

SUMMARY
Sin3/HDAC complexes function by deacetylating histones, condensing chromatin, and modulating gene expression. Although components used to build these complexes have been well defined, we still have only a limited understanding of the structure of the Sin3/HDAC subunits assembled around the scaffolding protein SIN3A. To characterize the spatial arrangement of Sin3 subunits, we combined Halo affinity capture, chemical crosslinking, and high-resolution mass spectrometry (XL-MS) to determine intersubunit distance constraints, identifying 66 interprotein and 63 self-crosslinks for 13 Sin3 subunits. Having assessed crosslink authenticity by mapping self-crosslinks onto existing structures, we used distance restraints from interprotein crosslinks to guide assembly of a Sin3 complex substructure. We identified the relative positions of subunits SAP30L, HDAC1, SUDS3, HDAC2, and ING1 around the SIN3A scaffold. The architecture of this subassembly suggests that multiple factors have space to assemble to collectively influence the behavior of the catalytic subunit HDAC1.
Graphical Abstract

In Brief
Banks et al. capture positional information for subunits within Sin3/HDAC complexes by combining crosslinking and high-resolution mass spectrometry. This information is then used to guide docking of Sin3 subunit structures to develop a model of a Sin3/HDAC complex sub-structure.
INTRODUCTION
Although solution NMR and crystallographic studies have provided insight into the structure of components of macromolecular complexes, it is often challenging to determine the architecture of subunits when assembled into higher-order structures. Crystallographic studies are limited by the requirement that the isolated molecules can form rigid crystals suitable for structure determination (Smialowski and Wong, 2016). In addition, NMR studies of larger proteins and complexes are hindered by the large number of NMR signals that cause spectral crowding (Frueh et al., 2013). Developments in crosslinking techniques combined with advances in high-resolution mass spectrometry (MS) have provided valuable tools to address these limitations (Leitner et al., 2016). Recently, Kao et al. (2011) developed an MS-cleavable crosslinker, disuccinimidyl sulfoxide (DSSO), which combined with high-resolution MS improves unambiguous identification of crosslinked peptides (Wang et al., 2017). Here, we combine this approach with Halo affinity purification (Los et al., 2008) to capture positional information for Sin3 complex subunits in solution.
Sin3/HDAC complexes influence gene transcription by modulating the chromatin environment, orchestrating lysine deacetylation on N-terminal histone tails using the catalytic subunits HDAC1 and HDAC2. This results in chromatin compaction and transcriptional repression as genes become inaccessible to the transcriptional machinery. The precise targeting of gene repression by Sin/HDAC-mediated histone deacetylation is likely controlled by the non-catalytic Sin3 subunits (Kelly and Cowley, 2013) because HDAC1/2 are not unique to Sin3 and are used by other histone deacetylase complexes, including NuRD (Zhang et al., 1999) and CoREST (Lee et al., 2005). Although the subunit composition of Sin3/HDAC complexes has been established (Banks et al., 2018), how subunits organize around SIN3A to accomplish HDAC1/2-mediated deacetylation of specific residues at specific genomic loci remains unclear.
Uncovering the architecture of Sin3/HDAC complexes is essential in understanding the contribution of subunits to complex function, which, in turn, is vital in understanding how mis regulated Sin3 complexes contribute to human disease. SIN3A, the scaffolding protein around which the complex assembles, is frequently mutated in human cancers (Kandoth et al., 2013), and Sin3 complexes offer likely therapeutic targets for a variety of diseases (Kandoth et al., 2013), including triple-negative breast cancer (Kwon et al., 2015) and pancreatic cancer (Rielland et al., 2014). Current therapeutic strategies using HDAC inhibitors, such as vorinostat, are not specific, targeting a variety of HDAC-containing complexes (Marks and Breslow, 2007). Targeting HDAC activity within the context of Sin3 complexes more specifically will require a more sophisticated understanding of how Sin3 subunits cooperatively control HDAC1/2 recruitment and function.
Here, we isolated Sin3/HDAC complexes using a Halo-tagged SAP30L subunit and captured positional information for individual Sin3 subunit residues using the crosslinker DSSO. After high-resolution MS, we identified 63 self- and 66 interprotein Sin3 subunit crosslinks. We next used previously determined structures to confirm that the distances between crosslinked subunits were consistent with the distance limits required by the ~10-Å DSSO crosslinker. We further judged the validity of our crosslinking data by asking whether SIN3A crosslink hotspots were required for capturing crosslinked subunits. Finally, we used intersubunit crosslinks, together with recent evidence for a SAP30/HDAC1 interface (Marcum and Radhakrishnan, 2019), to dock SAP30L, SIN3A, and HDAC1 structures and to map the relative locations of SUDS3, SAP130, HDAC2, and ING1 on the resulting structure. Importantly, this reveals the position of the HDAC1 active site, relative to other subunits. Using molecular modeling to integrate a comprehensive map of crosslinks between Sin3 subunits with existing structural data has revealed the arrangement of subunits at the core of the Sin3 complex, illuminating how they might function collectively to regulate chromatin accessibility and gene transcription.
RESULTS
A Foundation for Developing a High-Resolution Sin3 Interaction Network
Several important studies have enabled a progressively more detailed picture of Sin3 subunit interactions to emerge (Figure S1A). Laherty et al. (1997) defined a ~375-amino-acid conserved domain within the Sin3-scaffolding protein mSin3A, important for its interaction with the catalytic subunit HDAC2. They named this region the HDAC interaction domain (HID). Later studies determined further interactions between SIN3A and other subunits. In particular, Xie et al. (2011) determined a structure of part of the C terminus of SAP30 with the PAH3 domain of SIN3A, and Clark et al. (2015a) determined a structure explaining an interaction between part of SUDS3 with part of the SIN3A HID. Thus, the PAH3/HID region within SIN3A was established as a central organizing platform around which several other Sin3 components (HDAC1/2, SAP30, and SUDS3) might assemble. Despite these advances, it remains unclear whether there is space for these components to dock together on this SIN3A platform.
From AP-MS to XL-MS: Mapping Proximal Amino Acids among Sin3 Subunits
To address how Sin3 subunits might organize around the SIN3A HID, we used a crosslinking mass spectrometry (XL-MS) approach to determine proximity constraints for pairs of amino acids within Sin3 complexes. Previously, we had determined a set of Sin3 subunits co-purifying with SAP30L, a SAP30 homolog, using affinity purification mass spectrometry (AP-MS) (Banks et al., 2018; Figure S1B). To extend this analysis, we treated purified SAP30L containing complexes with the MS-cleavable crosslinker DSSO before liquid chromatography mass spectrometry (LC/MS) analysis, capturing additional structural information by highlighting pairs of residues within Sin3 assemblies that likely reside within a distance of <30Å (Figures 1A–1C and S1C).
Figure 1. MS Crosslink Analysis of Sin3/HDAC Complexes.
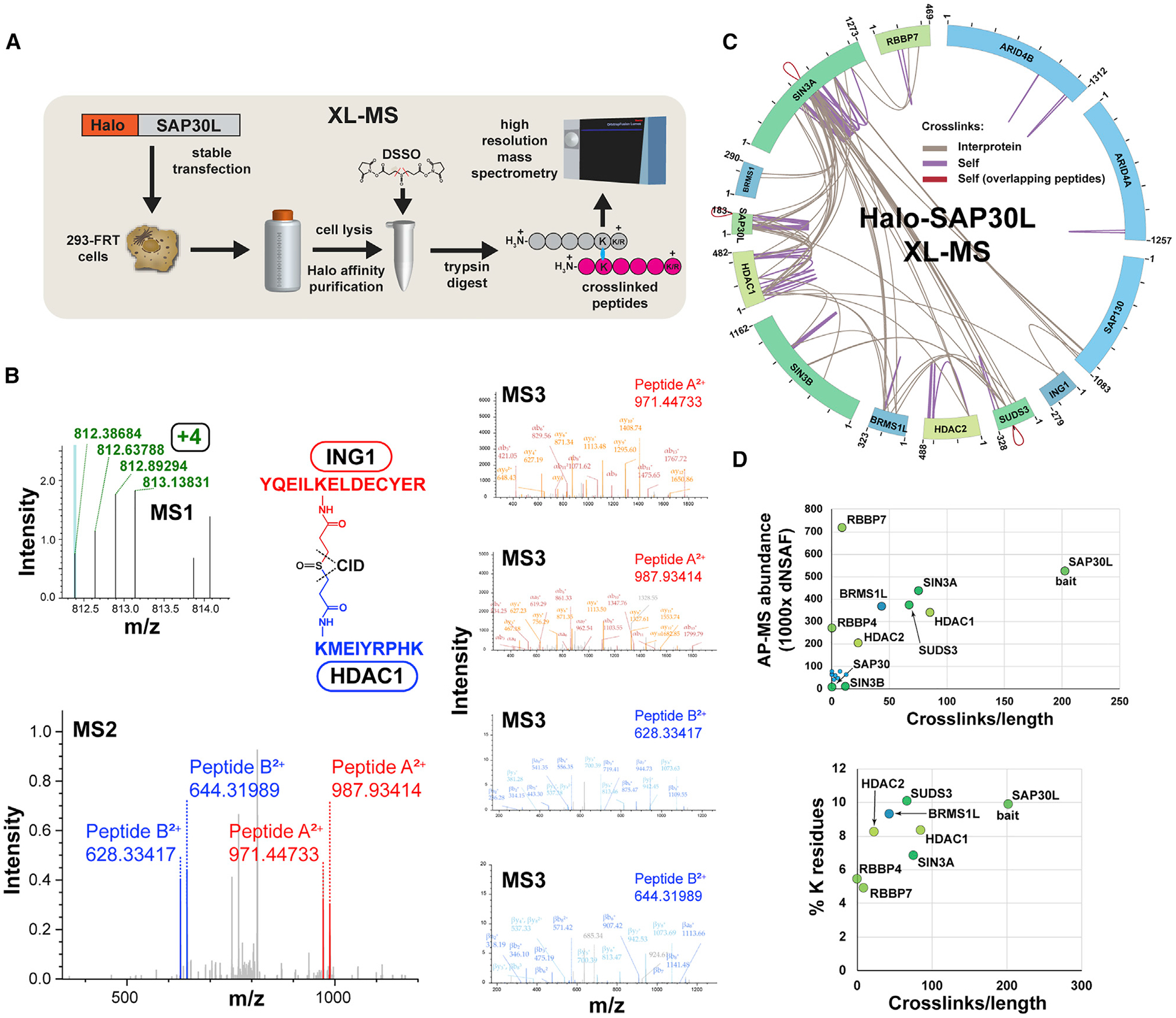
(A) Workflow for XL-MS experiments. In contrast to AP-MS experiments, Halo-purified samples were treated with DSSO before analysis by high-resolution mass spectrometry.
(B) High-resolution MS2 and MS3 spectra used to identify the ING1-HDAC1 crosslinked peptide. Putative crosslinked peptides with charge ≥ +4 were selected during MS1 analysis and low-energy collision-induced dissociation (CID) was used to cleave the DSSO crosslinker, generating a pair of fragments for each peptide (MS2-ING1 peptides shown in red; HDAC1 in blue). The four fragments were sequenced using MS3.
(C) Crosslink map for the Sin3/HDAC complex. Crosslink identifications are from three XL-MS experiments. Values indicate protein length (amino acids). Details of crosslinks are in Table S2.
(D) Relationship between observed crosslink abundance and either protein abundance or lysine content. Crosslink abundance is 1,000 × (semi-crosslinks/protein length [aa]), with two semi-crosslinks counted for each of the protein’s self-crosslinks and one for each of the protein’s interprotein crosslinks. Protein abundance distributed normalized spectral abundance factor (dNSAF) values for SIN3 subunits co-purifying with Halo-SAP30L (four biological replicates) were published previously (refer to Table S3 in Banks et al., 2018).
We identified 66 Sin3 subunit interprotein crosslinks and 63 self-crosslinks (Figure 1C; Table S2). It is not possible to tell whether these self-crosslinks result from crosslinks within a single molecule or from crosslinks between two identical molecules (from a homodimer, for example). However, for some self-crosslinks within the subunits SIN3A, SAP30L, and SUDS3, the sequences of crosslinked peptides overlap, suggesting that these subunits might form homodimers (Figure 1C, red crosslinks).
The crosslinks did not appear to be distributed evenly among the 13 Sin3 subunits (Figure 1C). Although some quite large proteins had few crosslinks (e.g., one self-crosslink for ARID4A), most crosslinks were distributed among five proteins: SIN3A, SAP30L, HDAC1, SUDS3, and BRMS1L. In addition, there appeared to be “hotspots” of crosslinks within proteins. We have defined hotspots as regions containing residues with two or more unique crosslinks within a 21-residue window centered on the residue (Figure S2). To explain the uneven distribution of crosslinks, we first assessed whether the paucity of crosslinks on some subunits reflected a low abundance of these subunits in our purifications. We compared protein abundances for the various Sin3 subunits captured by AP-MS with a factor reflecting the number of crosslinks per unit length of the protein (Figure 1D). Although low abundance can explain the deficit of crosslinks identified for some proteins (SIN3B, SAP130, ARID4A/B, SAP30, BRMS1, and FAM60A), it did not explain the deficit of crosslinks for the relatively abundant RBBP4/7. A second possibility was that the crosslink deficit for RBBP4/7 might be explained by a low number of lysine residues. Calculating the lysine percentage for the eight most abundant subunits, we found that RBBP4/7 did have the lowest percentage of lysines. This might partially explain their lack of crosslinks. In addition to their low lysine content, RBBP4 and RBBP7 are also largely formed from β-sheets, and previous studies have proposed that these structures often correlate with low levels of crosslinks (Schneider et al., 2016).
Distances between Crosslinked Residues Are Consistent with Crosslinker Length
To further assess our crosslinking data, we tested whether the distances between crosslinked residues, which mapped to experimentally determined Sin3 tridimensional structures, were consistent with the length of the DSSO crosslinker. Although the spacer length of DSSO is 10.1Å, Merkley et al. (2014) had previously determined that distances of up to 30Å between α-carbon (Cα) atoms of crosslinked residues were appropriate in their analysis of the similarly sized crosslinker DSS. We first assessed 11 crosslinks that mapped within the SIN3A partial structure PDB: 2N2H (Clark et al., 2015a; Figure 2A) and determined that 10 of these corresponded to Cα-Cα distances of <30Å (Figure 2B). Curiously, we found one crosslink between two residues within the structure with a much longer Cα-Cα distance of 44-Å. It is possible that either this 44-Å crosslink is between two different SIN3A molecules or that other conformations of this region exist in solution. Indeed, one of the two linked lysines is located at the end of the C-terminal α-helix in the PDB: 2N2H partial structure that could be folded differently in the context of full-length SIN3A and the assembled SIN3 complex. Four additional tridimensional structures map to regions of Sin3 subunits containing self-crosslinks. Mapping to SAP30L were PDB: 2N1U (Laitaoja et al., 2016) and PDB: 2LD7 (Xie et al., 2011), to HDAC1 was PDB: 5ICN (Watson et al., 2016), and to RBBP7 was PDB: 3CFV (Murzina et al., 2008; Figure 2C). Of the 23 crosslinks that mapped within these structures, 22 (96%) had corresponding Cα-Cα distances of <30Å, confirming that the self-crosslinks that we identified likely originate from intact Sin3 structures.
Figure 2. Cα-Cα Distance Distributions for Crosslinks Mapping to Sin3 Subunit Structures.
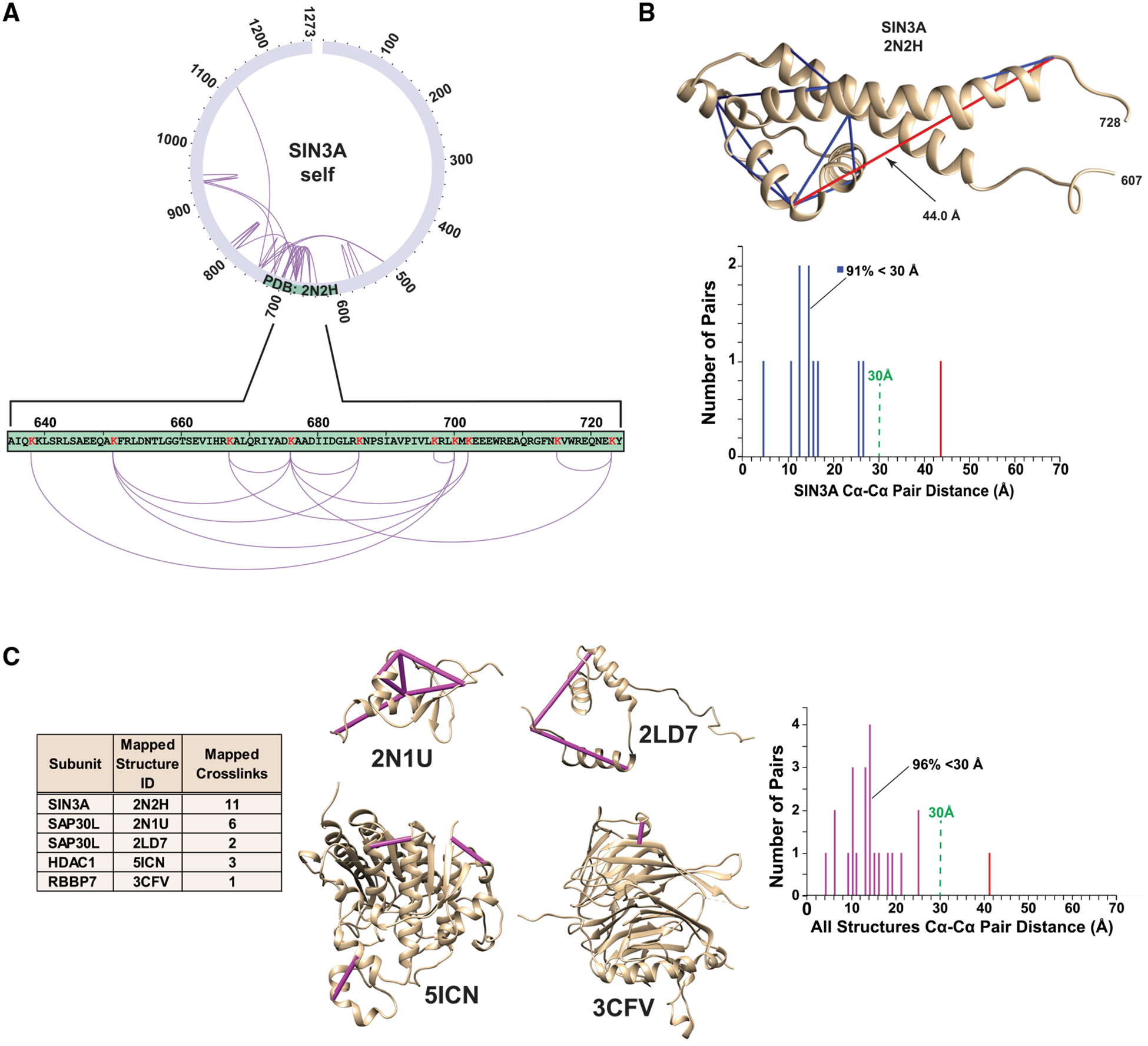
(A) PDB: 2N2H (Clark et al., 2015a) maps to a SIN3A region containing 11 self crosslinks.
(B) Distribution of Cα-Cα crosslink distances mapping to PDB: 2N2H (distances <30-Å,blue bars; distance >30Å, red bar).
(C) All regions of Sin3 subunits with both structural data and self-crosslinks.
Deleting Crosslinking Hotspots Disrupts Sin3 Complex Stability
Having observed crosslink hotspots within SIN3A, we reasoned that if a hotspot resulted from an important structural interface between SIN3A and other subunits, then, deleting regions over-lapping these hotspots would result in the loss of subunit binding. Therefore, we tested SIN3A deletion mutants for their ability to capture other Sin3 subunits (Figure 3A). The HID 688–829 region appears to be a major interaction interface and crosslinks to 5 subunits (SAP30L, SUDS3, BRMS1L, HDAC1, and SAP130; Figure 3B, red lines), whereas the PAH4 region crosslinks to three subunits (HDAC1, SAP30, and BRMS1L; Figure 3B, green lines) and the PAH3 region to only two subunits (SAP30L and HDAC1; Figure 3B, blue lines).
Figure 3. Deletion Analysis of SIN3A Crosslink Hotspots.
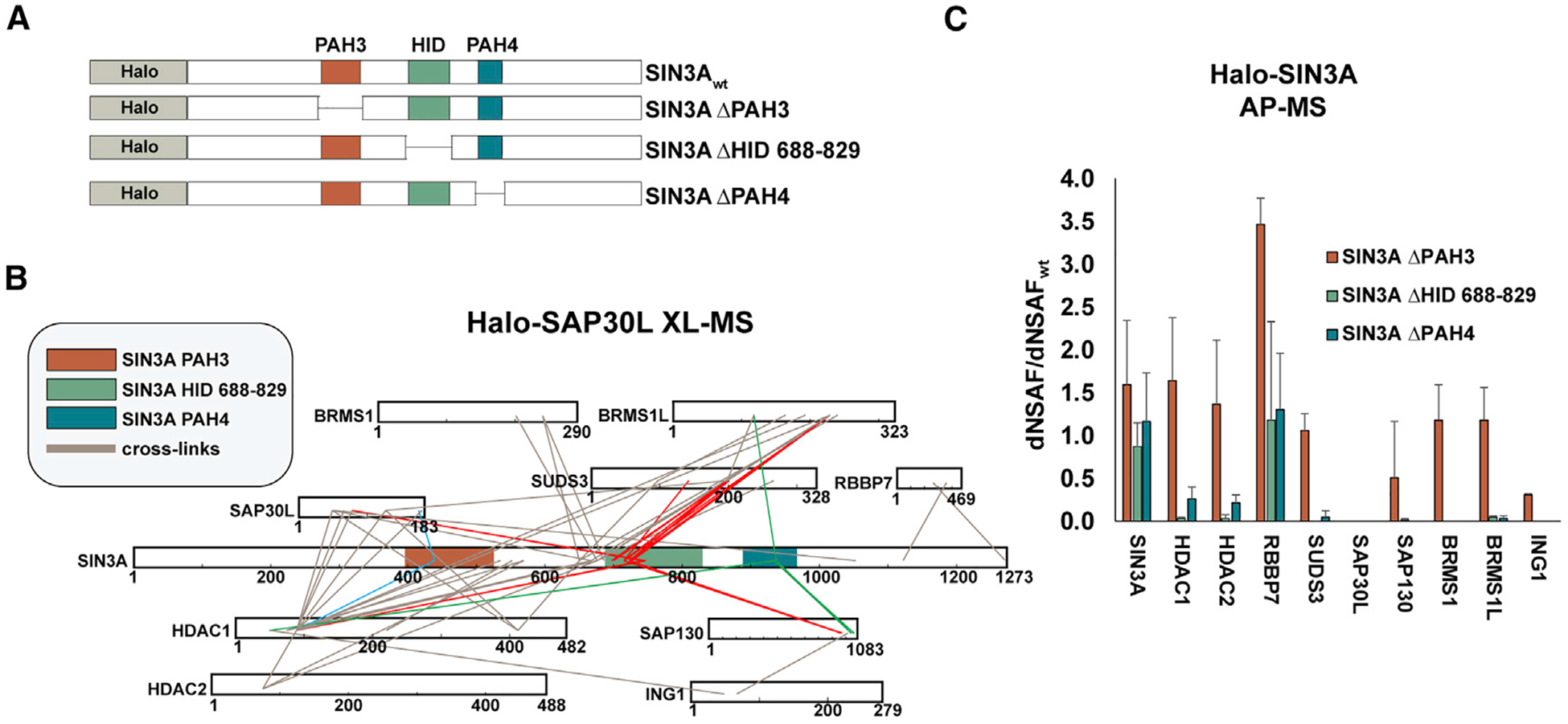
(A) Regions of SIN3A deleted.
(B) Crosslink map for Sin3 subunit interprotein crosslinks. Crosslinks to PAH3 (blue), HID 688–829 (red), and PAH4 (green) are highlighted.
(C) Relative abundance of the crosslinked Sin3 subunits shown in (B) co-purifying with the SIN3A deletion mutants in AP-MS experiments. Error bars represent standard deviation (Table S3).
Subunits crosslinking to SIN3A HID 688–829 (or close to this region—BRMS1) were lost upon its deletion, as were HDAC2 and ING1, which are linked to this region via other subunits (HDAC1 and SAP130) and may require these proteins for capture by SIN3A (Figure 3C; Table S3). In contrast, HID 688–829 removal does not disrupt capture of RBBP7, which crosslinks to a distal region at the C terminus of SIN3A. Unlike HID disruption, removal of the PAH3 domain only results in the loss of SAP30L. Although SAP30L crosslinks to other regions within SIN3A, to HDAC1 and to BRMS1L, it seems likely that its interaction with PAH3 is required for its stable integration into Sin3 complexes. Curiously, PAH3 deletion also results in a modest increase in levels of RBBP7 captured by SIN3A, but the mechanism underlying this remains unclear. Disruption of PAH4 had a similar, but more modest, effect than disruption of the HID, suggesting that this region is also involved in stabilizing SIN3A interactions with multiple Sin3 subunits. Taken together, the results of Figure 3 confirm that the interprotein crosslinks that we identified are mapped to important regions involved in Sin3 complex stability.
Building a Sin3 Complex Subassembly
Having assessed the validity of our crosslinking data, we next used information from the interprotein crosslinks to dock Sin3 structures and better understand the three-dimensional (3D) architecture of Sin3 complexes. We initially considered how to use the two structures that mapped to SAP30L, PDB: 2N1U and PDB: 2LD7. These covered most of the N-and C-terminal halves of SAP30L, respectively. There were three crosslinks that bridged these structures (Figure S3, shown in blue). Because we already had evidence that SAP30L might exist as a homodimer, we did not know whether these crosslinks bridge the N-and C-terminal halves of one SAP30L molecule or, alternatively, bridge the N terminus of one SAP30L molecule to the C terminus of a second molecule of SAP30L (Figure S3). In total, we used 11 interprotein crosslinks to guide docking of SIN3A, HDAC1, and SAP30L structures using the HADDOCK platform (Merkley et al., 2014; Figure 4A). We further refined our model by using additional docking restraints based on recent evidencefor a SAP30/HDAC1 interaction (Marcum and Radhakrishnan, 2019).
Figure 4. Architecture of the SIN3A/SAP30L/HDAC1 Complex.
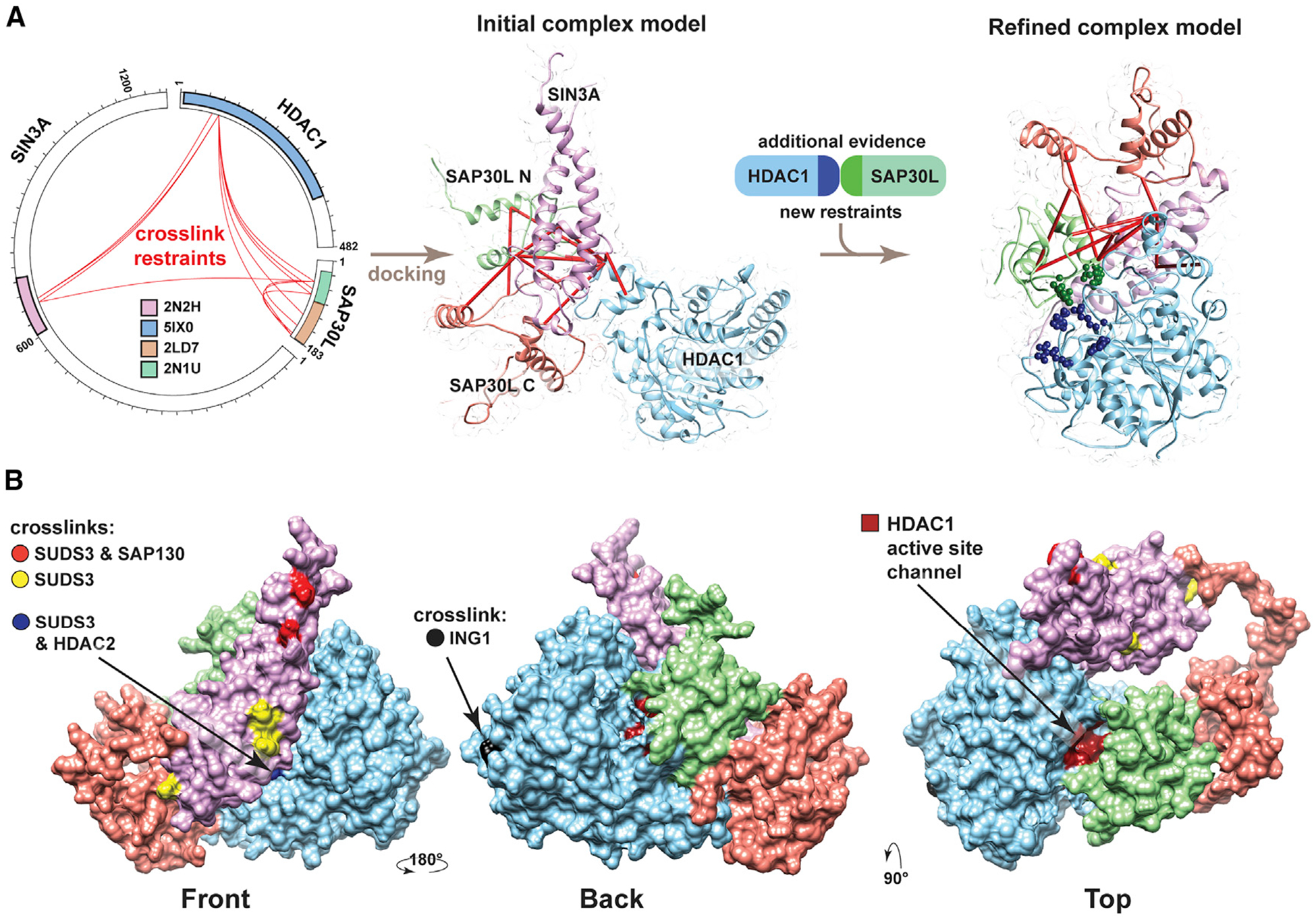
(A) Sin3 structures modeled using SWISS-Model (based on indicated PDB structures) were docked using HADDOCK (de Vries et al., 2010) guided by docking restraints from the indicated crosslinks (red lines) to generate an initial complex model. Additional evidence supporting an interaction between SAP30 and HDAC1 (Marcum and Radhakrishnan, 2019) was then used with the crosslinking restraints to generate a second refined-complex model.
(B) Refined model of the SAP30L/SIN3A/HDAC1 sub-structure, showing the SIN3A residues crosslinked to SUDS3 (yellow), to both SUDS3 and SAP130 (red), and to both SUDS3 and HDAC2 (blue) or showing the HDAC1 residue crosslinked to ING1 (black). The position of the HDAC1 active site channel is also shown in dark red (Video S1).
We observed important features in the resulting subassembly (Figure 4B). After we had docked SAP30L and HDAC1 structures with SIN3A 607–728 HID, there was still access to a large surface on this SIN3A platform onto which other subunits could assemble. Indeed, other key Sin3 subunits—SUDS3, SAP130, and HDAC2—crosslinked to the remaining exposed front surface of the SIN3A HID (Figure 4B, residues highlighted in yellow, red, and blue). In addition, docking both SAP30L N- and C-terminal structures to the SIN3A 667–728 HID does not obstruct SAP30L dimerization (Figure S3B) or interaction with the SIN3A PAH3 structure (Figure S3C). The probable position of the SIN3A PAH3 domain relative to SAP30L can be inferred from the structure of SAP30 (a SAP30L homolog) bound to the mSin3A PAH3 domain (Xie et al., 2011).
DISCUSSION
We have combined Halo affinity purification with MS-cleavable crosslinking techniques to map interface points between or within Sin3 complex subunits. A detailed map of Sin3 complex architecture facilitates our understanding of how Sin3 complex subunits function in concert to first recruit the HDAC1/2 deacetylases to genomic loci and then to orient them to coordinate timely deacetylation of histone tails.
The SIN3A HID Provides a Platform for Docking Other Sin3 Subunits
Crosslinking hotspots on the scaffolding protein SIN3A were centered on the HID, which is sufficient for transcriptional repression when recruited to promoters in reporter assays (Laherty et al., 1997). The region SIN3A 607–728 within the HID forms a platform around which the subunits SUDS3, SAP30/30L, SAP130, HDAC2, and HDAC1 congregate, conceivably to correctly position HDAC1/2 for histone tail de acetylation. These subunits may have subtly different roles in enabling the proper HDAC function. SUDS3 forms homo dimers and might be involved in tethering two Sin3 complexes together (Clark et al., 2015a). SAP30L appears to make direct contact with HDAC1 itself and could either guide HDAC1 positioning relative to the SIN3A platform or could influence HDAC1 conformation and, hence, activity. SAP30 and SAP30L also interact with both core histones and DNA (Viiri et al., 2009), and this interaction might help stabilize Sin3 complexes on nucleosomes. SAP130 has been shown to interact with co-repressors and coactivators and likely to co-ordinate Sin3 interactions with other coregulatory complexes (Fleischer et al., 2003). ING1, although not associated with the SIN3A HID platform, crosslinks to HDAC1 (Figure 4B). Here, ING1 might help position the HDAC1 correctly, relative to histone tail substrate. Indeed, ING proteins do associate with H3K4 trimethylated histone tails (Peña et al., 2006; Shi et al., 2006), and, adjacent to HDAC1, ING1 could offer other acetylated lysines in the histone tail to HDAC1 for deacetylation. Other evidence supports a model in which a direct, controlled interaction between ING proteins and HDAC1 could first direct HDAC1/substrate engagement and then ING protein disengagement. In particular, Sin3/HDAC complexes can lose binding of ING1/2 when the active site channel binds vorinostat (Sardiu et al., 2014; Smith et al., 2010), an HDAC inhibitor that binds the active site channel in class I HDACs (Lauffer et al., 2013). Loss of ING protein binding might be explained by inhibitor (or substrate) binding causing HDAC1 conformational changes, which in turn, abrogate the HDAC1/ING interaction.
Distribution of Crosslinks among Sin3 Subunits
The appearance of crosslinking hotspots, rather than crosslinks evenly distributed among co-purified subunits (Figures 1C and S2), could have several explanations. The lowest-abundance Sin3 proteins always generated low numbers of crosslinks (Figure 1D), and low numbers of crosslinks in higher-abundance proteins could originate from a dearth of lysine residues in these proteins. The absence of crosslinks might also originate from the structural arrangement of Sin3 complexes, with crosslinks confined to structured regions rich in α-helices (there is evidence that β-sheets do not yield crosslinks [Schneider et al., 2016]) and, with more-dynamic unstructured regions, not providing opportunities for crosslinking.
Subunit Interfaces
Some crosslinks support points of contact within and between Sin3 proteins. Self-crosslinks within SIN3A suggest that regions that are distant in the amino acid chain might be close in Euclidian space (e.g., residue 1,122 near the SIN3A C terminus is proximal to residue 747). Surprisingly, we found that RBBP7 also crosslinked to the SIN3A C terminus distal to the HID. An independent interaction between RBBP7 and the C terminus of SIN3A might explain why RBBP7 is not lost with other subunits when the SIN3A HID 688–829 is deleted (Figure 3C).
Crosslinks with Overlapping Peptides Support a Dimeric Sin3 Complex Model
The detection of self-crosslinks with overlapping peptides is consistent with previous evidence for SUDS3 dimerization and a model (Clark et al., 2015a, 2015b) with two Sin3 complex assemblies operating between adjacent nucleosomes, tethered together by SUDS3 dimers. Sin3 complex dimerization is also consistent with SAP30L and SIN3A self-crosslinks with overlapping peptides (Figure 1C). Further investigation of the oligomerization status of Sin3 complexes, possibly using native MS approaches (Leney and Heck, 2017), will provide valuable information for developing a more complete understanding of Sin3 complex structure and function. For example, deciphering the oligomerization status of Sin3/HDAC complexes will be essential, both for correct interpretation of structural models based on cryo-electron microscope (EM) microscopy and for evaluating the effect of oligomerization status on Sin3 biological function.
Crosslinks Likely Originate from Diverse Sin3 Complexes
It is likely that our dataset reflects crosslinks from a heterogeneous population of Sin3 complexes. Previously, we observed that some pairs of Sin3 subunits are mutually exclusive (SAP30/SAP30L and BRMS1/BRMS1L [Sardiu et al., 2014]). Consistent with this, we do not observe SAP30 in our SAP30L purifications and expect that BRMS1 and BRMS1L crosslink to different SIN3A containing complexes.
In conclusion, by combining Halo affinity capture with XL-MS using the MS-cleavable DSSO crosslinker, we have been able to gain valuable insight into the relative positioning of subunits within the quaternary structure of the Sin3 complex. Our high-confidence crosslink identifications are consistent with existing structural data and known subunit interactions. They highlight subunit interfaces among Sin3 subunits and facilitate docking of existing structures, providing unique perspectives on Sin3 complex architecture and function.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Washburn (mpw@stowers.org). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe Strains
NEB® 5-alpha Competent E. coli cells were used for cloning. Cells were grown in LB medium at 37°C supplemented with Ampicillin (100 μg/ml final concentration).
Cell lines
HEK293T cells (used for transient expression of proteins) and Flp-In-293 cells (used to generate the cell line stably expressing Halo-SAP30L) were cultured in DMEM (supplemented with 1x GlutaMAX and 10% Fetal Bovine Serum (FBS) (HEK293T cells) or 10% Calf Serum (Flp-In-293 cells)) at 37°C with 5% CO2. Both cell lines are derived from HEK293 cells which contain three copies of the X chromosome, no Y chromosome and are presumed to have been derived from a female fetus. Cell lines were authenticated by STR profiling using the Cell Line Authentication Service (Promega) on 26th June 2014.
METHOD DETAILS
Cloning Halo-SIN3A wt and deletion mutants
Plasmid FHC11647 (Promega, Madison, WI) coding for Halo-SIN3A in pFN21A was altered by site directed mutagenesis (A109V) to code for human SIN3A (Q96ST3/NP_001138830) and to insert a stop codon immediately upstream of the PmeI restriction site at the 3′ end of the ORF. This plasmid was then used as a template, together with the primers listed in Key Resources, to clone the SIN3A deletion mutants as follows. First, PCR products were generated corresponding to the portion of the SIN3A ORF 5′ to the deletion site. These N-terminal fragments of SIN3A, flanked by SgfI and KpnI restriction sites were then cloned between the PacI and KpnI sites in plasmid pcDNA5/FRT PacI PmeI previously described (Banks et al., 2014). C-terminal fragments of SIN3A downstream of the deletion site flanked by KpnI and PmeI sites were then generated by PCR and inserted between the KpnI and PmeI sites immediately downstream of the N-terminal fragments. This resulted in deletion versions of SIN3A with the deleted region replaced by the six base-pair KpnI sequence GGT ACC coding for Gly-Thr.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | Cat# C2987H |
| Biological Samples | ||
| Fetal Bovine Serum (FBS) | PEAK Serum | Cat# PS-FB1 |
| Calf Serum | Sigma | Cat# 12133C |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat# 35050061 |
| Lipofectamine LTX Reagent | Thermo Fisher Scientific | Cat# 15338500 |
| PLUS Reagent | Thermo Fisher Scientific | Cat# 11514015 |
| Magne® HaloTag® Beads | Promega | Cat# G7282 |
| AcTEV Protease | Thermo Fisher Scientific | Cat# 12575015 |
| Salt Active Nuclease High Quality (Bioprocessing grade) | ArcticZymes | Cat# 70920-202 |
| rLys-C, Mass Spec Grade | Promega | Cat# V1671 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5117 |
| 2-Chloroacetamide | Sigma | Cat# C0267 |
| Bestatin hydrochloride | ApexBio | Cat# A8621 |
| Leupeptin, Microbial | ApexBio | Cat# A2570 |
| Pepstatin A | ApexBio | Cat# A2571 |
| Phenylmethanesulfonyl fluoride solution (PMSF) | Sigma | Cat# 93482-50ML-F |
| Phenanthroline | Sigma | Cat# P9375 |
| Triton X-100 | Sigma | Cat# T8787 |
| Urea | Sigma | Cat# U1250-1KG |
| DSSO (disuccinimidyl sulfoxide) | Thermo Scientific | Cat# A33545 |
| Sodium Deoxycholate | Sigma | Cat# D6750 |
| Trichloroacetic acid solution | Sigma | Cat# T0699 |
| Pierce TCEP-HCI | Thermo Fisher Scientific | Cat# 20490 |
| Formic Acid, 90%, BAKER ANALYZED Reagent, J.T.Baker | Fisher Scientific | Cat# 02-002-910 |
| Deposited Data | ||
| AP-MS Halo-SIN3A | This paper | MassIVE MSV000084254 |
| AP-MS Halo-SIN3A DPAH3 | This paper | MassIVE MSV000084255 |
| AP-MS Halo-SIN3A DHID | This paper | MassIVE MSV000084256 |
| AP-MS Halo-SIN3A DPAH4 | This paper | MassIVE MSV000084257 |
| XL-MS Halo-SAP30 | This paper | MassIVE MSV000084311 |
| AP-MS control (HEK293T cells) | Banks et al., 2014 | Peptide Atlas: PASS00598 / GZ5438hrm |
| AP-MS control (FIp-In-293 cells) | Banks et al., 2018 | MassIVE MSV000081360 |
| AP-MS Halo-SAP30L | Banks et al., 2018 | MassIVE MSV000081352 |
| SIN3A structure | Clark et al., 2015a | PDB: 2N2H |
| SAP30L structure | Laitaoja et al., 2016 | PDB: 2N1U |
| SAP30 structure | Xieetal., 2011 | PDB: 2LD7 |
| HDAC1 structure | Watson et al., 2016 | PDB: 5ICN |
| HDAC2 structure | Watson et al., 2016 | PDB: 5IX0 |
| RBBP7 structure | Murzina et al., 2008 | PDB: 3CFV |
| Refined Model of SIN3A/HDAC1/SAP30L | This Paper | PDB-Dev PDBDEV_00000043 |
| Experimental Models: Cell Lines | ||
| FIp-In-293 | Thermo Fisher Scientific | Cat# R75007, RRID:CVCL_U421 |
| HEK293T | ATCC | Cat# CRL-11268, RRID:CVCL_1926 |
| Oligonucleotides | ||
| Primer: SIN3A SgfI F: 5’- CAG GCG ATC GCC ATG AAG CGG CGT TTG GATGAC C- 3’ | This paper | N/A |
| Primer: SIN3A PmeI R: 5’- CAG GTT TAA ACT TAA GGG GCT TTG AAT ACT GTG CCG TAT TTG - 3’ | This paper | N/A |
| Primer: SIN3A ΔPAH3 F: 5’- CAT GGT ACC GAG TCT GTA CAT CTG GAA ACT TAT CCA - 3’ | This paper | N/A |
| Primer: SIN3A ΔPAH3 R: 5’- CAT GGT ACC CTC AGC AGT TGT TTT GCT TAA AAG C - 3’ | This paper | N/A |
| Primer: SIN3A ΔHID F: 5’- CAT GGT ACC GAT CTC TCA GAT GTG GAG GAA GAG GAA - 3’ | This paper | N/A |
| Primer: SIN3A ΔHID R: 5’- CAT GGT ACC ATT CTT TCT CAG ACC ATC AAT GAT G - 3’ | This paper | N/A |
| Primer: SIN3A ΔPAH4 F: 5’- CAT GGT ACC AGC CTG CTG GAT GGC AAC ATA GAC TCA - 3’ | This paper | N/A |
| Primer: SIN3A ΔPAH4 R: 5’- CAT GGT ACC GAC ATA GAA GAG GTT GTA TAC TTC ATC CA- 3’ | This paper | N/A |
| Recombinant DNA | ||
| SIN3A - HaloTag® human ORF in pFN21A | Promega | Cat# FHC11647 |
| Software and Algorithms | ||
| RAWDistiller v. 1.0 | Zhang et al., 2011 | Available on request |
| ProLuCID version 1.3.5 | Xuetal., 2015 | http://fields.scripps.edu/yates/wp/?page_id=17 |
| DTASelect/Contrast | Tabb et al., 2002 | http://fields.scripps.edu/yates/wp/?page_id=17 |
| NSAF7 | Zhang et al., 2010 | Available on request |
| Proteome Discoverer 2.2 with XlinkX nodes | Thermo Fisher Scientific | Cat# OPTON-30945 and OPTON-30946 |
| xiView web-based visualization tool | Graham et al., 2019 | https://xiview.org/xiNET_website/index.php |
| HADDOCK2.2 webserver | de Vries et al., 2010; van Zundert et al., 2016 | https://milou.science.uu.nl/services/HADDOCK2.2/haddock.php |
| SWISS-MODEL protein structure homology-modeling server | Waterhouse et al., 2018 | https://swissmodel.expasy.org/ |
| Chimera version 1.13.1 | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Adobe After Effects version 17.0.2 | Adobe | https://www.adobe.com/products/aftereffects.html?sdid=KKQOW&mv=search&ef_id=XK9mmAAAARVcantO:20200123180751:s |
| Xwalk (beta version) | Kahraman et al., 2011 | http://www.xwalk.org/cgi-bin/about.cgi |
| Other | ||
| Orbitrap Fusion LumosTribrid Mass Spectrometer | Thermo Fisher Scientific | Cat# IQLAAEGAAPFADBMBHQ |
| Dynabeads MPC-1 (Magnetic Particle Concentrator) | Thermo Fisher Scientific | Cat# 12001D |
| DynaMag-2 Magnet | Thermo Fisher Scientific | Cat# 12321D |
| UltiMate 3000 RSLCnano System | Thermo Fisher Scientific | Cat # ULTIM3000RSLCNANO |
| Acclaim PepMap 100 C18 HPLC Column | Thermo Fisher Scientific | Cat# 160454 |
Preparation of whole cell lysates
Flp-In-293 cells stably expressing Halo-SAP30L
A Flp-In-293 cell line stably expressing Halo-SAP30L expression using a CMV promoter was made essentially as described previously (Banks et al., 2018). Approximately 1 × 109 cells were harvested, washed twice in ice cold PBS and frozen at −80°C overnight. Cells were lysed by dounce homogenization in ice-cold lysis buffer containing 20 mM HEPES (pH 7.5), 1.5 mM MgCl2, 0.42 M NaCl, 10 mM KCl, 0.2% Triton X-100, 0.5 mM DTT, 0.1 mM benzamidine HCl, 55 μM phenanthroline, 10 μM bestatin, 20 μM leupeptin, 5 μM pepstatin A, 1 mM PMSF, and 500 units SAN (Salt Active Nuclease) and subsequently incubated for 2 hours at 4°C. Lysates were centrifuged at 40, 000 × g at 4°C for 30 minutes and the salt concentration of the resulting supernatants lowered to 0.3 M NaCl by adding an appropriate volume of ice-cold buffer containing 10 mM HEPES (pH 7.5), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT,0.1 mM benzamidine HCl, 55 μM phenanthroline, 10 μM bestatin, 20 μM leupeptin, 5 μM pepstatin A, 1 mM PMSF. Lysates were again centrifuged at 40,000 × g at 4°C for 30 minutes and the resulting supernatant harvested for Halo affinity purification of Sin3 complexes for XL-MS experiments.
Transiently transfected 293T cells
Approximately 1 × 107 293T cells were transfected using Lipofectamine LTX and Plus reagents with 7.5 μg of plasmid DNA expressing wt or deletion mutant versions of Halo-SIN3A. Approximately 48 hours after transfection, cells were harvested, washed twice in ice cold PBS, and frozen at −80°C for at least 30 minutes. Cell pellets were resuspended in lysis buffer containing 50 mM Tris·HCl (pH 7.5), 150 mM NaCl, 1% Triton®X-100, 0.1% sodium deoxycholate, 0.1 mM benzamidine HCl, 55 μM phenanthroline, 10 mμM bestatin, 20 μM leupeptin, 5 μM pepstatin A and 1 mM PMSF. Lysates were passed through a 26-gauge needle 5–10 times and centrifuged at 21,000 × g for 30 minutes at 4°C. The resulting supernatant was diluted with 700 μL Tris-buffered saline (25 mM Tris·HCl (pH 7.4), 137 mM NaCl, 2.7 mM KCl).
Halo purification of protein complexes
Flp-In-293 cells for XL-MS analysis
Lysates prepared from Halo-SAP30L expressing cells were incubated overnight at 4°C with MagneHalo magnetic beads prepared from 200 μL bead slurry according to the manufacturer’s instructions. Beads were isolated using a Dynabeads MPC-1 magnetic particle concentrator and washed 4 times in buffer containing 10 mM HEPES (pH 7.5), 1.5 mM MgCl2, 0.3 M NaCl, 10 mM KCl, and 0.2% Triton X-100. Bound proteins were eluted by incubating the beads with 200 μL buffer containing 50 mM HEPES (pH 7.5), 0.5 mM EDTA, 1 mM DTT and 30 units AcTEV for at least 2 hours at 4°C. The resulting eluate was recovered, and proteins crosslinked in 5 mM DSSO for 40 minutes at room temperature. The crosslinking reaction was quenched with 50 mM NH4HCO3 for 15 minutes at room temperature.
293T cells for AP-MS analysis
Lysates prepared from 1 × 107 293T cells as described above were incubated for 2 hours at 4°C with MagneHalo magnetic beads prepared from 100 mL bead slurry according to the manufacturer’s instructions. Beads were isolated using a DynaMag-2 Magnet and washed 4 times in buffer containing 50 mM Tris●HCl (pH 7.4), 137 mM NaCl, 2.7 mM KCl and 0.05% Nonidet® P40. Beads were incubated in elution buffer containing 50 mM Tris●HCl (pH 8.0), 0.5 mM EDTA, 1 mM DTT, and 2 Units AcTEV for 2h at 25°C to elute bound proteins.
Digestion of proteins for mass spectrometry
Halo purified protein complexes were precipitated by incubation with ice-cold trichloroacetic acid (20% final concentration) overnight at 4°C. Precipitated protein pellets were isolated by centrifugation at 21,000 × g for 30 minutes at 4°C, washed twice in ice-cold acetone, dried, and resuspended in buffer containing 100 mM Tris●HCl (pH 8.5), and 8 M urea. Disulfide bonds were reduced with Tris(2-carboxylethyl)-phosphine hydrochloride. Samples were then treated with chloroacetamide to prevent di-sulfide bond reformation. Denatured proteins were then treated with 0.1 μg Lys-C for 6 hours at 37°C. The urea concentration was reduced to 2 M by adding an appropriate volume of 100 mM Tris●HCl (pH 8.5) and CaCl2 added to a final concentration of 2 mM. Proteins were further digested overnight with 0.5 μg trypsin, after which formic acid was added to a final concentration of 5%.
Mass spectrometry analysis
AP-MS mass spectrometry analysis
For AP-MS experiments, digested samples were loaded onto microcapillary columns containing three phases of chromatography resin (reverse phase, strong cation exchange, reverse phase) and eluted into LTQ mass spectrometers (Thermo Scientific, San Jose, CA) for MudPIT analysis with ten 2-hour chromatography steps (Banks et al., 2012). Processing of the resulting .raw files is described in Quantification and Statistical Analysis below.
XL-MS mass spectrometry analysis
Crosslinked peptides were resolved for mass spectrometry analysis using a Dionex UltiMate 3000 RSLCnano liquid chromatography system. Peptides were initially loaded from the autosampler onto an Acclaim PepMap 100 C18 LC Trap Cartridge (0.3 mm inside diameter, 5 mm length) (Thermo Scientific, San Jose, CA) using a loading pump flow rate of 2 μl/minute. Peptides were subsequently resolved for mass spectrometry analysis using an analytical column (50 μm inside diameter, 150 mm length) packed in-house with ReproSil®-Pur C18-aQ 1.9 μm resin (Dr. Masch GmbH, Germany). Chromatography was performed using combinations of buffer A (95% water, 5% acetonitrile, and 0.1% formic acid (v/v/v), pH 2.6), and buffer B (20% water, 80% acetonitrile, and 0.1% formic acid (v/v/v), pH 2.6). The following chromatography steps were performed using a flow rate of 120 nl/minute: (1) 2% B for 20 minutes (column equilibration); (2) a linear gradient from 2% to 10% B over 10 minutes; (3) a linear gradient from 10% to 40% B over either 120 minutes or 240 minutes; (4) a linear gradient from 40% to 95% B over 5 minutes; (5) 95% B for 14 minutes (column wash); (6) a linear gradient from 95% B to 2% B over 1 minute; (7) 2% B for 10 minutes (column re-equilibration).
Eluted peptides were analyzed by mass spectrometry using an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific, San Jose, CA). An MS3 based method was used for identification of DSSO crosslinked peptides as follows: Full MS scans were performed using the Orbitrap mass analyzer (60,000 m/z resolution, 1.6 m/z isolation window, and 375–1500 m/z scan range); The top 3 peptides identified with charge state 4 to 8 were selected for MS2 fragmentation (20% CID energy) and subsequent detection with the Orbitrap mass analyzer (30,000 m/z resolution and a dynamic exclusion time of 40 s); Pairs of MS2 fragments with a mass difference of 31.9720 (20 ppm mass tolerance) were selected for MS3 fragmentation (CID energy 35%) and detection using the Linear Ion Trap mass analyzer (rapid scan, 3 m/z isolation window, maximum ion injection time 200 ms); Each MS2 scan was followed by a maximum of 4 MS3 scans. Processing of the resulting .raw files to identify DSSO crosslinked peptides is described in Quantification and Statistical Analysis below.
Downstream analysis of crosslinking data
A summary of the high-confidence crosslinks identified using Proteome Discoverer 2.2 that were used for further analysis is presented in Table S2. The xiView platform (Graham et al., 2019) was used for crosslink visualization, for mapping crosslinks to the PDB structures, and for calculating distances between alpha carbon atoms. The following structures were analyzed (Figure 2): PDB: 2N2H mSIN3A (Clark et al., 2015a); PDB: 2N1U SAP30L (Laitaoja et al., 2016); PDB: 2LD7 SAP30 (Xie et al., 2011); PDB: 5ICN HDAC1 (Watson et al., 2016); PDB: 3CFV RBBP7 (Murzina et al., 2008).
For structure modeling, the SWISS-MODEL platform (Waterhouse et al., 2018) was first used for homology-modeling the PDB files corresponding to human SIN3A (using PDB 2N2H corresponding to mSin3A), the C terminus of SAP30L (using PDB 2LD7 corresponding to human SAP30), the N-terminus of SAP30L (using PDB: 2N1U corresponding to human SAP30L), and to HDAC1 (using PDB: 5IX0 (Watson et al., 2016) corresponding to human HDAC2) used for protein docking in Figure 4. Note, SWISS-MODEL Auto-model built the HDAC1 model based on PDB: 5IX0 rather than PDB: 5ICN as this template had a higher GMQE (Global Model Quality Estimation) score (0.79) than template 5ICN (0.75). Protein structures were initially docked using the HADDOCK server (Dominguez et al., 2003; de Vries et al., 2010; van Zundert et al., 2016) using default settings together with the unambiguous crosslinking restraints indicated in Figure 4A. HADDOCK generated 200 water-refined models of which 24.5% were clustered in 22 clusters with a minimum cluster size adjusted to 3. The top cluster of HADDOCK models (with the lowest HADDOCK score) was identified as Cluster 2 (3 models) and a HADDOCK score of −143.4 ± 39.6. The top model within this cluster was then identified using established guidelines to calculate normalized XL scores (Orbán-Németh et al., 2018) from Euclidean distances calculated using Xwalk (Kahraman et al., 2011). This analysis is presented in Table S4. In addition to this initial model, a refined model was generated by using additional interaction restraints based on recent evidence for a SAP30/HDAC1 interface (Marcum and Radhakrishnan, 2019). The additional active residues used for docking here were S46, S48, R50 and Y72 in SAP30L and K31, H33, R270, and R306 in HDAC1. Of the 200 water-refined HADDOCK models generated, 7.5% were clustered into 2 clusters. The top cluster, Cluster1, contained 8 models and had a HADDOCK score of −160.6 ± 18.2. Again, the top model within this cluster was identified using normalized XL scores (Table S4). Protein structures were processed for visualization with Chimera (Pettersen et al., 2004). Video S1 was produced using Chimera and Adobe After Effects 2020. Models generated by SWISS-MODEL and the refined model generated by docking subunits using HADDOCK are available from the PDB-Dev repository (https://pdb-dev.wwpdb.org/) with accession number PDBDEV_00000043. Models can also be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/LIBPB-1465.
The bar chart in Figure 3C shows mean values calculated from three biological replicates (SIN3A ΔPAH3 and SIN3A ΔHID 688–829) or from four biological replicates (SIN3A ΔPAH4) using Microsoft Excel. Error bars represent standard deviation.
XL-MS mass spectrometry data analysis
Proteome Discoverer 2.2 with the add on XlinkX crosslinking nodes (Liu et al., 2017) (Thermo Scientific, San Jose, CA) was used to identify crosslinked peptides from .raw files from three experiments as follows. For each .raw file, the Xlinkx Detect processing node was used to identify MS2 fragmentation scans with reporter ions characteristic of DSSO crosslinked peptides using DSSO lysine crosslink modifications of 158.00376 Da (monoisotopic mass) and 158.17636 Da (average mass), with cleaved DSSO lysine crosslink modifications of 54.01056 Da (alkene, monoisotopic mass), 54.04749 Da (alkene, average mass), 85.98264 Da (thiol, monoisotopic mass), and 86.11358 Da (thiol, average mass). Subsequently, a version of the database used for AP-MS searches, but without shuffled sequences, was searched using either the Xlinkx Search node (fragmentation scans with crosslink reporter ions) or with the Sequest HT node (scans without crosslink reporter ions). Both search strategies searched for peptides with 57.021 Da fixed modifications on cysteine residues (carbamidomethylation) and 15.995 Da variable modifications on methionine residues (oxidation). In addition, the Sequest HT node searched for the variable lysine modifications 176.014 Da (water quenched DSSO monoadducts) and 279.078 Da (Tris quenched DSSO monoadducts). For Sequest HT node searches, a precursor ion mass tolerance of 10 ppm and a fragment ion mass tolerance of 0.6 Da were used; for Xlinkx Search node searches, a precursor ion mass tolerance of 10 ppm and fragment ion mass tolerances of 20 ppm (FTMS) or 0.5 Da (ITMS) were used. The maximum number of equal dynamic modifications was 3 (Sequest HT searches). The protein FDR was set at 0.01 using the Xlinkx Validator node for Xlinkx searches, and the target FDR (Strict) was set at 0.01 using the Percolator node for Sequest HT searches. Table S2 contains comprehensive details of crosslink identifications, including crosslink-spectrum matches (CSMs). Raw data and Proteome Discoverer results files for XL-MS experiments has been deposited in the MassIVE repository with the identifier MSV000084311 (see Table S1).
DATA AND CODE AVAILABILITY
Data Availability Statement
The mass spectrometry datasets generated for this study are available from the Massive data repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) using the identifiers listed in Table S1. Models generated by SWISS-MODEL and the refined model generated by docking subunits using HADDOCK are available from the PDB-Dev repository (https://pdb-dev.wwpdb.org/) with accession number PDBDEV_00000043. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/LIBPB-1465.
Code Availability
Code for the software RAWDistiller v. 1.0 and NSAF7 is available on request.
Supplementary Material
Highlights.
66 interprotein and 63 self cross-links for 13 Sin3 subunits
Crosslink-guided docking of SIN3A, SAP30L, and HDAC1 structures
Positions of subunits SAP30L, HDAC1, SUDS3, HDAC2, and ING1 around SIN3A
ACKNOWLEDGMENTS
Research reported in this publication was supported by the Stowers Institute for Medical Research and the National Institute of General Medical Sciences of the National Institutes of Health under award RO1GM112639 to M.P.W. and F32GM122215 to M.K.A. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/LIBPB-1465.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.080.
REFERENCES
- Banks CA, Kong SE, and Washburn MP (2012). Affinity purification of protein complexes for analysis by multidimensional protein identification technology. Protein Expr. Purif 86, 105–119. [DOI] [PubMed] [Google Scholar]
- Banks CAS, Lee ZT, Boanca G, Lakshminarasimhan M, Groppe BD, Wen Z, Hattem GL, Seidel CW, Florens L, and Washburn MP (2014). Controlling for gene expression changes in transcription factor protein networks. Mol. Cell. Proteomics 13, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks CAS, Thornton JL, Eubanks CG, Adams MK, Miah S, Boanca G, Liu X, Katt ML, Parmely TJ, Florens L, and Washburn MP (2018). A structured workflow for mapping human Sin3 histone deacetylase complex interactions using Halo-MudPIT affinity-purification mass spectrometry. Mol. Cell. Proteomics 17, 1432–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MD, Marcum R, Graveline R, Chan CW, Xie T, Chen Z, Ding Y, Zhang Y, Mondragón A, David G, and Radhakrishnan I (2015a). Structural insights into the assembly of the histone deacetylase-associated Sin3L/Rpd3L corepressor complex. Proc. Natl. Acad. Sci. USA 112, E3669–E3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MD, Zhang Y, and Radhakrishnan I (2015b). Solution NMR Studies of an Alternative mode of Sin3 engagement by the Sds3 subunit in the histone deacetylase-associated Sin3L/Rpd3L corepressor complex. J. Mol. Biol 427, 3817–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk M, and Bonvin AMJJ (2010). The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc 5, 883–897. [DOI] [PubMed] [Google Scholar]
- Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, and Aebersold R (2006). The Peptide Atlas project. Nucleic Acids Res. 34, D655–D658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, and Bonvin AMJJ (2003). HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- Fleischer TC, Yun UJ, and Ayer DE (2003). Identification and characterization of three new components of the mSin3A corepressor complex. Mol. Cell. Biol 23, 3456–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frueh DP, Goodrich AC, Mishra SH, and Nichols SR (2013). NMR methods for structural studies of large monomeric and multimeric proteins. Curr. Opin. Struct. Biol 23, 734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham MJ, Combe C, Kolbowski L, and Rappsilber J (2019). xiView: a common platform for the downstream analysis of crosslinking mass spectrometry data. bioRxiv. 10.1101/561829. [DOI] [Google Scholar]
- Kahraman A, Malmström L, and Aebersold R (2011). Xwalk: computing and visualizing distances in cross-linking experiments. Bioinformatics 27, 2163–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, and Huang L (2011). Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics 10, 002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RDW, and Cowley SM (2013). The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem. Soc. Trans 41, 741–749. [DOI] [PubMed] [Google Scholar]
- Kwon YJ, Petrie K, Leibovitch BA, Zeng L, Mezei M, Howell L, Gil V, Christova R, Bansal N, Yang S, et al. (2015). Selective inhibition of SIN3 corepressor with avermectins as a novel therapeutic strategy in triple-negative breast cancer. Mol. Cancer Ther 14, 1824–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, and Eisenman RN (1997). Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 89, 349–356. [DOI] [PubMed] [Google Scholar]
- Laitaoja M, Tossavainen H, Pihlajamaa T, Valjakka J, Viiri K, Lohi O, Permi P, and Jänis J (2016). Redox-dependent disulfide bond formation in SAP30L corepressor protein: Implications for structure and function. Protein Sci. 25, 572–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer BEL, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, et al. (2013). Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, and Shiekhattar R (2005). An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437, 432–435. [DOI] [PubMed] [Google Scholar]
- Leitner A, Faini M, Stengel F, and Aebersold R (2016). Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci 41, 20–32. [DOI] [PubMed] [Google Scholar]
- Leney AC, and Heck AJR (2017). Native mass spectrometry: what is in the name? J. Am. Soc. Mass Spectrom 28, 5–13. [DOI] [PubMed] [Google Scholar]
- Liu F, Lössl P, Scheltema R, Viner R, and Heck AJR (2017). Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun 8, 15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- Marcum RD, and Radhakrishnan I (2019). Inositol phosphates and core subunits of the Sin3L/Rpd3L histone deacetylase (HDAC) complex up-regulate deacetylase activity. J. Biol. Chem 294, 13928–13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA, and Breslow R (2007). Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Bio-technol 25, 84–90. [DOI] [PubMed] [Google Scholar]
- Merkley ED, Rysavy S, Kahraman A, Hafen RP, Daggett V, and Adkins JN (2014). Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 23, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzina NV, Pei XY, Zhang W, Sparkes M, Vicente-Garcia J, Pratap JV, McLaughlin SH, Ben-Shahar TR, Verreault A, Luisi BF, and Laue ED (2008). Structural basis for the recognition of histone H4 by the histone chaperone RbAp46. Structure 16, 1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orbán-Németh Z, Beveridge R, Hollenstein DM, Rampler E, Stranzl T, Hudecz O, Doblmann J, Schlögelhofer P, and Mechtler K (2018). Structural prediction of protein models using distance restraints derived from cross-linking mass spectrometry data. Nat. Protoc 13, 478–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, and Kutateladze TG (2006). Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Rielland M, Cantor DJ, Graveline R, Hajdu C, Mara L, Diaz Bde.D., Miller G, and David G (2014). Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression. J. Clin. Invest 124, 2125–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiu ME, Smith KT, Groppe BD, Gilmore JM, Saraf A, Egidy R, Peak A, Seidel CW, Florens L, Workman JL, and Washburn MP (2014). Suberoylanilide hydroxamic acid (SAHA)-induced dynamics of a human histone deacetylase protein interaction network. Mol. Cell. Proteomics 13, 3114–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Belsom A, Rappsilber J, and Brock O (2016). Blind testing of cross-linking/mass spectrometry hybrid methods in CASP11. Proteins 84 (Suppl 1), 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Peña P, Lan F, Kaadige MR, et al. (2006). ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smialowski P, and Wong P (2016). Protein Crystallizability In Data Mining Techniques for the Life Sciences, Carugo O and Eisenhaber F, eds. (Springer; ), pp. 341–370. [Google Scholar]
- Smith KT, Martin-Brown SA, Florens L, Washburn MP, and Workman JL (2010). Deacetylase inhibitors dissociate the histone-targeting ING2 subunit from the Sin3 complex. Chem. Biol 17, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, and Yates JR 3rd. (2002). DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, and Bonvin AMJJ (2016). The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol 428, 720–725. [DOI] [PubMed] [Google Scholar]
- Viiri KM, Jänis J, Siggers T, Heinonen TYK, Valjakka J, Bulyk ML, Mäki M, and Lohi O (2009). DNA-binding and -bending activities of SAP30L and SAP30 are mediated by a zinc-dependent module and mono-phosphoinositides. Mol. Cell. Biol 29, 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cimermancic P, Yu C, Schweitzer A, Chopra N, Engel JL, Greenberg C, Huszagh AS, Beck F, Sakata E, et al. (2017). Molecular details underlying dynamic structures and regulation of the human 26S proteasome. Mol. Cell. Proteomics 16, 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BVL, and Schwabe JWR (2016). Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun 7, 11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, He Y, Korkeamaki H, Zhang Y, Imhoff R, Lohi O, and Radhakrishnan I (2011). Structure of the 30-kDa Sin3-associated protein (SAP30) in complex with the mammalian Sin3A corepressor and its role in nucleic acid binding. J. Biol. Chem 286, 27814–27824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015). ProLuCID: An improved SE-QUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, and Rein berg D (1999). Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wen Z, Washburn MP, and Florens L (2010). Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal. Chem 82, 2272–2281. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wen Z, Washburn MP, and Florens L (2011). Improving proteomics mass accuracy by dynamic offline lock mass. Anal. Chem 83, 9344–9351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Availability Statement
The mass spectrometry datasets generated for this study are available from the Massive data repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) using the identifiers listed in Table S1. Models generated by SWISS-MODEL and the refined model generated by docking subunits using HADDOCK are available from the PDB-Dev repository (https://pdb-dev.wwpdb.org/) with accession number PDBDEV_00000043. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/LIBPB-1465.
Code Availability
Code for the software RAWDistiller v. 1.0 and NSAF7 is available on request.
The mass spectrometry datasets generated for this study are available from the Massive data repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) using the identifiers listed in Table S1. Models generated by SWISS-MODEL and the refined model generated by docking subunits using HADDOCK are available from the PDB-Dev repository (https://pdb-dev.wwpdb.org/) with accession number PDBDEV_00000043. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/LIBPB-1465.