the fact that mRNA data were obtained using homogenized cortical specimens, while protein expression was studied at the cellular level using immunohistochemistry which provides a better resolution as compared to the use of homogenates.
The importance of ECM alterations in the brain of TSC patients has been suggested by sequencing studies investigating the transcriptional landscape of cortical tubers. It has been shown that the ECM was among the most significantly changed GO terms related to increased gene expression in cortical tubers 42. Moreover, out of all 438 differentially expressed genes in TSC tubers found by high‐throughput RNA sequencing, the majority of the elevated genes was associated with ECM organization along with the innate immune response 43. Only in few studies, the expression of a limited number of MMPs was investigated in resected brain tissue of patients with epilepsy‐related disorders. Li and colleagues studied MMP9 expression in TSC and focal cortical dysplasia (FCD), another malformation of cortical development and a well‐known cause of pharmacoresistant epilepsy. They found that MMP9 protein expression was higher in TSC and FCD brain compared to controls and observed high expression of MMP9 in giant cells and dysmorphic neurons 44. Konopka et al. observed strong MMP9 expression in dysmorphic neurons and balloon cells in the cortex of FCD patients and showed that neuronal protein expression of MMP9 was higher in FCD patients compared to control brain 45. Besides MMP9, they showed that MMP2, MMP3 and TIMP2 expressions were higher in the dysplastic cortex of adult FCD patients vs. controls. In hippocampi of patients with temporal lobe epilepsy (TLE), MMP9 expression was higher compared to controls 44. Furthermore, higher MMP2 and MMP9 activity was found in the epileptogenic and perilesional area of patients with TLE 46. Only one study investigated TIMP expression in a human epilepsy‐related pathology. Acar and colleagues observed pronounced protein expression of TIMP1 and TIMP2 in a small number of TLE patients 47. In the present study, co‐localization of several MMPs and TIMPs was found in glial cells as well as in tuberous cells. However, MMP3 was not found to be co‐localized with TIMP3 in glia and a similar pattern was seen for some, but not all, MMP2‐positive glial cells. Specific MMP and TIMP combinations were chosen based on the interaction between MMPs and TIMPs as described in literature 35. The fact that MMPs and TIMPs can be produced by the same cells and that their expressions are regulated by these cells can have important functional implications. As TIMPs are generally thought to be MMP inhibitors 5, the higher expression of TIMPs in tubers brain may occur in response to higher MMP expression, as a compensatory mechanism. Furthermore, TIMPs may protect the BBB 48. However, knowledge about the precise function and spatio‐temporal expression patterns of MMPs and TIMPs is still somewhat limited and it should be noted that some TIMPs might even be involved in enhancing MMP activity. Controversially, TIMP2, an effective inhibitor of all MMPs, is also responsible for the activation of pro‐MMP2 with the use of two MMP14 molecules 49, 50, leading to cleavage of the propeptide which activates MMP2. Furthermore, TIMP2, TIMP3 and TIMP4 are able to interact with propeptide of MMP2 and TIMP1 and TIMP3 is able to interact with pro‐MMP9 51. Future studies, in which the cellular distribution and expression in mutant animals are taken into account, may provide more answers.
Although the mechanisms underlying the regulation of MMPs and TIMPs in TSC tubers are still unclear, a potential link with the mTOR pathway has been suggested, as both MMPs, TIMPs and mTOR are downstream targets of the PI3K/Akt pathway 52, 53, 54, 55. A recent study showed that inhibition of the mTOR pathway by rapamycin leads to the downregulation of MMP9 activity in models of Alzheimer's disease and vascular cognitive impairment 56. Given the increased activation of the mTOR pathway due to TSC1/TSC2 mutation, the link between mTOR and MMPs deserves further investigation in relation to cortical tubers.
In the present study, a dysregulation of MMPs and TIMPs was associated with BBB dysfunction, which was shown by abundant albumin extravasation and the presence of CD163‐positive perivascular macrophages in cortical tubers. Increased expression and activation of MMPs are often observed in pathology where the BBB is compromised, such as cerebral ischaemia 57, 58 and traumatic brain injury 59. Indeed, studies using animal models of stroke have shown increased MMP expression and activity along with BBB dysfunction 60. In our previous studies, BBB dysfunction was also reported in both cortical tubers as well as in subependymal giant cell astrocytomas of TSC patients 28, 30.
The basement membrane of brain blood vessels consists of ECM secreted by the surrounding cells and plays an important role in vascular integrity. MMPs are able to degrade several components of this basement membrane such as laminin, fibronectin, heparan sulphate and type IV collagen 61, 62, 63, thereby influencing barrier permeability. Furthermore, several MMPs target tight junction and adherens junction proteins that control cell‐cell and cell‐matrix interactions at the barrier 64. Yang et al. showed that MMP2, which is bound and activated by MMP14, targets claudin‐5 and occludin and that fragmentation of these tight junction proteins can be prevented by inhibiting a broad‐spectrum of MMPs 65. As a consequence of basal lamina and tight junction degradation, BBB permeability increases which can allow the extravasation of leucocytes. Interestingly, leucocytes are an important source of MMP9 66, 67 and extravasation can lead to increased MMP2 activation, thereby providing a positive feedback loop on proteolysis 68. Furthermore, by opening the BBB, neuroinflammation can lead to increased expression and activity of MMPs 69, 70. In the present study, we showed that abundant perivascular macrophages were present in the TSC brain. We previously showed that the number of perivascular macrophages is positively correlated with BBB permeability 37. Furthermore, glial cells such as microglia and astrocytes, often with reactive morphology, had higher expression of MMPs in the TSC brain as compared to autopsy control or perituberal brain tissue. In turn, MMPs can activate cytokines such as tumour necrosis factor‐α, IL‐1β and IL‐8 by cleavage of the propeptide 71, 72, 73. Recently, it was shown that besides chemokines and cytokines, cytokine‐induced activity of MMP2 and MMP9 at the BBB also result in increased leucocyte infiltration 74. These studies, together with our observations, suggest that increased MMP activation, BBB disruption and increased neuroinflammation coincide and are able to enhance one another.
Taken together, we showed that dysregulation of MMPs/TIMPs and BBB dysfunction specifically occurs in epileptogenic tubers of TSC patients with recurrent seizures, but not in perilesional tissue. This indicates that, rather than just being a consequence of seizure activity, MMPs may play a crucial role in epilepsy pathology.
One of the main characteristics of TSC tubers is the presence of brain inflammation, in particular a prominent activation of the IL‐1β signalling pathway 28, 42. Several miRNAs have been shown to be important during inflammatory processes in epilepsy‐associated pathologies 75, 76, 77, 78 and have been proposed to have beneficial effects on MMP expression in in vitro studies 33. Furthermore, a recent meta‐analysis has shown the involvement of differentially expressed miRNAs in both human and experimental TLE in pathways of inflammation, gliosis and ECM dysregulation 79.
We wondered whether MMP expression could be modulated in vitro by anti‐inflammatory miR146a or miR147b. Stimulation of tuber‐derived cells with IL‐1β resulted in higher mRNA expression of MMP3, though it did not affect the mRNA expression of MMP2, MMP9 and MMP14. This might be explained by the fact that the cells, taken from the cortical tubers of TSC patients, already exhibit a high amount of MMP2, MMP9 and MMP14 transcripts and are therefore not sensitive to further induction by pro‐inflammatory molecules. Moreover, mRNA expression of TIMP2, TIMP3 and TIMP4 was lower after IL‐1β stimulation compared to controls, indicating a misbalance in the MMP/TIMP proteolytic system after this inflammatory stimulus. We found that IL‐1β‐induced upregulation of MMP3 and downregulation of TIMPs could be attenuated by miR146a or miR147b in tuber‐derived cell cultures, thereby re‐establishing the balance. Previously, we have shown that inhibition of pro‐inflammatory miR155 attenuated IL‐1β‐induced MMP3 overexpression in human foetal astrocytes 33. miR146a has been shown to inhibit MMP2 expression in prostate cancer tissue 80 and the activity of MMP9 in human cardiac cells 81. In breast cancer cells, miR146a negatively regulates the activity of NFκB, thereby suppressing the expression of NFκB target gene MMP9 82. Furthermore, it was shown that transient treatment with miR146a mimic molecules after kainic acid‐induced epilepsy in mice had disease‐modifying effects as it decreased seizure recurrence and prevented disease progression 83. As it has been shown that both miR146a and miR147b are negative‐feedback regulators of astrocyte‐mediated inflammatory response 34, 77, 84, it is tempting to speculate that these miRNAs inhibit MMP expression in pathological tissue, but this needs to be further investigated.
This study shows that the expression of MMPs and TIMPs is dysregulated and associated with BBB dysfunction in cortical tubers which may contribute to the pathological brain networks in TSC. Dysregulated MMP and TIMP expression can be ameliorated in vitro by the anti‐inflammatory miR146a or miR147b. Thus, these miRNAs deserve further investigation as a novel therapeutic approach to restore the BBB and the ECM.
Author contributions
DWMB, JvS, EA and EAvV conceived and designed the experiments. FEJ, WGS, PCvR have provided human brain tissue which was analysed and diagnosed by EA. DWMB, JvS and JJA processed human brain tissue. DWMB performed immunohistochemistry and analysed the data. DWMB and JvS performed RT‐qPCR and analysed the data. DWMB, JvS, LW, BvhH, WWK and HEdV were involved in the in vitro experiments. DWMB, JvS, EA and EAvV contributed to the data interpretation and writing of the manuscript. All authors read, revised and approved the final manuscript.
Disclosure
The authors have no conflicts of interest to report. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1. Co‐localization of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinase (TIMPs) in cortical tubers. Co‐localization of MMP2 and TIMP2 was observed in giant cells and dysmorphic neurons, while glial cells were only MMP2‐positive (A). MMP3 co‐localized with TIMP1 in all cells and with TIMP3 in some giant cells, but not in glia (B,C). Co‐localization was observed between MMP9 and TIMP1 (D) and MMP14 with both TIMP2 and TIMP4 (E,F). Arrowheads depict glial cells and asterisk depict giant cell. Scale bar A–F: 25 μm.
Table S1. Clinical findings of patients with tuberous sclerosis complex (TSC) (1–20) and autopsy controls (21–43).
Acknowledgements
The research leading to these results has received funding from the European Union's Seventh Framework Program (FP7/2007–2013) under grant agreement 602102 (EPITARGET; EAvV, EA) and 602391 (EPISTOP; JvS, EA, FEJ) and the Dutch Epilepsy Foundation, project 16‐05 (DWMB, EAvV).
Broekaart D. W. M., van Scheppingen J., Anink J. J., Wierts L., van het Hof B., Jansen F. E., Spliet W. G., van Rijen P. C., Kamphuis W. W., de Vries H. E., Aronica E. and van Vliet E. A. (2020) Neuropathology and Applied Neurobiology 46, 142–159 Increased matrix metalloproteinases expression in tuberous sclerosis complex: modulation by microRNA 146a and 147b in vitro
Contributor Information
E. Aronica, Email: e.aronica@amc.uva.nl.
E. A. van Vliet, Email: e.a.vanvliet@uva.nl.
References
- 1. Rivera S, Khrestchatisky M, Kaczmarek L, Rosenberg GA, Jaworski DM. Metzincin proteases and their inhibitors: foes or friends in nervous system physiology? J Neurosci 2010; 30: 15337–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res 2003; 92: 827–39 [DOI] [PubMed] [Google Scholar]
- 3. Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost 2001; 86: 324–33 [PubMed] [Google Scholar]
- 4. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochem Biophys Acta 2000; 1477: 267–83 [DOI] [PubMed] [Google Scholar]
- 5. Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol 2015; 44–46: 247–54 [DOI] [PubMed] [Google Scholar]
- 6. Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005; 50: 329–39 [DOI] [PubMed] [Google Scholar]
- 7. Cauwe B, Opdenakker G. Intracellular substrate cleavage: a novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit Rev Biochem Mol Biol 2010; 45: 351–423 [DOI] [PubMed] [Google Scholar]
- 8. Pauly T, Ratliff M, Pietrowski E, Neugebauer R, Schlicksupp A, Kirsch J, et al Activity‐dependent shedding of the NMDA receptor glycine binding site by matrix metalloproteinase 3: a PUTATIVE mechanism of postsynaptic plasticity. PLoS ONE 2008; 3: e2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michaluk P, Kolodziej L, Mioduszewska B, Wilczynski GM, Dzwonek J, Jaworski J, et al Beta‐dystroglycan as a target for MMP‐9, in response to enhanced neuronal activity. J Biol Chem 2007; 282: 16036–41 [DOI] [PubMed] [Google Scholar]
- 10. Conant K, Wang Y, Szklarczyk A, Dudak A, Mattson MP, Lim ST. Matrix metalloproteinase‐dependent shedding of intercellular adhesion molecule‐5 occurs with long‐term potentiation. Neuroscience 2010; 166: 508–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vaillant C, Meissirel C, Mutin M, Belin MF, Lund LR, Thomasset N. MMP‐9 deficiency affects axonal outgrowth, migration, and apoptosis in the developing cerebellum. Mol Cell Neurosci 2003; 24: 395–408 [DOI] [PubMed] [Google Scholar]
- 12. Barkho BZ, Munoz AE, Li X, Li L, Cunningham LA, Zhao X. Endogenous matrix metalloproteinase (MMP)‐3 and MMP‐9 promote the differentiation and migration of adult neural progenitor cells in response to chemokines. Stem Cells 2008; 26: 3139–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Hove I, Lemmens K, Van de Velde S, Verslegers M, Moons L. Matrix metalloproteinase‐3 in the central nervous system: a look on the bright side. J Neurochem 2012; 123: 203–16 [DOI] [PubMed] [Google Scholar]
- 14. Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends Immunol 2001; 22: 571–9 [DOI] [PubMed] [Google Scholar]
- 15. Rosenberg GA, Mun‐Bryce S. Matrix metalloproteinases in neuroinflammation and cerebral ischemia In Neuroinflammation in Stroke. Eds Dirnagl U, Elger B. Berlin, Heidelberg: Springer, 2004; 1–16. [DOI] [PubMed] [Google Scholar]
- 16. Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus 2007; 22: E4 [DOI] [PubMed] [Google Scholar]
- 17. van Vliet EA, da Costa AS, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood‐brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007; 130(Pt. 2): 521–34 [DOI] [PubMed] [Google Scholar]
- 18. Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, et al Lasting blood‐brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci 2004; 24: 7829–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marchi N, Granata T, Ghosh C, Janigro D. Blood‐brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia 2012; 53: 1877–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedman A. Blood‐brain barrier dysfunction, status epilepticus, seizures, and epilepsy: a puzzle of a chicken and egg? Epilepsia 2011; 52(Suppl. 8): 19–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet 2008; 372: 657–68 [DOI] [PubMed] [Google Scholar]
- 22. Crino PB. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol 2013; 125: 317–32 [DOI] [PubMed] [Google Scholar]
- 23. Curatolo P, Moavero R, van Scheppingen J, Aronica E. mTOR dysregulation and tuberous sclerosis‐related epilepsy. Expert Rev Neurother 2018; 18: 185–201 [DOI] [PubMed] [Google Scholar]
- 24. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006; 355: 1345–56 [DOI] [PubMed] [Google Scholar]
- 25. Curatolo P, Aronica E, Jansen A, Jansen F, Kotulska K, Lagae L, et al Early onset epileptic encephalopathy or genetically determined encephalopathy with early onset epilepsy? Lessons learned from TSC. Eur J Paediatr Neurol 2016; 20: 203–11 [DOI] [PubMed] [Google Scholar]
- 26. Curatolo P, D'Argenzio L, Cerminara C, Bombardieri R. Management of epilepsy in tuberous sclerosis complex. Expert Rev Neurother 2008; 8: 457–67 [DOI] [PubMed] [Google Scholar]
- 27. Kannan L, Vogrin S, Bailey C, Maixner W, Harvey AS. Centre of epileptogenic tubers generate and propagate seizures in tuberous sclerosis. Brain 2016; 139(Pt. 10): 2653–67 [DOI] [PubMed] [Google Scholar]
- 28. Boer K, Jansen F, Nellist M, Redeker S, van den Ouweland AM, Spliet WG, et al Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res 2008; 78: 7–21 [DOI] [PubMed] [Google Scholar]
- 29. Prabowo AS, Anink JJ, Lammens M, Nellist M, van den Ouweland AM, Adle‐Biassette H, et al Fetal brain lesions in tuberous sclerosis complex: TORC1 activation and inflammation. Brain Pathol 2013; 23: 45–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Vliet EA, Aronica E, Gorter JA. Blood‐brain barrier dysfunction, seizures and epilepsy. Semin Cell Dev Biol 2015; 38: 26–34 [DOI] [PubMed] [Google Scholar]
- 31. Prabowo AS, Iyer AM, Anink JJ, Spliet WG, van Rijen PC, Aronica E. Differential expression of major histocompatibility complex class I in developmental glioneuronal lesions. J Neuroinflammation 2013; 10: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ha TY. MicroRNAs in human diseases: from cancer to cardiovascular disease. Immune Netw 2011; 11: 135–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Korotkov A, Broekaart DWM, Van Scheppingen J, Anink JJ, Baayen JC, Idema S, et al Increased expression of matrix metalloproteinase 3 can be attenuated by inhibition of microRNA‐155 in cultured human astrocytes. J Neuroinflammation 2018. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Scheppingen J, Mills JD, Zimmer TS, Broekaart DWM, Iori V, Bongaarts A, et al miR147b: a novel key regulator of interleukin 1 beta‐mediated inflammation in human astrocytes. Glia 2018; 66: 1082–97 [DOI] [PubMed] [Google Scholar]
- 35. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochem Biophys Acta 2010; 1803: 55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Broekaart DWM, van Scheppingen J, Geijtenbeek KW, Zuidberg MRJ, Anink JJ, Baayen JC, et al Increased expression of (immuno)proteasome subunits during epileptogenesis is attenuated by inhibition of the mammalian target of rapamycin pathway. Epilepsia 2017; 58: 1462–72 [DOI] [PubMed] [Google Scholar]
- 37. Broekaart DWM, Anink JJ, Baayen JC, Idema S, de Vries HE, Aronica E, et al Activation of the innate immune system is evident throughout epileptogenesis and is associated with blood‐brain barrier dysfunction and seizure progression. Epilepsia 2018; 59: 1931–44 [DOI] [PubMed] [Google Scholar]
- 38. Aronica E, Gorter JA, Ijlst‐Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, et al Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci 2003; 17: 2106–18 [DOI] [PubMed] [Google Scholar]
- 39. Aronica E, Gorter JA, Rozemuller AJ, Yankaya B, Troost D. Interleukin‐1 beta down‐regulates the expression of metabotropic glutamate receptor 5 in cultured human astrocytes. J Neuroimmunol 2005; 160: 188–94 [DOI] [PubMed] [Google Scholar]
- 40. van Scheppingen J, Broekaart DW, Scholl T, Zuidberg MR, Anink JJ, Spliet WG, et al Dysregulation of the (immuno)proteasome pathway in malformations of cortical development. J Neuroinflammation 2016; 13: 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, et al Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 2009; 37: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boer K, Crino PB, Gorter JA, Nellist M, Jansen FE, Spliet WG, et al Gene expression analysis of tuberous sclerosis complex cortical tubers reveals increased expression of adhesion and inflammatory factors. Brain Pathol 2010; 20: 704–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mills JD, Iyer AM, van Scheppingen J, Bongaarts A, Anink JJ, Janssen B, et al Coding and small non‐coding transcriptional landscape of tuberous sclerosis complex cortical tubers: implications for pathophysiology and treatment. Sci Rep 2017; 7: 8089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li S, Yu S, Zhang C, Shu H, Liu S, An N, et al Increased expression of matrix metalloproteinase 9 in cortical lesions from patients with focal cortical dysplasia type IIb and tuberous sclerosis complex. Brain Res 2012; 1453: 46–55 [DOI] [PubMed] [Google Scholar]
- 45. Konopka A, Grajkowska W, Ziemianska K, Roszkowski M, Daszkiewicz P, Rysz A, et al Matrix metalloproteinase‐9 (MMP‐9) in human intractable epilepsy caused by focal cortical dysplasia. Epilepsy Res 2013; 104: 45–58 [DOI] [PubMed] [Google Scholar]
- 46. Quirico‐Santos T, Nascimento Mello A, Casimiro Gomes A, de Carvalho LP, de Souza JM, Alves‐Leon S. Increased metalloprotease activity in the epileptogenic lesion–Lobectomy reduces metalloprotease activity and urokinase‐type uPAR circulating levels. Brain Res 2013; 1538: 172–81 [DOI] [PubMed] [Google Scholar]
- 47. Acar G, Tanriover G, Acar F, Demir R. Increased expression of matrix metalloproteinase‐9 in patients with temporal lobe epilepsy. Turk Neurosurg 2015; 25: 749–56 [DOI] [PubMed] [Google Scholar]
- 48. Rosenberg GA, Kornfeld M, Estrada E, Kelley RO, Liotta LA, Stetler‐Stevenson WG. TIMP‐2 reduces proteolytic opening of blood‐brain barrier by type IV collagenase. Brain Res 1992; 576: 203–7 [DOI] [PubMed] [Google Scholar]
- 49. Atkinson SJ, Crabbe T, Cowell S, Ward RV, Butler MJ, Sato H, et al Intermolecular autolytic cleavage can contribute to the activation of progelatinase A by cell membranes. J Biol Chem 1995; 270: 30479–85 [DOI] [PubMed] [Google Scholar]
- 50. Itoh Y, Takamura A, Ito N, Maru Y, Sato H, Suenaga N, et al Homophilic complex formation of MT1‐MMP facilitates proMMP‐2 activation on the cell surface and promotes tumor cell invasion. EMBO J 2001; 20: 4782–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nagase H. Tailoring TIMPs for selective metalloproteinase inhibition In The Cancer Degradome, Ed Edwards D. Berlin: Springer, 2008; 787–810. [Google Scholar]
- 52. Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, et al Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP‐9. Hepatol Res 2009; 39: 177–86 [DOI] [PubMed] [Google Scholar]
- 53. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129: 1261–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qureshi HY, Ahmad R, Sylvester J, Zafarullah M. Requirement of phosphatidylinositol 3‐kinase/Akt signaling pathway for regulation of tissue inhibitor of metalloproteinases‐3 gene expression by TGF‐beta in human chondrocytes. Cell Signal 2007; 19: 1643–51 [DOI] [PubMed] [Google Scholar]
- 55. Valacca C, Tassone E, Mignatti P. TIMP‐2 interaction with MT1‐MMP activates the AKT pathway and protects tumor cells from apoptosis. PLoS ONE 2015; 10: e0136797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari Z, et al Inhibition of mTOR protects the blood‐brain barrier in models of Alzheimer's disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol 2018; 314: H693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chaturvedi M, Kaczmarek L. Mmp‐9 inhibition: a therapeutic strategy in ischemic stroke. Mol Neurobiol 2014; 49: 563–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lenglet S, Montecucco F, Mach F. Role of matrix metalloproteinases in animal models of ischemic stroke. Curr Vasc Pharmacol 2015; 13: 161–6 [DOI] [PubMed] [Google Scholar]
- 59. Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, et al The role of hypoxia‐inducible factor‐1alpha, aquaporin‐4, and matrix metalloproteinase‐9 in blood‐brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg 2011; 114: 92–101 [DOI] [PubMed] [Google Scholar]
- 60. Gu Y, Zheng G, Xu M, Li Y, Chen X, Zhu W, et al Caveolin‐1 regulates nitric oxide‐mediated matrix metalloproteinases activity and blood‐brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem 2012; 120: 147–56 [DOI] [PubMed] [Google Scholar]
- 61. Lee H, Park JW, Kim SP, Lo EH, Lee SR. Doxycycline inhibits matrix metalloproteinase‐9 and laminin degradation after transient global cerebral ischemia. Neurobiol Dis 2009; 34: 189–98 [DOI] [PubMed] [Google Scholar]
- 62. Giannelli G, Falk‐Marzillier J, Schiraldi O, Stetler‐Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease‐2 cleavage of laminin‐5. Science 1997; 277: 225–8 [DOI] [PubMed] [Google Scholar]
- 63. Kelly MA, Shuaib A, Todd KG. Matrix metalloproteinase activation and blood‐brain barrier breakdown following thrombolysis. Exp Neurol 2006; 200: 38–49 [DOI] [PubMed] [Google Scholar]
- 64. Feng S, Cen J, Huang Y, Shen H, Yao L, Wang Y, et al Matrix metalloproteinase‐2 and ‐9 secreted by leukemic cells increase the permeability of blood‐brain barrier by disrupting tight junction proteins. PLoS ONE 2011; 6: e20599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase‐mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab 2007; 27: 697–709 [DOI] [PubMed] [Google Scholar]
- 66. Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, et al Neutrophil infiltration increases matrix metalloproteinase‐9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab 2003; 23: 1430–40 [DOI] [PubMed] [Google Scholar]
- 67. Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, et al Leukocyte‐derived matrix metalloproteinase‐9 mediates blood‐brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 2005; 289: H558–68 [DOI] [PubMed] [Google Scholar]
- 68. Krizanac‐Bengez L, Mayberg MR, Cunningham E, Hossain M, Ponnampalam S, Parkinson FE, et al Loss of shear stress induces leukocyte‐mediated cytokine release and blood‐brain barrier failure in dynamic in vitro blood‐brain barrier model. J Cell Physiol 2006; 206: 68–77 [DOI] [PubMed] [Google Scholar]
- 69. Rempe RG, Hartz AM, Bauer B. Matrix metalloproteinases in the brain and blood‐brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab 2016; 36: 1481–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mun‐Bryce S, Rosenberg GA. Gelatinase B modulates selective opening of the blood‐brain barrier during inflammation. Am J Physiol 1998; 274(5 Pt. 2): R1203–11 [DOI] [PubMed] [Google Scholar]
- 71. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin‐8 tenfold by aminoterminal processing, whereas it degrades CTAP‐III, PF‐4, and GRO‐alpha and leaves RANTES and MCP‐2 intact. Blood 2000; 96: 2673–81 [PubMed] [Google Scholar]
- 72. Schonbeck U, Mach F, Libby P. Generation of biologically active IL‐1 beta by matrix metalloproteinases: a novel caspase‐1‐independent pathway of IL‐1 beta processing. J Immunol 1998; 161: 3340–6 [PubMed] [Google Scholar]
- 73. Mohan MJ, Seaton T, Mitchell J, Howe A, Blackburn K, Burkhart W, et al The tumor necrosis factor‐alpha converting enzyme (TACE): a unique metalloproteinase with highly defined substrate selectivity. Biochemistry 2002; 41: 9462–9 [DOI] [PubMed] [Google Scholar]
- 74. Song Y, Sanganahalli BG, Hyder F, Lin WC, Riera JJ. Distributions of irritative zones are related to individual alterations of resting‐state networks in focal epilepsy. PLoS ONE 2015; 10: e0134352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aronica E, Fluiter K, Iyer A, Zurolo E, Vreijling J, van Vliet EA, et al Expression pattern of miR‐146a, an inflammation‐associated microRNA, in experimental and human temporal lobe epilepsy. Eur J Neurosci 2010; 31: 1100–7 [DOI] [PubMed] [Google Scholar]
- 76. Gorter JA, Iyer A, White I, Colzi A, van Vliet EA, Sisodiya S, et al Hippocampal subregion‐specific microRNA expression during epileptogenesis in experimental temporal lobe epilepsy. Neurobiol Dis 2014; 62: 508–20 [DOI] [PubMed] [Google Scholar]
- 77. Iyer A, Zurolo E, Prabowo A, Fluiter K, Spliet WG, van Rijen PC, et al MicroRNA‐146a: a key regulator of astrocyte‐mediated inflammatory response. PLoS ONE 2012; 7: e44789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kan AA, van Erp S, Derijck AA, de Wit M, Hessel EV, O'Duibhir E, et al Genome‐wide microRNA profiling of human temporal lobe epilepsy identifies modulators of the immune response. Cell Mol Life Sci 2012; 69: 3127–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Korotkov A, Mills JD, Gorter JA, van Vliet EA, Aronica E. Systematic review and meta‐analysis of differentially expressed miRNAs in experimental and human temporal lobe epilepsy. Sci Rep 2017; 7: 11592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xu B, Wang N, Wang X, Tong N, Shao N, Tao J, et al MiR‐146a suppresses tumor growth and progression by targeting EGFR pathway and in a p‐ERK‐dependent manner in castration‐resistant prostate cancer. Prostate 2012; 72: 1171–8 [DOI] [PubMed] [Google Scholar]
- 81. Palomer X, Capdevila‐Busquets E, Botteri G, Davidson MM, Rodriguez C, Martinez‐Gonzalez J, et al miR‐146a targets Fos expression in human cardiac cells. Dis Model Mech 2015; 8: 1081–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA‐146 suppresses NF‐kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene 2008; 27: 5643–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Iori V, Iyer AM, Ravizza T, Beltrame L, Paracchini L, Marchini S, et al Blockade of the IL‐1R1/TLR4 pathway mediates disease‐modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis 2017; 99: 12–23 [DOI] [PubMed] [Google Scholar]
- 84. van Scheppingen J, Iyer AM, Prabowo AS, Muhlebner A, Anink JJ, Scholl T, et al Expression of microRNAs miR21, miR146a, and miR155 in tuberous sclerosis complex cortical tubers and their regulation in human astrocytes and SEGA‐derived cell cultures. Glia 2016; 64: 1066–82 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Co‐localization of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinase (TIMPs) in cortical tubers. Co‐localization of MMP2 and TIMP2 was observed in giant cells and dysmorphic neurons, while glial cells were only MMP2‐positive (A). MMP3 co‐localized with TIMP1 in all cells and with TIMP3 in some giant cells, but not in glia (B,C). Co‐localization was observed between MMP9 and TIMP1 (D) and MMP14 with both TIMP2 and TIMP4 (E,F). Arrowheads depict glial cells and asterisk depict giant cell. Scale bar A–F: 25 μm.
Table S1. Clinical findings of patients with tuberous sclerosis complex (TSC) (1–20) and autopsy controls (21–43).