

Abstract
While the recently developed antiandrogen Enzalutamide (Enz) can extend survival for 4.8 months in castration-resistant prostate cancer (CRPC) patients, eventually most of these CRPC patients may develop resistance to the Enz without a clear mechanism. Here we found the expression of Beclin 1 was decreased in both Enz-resistant (EnzR) cell lines (EnzR1-C4-2 and EnzR2-C4-2B) as compared to their parental Enz-sensitive (EnzS) (EnzS1-C4-2 and EnzS2-C4-2B) cells, and targeting the Beclin 1 could lead to increase the Enz-sensitivity in these two CRPC cell lines. Mechanism dissection revealed that Enz might function via altering the interaction between Beclin 1 and the androgen receptor (AR) to decrease the activity of Beclin 1-Vps15-Vps34 complex thus increasing the ERK-mediated growth factor signaling to alter the Enz sensitivity. Interrupting the AR-Beclin 1/ERK signaling with ectopic BECN1 or ERK inhibitor led to alter the Enz sensitivity in both EnzR1/S1-C4-2 and EnzR2/S2-C4-2B cells. Together, these results suggest that targeting this newly identified AR-Beclin 1 complex-mediated ERK growth factor signaling with small molecule inhibitor may help potentially develop new therapies to better suppress the EnzR CRPC.
Keywords: Prostate cancer, Beclin 1 (BECN1), enzalutamide, growth factor signaling
1. Introduction
Prostate cancer (PCa) is the most commonly diagnosed cancer in males in the western world and causes over 80 deaths in America per day [34]. Although the androgen-deprivation therapy (ADT) with the recently developed antiandrogen Enzalutamide (Enz) could extend patients survival an extra 4.8 months [6, 31, 38], most of these patients still succumb to the disease following development of Enz-resistance.
Recently, accumulating evidence has suggested that the androgen receptor (AR) splice variants, such as AR-v7, that lacks the androgen-binding domain, may play critical roles for the development of Enz-resistance [3, 20, 22, 25, 26]. However, results from human clinical sample surveys [23, 32] and in vitro cell lines [35, 37] indicated other non-AR-v7 mechanisms may also play roles in Enz-resistance. In particular, enhanced growth factor signals, including AKT activation, might also promote the development of Enz-resistance [18, 27, 37]. Indeed AKT signaling is found to be constitutively activated after Enz treatment [14, 17, 37]. Results from Brett S. Carver et al. [8] indicated that AR could negatively regulate AKT activation through transcriptionally increasing PHLPP expression, and suppressing the AR via Enz could lead to reduced PHLPP expression, thus enhanced AKT activation. Separately, beside the function in regulating macro-autophagy, Rohatgia et al. [30] found suppressing Beclin 1 led to sustain the growth factor-stimulated activation of AKT and ERK signals via altering the PI3P (+) lipid levels, thus likely contributing to breast cancer cell progression [8].
Here, we found Enz might function via promoting AR interaction with the Beclin 1-Vps15-Vps34 complex to suppress the latter’s activity thus enhanced ERK signaling. Targeting this newly identified AR-Beclin 1 complex-mediated ERK growth factor signaling with ectopic BECN1 expression or ERK inhibitor could lead to increase Enz efficacy.
2. Materials and Methods
2.1. Cell lines, inhibitors and antibodies.
EnzR1-C4-2 cells were generated by culturing EnzS1-C4-2 cells under increasing Enz concentrations from 10 μM to 40 μM (every 20 days) for 3 months. The EnzS2-C4-2B and EnzR2-C4-2B resistant (EnzR2-C4-2B) cells were a gift from Dr. Allen Gao (University of California, Davis, CA, USA). Both EnzR1-C4-2 and EnzR2-C4-2B cells were maintained in RPMI media supplied with 10 μM and 20 μM Enz, respectively. Antibodies used for immunoblotting including GAPDH (sc-47724), AR (sc-816), ATG5 (sc-133158), ERK 1/2 (sc-514302), EGFR (sc-71034), VPS34 (sc-365404), cleaved PARP (h215), BECN1 (sc-48341) and IGF-1Rβ (sc-9038) were purchased from Santa Cruz Biotechnology. BECN1 (cst-3495), AKT (cst-9272), p-AKT-T308 (cst-13038) and p-AKT-S473 (cst-4060) antibodies were purchased from Cell Signaling Technology (USA). VPS15 (17894-1-AP) antibody was purchased from Proteintech company (USA). ERK inhibitor was purchased from Cayman (1049738-54-6, USA).
2.2. Plasmids and lentivirus packaging.
The pLKO.1-ShBECN1#1, pLKO.1-ShBECN1#2, pLKO.1-ShAR, pLVTHM-ShBECN1, pLVTHM-Sh-ATG5, pWPI-oeBECN1 and pWPI-oeAR, the pMD2G envelope and psAX2 packaging plasmids were transfected into 293T cells, respectively, using calcium-chloride transfection method for 48 h to generate the supernatant virus. Details of the plasmid sequencing were described in Table S1. For pLKO.1 vector-infected cells, puromycin (1 μg/mL) was used to select stable expression.
2.3. Growth factor stimulation assays and western blotting (WB).
Cells were cultured in serum-starved media overnight comprising 0.1% BSA and then stimulated with IGF-1 (100 ng/mL) or EGF (50 ng/mL) for the time periods indicated. To inhibit internalization, cells were incubated with chlorpromazine (10 μg/ml) for 1 h befoe the stimulation. Cells were infected with lentiviruses containing pLKO.1-shBECN1 or pWPI-oeBECN1 for knocking down and rescue experiments and stimulated after 48 h infection.
We used cell lysis buffer (10 mM Tris-HCl/pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.1% Nonidet P40, 1 mM DTT and 1 mM PMSF) to extracted the protein for each sample, which were then diluted and boiled with 5 x loading buffer for 10 mins. After that, these proteins were separated and run on 10% SDS/PAGE gel, and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The membranes will be blocked for 1 h in 5% milk, then they were incubated with specific primary antibodies overnight. Next day, the blots will be incubated with HRP-conjugated secondary antibodies (mouse or rabit, depended on primary antibodies), and then visualized using ECL system (Thermo Fisher Scientific, Rochester, NY, USA).
In addition, PI3P (+)levels were determined by using the PI3P Mass ELISA Kit (Catalog # K-3300, Echelon Biosciences, USA) according to the manufacturer's instructions.
2.4. MTT, Trypan blue and BrDU assays for determining cell proliferation.
For MTT assay, cell growth curve was determined by applying an MTT proliferation assay. Briefly, 2,000-5,000 cells were seeded on several 24-well plates containing 600 μL RPMI media. Cell growth was tested at the indicated time points. After Enz treatment, 50 μL of MTT reagent (Amresco Inc., Solon, OH, USA) was added to the medium and then the cells were incubated at the incubator for a further 2 h at 37°C. After that, the media was removed and 1 mL of dissolving reagent DMSO (Amresco Inc., USA) was added to dissolve the formazan crystals. The optical density value (OD) was determined at wavelength of 570 nm on a microplate reader. For Trypan blue staining assay, 200,000 cells were seeded on several 6-well plates containing 2 mL RPMI medium, after 5 and 6 days culturing (with Enz treatment), the cell number was determined by Trypan blue assay. Briefly, we resuspended the cells with 1 mL RPMI after spin, then got 50 μL cell suspension mixed with 50 μL Trypan blue solution. And then 20 μL mix was examined using automated cell counting slides (Bio-Rad, USA). For BrDU (BrdU; Sigma-Aldrich, St. Louis, MO, USA) staining assay, the detail steps were referring to BrdU staining and BrdU assay protocol on the official website of Abcam (USA) (https://www.abcam.com/protocols/brdu-staining-protocol). The staining was examined by fluorescence microscopic analysis (Olympus, Japan).
2.5. Co-Immunoprecipitation (CO-IP).
Confluent cells on 150-mm plates were harvested and solubilized in lysis buffer. Insoluble material was removed by centrifugation. The supernatants were incubated overnight at 4°C with 2 mg of anti-IgG, BECN1 or AR antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Following the addition of 10 μl Protein-A/G–agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mixtures were incubated for 2 h at 4°C with rotation. Immune complexes were washed 15 times with regular lysis buffer; the agarose beads were then boiled for 10 mins with loading buffer. Immunoprecipitates were run on 8% SDS/PAGE gel, followed by western blot assay with antibodies against AR, BECN1, Vps15, and Vps34.
2.6. Statistical Analysis.
Statistical analyses between two groups were performed using the two-tailed unpaired Student's t-test. Data are presented as mean ± standard error (SEM) unless mentioned otherwise. A P-value of 0.05 was considered statistically significant.
3. Results
3.1. Enzalutamide Regulates Growth Factor Signaling
Enzalutamide (Enz) may extend the survival of CRPC patients an extra 4.8 months before the development of Enz-resistance [7]. The underlying mechanisms, especially its linkage to the growth factor signals, however, remain unclear. To investigate whether Enz-resistance was partly due to dysregulation of growth factor signals, we focused on the insulin-like growth factor-1 receptor (IGF-1R) and the epidermal growth factor receptor (EGFR), as their higher expression have been frequently observed in PCa and their activations are often correlated with poor prognosis of PCa (Fig. S1A-D) [1, 12, 43].
We compared the EnzR1-C4-2 cells with their parental Enz-sensitive C4-2 cells (EnzS1-C4-2) for the activation of growth factor signals [10, 42], via assaying the phosphorylation of key downstream signaling effectors of the IGF-1R and EGFR, including serine/threonine kinases AKT and ERK, after treating with either EGF or IGF (Fig. 1 and Fig. S2), which could be used to activate these two signals. Before that, the basal phosphorylation levels of AKT and ERK were measured and described in Fig. S1E.
Figure 1. Enzalutamide enhances growth factor signals.
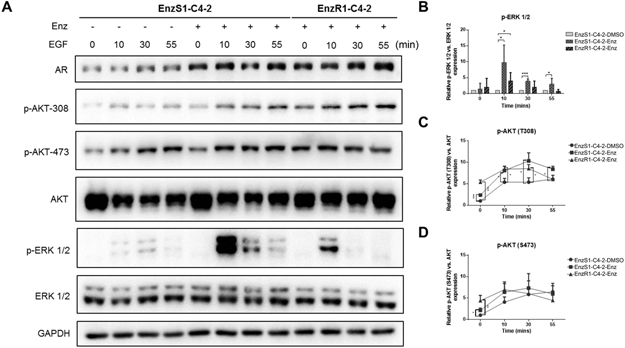
A. EnzS1-C4-2 cells were pre-treated with Enz for three days, and serum-starved overnight in media containing 0.1% BSA, then stimulated with 50ng/ml human EGF for the indicated time periods. P-AKT-S473, p-AKT-T308 and p-ERK1/2 (T202/Y204) levels were detected by western blot. B-D. Quantification results of p-ERK 1/2, p-AKT-S473, and p-AKT-T308. The data shown represent the mean ± SEM of three independent experiments. *p < 0.05, ***p < 0.001.
The results revealed that AKT and ERK were phosphorylated in a transient manner in response to EGF stimulation after pretreated with Enz for three days in EnzS1-C4-2 and EnzR1-C4-2 cells (Fig. 1A). We found the basal phosphorylation levels of AKT (S473 and T308) were increased after pretreated with 10 μM Enz for three days. After EGF stimulation, the phosphorylation levels of AKT (S473 and T308) could still be increased in Enz-treated and Enz-resistant groups compared to control group, but could not be sustained for an extended time, while the phosphorylation level of ERK was significantly increased and also sustained in response to EGF stimulation (quantified in Fig. 1B-D). Meanwhile, IGF was also used to stimulate the signaling cascade, and similar results for the AKT signal were found, but not ERK signal (Fig. S2A, quantified in Fig. S2B-C).
We then replaced the EnzS1-C4-2 cells with EnzS2-C4-2B cells, and used EGF to activate the growth factor signaling. We found the EGF stimulation could further increase the enhanced phosphorylation level of AKT (308T) by Enz, but still could not sustain this signal (Fig. S2D, quantified in Fig. S2E-F), while the ERK signal could be both increased and sustained in response to EGF after pretreated with Enz for three days, consistent with the earlier findings (Fig. S2D, quantified in Fig. S2E-F).
Although IGF and EGF both could activate AKT signal, only EGF could increase and sustained the ERK signal, and these results were consistent with previous findings that the ERK phosphorylation was more significant downstream of EGFR, while AKT phosphorylation were more significant downstream of the IGF-1R [30]. Together, these data suggest that Enz may increase the AKT and ERK growth factor signals, and Enz-resistance may be associated with enhanced growth factor signals in the PCa cells.
3.2. Beclin 1 may function via suppressing the ERK growth factor signaling to alter the Enz-resistance in PCa cells
Next, to dissect the molecular mechanism of how Enz alters these growth factor signals, we focused on the role of Beclin 1, as recent studies indicated that Beclin 1 complex might function via regulating cellular macro-autophagy to control multiple growth factor signals [28, 30, 39]. For example, Beclin 1 complex may promote the conversion of PI3P to PI4P to terminate the signaling capacity of endosome-bound growth factor receptors to reduce intracellular signaling transmission [28], thus loss of Beclin 1 could sustain growth factor stimulated activation of AKT and ERK signals resulting in cancer progression [30].
We first silenced the BECN1 expression via stably infecting BECN1-shRNAs (shBECN1#1 & shBECN1#2) in the EnzS1-C4-2 cells (Fig. 2A), and results revealed that the phosphorylation levels of AKT (both T308 and S473) and ERK were significantly increased and sustained longer in the shBECN1-EnzS1-C4-2 cells compared with vector control in response to EGF stimulation (Fig. 2B, and quantification in Fig. 2C-E). However, ectopic Beclin1 expression (Fig. 2A) could only reverse the Enz-enhanced ERK growth factor signal rather than the AKT signal in response to EGF stimulation after pretreatment with Enz for three days (Fig. 2F, and quantification in Fig. 2G), suggesting that Beclin 1 loss could specifically regulate ERK growth signal to influence Enz-sensitivity.
Figure 2. Beclin 1 regulates ERK-growth factor signaling.
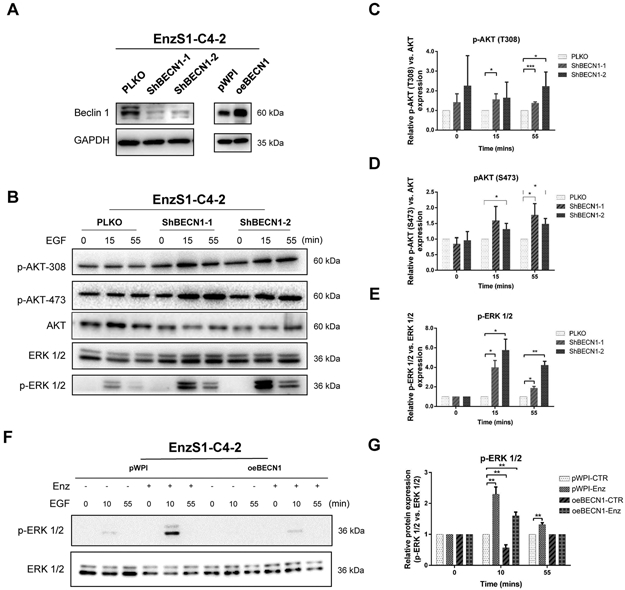
A. Knock down and overexpression BECN1 efficiencies in EnzS1-C4-2 cells. B. EnzS1-C4-2 cells were infected with BECN1 shRNAs (ShBECN1-1/2), serum-starved overnight in media containing 0.1% BSA, then stimulated with 50 ng/ml human EGFfor the indicated time periods. P-AKT-S473, p-AKT-T308 and p-ERK1/2 (T202/Y204) levels were detected by western blot. C-E. Quantification data for p-AKT (S473), p-AKT (T308) and p-ERK1/2 (T202/Y204). F. Ectopic BECN1 expression could reverse Enz enhanced ERK signaling, but not AKT signaling in EnzS1-C4-2 cells (The expression of Beclin 1 in EnzS1-C4-2 cells were slightly knocked down by lenti-virus, then treated them as the parental cells for viral overexpression of BECN1). We pretreated with Enz for three days, and serum-starved cells overnight in media containing 0.1% BSA, then stimulated with 50 ng/ml human EGF for the indicated time periods. G. Quantification data for p-ERK 1/2. The data shown represent the mean ± SEM of three independent experiments.*p < 0.05, **p < 0.01, ***p < 0.001.
We also found that Beclin 1 expression was significantly lower in EnzR1-C4-2 and EnzR2-C4-2B cells compared with EnzS1-C4-2 and EnzS2-C4-2, respectively (Fig. 3A), and Enz-sensitivity was significantly decreased after silencing BECN1 expression in EnzS1-C4-2 and EnzS2-C4-2B cells (Fig. 3B-D). In addition, interruption approaches using ERK inhibitor (ERKI) treatment also demonstrated that inhibition of ERK activation could partially rescue the Enz-sensitivity in the shBECN1-EnzS1-C4-2 parental cells (Fig. 3E), as well as EnzR1-C4-2/EnzR2-C4-2B cells (Fig. S3A-B). Furthermore, we ectopically expressed BECN1 in EnzS1/R1-C4-2 and EnzR2-C4-2B cells, and found Enz-sensitivity was significantly increased (Fig. 3F-G and Fig. S3C).
Figure 3. Beclin 1 regulates Enzalutamide sensitivity.
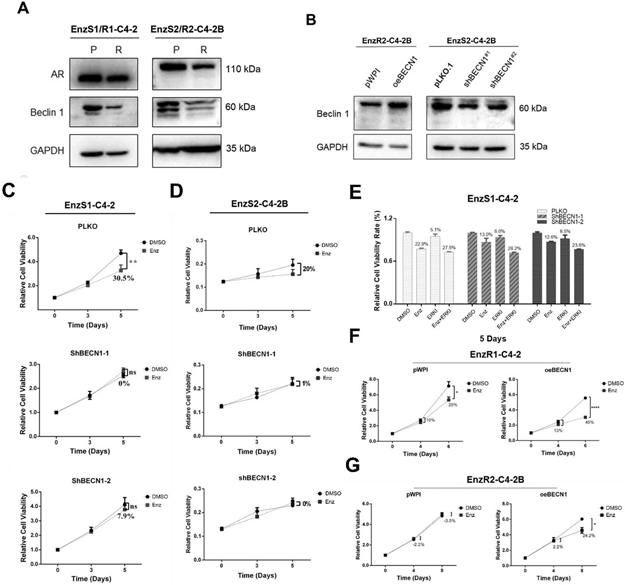
A. Comparing Beclin 1 expression between EnzS1-C4-2 & EnzS2-C4-2B and EnzR1-C4-2 & EnzR2-C4-2B cells (P, parental; R, resistant). B. Knock down and overexpression of BECN1 efficiencies in EnzS2-C4-2B and EnzR2-C4-2B cells, respectively. C-D. MTT assay demonstrated knocking down BECN1 expression (with 2 Sh-BECN1s) decreased Enz sensitivity in EnzS1-C4-2 (C) and EnzS2-C4-2B (D) cells. E. MTT assay demonstrated that combining Enz with ERK inhibitor treatment could partially reverse ShBECN1 decreased Enz-sensitivity in EnzS1-C4-2 cells. F-G. MTT assay demonstrated ectopic BECN1 expression in EnzR1-C4-2 (F) and EnzR2-C4-2B (G) cells re-sensitized Enzalutamide resistance.
In addition to MTT assay, we also conducted the Trypan blue, BrDU staining and western blotting assays to determine the effects of Enz on cell fate, and the results were consistent with the findings above that Enz mainly affect cell proliferation and not cell apoptosis (Fig. S3D-J).
Together, results from Fig. 2A-G, Fig. 3A-G, and Fig. S3A-J suggest that Enz may function via altering Beclin 1 to modulate the ERK-mediated growth factor signal and targeting these newly identified Beclin 1-modulated ERK growth factor signal with small molecule ERK inhibitor may increase the efficacy of Enz-treatment in PCa cells.
3.3. Mechanism dissection of how Enz can function via altering the AR-Beclin 1 interaction to modulate the ERK activation
Beclin 1 functions in a complex containing Vps34/Class III PI3K (PI3KC3), and Vps15/p150 to regulate the formation of endosome maturation via lipid phosphorylation in the transition of APPL1-containing phosphatidylinositol 3-phosphate-negative (PI3P−) endosomes to PI3P positive (PI3P+) endosomes [30]. Interestingly, early studies indicated that proteins containing PolyQ, for example, Ataxin-3, might interact with Beclin 1 to promote the autophagy [5], and AR proteins with different PolyQ lengths have been reported to have different transactivation capacity to modulate AR target genes [9, 11, 45]. Therefore, we hypothesized that Enz might promote AR interaction with Beclin 1 to influence the activity of Beclin 1 complex to regulate the intracellular ERK-growth factor signal.
We first examined the potential interaction between Beclin 1 and AR via co-immunoprecipitation (Co-IP) assay in the 293T cells, and results revealed that AR could directly interact with Beclin 1 (Fig. 4A). Next, to test the effects of Enz on the AR-Beclin 1 interaction, we treated EnzS1-C4-2 cells with 10 μM Enz and results revealed that Enz treatment increased the interaction between AR and Beclin 1, as well as the formation of Beclin 1-Vps15-Vps34 complex (Fig. 4B). Consistent with the role of AR, there is no increased Beclin 1-Vps15-Vps34 complex formation in AR-negative cell line Du145 in response to Enz treatment (Fig. 4C). Furthermore, the Co-IP assay using AR antibody also found that AR could interact with Beclin 1 and demonstrably more so with Vps34 in EnzS1-C4-2 in response to Enz (Fig. 4D).
Figure 4. Mechanism (s) why Enzalutamide can maintain growth factor signaling pathway.
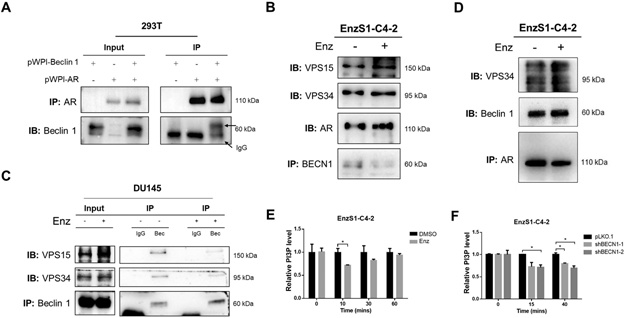
A. Exogenous Co-Immunoprecipitation (Co-IP) data proved the interaction between AR and Beclin 1 in 293T cells. B. Endogenous immunoprecipitation (IP) by pull-down of Beclin 1 suggested that Enz increased the interaction between Beclin 1 and AR, as well as the complex formation of Beclin 1-Vps15-Vps34 in EnzS1-C4-2 cells. C. Endogenous IP by pull-down of Beclin 1 suggested that Enz failed to increase the Beclin 1-Vps15-Vps34 complex formation in AR-negative DU145 cells. D. Endogenous IP by pull-down AR suggested that AR could interact with Beclin 1 and Vps34 in EnzS1-C4-2 cells. E and F. ELISA data suggested that Enz (E) or ShBECN1 (F) expression could suppressed PI3P+ generation in EnzS1-C4-2 cells. For E-F, the data shown represent the mean ± SEM of three independent experiments.*p < 0.05.
To examine whether Enz-regulated AR-Beclin 1 interaction could lead to the modulation of Beclin 1 complex’s function on ERK growth factor signal activation, we applied the ELISA assay to measure the PI3P generation, as the total cellular PI3P lipid level may represent the degree of growth factor activation [30]. We found that Enz significantly decreased PI3P generation (Fig. 4E), which was consistent with the ELISA data (Fig. 4F) showing a prolonged suppression of PI3P in cells with BECN1 knock down, supporting the conclusion that AR may interact with Beclin 1 complex to negatively impact the latter’s activity. Data from the Cancer Genome Atlas (TCGA) database suggested that AR was negatively associated with Beclin 1 protein expression, which was also negatively associated with phosphorylation level of EGFR (Fig. S4A-B).
To distinguish this AR-Beclin 1 interaction from the regulation of AR/Enz on cellular macroautophagy where Beclin 1 plays a significant role, we knocked down ATG5 expression in EnzS1-C4-2 cells. The results suggested that the early endosome formation involved Beclin 1’s function that was independent of its role on autophagy, a result consistent with previous work in breast cancer [30], further suggesting that AR-Beclin 1 interaction for endosome maturation is a distinct regulatory step in response to Enz treatment, separate from the potential regulation of Enz on cellular autophagy (Fig. S5A). Moreover, we also analyzed our unpublished whole genome transcript sequencing work related to Enz treatment as well as other publicly available GEO datasets, and found that genes involved in cellular autophagy were not significantly changed in response to Enz (Fig. S5 B-F).
Together, results from Fig. 4A-G and Fig. S5A-F suggest that Enz may function via promoting the AR-Beclin 1 interaction to influence the activity of Beclin 1 complex, then positively regulate the growth factor signaling activation. The consequences of such alteration in the AR-Beclin 1 complex-modulated the ERK growth factor signaling may lead to compromised Enz sensitivity as illustrated in Fig. 5.
Figure 5.

Hypothesis. Enzalutamide promotes AR interacting with Beclin 1 complex, to inactive its activity in mediating growth factor ERK signaling transit via decreasing P13P generation. In the upper panel, Enzalutamide could increase AR interaction with Beclin 1 complex. In the lower panel of means Beclin 1 complex could transfer growth factor from activation to inactivation status. Because Enzalutamide suppresses Beclin 1 complex’s activity, therefore, the ERK growth factor signal could be continuely activated, and cells become more “resistant”, while ERKI combined with Enzalutamide could re-sensitize cell resistant. ERKI: ERK Inhibitor.
Discussion
Several mechanisms have been involved in the development of Enz resistance in CRPC, including induction of altered glucocorticoid receptor [4], AKT signal [24, 36, 37], AR-v7 [2] and ARF876L mutation [15, 19, 41]. Although AR-v7 expression has the strongest clinical supports showing CRPC patients with detectable AR-v7 in circulating tumor cells had poorer responses to ADT-Enzalutamide [3], there are still many patients, who are resistant to Enz, but were found AR-v7-negative.
Previous publications indicated the growth factor signals, such as PI3K/AKT pathway, play critical roles during Enz-resistance development and serves as key potential therapeutic targets for CRPC patients [3] [8, 17]. In this study, we found that Enz could induce and maintain the phosphorylation of ERK in response to growth factor stimulation, which likely contributed to the development of Enz-resistance. Mechanism dissection indicated Enz could enhance the interaction between AR and Beclin 1 complex thus decreasing Beclin1 complex’s activity in mediating ERK-growth factor signaling inactivation. This post-translational mechanism was different from a transcriptional regulation by AR of the PHLPP phosphatase that inactivates AKT by removing activating phosphorylation [8]. Indeed, this difference might also underlie the different response towards the rescue by Beclin 1 overexpression, which appeared only for ERK signal, but not for AKT signal. These findings were consistent with a similar mechanistic role of Beclin 1 in enhancing growth factor signaling in breast cancer [30]. Moreover, these results were consistent with our finding that Beclin 1 was expressed lower in both EnzR1-C4-2 and EnzR2-C4-2B cells compared with their parental cells while knocking down BECN1 expression significantly decreased Enz sensitivity by increasing and maintaining ERK growth factor signal. Therefore, increasing Beclin 1 expression, or targeting its downstream ERK growth factor signal with small molecule ERK inhibitor may reduce Enz-resistance in PCa cells and enhance its efficacy.
Beclin 1 is the mammalian homolog of the yeast Atg6/Vacuolar protein sorting 30 (Vps30) protein that plays an essential role in macroautophagy and vacuolar protein sorting [16, 33]. Beclin 1 is required for normal mammalian development and it has been shown to play a critical role in pathogenesis, including cancers [29, 46]. Reduction of Beclin 1 was observed in many tumors [13, 40], including PCa [21]. In addition, there is reciprocal regulation between Beclin 1 and growth factor receptors such that posttranslational modifications of Beclin 1 result in its degradation while its loss can impact growth factor trafficking and signaling. Wei et al. [44] found that EGFR activation promotes the receptor binding to Beclin 1, leading to its multisite tyrosine phosphorylation, and decreased Beclin 1-associated Vps34 lipid kinase activity. Hyperactivation of growth factor signaling pathways in cancers likely enhance their oncogenic signaling potential through regulating Beclin 1-mediated regulation of receptor trafficking and duration of signal activation. In our work, we found that AR-Beclin 1 complex interaction inactivated the complex’s activity in mediating PI3P (+) generation, and therefore, suppressed ERK growth factor transit, consequently contributing to Enz sensitivity decreasing.
Our study reveals an alternative mechanism for how Beclin 1 loss, in addition to its function in regulating autophagy, may impact PCa cell Enz-resistance that involves enhancing the magnitude and duration of ERK growth factor signal. This study also suggests that targeting Beclin 1 or downstream ERK growth factor signal by ERK inhibitor could effectively suppress Enz resistant CRPC cell growth, serving as potential therapeutic targets for PCa patients.
Supplementary Material
Figure S1. A-B. Abnormal status of IGF-1R (A) and EGFR (B) in PCa patients. Mutation, amplification, and deletion of IGF-1R and EGFR in a series of PCa cohort studies. C-D. IGF-1R (C) and EGFR (D) expression and PCa progression derived from two GEO datasets (GDS2545 and GDS1439). E. The basal phosphorylation levels of AKT and ERK were described, and the phosphorylation levels of AKT (T308 and S473) were increased, but the p-ERK 1/2 was not.
Figure S2. Enzalutamide enhances growth factor signals. A. EnzS1-C4-2 cells were pretreated with Enz for three days, and serum-starved overnight in media containing 0.1% BSA, then stimulated with 100 ng/ml human IGF for the indicated time periods. P-AKT-S473, p-AKT-T308, and p-ERK (T202/Y204) levels were detected by western blot. B-C. Quantification results of p-AKT-T308 (B) and p-AKT-S473 (C). D. EnzS2-C4-2B cells were pretreated with Enz for three days, and serum-starved overnight in media containing 0.1% BSA, then stimulated with 50 ng/ml human EGF for the indicated time periods. P-AKT-S473, p-AKT-T308, and p-ERK1/2 (T202/Y204) levels were detected by western blot. E-F. Quantification results of p-AKT-T308 and p-ERK (T202/Y204). The data shown represent the mean of three independent experiments. *, p < 0.05; **, p < 0.01, ***, p < 0.001.
Figure S3. Beclin 1 had lower expression in resistant cells relative to parental prostate cancer cells. A. MTT assay revealed that Ectopic expression of BECN1 increased Enzalutamide sensitivity in EnzS1-C4-2 cells. B-C. MTT assay revealed that ERKI inhibitor suppressed EnzR1-C4-2 (B) and EnzR2-C4-2B (C) cell growth (Both group maintained with Enz when treated with the inhibitor). D. Western blotting assay showed Enz has no obvious effect on cell apoptosis (The last column was the positive control). E-H. Trypan blue assay showed the cell viability changes in EnzS1-C4-2-shBECN1 (E), EnzS2-C4-2B-shBECN1 (F), EnzR1-C4-2-oeBECN1 (G), EnzR2-C4-2B-oeBECN1 (H) cells treated with or without Enz for five or six days. I-J. EnzS1-C4-2-shBECN1 and EnzR1-C4-2R-oeBECN1 cells were treated with BrdU along with Enz, and the results showed in EnzS1-C4-2-shBECN1 cells has higher BrDu staining rate compared to vector control, while lower BrDu staining rate was uncovered in EnzR1-C4-2-oeBECN1 group compared to vector control.
Figure S4. Association between Beclin 1 and AR or EGFR. A. AR was negatively associated with Beclin1. B. Beclin 1 was negatively associated with EGFR.
Figure S5. Beclin 1 mediated ERK growth factor signaling was not influenced by autophagy. A. Knocking down ATG5 did not influence growth factor signaling in response to growth factor stimulation. Cells were serum-starved overnight in media containing 0.1% BSA, then stimulated with 50 ng/ml human EGF for the indicated time periods. P-AKT-S473, p-AKT-T308, and p-ERK1/2 (T202/Y204) levels were detected by western blot. B-F. Data generated from our own RNA-seq, and GEO dataset (GSE78201) suggested that Enz did not significantly influence the expression of genes related to Autophagosome formation in different PCa cell lines.
Highlights:
Novel relationship between Beclin 1 and Enz sensitivity in CRPC cell lines.
Molecular mechanisms of Beclin 1 function independent of macroautophagy in regulating growth factor signaling.
Potential novel therapies for CRPC with interrupting the AR-Beclin 1 complex/ERK signal to alter the Enz sensitivity in CRPC PCa cells.
Acknowledgments.
This work was supported by NIH Grant (CA156700), George Whipple Professorship Endowment, Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence (MOHW104-TDU-B-212-113002), The National Natural Science Foundation of China 81630019, and the Natural Science Foundation of Guangdong Province, China (2017A030313800).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare that they have no conflicts of interest.
References:
- [1].Aleksic T Verrill C, Bryant RJ, Han C, Worrall AR, Brureau L, Larre S, Higgins GS, Fazal F, Sabbagh A, Haider S, Buffa FM, Cole D, Macaulay VM, IGF-1R associates with adverse outcomes after radical radiotherapy for prostate cancer, Br J Cancer, 117 (2017) 1600–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y Mohammad TA, Chen Y, Fedor HL, AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer, N Engl J Med, 371 (2014) 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J, AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer, N Engl J Med, 371 (2014) 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, Zheng D, Sawyers CL, Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade, Cell, 155 (2013) 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM, Rubinsztein DC, Polyglutamine tracts regulate beclin 1-dependent autophagy, Nature, 545 (2017) 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, Davis ID, de Bono JS, Evans CP, Fizazi K, Joshua AM, Kim CS, Kimura G, Mainwaring P, Mansbach H, Miller K, Noonberg SB, Perabo F, Phung D, Saad F, Scher HI, Taplin ME, Venner PM, Tombal B, P. Investigators, Enzalutamide in metastatic prostate cancer before chemotherapy, N Engl J Med, 371 (2014) 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bianchini D, Lorente D, Rodriguezvida A, Omlin A, Pezaro C, Ferraldeschi R, Zivi A, Attard F, Chowdhury S, de Bono JS, Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration-resistant prostate cancer (CRPC) pre-treated with docetaxel and abiraterone, European Journal of Cancer, 50 (2014) 78. [DOI] [PubMed] [Google Scholar]
- [8].Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL, Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer, Cancer Cell, 19 (2011) 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chamberlain NL, Driver ED, Miesfeld RL, The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function, Nucleic Acids Res, 22 (1994) 3181–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen J, Li L, Yang Z, Luo J, Yeh S, Chang C, Androgen-deprivation therapy with enzalutamide enhances prostate cancer metastasis via decreasing the EPHB6 suppressor expression, Cancer letters, 408 (2017) 155–163. [DOI] [PubMed] [Google Scholar]
- [11].Chua JP, Reddy SL, Yu Z, Giorgetti E, Montie HL, Mukherjee S, Higgins J, McEachin RC, Robins DM, Merry DE, Iniguez-Lluhi JA, Lieberman AP, Disrupting SUMOylation enhances transcriptional function and ameliorates polyglutamine androgen receptor-mediated disease, J Clin Invest, 125 (2015) 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Di Lorenzo G, Tortora G, D'Armiento FP, De Rosa G, Staibano S, Autorino R, D'Armiento M, De Laurentiis M, De Placido S, Catalano G, Bianco AR, Ciardiello F, Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer, Clin Cancer Res, 8 (2002) 3438–3444. [PubMed] [Google Scholar]
- [13].Dong M, Wan XB, Yuan ZY, Wei L, Fan XJ, Wang TT, Lv YC, Li X, Chen ZH, Chen J, Lin Q, Wen JY, Ma XK, Liu Q, Wu XY , Low expression of Beclin 1 and elevated expression of HIF-1alpha refine distant metastasis risk and predict poor prognosis of ER-positive, HER2-negative breast cancer, Med Oncol, 30 (2013) 355. [DOI] [PubMed] [Google Scholar]
- [14].Edlind MP, Hsieh AC, PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance, Asian J Androl, 16 (2014) 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509, Cancer Discovery, 3 (2013) 1020–1029. [DOI] [PubMed] [Google Scholar]
- [16].Kametaka S, Okano T, Ohsumi M, Ohsumi Y, Apg14p and Apg6/Vps30p Form a Protein Complex Essential for Autophagy in the Yeast, Saccharomyces cerevisiae, Journal of Biological Chemistry, 273 (1998) 22284–22291. [DOI] [PubMed] [Google Scholar]
- [17].Kato M, Banuelos CA, Imamura Y, Leung JK, Caley DP, Wang J, Mawji NR, Sadar MD, Cotargeting Androgen Receptor Splice Variants and mTOR Signaling Pathway for the Treatment of Castration-Resistant Prostate Cancer, Clin Cancer Res, 22 (2016) 2744–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kaushik AK, Shojaie A, Panzitt K, Sonavane R, Venghatakrishnan H, Manikkam M, Zaslavsky A, Putluri V, Vasu VT, Zhang Y, Khan AS, Lloyd S, Szafran AT, Dasgupta S, Bader DA, Stossi F, Li H, Samanta S, Cao X, Tsouko E, Huang S, Frigo DE, Chan L, Edwards DP, Kaipparettu BA, Mitsiades N, Weigel NL, Mancini M, McGuire SE, Mehra R, Ittmann MM, Chinnaiyan AM, Putluri N, Palapattu GS, Michailidis G, Sreekumar A, Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer, Nat Commun, 7 (2016) 11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide), Cancer Discovery, 3 (2013) 1030–1043. [DOI] [PubMed] [Google Scholar]
- [20].Lev A, Lulla AR, Ross BC, Ralff MD, Makhov PB, Dicker DT, El-Deiry WS, ONC201 Targets AR and AR-V7 Signaling, Reduces PSA, and Synergizes with Everolimus in Prostate Cancer, Mol Cancer Res, 16 (2018) 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu C, Xu P, Chen D, Fan X, Xu Y, Li M, Yang X, Wang C, Roles of autophagy-related genes Beclin-1 and LC3 in the development and progression of prostate cancer and benign prostatic hyperplasia, Biomedical reports, 1 (2013) 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu X, Ledet E, Li D, Dotiwala A, Steinberger A, Feibus A, Li J, Qi Y, Silberstein J, Lee B, Dong Y, Sartor O, Zhang H, A Whole Blood Assay for AR-V7 and AR(v567es) in Patients with Prostate Cancer, The Journal of urology, 196 (2016) 1758–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lokhandwala PM, Riel SL, Haley L, Lu C, Chen Y, Silberstein J, Zhu Y, Zheng G, Lin MT, Gocke CD, Partin AW, Antonarakis ES, Luo J, Eshleman JR, Analytical Validation of Androgen Receptor Splice Variant 7 Detection in a Clinical Laboratory Improvement Amendments (CLIA) Laboratory Setting, J Mol Diagn, 19 (2017) 115–125. [DOI] [PubMed] [Google Scholar]
- [24].Luo Y, Azad AK, Karanika S, Basourakos SP, Zuo X, Wang J, Yang L, Yang G, Korentzelos D, Yin J, Park S, Zhang P, Campbell JJ, Schall TJ, Cao G, Li L, Thompson TC, Enzalutamide and CXCR7 inhibitor combination treatment suppresses cell growth and angiogenic signaling in castration-resistant prostate cancer models, Int J Cancer, 142 (2018) 2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nadiminty N, Tummala R, Liu C, Lou W, Evans CP, Gao AC, NF-kappaB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer, Mol Cancer Ther, 14 (2015) 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Okegawa T, Ninomiya N, Masuda K, Nakamura Y, Tambo M, Nutahara K, AR-V7 in circulating tumor cells cluster as a predictive biomarker of abiraterone acetate and enzalutamide treatment in castration-resistant prostate cancer patients, Prostate, 78 (2018) 576–582. [DOI] [PubMed] [Google Scholar]
- [27].Qi W, Morales C, Cooke LS, Johnson B, Somer B, Mahadevan D, Reciprocal feedback inhibition of the androgen receptor and PI3K as a novel therapy for castrate-sensitive and -resistant prostate cancer, Oncotarget, 6 (2015) 41976–41987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang Y, Lee JH, Hawke D, Wang Y, Xia Y, Zheng Y, Jiang BH, Liu DX, Jiang T, Lu Z, Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy, Molecular cell, 65 (2017) 917–931 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B, Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene, J Clin Invest, 112 (2003) 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rohatgi RA, Janusis J, Leonard D, Bellve KD, Fogarty KE, Baehrecke EH, Corvera S, Shaw LM, Beclin 1 regulates growth factor receptor signaling in breast cancer, Oncogene, 34 (2015) 5352–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ryan CJ, M.D, Smith MR, M.D, D. Ph., Bono JSD, M.B, B. Ch., D. Ph., Molina A, Abiraterone in Metastatic Prostate Cancer without Previous Chemotherapy, N Engl J Med, 368 (2013) 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D, McLaughlin B, Wahl J, Greene SB, Heller G, Marrinucci D, Fleisher M, Dittamore R, Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer, JAMA Oncol, 2 (2016) 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Seaman MNJ, Marcusson EG, Cereghino JL, Emr SD, Endosome to Golgi Retrieval of the Vacuolar Protein Sorting Receptor, Vps10p, Requires the Function of the VPS29, VPS30, and VPS35 Gene Products, Journal of Cell Biology, 137 (1997) 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Siegel RL, Miller KD, Jemal A, Cancer Statistics, 2017, Ca A Cancer Journal for Clinicians, 67 (2017) 5. [DOI] [PubMed] [Google Scholar]
- [35].Thomas C, Lamoureux F, Crafter C, Davies BR, Beraldi E, Fazli L, Kim S, Thaper D, Gleave ME, Zoubeidi A, Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo, Mol Cancer Ther, 12 (2013) 2342–2355. [DOI] [PubMed] [Google Scholar]
- [36].Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, Gleave ME, Zoubeidi A, Combination AZD5363 with Enzalutamide Significantly Delays Enzalutamide-resistant Prostate Cancer in Preclinical Models, Eur Urol, 67 (2015) 986–990. [DOI] [PubMed] [Google Scholar]
- [37].Toren P, Kim S, Johnson F, Zoubeidi A, Combined AKT and MEK Pathway Blockade in Pre-Clinical Models of Enzalutamide-Resistant Prostate Cancer, PloS one, 11 (2016) e0152861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Development of a second-generation antiandrogen for treatment of advanced prostate cancer, Science, 324 (2009) 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vega-Rubin-de-Celis S, Zou Z, Fernandez AF, Ci B, Kim M, Xiao G, Xie Y, Levine B, Increased autophagy blocks HER2-mediated breast tumorigenesis, Proceedings of the National Academy of Sciences of the United States of America, 115 (2018) 4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang J, Pan XL, Ding LJ, Liu DY, Da-Peng L, Jin T, Aberrant expression of Beclin-1 and LC3 correlates with poor prognosis of human hypopharyngeal squamous cell carcinoma, PloS one, 8 (2013) e69038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang R, Lin W, Lin C, Lei L, Yin S, Chang C, ASC-J9 ®; suppresses castration resistant prostate cancer progression via degrading the enzalutamide-induced androgen receptor mutant AR-F876L, Cancer Letters, 379 (2016) 154–160. [DOI] [PubMed] [Google Scholar]
- [42].Wang R, Sun Y, Li L, Niu Y, Lin W, Lin C, Antonarakis ES, Luo J, Yeh S, Chang C, Preclinical Study using Malat1 Small Interfering RNA or Androgen Receptor Splicing Variant 7 Degradation Enhancer ASC-J9((R)) to Suppress Enzalutamide-resistant Prostate Cancer Progression, European urology, 72 (2017) 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Weber DC, Tille JC, Combescure C, Egger JF, Laouiti M, Hammad K, Granger P, Rubbia-Brandt L, Miralbell R, The prognostic value of expression of HIF1alpha, EGFR and VEGF-A, in localized prostate cancer for intermediate- and high-risk patients treated with radiation therapy with or without androgen deprivation therapy, Radiat Oncol, 7 (2012) 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, Grishin NV, Peyton M, Minna J, Bhagat G, Levine B, EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance, Cell, 154 (2013) 1269–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yang Z, Chang YJ, Yu IC, Yeh S, Wu CC, Miyamoto H, Merry DE, Sobue G, Chen LM, Chang SS, Chang C, ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor, Nat Med, 13 (2007) 348–353. [DOI] [PubMed] [Google Scholar]
- [46].Yue Z, Jin S, Yang C, Levine AJ, Heintz N, Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor, Proceedings of the National Academy of Sciences of the United States of America, 100 (2003) 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A-B. Abnormal status of IGF-1R (A) and EGFR (B) in PCa patients. Mutation, amplification, and deletion of IGF-1R and EGFR in a series of PCa cohort studies. C-D. IGF-1R (C) and EGFR (D) expression and PCa progression derived from two GEO datasets (GDS2545 and GDS1439). E. The basal phosphorylation levels of AKT and ERK were described, and the phosphorylation levels of AKT (T308 and S473) were increased, but the p-ERK 1/2 was not.
Figure S2. Enzalutamide enhances growth factor signals. A. EnzS1-C4-2 cells were pretreated with Enz for three days, and serum-starved overnight in media containing 0.1% BSA, then stimulated with 100 ng/ml human IGF for the indicated time periods. P-AKT-S473, p-AKT-T308, and p-ERK (T202/Y204) levels were detected by western blot. B-C. Quantification results of p-AKT-T308 (B) and p-AKT-S473 (C). D. EnzS2-C4-2B cells were pretreated with Enz for three days, and serum-starved overnight in media containing 0.1% BSA, then stimulated with 50 ng/ml human EGF for the indicated time periods. P-AKT-S473, p-AKT-T308, and p-ERK1/2 (T202/Y204) levels were detected by western blot. E-F. Quantification results of p-AKT-T308 and p-ERK (T202/Y204). The data shown represent the mean of three independent experiments. *, p < 0.05; **, p < 0.01, ***, p < 0.001.
Figure S3. Beclin 1 had lower expression in resistant cells relative to parental prostate cancer cells. A. MTT assay revealed that Ectopic expression of BECN1 increased Enzalutamide sensitivity in EnzS1-C4-2 cells. B-C. MTT assay revealed that ERKI inhibitor suppressed EnzR1-C4-2 (B) and EnzR2-C4-2B (C) cell growth (Both group maintained with Enz when treated with the inhibitor). D. Western blotting assay showed Enz has no obvious effect on cell apoptosis (The last column was the positive control). E-H. Trypan blue assay showed the cell viability changes in EnzS1-C4-2-shBECN1 (E), EnzS2-C4-2B-shBECN1 (F), EnzR1-C4-2-oeBECN1 (G), EnzR2-C4-2B-oeBECN1 (H) cells treated with or without Enz for five or six days. I-J. EnzS1-C4-2-shBECN1 and EnzR1-C4-2R-oeBECN1 cells were treated with BrdU along with Enz, and the results showed in EnzS1-C4-2-shBECN1 cells has higher BrDu staining rate compared to vector control, while lower BrDu staining rate was uncovered in EnzR1-C4-2-oeBECN1 group compared to vector control.
Figure S4. Association between Beclin 1 and AR or EGFR. A. AR was negatively associated with Beclin1. B. Beclin 1 was negatively associated with EGFR.
Figure S5. Beclin 1 mediated ERK growth factor signaling was not influenced by autophagy. A. Knocking down ATG5 did not influence growth factor signaling in response to growth factor stimulation. Cells were serum-starved overnight in media containing 0.1% BSA, then stimulated with 50 ng/ml human EGF for the indicated time periods. P-AKT-S473, p-AKT-T308, and p-ERK1/2 (T202/Y204) levels were detected by western blot. B-F. Data generated from our own RNA-seq, and GEO dataset (GSE78201) suggested that Enz did not significantly influence the expression of genes related to Autophagosome formation in different PCa cell lines.