

Abstract
The splitting of N2 into well‐defined terminal nitride complexes is a key reaction for nitrogen fixation at ambient conditions. In continuation of our previous work on rhenium pincer mediated N2 splitting, nitrogen activation and cleavage upon (electro)chemical reduction of [ReCl2(L2)] {L2 = N(CHCHPtBu2)2 –} is reported. The electrochemical characterization of [ReCl2(L2)] and comparison with our previously reported platform [ReCl2(L1)] {L1 = N(CH2CH2PtBu2)2 –} provides mechanistic insight to rationalize the dependence of nitride yield on the reductant. Furthermore, the reactivity of N2 derived nitride complex [Re(N)Cl(L2)] with electrophiles is presented.
Keywords: Nitrogen fixation, Rhenium, Pincer complexes, Electrochemistry, Cyclic voltammetry
N2 splitting into terminal nitrides by chemical and electrochemical reduction of [ReCl2{N(CHCHPtBu2)2}] is presented. Comparison of electrochemical data with that of the previously reported, related pincer complex [ReCl2{N(CH2CH2PtBu2)2}] allowed for identifying key parameters that control the efficiency of the reaction sequence, which defines reductive N2 splitting.
Introduction
Industrial ammonia synthesis by the Haber–Bosch process is carried out at a scale of 150 Mt/a, using hydrogen produced via steam reforming of fossil fuels that accounts for massive energy consumption and CO2 emission.1 The replacement of H2 as reductant is therefore highly desirable to enhance the sustainability of nitrogen fixation. The electrochemically driven nitrogen reduction reaction (NRR) is an appealing alternative to feed renewable energy from photovoltaic harvesting.2 Electrocatalytic NRR has seen tremendous progress in recent years. Faradaic yields up to 73.3 % have been reported, yet with current densities far below the US Department of Energy targets.3, 4 Furthermore, the mechanistic basis of heterogeneous electrocatalysts remains comparatively ill‐defined. Homogeneous (model) NRR catalysts could give detailed insight on key reaction steps and thermochemical and kinetic parameters.5 However, molecular NRR electrocatalysts are highly limited.6
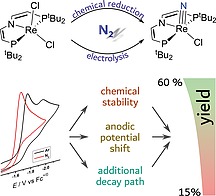
Two general mechanisms have been proposed for the NRR with molecular catalysts. The “bio‐inspired” route is comprised of successive proton coupled electron transfer (PCET) steps at terminally coordinated N2, in analogy to the mechanism proposed for the [Fe,Mo]‐nitrogenase enzyme.7, 8 Initial full cleavage of the N≡N triple bond via N2‐bridged, multinuclear complexes and subsequent PCET of the resulting nitrides, as in the Fe‐catalyzed Haber–Bosch process, has been alternatively considered.9 The splitting of dinitrogen into well‐defined nitride complexes was pioneered by Laplaza and Cummins 25 years ago and several examples are known by now.10, 11 Recently, group 6 and 7 pincer platforms attracted particular attention (Scheme 1).9, 12 Our group reported N2 splitting upon chemical reduction [Na/Hg, Co(Cp*)2] of the rhenium(III) PNP pincer complex [ReCl2(L1)] {(1L1; L1 = N(CH2CH2PtBu2)2)–} to the nitrido complex [Re(N)Cl(L1)] (2L1; Scheme 2).[12b] Miller, Siewert, Schneider and co‐workers jointly examined electrochemically driven N2 cleavage for this platform, which allowed for detailed mechanistic study by cyclic voltammetry (CV).[12g] The reaction goes through rate determining splitting of the N2‐bridged dirhenium complex [{ReCl(L1)}2(µ‐N2)] (3L1; t 1/2 298K ≈ 35 s). Intermediate 3L1 is formed within a complex EC N2 C C lEC dim pathway via (electro)chemical ReIII/ReII reduction (E 1) of 1L1, followed by N2 binding (C N2), chloride loss (C Cl), ReII/ReI reduction (E 2) and subsequent comproportionation with parent 1L1 (C dim). Besides mechanistic insight, this study provided the first example of N2 splitting into nitrido complexes by controlled potential electrolysis (CPE at –1.90 V vs. Fc+/0) with yields around 60 %. Recently, Masuda and co‐workers demonstrated electrochemically driven N2 splitting upon anodic oxidation of trans‐[Mo(N2)2(depe)2] (depe = Et2PCH2CH2PEt2).13 However, further systematic studies are required to identify the key parameters that control the N2 splitting reaction.
Scheme 1.

Selected examples for N2‐splitting into terminal nitride complexes with transition metal pincer platforms.
Scheme 2.

(Electro)chemical N2‐activation from [ReCl2(L1)] (1L1) into [Re(N)Cl(L1)] (2L1) via the N2‐bound dimeric intermediate [{ReCl(L1)}2(µ‐N2)] (3L1) formed via an EC N2 C Cl EC dim type mechanism.
Here, (electro‐)chemical N2 splitting with a modified Re pincer platform is reported. The divinylamide ligand N(CHCHPtBu2)2 – (L2–) was previously utilized for the stabilization of a wide variety of transition metal complexes.14, 15 The enhanced rigidity resembles that of “archetypical” amide pincer ligands, like Milstein's pyridine‐based dearomatized ligand NC5H3(2‐CHPtBu2)(6‐CHPtBu2)–, with increased steric protection, as compared to phenylene‐bridged diphosphinoamide N(C6H4PiPr2)2 –.16 In comparison to parent L1, backbone unsaturation leads to significant reduction of N→M π‐donation, as reflected in CO and N2 stretching frequencies of Ru and Ir complex series.14, [15e], [15k] Starting from [ReCl2(L2)] (1L2), the effects of backbone unsaturation on the reduction potential, N2 splitting yields and functionalization of the nitride product are discussed.
Results and Discussion
Synthesis and Characterization of 1L2
Complex 1L2 was synthesized starting from 1L1 by templated ligand modification via hydrogen atom abstraction with excess 2,4,6‐tert‐butylphenoxy radical (TBP) at 50 °C (Scheme 3), as similarly reported for other L2 complexes.15 Small amounts of a paramagnetic side‐product found by 1H NMR spectroscopy could be identified as overoxidized rhenium(IV) complex [ReCl3(L2)] (4L2) upon comparison with an original sample that was independently synthesized. Facile conversion of the side product 4L2 to 1L2 is accomplished by in situ reduction with Co(Cp)2, providing the analytically pure product in 63 % isolated yield. The 1H NMR spectrum of 1L2 indicates C 2v symmetry in solution. In the 31P{1H} NMR spectrum, a sharp singlet resonance was found at δ 31P = –275 ppm (Figure S3). In analogy to other rhenium(III) phosphine complexes and 1L1,[12h], 17 the high‐field shift is attributed to mixing of the ground‐state with low‐lying excited states leading to temperature independent paramagnetism (TIP),19 as substantiated for 1L1 and 1L2 by SQUID magnetometry {χM[10–6 × cm3 mol–1] = 280 (1L1), 300 (1L2); Figure S26}. Despite several attempts, single crystals of 1L2 suitable for X‐ray analysis could not be obtained.
Scheme 3.
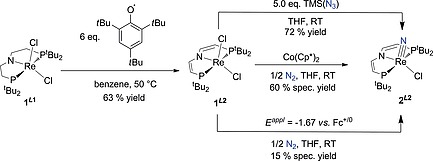
Synthesis of complex 1L2 by ligand oxidation starting from 1L1 using the 2,4,6‐tert‐butylphenoxy radical and different routes for the synthesis of complex 2L2 by either (electro)chemical N2‐splitting, or via reaction with TMS(N3).
N2 Splitting by (Electro‐)Chemical Reduction
Reduction of 1L2 with an equimolar amount of Co(Cp*)2 in THF under 1 atm N2 results in rapid conversion to a mixture of several diamagnetic products, according to 1H and 31P{1H} NMR spectroscopy. The rhenium(V) nitride [Re(N)Cl(L2)] (2L2) was identified as the major species (60 % yield by NMR spectroscopy, see Figure S7) by comparison to an authentic sample prepared by reaction of 1L2 with trimethylsilyl azide (Scheme 3). All attempts to identify intermediates by NMR monitoring at low temperatures were unsuccessful. The yield in 2L2 is slightly lower compared with parent 2L1 [75 % with Co(Cp*)2] and notably depends on the reductant. Considerably lower spectroscopic nitride yields are obtained with alkali metal reductants, such as Na/Hg (approx. 30 %) or KC8 (approx. 20 %), under otherwise identical conditions. In comparison, 80 % yield in 2L1 was obtained upon reducing 1L1 with Na/Hg under N2. Notably, with Na/Hg or KC8 as reductant, yet not with Co(Cp*)2, the liberation of isobutene was detected spectroscopically for 1L2 (Figure S14), as previously observed for the thermal decomposition of [OsCl(L2)],[15j] suggesting fragmentation of the L2 ligand platform upon overreduction. Strong dependence of N2 splitting yields on the nature of the reductant has been previously reported.21 However, in most cases these effects are poorly understood.
Multinuclear NMR spectroscopic characterization of 2L2 indicates C s symmetry with a peak at δ 31P = 71.8 ppm in the 31P{1H} NMR spectrum. Single crystal X‐ray characterization (Figure 1) reveals a slightly distorted square pyramidal geometry (τ 5 = 0.15)22 with the nitride [Re–N2 1.647(18) Å] in apical position. These bond metrics are close to those of 2L1 [Re≡N = 1.643(6) Å, τ 5 = 0.14], which was recently characterized crystallographically.23 The planar ligand backbone with shortened C=C bonds [2L2: 1.35(2) Å; 2L1: 1.545(10)/1.526(10) Å] confirms the presence of vinylene linkers in the pincer ligand backbone. Electrochemical characterization of the nitrido species was carried out by cyclic voltammetry (CV) in THF (Figure S21). A reversible oxidation at +0.21 V (vs. Fc+/0)24 was assigned to the ReV/ReVI couple and is significantly anodically shifted with respect to 2L1 (E 1/2 = –0.086 V).[12g] This potential shift is consistent with reduced electron density at the rhenium ion of 2L2 due to weaker donation by pincer ligand L2. 2L2 features an additional, irreversible reduction feature at low potential (E p,c = –3.3 V vs. Fc+/0).
Figure 1.
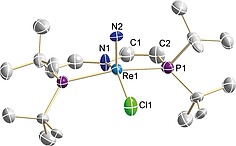
Molecular structure of 2L2 from single‐crystal X‐ray diffraction with anisotropic displacement parameters drawn at the 50 % probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Re1–N1 2.106(3), Re1–Cl1 2.395(7), Re1–N2 1.647(18), Re1–P1 2.447(3), C1–C2 1.35(2), N1–Re1–Cl1 145.9(2), N1–Re1–N2 109.2(5), Cl1–Re1–N2 104.8(6), P1–Re–P2 155.1(1).
CPE of 1L2 under 1 atm N2 was carried out in THF at E = –1.67 V, i.e. the cathodic peak potential of the first reductive feature (Figure 2, top left; vide infra for discussion). Thus, the use of ligand L2 enables electrolysis at approx. 230 mV less negative potential with respect to 1L1, presumably due to the poorer π‐donor properties of the unsaturated pincer. Transfer of approximately 1.2 electrons per Re over the course of 2 h was accompanied by a gradual color change from brown to light brown/green. Spectroscopic yields of nitride 2L2 of approx. 15 % were obtained (Figure S8), which are significantly lower than the electrolysis yields of nitride 2L1 (60 %). The low electrolysis yield in 2L2 is in stark contrast with Co(Cp*)2, and closer to other heterogeneous reductants (Na/K, KC8).
Figure 2.
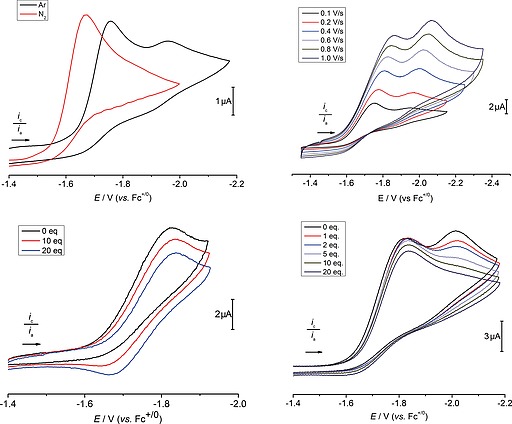
CVs of 1L2. Top Left: Ar (black) and N2 (red) at ν = 0.1 V s–1. Top Right: Scan rate dependence under Ar. Bottom Left: Under Ar, in the presence of varying amounts of (nBu4N)Cl (v = 0.5 V s–1). Bottom Right: Under Ar, in the presence of varying amounts of (nBu4N)Cl (v = 0.5 V s–1). General conditions: 1.0 mm 1L2 in THF, 0.2 m (nBu4N)PF6.
In order to rationalize the lower N2 splitting yields, the stability of 1L2 in THF in the presence of N2 and chloride ions was assessed. NMR spectroscopic monitoring under 1 atm N2 reveals partial conversion to several unidentified new species in the spectral range δ 31P = 20–60 ppm (Figure S15). CV characterization at higher N2‐pressure initially shows a slight rise in the current of the reduction feature by around 5 % upon increasing pressure from 1 to up to 11 bars (Figure S20). However, over the course of 45 min at 11 bars of N2 (see Experimental Section), the current drops by about 20 % suggesting chemical instability of 1L2 under these conditions. 31P{1H} NMR spectroscopic analysis after this experiment shows complete conversion of 1L2 to an intractable reaction mixture (Figure S16). More rapid decay was even found upon addition of a chloride source, suggesting that accumulation of chloride ions released during electrolysis may accelerate decomposition. A mixture of 1L2 and (nBu4N)Cl under 1 atm N2 gradually changes color from light brown to green over the course of a couple of hours, with concomitant formation of a mixture of diamagnetic and paramagnetic species (Figure S17). Comparison with 1H NMR spectra of mixtures of authentic 1L2, (nBu4N)Cl, and 4L2 (Figure S18) supports the assignment of a broad signal at +12 ppm to rhenium(IV) complex 4L2. This observation suggests that the chloride‐induced decay of 1L2 proceeds via disproportionation of [ReIIICl3(L2)]– to 4L2 and further, unstable rhenium(II) species as outlined in Scheme 4.
Scheme 4.

Proposed chloride‐induced disproportionation of 1L2.
The relevance of the decay pathway shown in Scheme 4 for the electrochemical transformations was evaluated from available thermochemical data (see also Electronic Supporting Information, Section 5). The invariance of δ 31P(1L2) and inability to detect a new signal for [ReCl3(L2)]– in the presence of added chloride (5 equiv.) allows for estimating an upper limit of the chloride association constant (K Cl ≤ 0.015 m –1; ΔG 0 Cl ≥ +2.5 kcal mol–1). Subsequent disproportionation of [ReIIICl3(L2)]– with 1L2 to [ReIICl3(L2)]– and 4L2 is defined by the reduction potentials of 4L2 (E 1/2 ≈ –0.9 V vs. Fc+/0; Figure S22) and 1L2 (E 1/2 = –1.75 V vs. Fc+/0; vide infra), giving K Disp ≈ 4 × 10–15 and ΔG 0 Disp ≈ +20 kcal mol–1. The chloride‐induced decomposition pathway outlined in Scheme 4 would therefore have to be driven by the decay of [ReCl2(L2)]–. However, the overall effective kinetic barrier needs to be larger than ΔG ‡ ≥ 22.5 kcal mol–1. In consequence, chloride induced decomposition is irrelevant on the CV timescale but might reduce electrolysis yields, which goes over hours.
In comparison, parent 1L1 proved stable under these conditions over an extended period of time. Structural comparison of 2L1 and 2L2 shows only minor differences, like the steric shielding as expressed by the pincer bite angle [P–Re–P: 156.16(7)° (2L1), 155.11(13)° (2L2)]. We therefore tentatively associate the reduced stability to electronic reasons. Backbone unsaturation changes the donor properties (poorer π‐donation) and increases the metal Lewis acidity. Furthermore, ligand L2 is potentially non‐innocent and can undergo versatile proton/electron transfer at the vinyl groups.[15i] The reduced stability of 1L2 in the presence of N2 and chloride will contribute to lowering the electrolysis yields. Electrochemical reduction occurs on a longer time scale (2 h) than chemical N2‐splitting, e.g. with Co(Cp*)2 as reductant (5 min). Thus, 1L2 will be exposed to N2 and released free chloride during electrolysis for a longer time. However, the estimated decay rates suggest that further processes contribute to the low nitride electrolysis yields. Therefore, the reduction of 1L2 was examined in depth by CV, which is presented in the next section.
CV Examinations
The CV of 1L2 under Ar (Figure 2) reveals two irreversible, reductive features at E p,c,1 = –1.75 V and E p,c,2 = –1.95 V (vs. Fc+/0; ν = 0.1 V s–1), respectively. The peak currents i p,c,1 and i p,c,2 scale linearly with v 1/2, indicating diffusion‐controlled electron transfer. Both reductions exhibit distinct cathodic potential shifts with rising current ratio i p,c,1/i p,c,2 at increasing scan rates (Figure 2, top right). The current characteristics suggest the presence of competing chemical reaction pathways after initial reduction of 1L2 including decay to a redox‐inactive species.
Changing from Ar to N2 (1 bar), the irreversible first reduction of 1L2 shifts anodically by about 85 mV to E p,c = –1.67 V (Figure 2, top left) accompanied by a small peak current increase (approx. 5 %). The second reduction feature present under Ar vanishes under N2 without appearance of new reductive events. The anodic potential shift and the disappearance of the ReII/ReI reduction are in agreement with N2‐activation at the rhenium(II) stage, as proposed for 1L1.[12g] The anodic shift with respect to 1L1 (approx. 230 mV) compares well with the shift found for the corresponding nitrides 2L1 and 2L2 (vide infra) and is therefore associated with weaker π‐donation by pincer ligand L2. Besides the first reduction (ReIII/ReII), the second reduction feature (ReII/ReI) that is obtained in the absence of N2 is even more anodically shifted, leading to decreased peak separation for 1L2 (ΔE = 0.17 V) as compared to 1L1 (ΔE = 0.29 V). In consequence, strong reductants, like Na/Hg (E° < –2.3 V),25 have potentials that are well beyond the ReII/ReI couple of 1L2. Unproductive overreduction in case of incomplete trapping of the rhenium(II) intermediate by dinitrogen might therefore be a contributing factor to the lower nitride yields obtained with Na/Hg or KC8, respectively, vs. Co(Cp*)2 (E° = –1.84 V).26
Further insight was obtained by electrochemical evaluation at varying conditions. Due to the limited chemical stability of 1L2 in the presence of N2 and low electrolytic Faradaic yields, we focused on the decay kinetics under argon to identify pathways that could lead to the reduced nitride yields with respect to 1L1. The effect of added (nBu4N)Cl on the CV response was examined to probe for coupled chloride loss. Modest increase in reversibility and a slight cathodic shift are obtained for the first reduction event E 1 with rising chloride concentrations (Figure 2, bottom left), in line with coupled, fast and reversible chloride dissociation after reduction of 1L2. The peak current decrease is attributed to slow decomposition of 1L2 in presence of excess chloride (vide supra). Scanning both reduction events E 1 and E 2 (Figure 2, bottom right), the second feature drops in current and shifts cathodically with increasing chloride ion concentration. The concentration dependence in 1L2 (0.5–4.0 mm) shows increasing i p,c,1/i p,c,2 current ratio at higher c Re (Figure S20), indicating a bimolecular decay route between the two reduction events.
Our previous electrochemical study for the reduction of parent 1L1 allowed for rationalization of the CV data under Ar by an EC Cl E minimum model with ReIII/ReII and ReII/ReI redox couples that are connected by chloride dissociation between electron transfers.[12g] Quantitative kinetic modelling by digital simulation of the CV data further required the introduction of a unimolecular decay step at the rhenium(II) stage after chloride loss. For 1L2, the data indicates at least two coupled chemical reactions after the first reduction event: chloride dissociation that forms [ReCl(L2)] (as proposed for 1L1) and competing bimolecular decay of [ReCl2(L2)]–, respectively. A best fit over all CV data of 1L2 under Ar was found for the kinetic model and simulation parameters presented in Scheme 5.
Scheme 5.
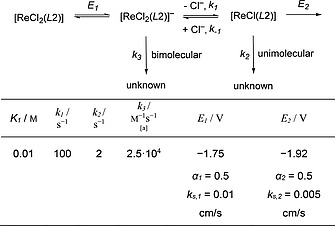
Minimum kinetic model for the digital simulation of the electrochemical reduction of 1L2 under Ar and thermodynamic and kinetic parameters (formal potentials, rate constants, and electron transfer parameters) obtained from CV data simulation; [a] for bimolecular decay of [ReCl2(L2)]–.
Typical simulation data are shown in Figure 3 and Figures S24/S25. Within the model, reduction of 1L2 (E 1) is succeeded by reversible chloride dissociation (K 1) and irreversible ReII/ReI reduction (E 2). Importantly, a satisfactory minimum model required two decay routes to account for the concentration dependence of i p,c,1/i p,c,2: unimolecular decay of [ReCl(L2)] (k 2) after chloride loss as proposed for 1L1, but also bimolecular decay before chloride dissociation (k 3). Assuming formation of electrochemically silent species, bimolecular decay of [ReCl2(L2)]– was modeled since an alternative reaction of [ReCl2(L2)]– with parent [ReCl2(L2)] would exhibit decreasing normalized i p,c,1 at increasing concentration, which is not observed.
Figure 3.

Experimental (black lines) and simulated (red dashed lines) CV data [0.2 m (nBu4N)PF6 in THF] of 1L2 under Ar; mechanism and simulation parameters according to Scheme 5. Left: Concentration dependent data, ν = 0.1 V s–1. Right: Chloride dependent data, ν = 0.5 V s–1.
It is tempting to assume disproportionation of ReII to ReIII and ReI as bimolecular pathway. However, simple disproportionation, e.g., of [ReCl2(L2)]– to [ReCl(L2)]– and parent 1L2 after chloride loss from [ReCl3(L2)]– should lead to increasing overall currents at higher chloride concentrations, which is not in agreement with the data. In consequence, disproportionation requires the introduction of additional decay routes, e.g. at the ReI stage, which was renounced to avoid overparameterization of the model. However, disproportionation cannot be fully excluded.
The quality of the simulations is quite sensitive with respect to doubling or halving the decay rate constants k 2 or k 3, respectively. However, the two parameters are correlated: a higher bimolecular rate constant k 3 could be partially compensated by lower k 2 (and vice versa), yet with poorer resemblance of reversibility. For the rate and equilibrium constants of chloride loss (k 1, K 1), the fit proved highly sensitive with respect to variations.
Rapid N2‐activation (k > 5 × 107 M–1s–1) by anionic [ReIICl2(L1)]– was demonstrated as key step for N2 splitting with 1L1.[12g] Thus, the lifetime of the rhenium(II) intermediate predetermines the N2 splitting yield. In case of 1L2, the chloride dissociation preequilibrium (K 1) is followed by unimolecular decay (k 2) that is about an order of magnitude faster as compared with 1L1. In addition, a bimolecular decay pathway (k 3) prior to chloride loss may further reduce the lifetime of rhenium(II) species. Besides lowering the electrosynthetic yield, the bimolecular decay may also be detrimental for heterogeneous reductants (Na/Hg, KC8). There, high local surface concentrations of reduced species are expected as opposed to homogeneous reduction, e.g. with Co(Cp*)2, which gave the highest N2 splitting yields for 1L2.
Nitride Functionalization
The functionalization of the nitride complex 2L2 derived from N2 splitting was investigated. No reactivity was found with ONMe3, PMe3, or CO, indicating that the weaker donor properties of the pincer ligand do not open up pathways for potential nucleophiles/ambiphiles. However, in analogy to 2L1, 2L2 readily reacts with strong electrophiles, such as triflic acid and methyl triflate (Scheme 6). With triflic acid in Et2O, almost quantitative protonation of a vinyl group in the pincer backbone and formation of [Re(N)Cl(HL2)]OTf (5HL2‐OTf) is evidenced by the NMR signature, such as the two 31P{1H}‐NMR signals with typical trans coupling constant (2 J PP = 148 Hz). The same reactivity of L2 complexes with Brønsted acids was previously found for nickel(II), cobalt(II), and ruthenium(II) complexes.[15h], [15i][15k] Electrochemical examination of 5HL2‐OTf in THF revealed a reversible oxidation at E 1/2 = +0.24 V (Figure S23), yet no reductive process within the potential window of THF.
Scheme 6.

Reactivity of 2L2 towards electrophiles. Reaction with HOTf results in backbone protonation, whereas MeOTf leads to C–N bond formation by methylation at the nitride.
In contrast to protonation, treatment of 2L2 with MeOTf in chlorobenzene at elevated temperatures results in functionalization of the N2 derived nitride group (Scheme 6). The imido complex [Re(NMe)Cl(L2)]OTf (6L2‐OTf) with a single 31P{1H} signal at δ = 88.8 ppm is obtained. Nitride methylation by the electrophile was confirmed by 1H‐1H NOESY spectroscopy, which shows cross‐peaks of the N–CH 3 group at δ = 2.70 pm with one of the two tert‐butyl signals but not with pincer backbone protons (Figure S12).
Conclusions
The unsaturated PNP complex 1L2 provides the second example of reductive, electrochemically driven N2 splitting. In analogy to parent 2L1, Brønsted acid protonates the pincer backbone of N2‐derived nitride 2L2, yet at a distinctly different site. However, this product may serve as starting platform for nitrogen incorporation into organic molecules as demonstrated by nitride methylation with MeOTf. A strong dependence of the nitrogen splitting yield on the nature of chemical reductants (CoCp*2: 60 %, Na/Hg: 30 %, KC8: 20 %) or electrolysis (15 %) was found, which markedly differs from parent 1L1 (CoCp*2: 75 %, Na/Hg: 80 %, electrolysis: 60 %). The unproductive decomposition pathways that diminish the yield in 2L2 were not examined in detail. However, detailed comparison of electrochemical data for 1L2 vs. parent 1L1 allowed for identifying three key differences that provide a qualitative basis to rationalize the trends in rhenium mediated N2 splitting yields with different pincer ligands and reductants:
a) Unlike 1L1, the starting complex 1L2 exhibits slow decomposition in the presence of N2 and chloride ions. The decreased stability against chloride is partly attributed to decay via chloride‐induced disproportionation. The reduced chemical stability should affect electrosynthetic vs. chemical reduction yields which proceed on much slower timescales with concomitant free chloride buildup.
b) Weaker N→M π‐donation by pincer ligand L2 results in an anodic shift of the ReIII/II and ReII/I redox couples and a smaller separation of their potentials. This allows for electrochemically driven N2 splitting at more desired, less negative potentials. However, unproductive ReII/I reduction prior to N2 activation and additional L2 ligand fragmentation pathways via isobutene liberation might be more accessible with strong chemical reductants, such as Na/Hg or KC8, leading to decay due to over‐reduction.
c) In addition to the kinetic model proposed for 1L2, a rapid bimolecular decay pathway was found for the key rhenium(II) species [ReCl2(L2)]– that can compete with productive N2 activation. This pathway will be particularly detrimental for heterogeneous chemical (Na/Hg, KC8) and electrochemical reduction where high local ReII concentrations are expected.
This study exemplifies the subtle interplay of the underlying thermodynamics and kinetics of electron transfer processes and coupled chemical steps, respectively, as determining parameters for the yields in reductive N2 splitting. Future work will have to focus on the nature of the decay pathways to design improved platforms for (electro‐)chemical N2 fixation.
Experimental Section
Materials and Synthetic Methods
All experiments were carried out under inert conditions using standard Schlenk and glove‐box techniques under Ar or N2. HPLC grade solvents (Sigma Aldrich/Merck) were dried using an MBRAUN Solvent Purification System. THF was additionally dried with Na/K and chlorobenzene over CaH2. Deuterated solvents were bought from Euriso‐Top GmbH and dried with Na/K ([D8]THF) or 4 Å molecular sieves (C6D6). 15N2, Si(CH3)3N3, Co(Cp)2, Co(Cp*)2, hexamethylbenzene, P[OSi(CH3)3]3, PPh3O were used as purchased. HOTf and MeOTf were distilled prior to use. Na/Hg (1 m) was prepared from elemental Na and Hg. Fe(Cp)2 and Fe(Cp*)2 were sublimed and (nBu4N)PF6, (nBu4N)Cl, and (nHe4N)Cl dried before use. KC8 was synthesized by layering metallic potassium (332 mg, 8.49 mmol, 1.15 equiv.) with graphite (mesh 335, 711 mg, 59.2 mmol, 8 equiv.) and heating under vacuum, until full intercalation and observation of the characteristic bronze color. 2,4,6‐tri‐tert‐butylphenoxy radical, 1L1, and [ReCl3(L1)] were prepared according to published procedures.[12c], [12f], [12g], 27
Analytical Methods
Elemental analyses were obtained from the “Analytisches Labor” at University of Goettingen using an Elementar Vario EL 3 analyzer. NMR spectra were recorded on a Bruker Avance III 300, Avance III 400, or Avance 500 spectrometer with broadband cryoprobe and calibrated to the residual solvent signals (C6D6: δ 1H = 7.16 ppm, δ 13C = 128.4 ppm, [D8]THF: δ 1H = 3.58 ppm, δ 13C = 67.6 ppm, CD2Cl2: δ 1H = 5.32 ppm, δ 13C = 53.84 ppm). 31P NMR and 15N NMR chemical shifts are reported relative to external phosphoric acid and nitromethane standard (δ 31P = 0.0 ppm, δ 15N = 0.0 ppm), respectively. Signal multiplicities are abbreviated as: s (singlet), d (doublet), m (multiplet). UV/Vis absorption spectra were measured on a CARY300 Scan Varian spectrometer using inert sealed cuvettes. Liquid injection field desorption mass spectrommetry (LIFDI‐MS, JEOL AccuTOF JMS‐T100GCV) was measured at the “Zentrale Massenabteilung” at University of Goettingen. Electrochemical experiments were carried out with Metrohm PGSTAT101 (data under Ar) and GAMRY 600 reference (N2 data) potentiostats using standard software. CV was measured using glassy carbon (1.6 mm diameter) working and Pt wire counter electrodes and a Ag wire pseudo‐reference electrode in a fritted sample holder compartment and referenced against the [Fe(Cp)2]+/0 couple. CPE was performed using reticulated vitreous carbon as working electrode, Pt‐wire counter electrode in a fritted compartment with Fe(Cp*)2 as sacrificial reductant and a Ag‐wire as pseudo‐reference electrode in a fritted sample holder. For all electrochemical experiments, a 0.2 m (nBu4N)PF6 solution in THF was used as electrolyte, with appropriate iR compensation. High‐pressure CV was carried out in a reactor as described previously.[12g] Magnetic susceptibility measurements were performed with a Quantum Design MPMS‐XL‐5 SQUID magnetometer in the temperature range from 295–2 K at 0.5 T applied field. Powdered samples were contained in Teflon buckets and fixed in a non‐magnetic sample holder. Each raw data point was corrected for diamagnetic contribution of the bucket by subtraction of its experimentally derived magnetic moment. The molar susceptibility data were corrected for the diamagnetic contribution using the Pascal constants and the increment method according to Haberditzl.26 Experimental data were modelled with the julX program.27 The diffraction data were obtained at 100 K on a Bruker D8 three‐circle diffractometer, equipped with a PHOTON 100 CMOS detector and an INCOATEC microfocus source with Quazar mirror optics (Mo‐K α radiation, λ= 0.71073 Å).
https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/ejic.201901278 1832926 (for 2L2) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/structures.
Synthetic and Electrochemical Experiments
ReCl2(L2) (1L2): 1L1 (120 mg, 194 µmol, 1.0 equiv.) and the 2,4,6‐tri‐tert‐butylphenoxy radical (305 mg, 1.17 mmol, 6.0 equiv.) are mixed in benzene (15 mL) and stirred for 24 h at 50 °C. The solvent is removed in vacuo and the product is washed with excess pentane, until the washing solution is colourless. Co(Cp)2 (7 mg, 37 µmol, 0.2 equiv.) is added and the product is dissolved in benzene and stirred for 2 h at r.t. The reaction mixture is filtered, the benzene phase is lyophilized and remaining CoCp2 and 2,4,6‐tri‐tert‐butylphenol are sublimed off overnight at 75 °C. 1L2 is obtained as a brown powder in 63 % yield. Anal. Calcd for C20H40Cl2NP2Re (%): C, 39.15; H, 6.57; N, 2.28; found C, 38.81, H, 6.63; N, 2.14. NMR (C6D6, [ppm]): 1H (300 MHz): δ = 0.90 (d, 3 J HH = 6.4 Hz, 2H, PCH), 2.61 (A18XX'A'18, N = |3 J HP+5 J HP| = 6.2 Hz, 36H, P(C(CH3)3)), 3.65 (A2B2XX'B2'A2', N = |3 J HP+4 J HP| = 16.4 Hz, 3 J HH = 6.4 Hz, 2H, NCH). 13C (75.5 MHz): δ = 34.7 (A6XX'A'6, N = |2 J CP+4 J CP| = 2.5 Hz, P(C(CH3)3)), 77.4 (A2XX'A'2, N = |1 J CP+3 J CP| = 9.5 Hz, P(C(CH3)3)), 147.6 (AXX'A', N = |1 J CP+3 J CP| = 15.4 Hz, PCH), 212.4 (AXX'A', N = |2 J CP+3 J CP| = 7.9 Hz, NCH). 31P{1H} (121.5 MHz): δ = –275.6 (s). LIFDI‐MS (toluene, [m/z]): 613.1 (100 %, [M]+).
Re(N)Cl(L2) (2L2): N2 route. Degassed THF (0.45 mL) is vacuum‐transferred to a mixture of 1L2 (5.0 mg, 8.1 µmol, 1.0 equiv.) and reductant [Co(Cp*)2: 3.0 mg, 9.0 µmol, 1.1 eq; NaHg (1 m): 121.3 mg, 9.0 µmol, 1.1 eq or KC8: 1.1 mg, 8.1 µmol, 1.0 equiv.] in a J‐Young NMR tube and placed under an N2‐atmosphere. After thawing of the solvent, the mixture is shaken vigorously with gradual colour change from dark brown to light brown. After 30 min at r.t., the solvent is removed, hexamethylbenzene (1 eq via a 0.08 m stock solution in THF) is added and the solvent is removed again. Spectroscopic yields of the title compound are obtained by integration of the L2 ligand backbone 1H NMR signals vs. the internal standard C6Me6 [60 % for Co(Cp*)2; 30 % for Na/Hg; 20 % for KC8]. 2L2 was not isolated via this route.
Azide route. 1L2 (25.0 mg, 40.7 µmol, 1.0 equiv.) is dissolved in THF (1 mL) and added dropwise over a period of 5 min to a stirring solution of Me3SiN3 (26.78 µL, 23.5 mg, 203 µmol, 5.0 equiv.) in THF (0.5 mL). The solution is stirred at r.t. for 1.5 h after which the solvent is removed in vacuo. After extraction with pentane (4 × 5 mL) and removal of the solvent, 2L2 is obtained as a light brown solid in 72 % yield. Anal. Calcd. for C20H40ClN2P2Re (%): C, 40.57; H, 6.81; N, 4.73; found C, 40.66; H, 6.73; N, 5.01. NMR (C6D6, ppm): 1H (400 MHz): δ = 1.18 (A9XX'A'9, N = |3 J HP+5 J HP| = 7.2 Hz, 18H, P(C(CH3)3)), 1.49 (A9XX'A'9, N = |3 J HP+5 J HP| = 7.0 Hz, 18H, P(C(CH3)3)), 4.29 (A2B2XX'B2'A2', N = |2 J HP+4 J HP| = 2.2 Hz, 3 J HH = 6.3 Hz, 2H, PCH), 7.00 (A2B2XX'B2'A2', N = |3 J HP+5 J HP| = 17.1 Hz, 3 J HH = 6.2 Hz, 2H, NCH). 13C{1H} (125.76 MHz): δ = 28.5 (br, 6C, P(C(CH3)3)), 29.4 (br, 6C, P(C(CH3)3)), 34.9 (AXX'A', N = |1 J CP+3 J CP| = 10.3 Hz, 2C, P(C(CH3)3)), 36.7 (AXX'A', N = |1 J CP+3 J CP| = 11.8 Hz, 2C, P(C(CH3)3)), 91.8 (AXX'A', N = |1 J CP+3 J CP| = 20.9 Hz, 2C, PCH), 170.3 (AXX'A', N = |2 J CP+4 J CP| = 6.8 Hz, 2C, NCH). 31P{1H} (161.25 MHz): δ = 71.8 (s). LIFDI‐MS (toluene, [m/z]): 592.1 (100 %, [M]+).
ReCl3(L2) (4L2): ReCl3(L1) (15.3 mg, 0.024 mmol) and 2,4,6‐tri‐tert‐butylphenoxy radical (33.9 mg, 0.12 mmol, 5.4 equiv.) are combined in C6H6 (dried with Na/K). After heating at 60 °C for 1.5 h, the solvents are evaporated in vacuo. After extensive washing with pentane, and lyophilization (C6H6), 4L2 is obtained in 70 % yield (10.6 mg; 0.016µmol). Anal. Calcd. for C20H40Cl3NP2Re (%): C, 37.01; H, 6.21; N, 2.16; found C, 36.66; H, 6.29; N, 1.96. 1H NMR (300 MHz, C6D6): 15.2 ppm (s, Δv1/2 = 7.5 Hz), –51.7 (s, Δv1/2 = 14.8 Hz), –194.6 (s, Δv1/2 = 30.3 Hz). LIFDI‐MS (Toluene, [m/z]): 648.1 (100 %, [M]+), calculated 648.1.
[ReNCl(HL2)]OTf (5HL2‐OTf): 2L2 (5.0 mg, 8.4 µmol, 1.0 equiv.) is dissolved in Et2O (1 mL) and HOTf (0.74 µL, 8.4 µmol, 1 equiv.) is added via an Eppendorf pipette. Upon stirring for 1 h, a red‐brownish precipitate forms which is collected by filtration, washed with pentanes (3 × 2 mL) and dried in vacuo to give 5HL2‐OTf in 82 % yield. NMR (CD2Cl2, ppm) 1H (500 MHz): δ = 1.28 (d, 3 J HP = 14.8 Hz, 9H, CH3), 1.32 (d, 3 J HP = 14.7 Hz, 9H, CH3), 1.57 (d, 3 J HP = 15.9 Hz, 9H, CH3), 1.60 (d, 3 J HP = 15.6 Hz, 9H, CH3), 3.79 (dd, 2 J HH = 21.2 Hz, 2 J HP = 7.7 Hz, 1H, P‐CH2‐CH), 4.40 (dd, 2 J HH = 21.2 Hz, 2 J HP = 7.4 Hz, 1H, P‐CH2‐CH), 6.66 (d, 3 J HH = 6.7 Hz, 1H, P‐CH), 8.02 (dd, 3 J HP = 27.8 Hz, 3 J HH = 6.6 Hz, 1H, N‐CH=CH), 9.35 (d, 3 J HP = 20.9 Hz, 1H, N=CH‐CH2). 13C{1H} (125.7 MHz): δ = 28.6 (d, 2 J CP = 3.5 Hz, CH3), 28.7 (d, 2 J CP = 4.1 Hz, CH3), 29.0 (d, 2 J CP = 3.9 Hz, CH3), 29.1 (d, 2 J CP = 3.3 Hz, CH3), 36.0 (d, 1 J CP = 18.1 Hz, C(CH3)3), 36.3 (d, 1 J CP = 15.1 Hz, C(CH3)3), 37.8 (dd, 1 J CP = 15.5 Hz, 3 J CP = 3.9 Hz, C(CH3)3), 38.2 (dd, 1 J CP = 18.7 Hz, 3 J CP = 3.7 Hz, C(CH3)3), 41.0 (d, 1 J CP = 22.5 Hz, CH2‐P), 127.9 (d, 1 J CP = 31.9 Hz, CH‐P), 163.9 (d, 2 J CP = 3.8 Hz, N‐CH=CH), 201.2 (s, N=CH‐CH2). 31P{1H} (121.5 MHz): δ = 70.0 (d, 2 J PP = 148.1 Hz), 73.0 (d, 2 J PP = 148.1 Hz).
[Re(NMe)Cl(L2)]OTf (6L2‐OTf): 2L2 (25.0 mg, 44.2 µmol, 1.0 equiv.) and MeOTf (5.26 µL, 46.4 µmol, 1.1 equiv.) are dissolved in chlorobenzene and heated to 80 °C for 12 h. After removal of all volatiles in vacuo, the product is washed with Et2O and 6L2‐OTf is obtained as brown solid in 80.5 % yield. Anal. Calcd. (%): C, 34.94; H, 5.71; N, 3.70; found C, 35.18; H, 5.71; N, 3.49. NMR (CD2Cl2, ppm): 1H (500 MHz): δ = 1.29 (A9XX'A'9, N = |3 J HP + 5 J HP| = 6.6 Hz, 18H, PC(CH3)3), 1.49 (A9XX'A'9, N = |3 J HP + 5 J HP| = 7.8 Hz, 18H, PC(CH3)3), 2.70 (sbr, 3H, NCH3), 5.34 (m, 2H, PCH), 7.99 (A2B2XX'B'2A'2, N = |3 J HP + 4 J HP| = 18.2 Hz, 3 J HH = 6.5 Hz, 2H, NCH). 13C{1H} (125.7 MHz): δ = 28.9 (s, PC(CH3)3), 30.2 (A3XX'A'3, N = |2 J CP + 4 J CP| = 2.0 Hz, PC(CH3)3), 39.6 (AXX'A', N = |1 J CP + 3 J CP| = 11.6 Hz, PC(CH3)3), 40.2 (AXX'A', N = |1 J CP + 3 J CP| = 9.7 Hz, PC(CH3)3), 61.9 (s, NCH3), 99.1 (AXX'A', 1 J CP = 22.6 Hz, 3 J CP = 20.4 Hz, PCH), 172.9 (AXX'A', N = |2 J CP + 3 J CP| = 5.3 Hz, NCH). 31P{1H}: (202.4 MHz) δ = 88.8 (s). LIFDI (toluene, m/z) = 607.2 (100 %, [M+]).
Chemical stability tests of 1L2: 1L2 (3.0 mg; 5.0 µmol) was dissolved in THF (0.6 mL) in a J‐Young tube under Argon and the stability was monitored by NMR spectroscopy. The sample was degassed by three freeze‐pump‐thaw cycles and backfilled with N2 and the stability was again monitored by NMR spectroscopy over time. To examine the stability in the presence of chloride, a sample with added (nBu4N)Cl (6.5 mg; 23.5 µmol; 5 equiv.) was monitored by NMR spectroscopy. NMR spectra are depicted as Figure S15 and Figure S17.
Controlled potential electrolysis: 1L2 (2.6 mg, 4.2 µmol) and 4 mL of 0.2 m (nBu4N)PF6 electrolyte solution in THF was added to the working electrode compartment of the electrolysis cell. The solution was electrolyzed for 2 h at the peak potential of the first reduction feature obtained by CV, resulting in a colour change from light brown to green. Integration of the current vs. time plot gave a charge corresponding to 1.2 mol e– per mol Re. The solvent was evaporated to give a light green solid, which was dissolved in 0.6 mL of THF. PPh3O (3.2 mg, 11.5 µmol) was added as internal standard, and the yield in Re(N)Cl(L2) (2L2) (17 %) was derived by 31P{1H} NMR spectroscopically in C6D6, see Figure S8.
Chloride concentration dependent CV under Ar: 1L2 (2.5 mg, 4.0 µmol) was dissolved in a 0.2 m solution of (nBu4N)PF6 in THF (4 mL) and a small amount of Fe(Cp*)2 was added as an electrochemical reference. In sequence, equivalents of (nBu4N)Cl (1.1 mg, 1 equiv.; 1.1 mg, 2 eq total; 3.3 mg, 5 eq total; 5.5 mg, 10 equiv. total; 11.1 mg, 20 equiv. total) were added. After each chloride addition, CV's were recorded quickly at 0.5, 1, 2, 3, 4, and 5 V s–1 only under Ar, before 1L2 shows substantial decomposition (see Figure 2).
Rhenium concentration dependent CV under Ar: A stock solution of 1L2 was prepared by dissolving 1L2 (15.3 mg, 25 µmol) in a 1.0 mL of solution of 0.2 m (nBu4N)PF6 in THF. Aliquots of this stock solution were added to a 5 mL of solution of 0.2 m (nBu4N)PF6 in THF, with a spatula tip of Fe(Cp)2 as an electrochemical reference, to afford solutions of 0.5, 1.0, 2.0, 3.0 and 4.0 mm 1L2. CVs for both the first two reduction features were recorded at 0.1 V s–1 (see Figure S20).
N2‐pressure dependent CV: 1L2 (2.5 mg, 4.0 µmol) was dissolved in a 0.2 m solution of (nBu4N)PF6 in THF (4 mL) and a small amount of Fe(Cp*)2 was added as an electrochemical reference. The solution was transferred to the Parr reactor and subsequently pressurized with N2 to obtain CVs at 1, 3, 5, 7, 9, 11 bars. At 11 bars, the system was allowed to stay for 45 minutes while regular CVs were measured (see Figures S20). After depressurzing, the reactor was transferred back in the glovebox and the reaction mixture was analysed by 31P{1H} NMR spectroscopy (see Figure S16).
Supporting information
Supporting Information
Acknowledgements
This work was supported by the European Research Council (ERC Consolidator Grant Agreement 646747, grant holder S.Sch.). Dr. J. Abbenseth and J. C. Becker are acknowledged for supporting synthetic work and Dr. E. Yuzik‐Klimova for recording high‐pressure CV data. A.J.M.M. acknowledges support from the National Science Foundation Chemical Catalysis program under Grant No. CHE‐1665135
Contributor Information
Inke Siewert, Email: inke.siewert@chemie.uni-goettingen.de, https://siewertlab.de/.
Sven Schneider, Email: sven.schneider@chemie.uni-goettingen.de, https://www.uni-goettingen.de/en/356646.html.
References
- 1. Patil B. S., Hessel V., Seefeldt L. C., Dean D. R., Hoffman B. M., Cook B. J. and Murray L. J., Nitrogen Fixation. In Ullmann's Encyclopedia of Industrial Chemistry; Wiley; 2017. [Google Scholar]
- 2.a) Foster S. L., Bakovic S. I. Perey, Duda R. D., Maheshwari S., Milton R. D., Minteer S. D., Janik M. J., Renner J. N. and Greenlee L. F., Nat. Catal, 2018, 1, 490–500; [Google Scholar]; b) Yan Z., Ji M., Xia J. and Yhu H., Adv. Energy Mater, 2019, 1902020. [Google Scholar]
- 3. Tan L., Yang N., Huang X., Peng L., Tong C., Deng M., Tang X., Li L., L Q. and Wei Z., Chem. Comm. 2019, Advance Article. DOI: 10.1039/C9CC06132K. [DOI] [PubMed] [Google Scholar]
- 4. McPherson I. J., Sudmeier T., Fellowes J. and Tsang S. C. E., Dalton Trans, 2019, 48, 1562–1568. [DOI] [PubMed] [Google Scholar]
- 5. Ostermann N. and Siewert I., Curr. Opin. Electrochem, 2019, 15, 97–101. [Google Scholar]
- 6.a) Chalkey M. J., Del Castillo T. J., Matson B. D. and Peters J. C., J. Am. Chem. Soc, 2018, 140, 6122–6129; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sherbow T. J., Thompson E. J., Arnold A., Sayler R. I., Britt R. D. and Berben L. A., Chem. Eur. J. 2019, 25K, 454–458. [DOI] [PubMed] [Google Scholar]
- 7. Schrock R. R., Angew. Chem. Int. Ed, 2008, 47, 5512–5522; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2008, 120, 5594. [Google Scholar]
- 8. Hoffman B. M., Lukoyanov D., Yang Z.‐Y., Dean D. R. and Seefeldt L. C., Chem. Rev, 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Arashiba K., Eizawa A., Tanaka H., Nakajima K., Yoshizawa K. and Nishibayashi Y., Bull. Chem. Soc. Jpn, 2017, 90, 1111–1118; [Google Scholar]; b) Ashida Y., Arashiba K., Nakajima K. and Nishibayashi Y., Nature, 2019, 568, 536–540. [DOI] [PubMed] [Google Scholar]
- 10. Laplaza C. E. and Cummins C. C., Science, 1995, 268, 861–863. [DOI] [PubMed] [Google Scholar]
- 11. Klopsch I., Yuzik‐Klimova E. Y. and Schneider S., Top. Organomet. Chem, 2017, 60, 71–112. [Google Scholar]
- 12.a) Hebden T. J., Schrock R. R., Takase M. K. and Müller P., Chem. Commun, 2012, 48, 1851–1853; [DOI] [PubMed] [Google Scholar]; b) Klopsch I., Finger M., Würtele C., Milde B., Werz D. B. and Schneider S., J. Am. Chem. Soc, 2014, 136, 6881–6883; [DOI] [PubMed] [Google Scholar]; c) Klopsch I., Kinauer M., Finger M., Würtele C. and Schneider S., Angew. Chem. Int. Ed, 2016, 55, 4786–4789; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2016, 128, 4864; [Google Scholar]; d) Liao Q., Cavaille A., Saffon‐Merceron N. and Mézailles N., Angew. Chem. Int. Ed, 2016, 55, 11212–11216; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2016, 128, 11378; [Google Scholar]; e) Silantyev G. A., Förster M., Schluschaß B., Abbenseth J., Würtele C., Volkmann C., Holthausen M. C. and Schneider S., Angew. Chem. Int. Ed, 2017, 56, 5872–5876; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2017, 129, 5966; [Google Scholar]; f) Klopsch I., Schenzielorz F., Volkmann C., Würtele C. and Schneider S., Z. Anorg. Allg. Chem, 2018, 644, 916–919; [Google Scholar]; g) Lindley B. M., van Alten R. S., Finger M., Schenzielorz F., Würtele C., Miller A. J. M., Siewert I. and Schneider S., J. Am. Chem. Soc, 2018, 140, 7922–7935; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Schendzielorz F., Finger M., Abbenseth J., Würtele C., Krewald V. and Schneider S., Angew. Chem. Int. Ed, 2019, 58, 830–834; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2019, 131, 840; [Google Scholar]; i) Schluschaß B., Abbenseth J., Demeshko S., Finger M., Franke A., Herwig C., Würtele C., Ivanovic‐Burmazovic I., Limberg C., Telser J. and Schneider S., Chem. Sci, 2019, 10, 10275–10282; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Bruch Q. J., Connor G. P., Chen C.‐H., Holland P. L., Mayer J. M., Hasanayn F. and Miller A. J. M., J. Am. Chem. Soc. 2019, DOI: 10.1021/jacs.9b10031. [DOI] [PubMed] [Google Scholar]
- 13. Katayama A., Ohta T., Wasada‐Tsutsui Y., Inomata T., Ozawa T., Ogura T. and Masuda H., Angew. Chem. Int. Ed, 2019, 58, 11279–11284; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2019, 131, 11401–11406. [Google Scholar]
- 14. Askevold B., Meiners J. and Schneider S., Eur. J. Inorg. Chem, 2012, 412–429. [Google Scholar]
- 15.a) Meiners J., Scheibel M., Lemée‐Cailleau M.‐H., Mason S. A., Boeddinghaus M. B., Fässler T. F., Herdtweck E., Khusniyarov M. M. and Schneider S., Angew. Chem. Int. Ed, 2011, 50, 8184–8187; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2011, 123, 8334; [Google Scholar]; b) Scheibel M., Askevold B., Heinemann F., Reijerse E. I., de Bruin B. and Schneider S., Nat. Chem, 2012, 4, 552–558; [DOI] [PubMed] [Google Scholar]; c) Scheibel M. G., Wu Y., Stückl A. C., Krause L., Carl E., Stalke D., de Bruin B. and Schneider S., J. Am. Chem. Soc, 2013, 135, 17719–17722; [DOI] [PubMed] [Google Scholar]; d) Kinauer M., Scheibel M., Abbenseth J., Heinemann F. W., Stollberg P., Würtele C. and Schneider S., Dalton Trans, 2014, 43, 4506–4513; [DOI] [PubMed] [Google Scholar]; e) Askevold B., Khusniyarov M. M., Kroener W., Gieb K., Müller P., Herdtweck E., Heinemann F. W., Diefenbach M., Holthausen M. C., Vieru V., Chibotaru L. F. and Schneider S., Chem. Eur. J, 2015, 21, 579–589; [DOI] [PubMed] [Google Scholar]; f) Scheibel M. G., Abbenseth J., Kinauer M., Heinemann F. W., Würtele C., de Bruin B. and Schneider S., Inorg. Chem, 2015, 54, 9290–9302; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Abbenseth J., Finger M., Würtele C., Kasanmascheff M. and Schneider S., Inorg. Chem. Front, 2016, 3, 469–477; [Google Scholar]; h) Lagaditis P. O., Schluschaß B., Demeshko S., Würtele C. and Schneider S., Inorg. Chem, 2016, 55, 4529–4536; [DOI] [PubMed] [Google Scholar]; i) Schneck F., Finger M., Tromp M. and Schneider S., Chem. Eur. J, 2017, 23, 33–37; [DOI] [PubMed] [Google Scholar]; j) Abbenseth J., Diefenbach M., Bete S. C., Würtele C., Volkmann C., Demeshko S., Holthausen M. C. and Schneider S., Chem. Commun, 2017, 53, 5511–5514; [DOI] [PubMed] [Google Scholar]; k) Bruch Q., Lindley B. M., Askevold B., Schneider S. and Miller A. J. M., Inorg. Chem, 2018, 57, 1964–1975. [DOI] [PubMed] [Google Scholar]
- 16. Khusnutdinova J. R. and Milstein D., Angew. Chem. Int. Ed, 2015, 54, 12236–12273; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2015, 127, 12406. [Google Scholar]
- 17. Chatt J., Leigh G. J. and Mingos D. M. P., J. Chem. Soc. A, 1969, 1674–1680. [Google Scholar]
- 18. Soncinia A. and Van der Heuvel W., J. Chem. Phys, 2013, 138, 021103. [DOI] [PubMed] [Google Scholar]
- 19.a) Figg T. M., Holland P. L. and Cundari T. R., Inorg. Chem, 2012, 51, 7546–7550; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) MacLeod K. V., Menges F. S., McWilliams S. F., Craig S. M., Mercado B. Q., Johnson M. A. and Holland P. L., J. Am. Chem. Soc, 2016, 138, 11185–11191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Addison A. W., Rao T. N., Reedijk J., van Rijn J. and Verschoor G. C. J., J. Chem. Soc., Dalton Trans, 1984, 1349–1356. [Google Scholar]
- 21. Connor G. P., Mercado B. Q., Lant H. M. C., Meyer J. M. and Holland P. L., Inorg. Chem, 2019, 58, 10791–10801. [DOI] [PubMed] [Google Scholar]
- 22.All potentials in this article are internally referenced to the ferrocenium/ferrocene reduction potential, Fc+/0.
- 23. Connelly N. G. and Geiger W. E., Chem. Rev, 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 24. Ritleng V., Yandulov D. V., Weare W. W., Schrock R. R., Hock A. S. and Davis W. M., J. Am. Chem. Soc, 2004, 126, 6150–6163. [DOI] [PubMed] [Google Scholar]
- 25. Manner V. W., Markle T. F., Freudenthal J. H., Roth J. P. and Mayer J. M., Chem. Commun, 2008, 256–258. [DOI] [PubMed] [Google Scholar]
- 26. Haberditzl W., Angew. Chem. Int. Ed. Engl, 1966, 5, 288–298; [Google Scholar]; Angew. Chem, 1966, 78, 277. [Google Scholar]
- 27. Bill E., “julX, Program for Simulation of Molecular Magnetic Data”, 2008.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information