

ABSTRACT
The recent announcement of marijuana legalization in Canada spiked many discussions about potential health benefits of Cannabis sativa. Cannabinoids are active chemical compounds produced by cannabis, and their numerous effects on the human body are primarily exerted through interactions with cannabinoid receptor types 1 (CB1) and 2 (CB2). Cannabinoids are broadly classified as endo-, phyto-, and synthetic cannabinoids. In this review, we will describe the activity of cannabinoids on the cellular level, comprehensively summarize the activity of all groups of cannabinoids on various cancers and propose several potential mechanisms of action of cannabinoids on cancer cells.
KEYWORDS: Cannabinoids, cancer, cannabidiol, tetrahydrocannabinol, anti-tumor activity
Introduction
Cannabinoids are compounds that exert numerous effects in the human body and include molecules that are structurally similar to Δ9-tetrahydrocannabinol (Δ9-THC) and interact with cannabinoid receptors [1]. Cannabinoids affect cell function through various cellular pathways mediated by two types of G-protein coupled receptors (GPCRs): the cannabinoid receptor type 1 (CB1) and the cannabinoid receptor type 2 (CB2).
The known types of cannabinoids
There are three major types of cannabinoids: endocannabinoids produced by the human body, phytocannabinoids produced primarily by Cannabis sativa, and synthetic cannabinoids that are synthesized artificially. All three groups of classical cannabinoids have a somewhat similar structure because they are decarboxylated from 2-carboxylic acids (2-COOH); the main structural difference is due to different methods of precursor cyclization [2].
Two major endocannabinoids with the defined activity in the human body are N-arachidonoylethanolamine (AEA-anandamide) and 2-arachidonoylglycerol (2-AG) [3]. Other endocannabinoid molecules with less clear roles include oleamide, O-arachidonoyl ethanolamine which is also known as virodhamine, 2-AG ether also known as noladin, pregnenolone, lipoxin A4, and N-arachidonoyl- dopamine [3] (Table 1). Endocannabinoids affect our pain sensation and response, stress response, inflammation response, appetite, memory, and mood, among other processes [1,3]. Immune response, neurotransmission, energy homeostasis, reproduction, and cell survival/death are all affected by the endocannabinoid system (ECS) [1]. Whereas anandamide functions as a ligand for CB1, 2-AG can bind with both CB1 and CB2 receptors [4].
Table 1.
Various types of cannabinoids.
| Endocannabinoids | Receptor/ transporter |
Synthetic cannabinoids | Receptor/ transporter |
Phytocannabinoids | Receptor/ transporter |
|---|---|---|---|---|---|
| N-arachidonoylethanolamine (AEA-anandamide) | CB1, CB2, M1, M4, AMPA, Glycine, Glycine α1, GPR18, GPR55, NCX1, NMDA | Dronabinol | CB1, CB2 | Cannabigerolic acid (CBGA) | CB1, CB2, TRPA1, TRPM8, TRPV1, TRPV2 |
| 2- arachidonoylglycerol (2-AG) | CB1, CB2, GABAA B2, Glycine, GPR55 | Nabilone | CB1, CB2 | Cannabigerol (CBG) | Anandamide transporter, TRPA1, TRPM8, TRPV1, TRPV2 |
| oleamide | CB1, CB2 | Rimonabant | CB1 | Δ9-tetrahydrocannabinolic acid (THCA) | CB1, CB2, TRPA1, TRPM8, TRPV1, TRPV2 |
| O-arachidonoyl ethanolamine (virodhamine) | CB1, CB2, GPR55 | CP55940 | CB1, CB2, GPR55, GABAA B2 | Cannabidiolic acid (CBDA) | 5-HT1A, TRPV1, TRPV3, TRPV4 and TRPA1 |
| 2-AG ether (noladin) | CB1, CB2 | R-(+)-WIN55212 | CB1, CB2, TRPA1, TRPV1, 5-HT3, Glycine, Glycine α2 | Cannabichromenic acid (CBCA) | CB1, CB2 |
| pregnenolone | CB1, CB2 | JWH-015 | CB2 | Δ9 – tetrahydrocannabinol (THC) | CB1, CB2, GPR55, GPR18, TRPM8, TRPA1, TRPV1, TRPV2, ENT1, TRPM8, Glycine |
| lipoxin A4 | CB1, CB2 | JWH-133 | CB2, TRPV1 | Cannabidiol (CBD) | CB1, CB2, GPR55, GPR18, TRPM8, TRPV1, TRPV2, 5-HT1A, 5-HT3, Anandamide transporter, ENT1 |
| N-arachidonoyl- dopamine | CB1, CB2 | JWH-018, −073, −122, −210, KM-233 O-1663 |
CB1, CB2 | Cannabichromene (CBC) | CB2, TRPA1, TRPM8, TRPV1, TRPV2 |
| Hu-210 | CB1, CB2, GPR55, 5-HT2, Glycine α2 | Cannabinolic acid (CBNA) | CB1, CB2 | ||
| Hu-308 | CB2, Glycine α2 | Cannabinol (CBN) | CB1, CB2, TRPA1, TRPV1, TRPV2, TRPV3, TRPV4, TRPM8 | ||
| ACEA | CB1 | Δ9- tetrahydrocannabivarin (THCV or THV) | CB1, CB2, TRPV1, TRPA1, TRPM8 | ||
| Δ9 – tetrahydrocannabivarin carboxylic acid (THCAV) | TRPA1, TRPM8 | ||||
| Δ8-THC | CB1, CB2 |
Synthetic cannabinoids (SCs) include dronabinol and its analogues nabilone and rimonabant which are used to treat pain, loss of appetite, obesity, and other conditions. Most of the SCs were developed to study the function of ECS, avoiding restrictions associated with the use of cannabis and endocannabinoids. SCs can be broadly divided into synthetic equivalents of endo- or phytocannabinoids (for example, dronabinol is similar to Δ9‐THC), analogs of endo- or phytocannabinoids (i.e., nabilone, HU‐210), derivatives of endocannabinoids (i.e., methanandamide), or completely new compounds of various chemical structure and affinity to cannabinoid receptors [5].
Phytocannabinoids are mainly found in Cannabis sativa and are represented by over 100 cannabinoids, although only a few of them are relatively abundant and believed to be active [6]. All these compounds are defined as cannabinoids because they are structurally similar to endocannabinoids; however, most of them either do not bind known cannabinoid receptors or do that very inefficiently. All cannabinoids derive from cannabigerolic acid (CBGA), a molecule produced from the combination of geranyl pyrophosphate and olivetolic acid catalyzed by GOT, a prenyltransferase group enzyme. CBGA is then converted into four major cannabinoids: cannabigerol (CBG), Δ9-tetrahydrocannabinolic acid (THCA), cannabidiolic acid (CBDA), and cannabichromenic acid (CBCA) (Figure 1). Whereas CBG is obtained by direct heat-, light-, or alkylation-induced decarboxylation events (loss of CO2 group), THCA, CBDA, and CBCA are obtained through the activity of THCA, CBDA, and CBCA synthase, respectively. Finally, THCA, CBDA, and CBCA are converted into the active compounds Δ9 – tetrahydrocannabinol (THC), cannabidiol (CBD), and cannabichromene (CBC) through the heat-induced decarboxylation. Prolonged exposure to the air also results in the conversion of THCA into cannabinolic acid (CBNA) and THC into cannabinol (CBN). Decarboxylising CBNA also results in CBN. The most prevalent cannabinoids are Δ9-THC, cannabidiol (CBD), and cannabinol (CBN) [7]. Furthermore, some plants have an active alternative pathway that generates an almost mirror image of major cannabinoids from divarinic acid instead of olivetolic acid, starting from the precursor CBGA variant (CBGVA) instead of CBGA and downstream molecules including CBCA (CBCVA), CBDA (CBDVA), and THCA (THCVA) variants, and other compounds (Figure 1).
Figure 1.
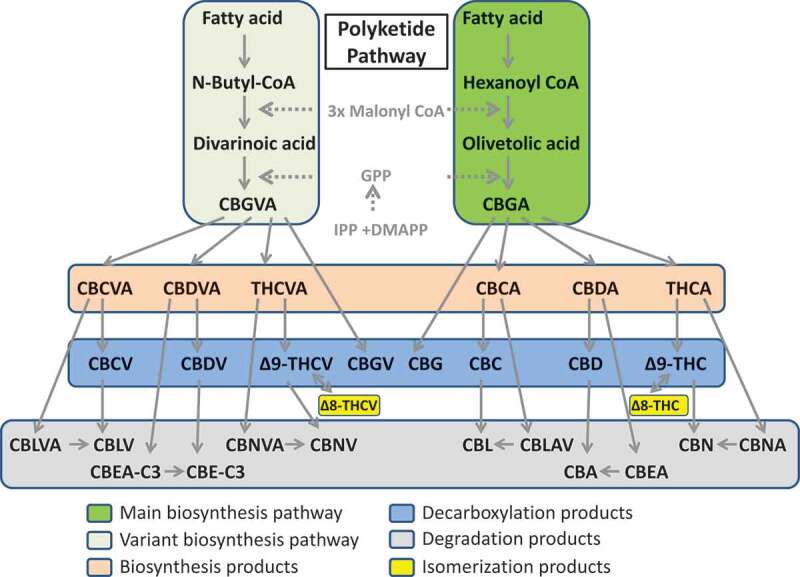
Polyketide pathway of cannabinoids synthesis. Dark green (right panel) shows the main biosynthesis pathway, whereas light green (left panel) shows the variant biosynthesis pathway. Biosynthesis products consist of CBCA, CBDA, THCA and their variants, CBCVA, CBDVA and THCVA. Decarboxylation products are shown in the blue box, whereas degradation products are in gray. Delta 8 isomerization products are shown in yellow.
The mode of action of cannabinoids
The main mode of action of cannabinoids is based on their neuromodulator activity through retrograde neurotransmission and binding to CB1 and CB2 receptors. The two main types of cannabinoid receptors vary mostly in their localization. CB1 receptors are more abundant and ubiquitously present in the body; they are mainly localized in the central nervous system (CNS) and secondarily in various extra-neural tissues and peripheral nerve terminals [8,9]. CB2 receptors are also expressed in most regions of the body but are in abundance in lymph nodes, spleen, bone marrow and appendix [10,11]. Certain level of CB2 receptors is also found in CNS, primarily in microglia but also in neurons [11].
The specific class of G-proteins with which CB1/CB2 receptors interact belongs to the heterotrimeric class that consists of three subunits, Gα, Gβ, and Gγ, with the latter two often forming a dimer. Gα subunits are represented by several classes including, but not limited to, Gαs (G stimulatory), Gαi (G inhibitory), Gαo (G other), Gα q/11, and Gα 12/13. Both Gα and Gβ/Gγ activate or inactivate different secondary pathways. Whereas Gαs activates the cyclic AMP (cAMP)-dependent pathway, Gαi inhibits it, thus preventing cAMP production from ATP; both reactions occur through the interaction with adenylate cyclase, an enzyme that converts ATP into cAMP. cAMP functions as a secondary messenger that activates protein kinase A (PKA) which in turn phosphorylates different targets. The Gq/11α subunit interacts with and activates phospholipase C beta (PCB). The active PCB form cleaves phosphoinositol PIP2 into inositol triphosphate (IP3) and diacylglycerol (DAG). Gα12/13 signals through the interaction with the RhoGEF domain of the Rho family GTPases, resulting in the regulation of cell cytoskeleton remodeling and the activation of cell migration [12].
Nogueras-Ortiz and Yudowski [13] proposed three waves of spatiotemporal responses of cannabinoid binding to CB receptors [13]. The first wave is initiated by cannabinoids binding to the CB1/CB2 receptors. The interaction with these receptors activates heterotrimeric G proteins, thus leading to their dissociation into Gαi and Gβ/Gγ subunits and resulting in three major events: a rapid decrease in cAMP levels due to the direct binding and inhibition of adenylate cyclase by a Gαi subunit, a decrease in Ca2+ conductance, and an increase in K+ conductance (Figure 2). The second wave consists of the events associated with the ligand-induced receptor phosphorylation followed by receptor desensitization through the phosphorylation of GPCR by the G protein-coupled receptor kinases. Arrestin proteins bind to the phosphorylated receptor which leads to its internalization and unavailability for further signaling, thus redirecting all downstream signaling to other receptors and activating pathways such as the mitogen-activated protein family of kinases [14,15].
Figure 2.
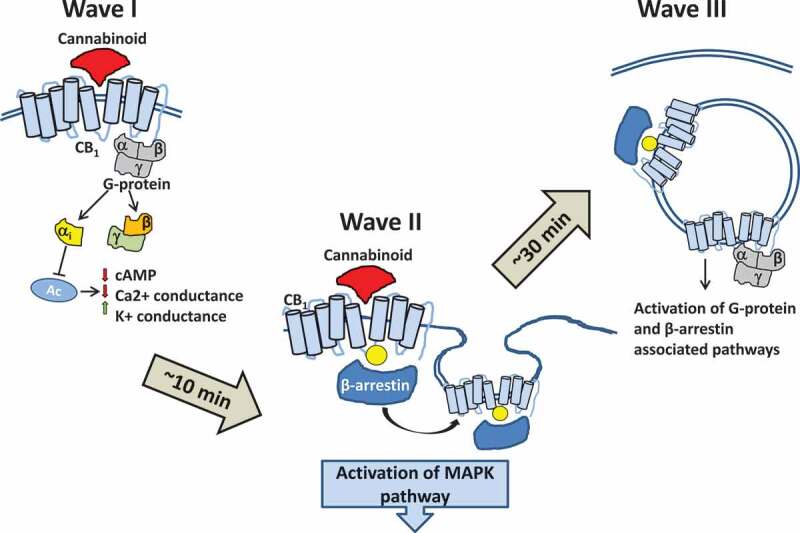
Three waves of responses to cannabinoid binding. The first wave is initiated by cannabinoids binding to the CB1/CB2 receptors. The interaction of cannabinoids with one of these receptors activates heterotrimeric G proteins, thus leading to their dissociation into Gαi and Gβ/Gγ subunits and resulting in three major events: a rapid decrease in cAMP levels due to the direct binding and inhibition of adenylate cyclase by a Gαi subunit, a decrease in Ca2+ conductance, and an increase in K+ conductance. The second wave consists of the events associated with the receptor phosphorylation followed by receptor desensitization. Arrestin proteins bind to the phosphorylated receptor leading to receptor internalization, thus redirecting all downstream signaling to other receptors and activating the pathways like mitogen-activated protein family of kinases. The third wave is initiated at the receptors in the intracellular compartments such as endosomes and lysosomes and can be initialized through either G proteins or β-arrestins.
The third wave is initiated at the receptors in the intracellular compartments such as endosomes and lysosomes and can be triggered through either G proteins or β-arrestins. The existence of this wave and its functionality in vivo are unclear, but several lines of evidence support its importance. Immunostaining a hippocampal Neuro2A cell culture demonstrates the enrichment of CB1 in the intracellular compartments and shows the co-immunoprecipitation of CB1 and G proteins isolated from the endosomal compartments [16]. Treating Neuro2A cells with the combination of an agonist that can cross the plasma membrane (WIN-55212-2) and the receptor blocker (hemopressin) that cannot cross the plasma membrane demonstrates the phosphorylation and activation of the extracellular signal-regulated protein kinase (ERK), presumably occurring through the activation of the receptor in the intracellular compartments.
CB1 receptor activation
CB1 receptors are primarily located in the CNS. In fact, the human CNS has more CB1 receptors than any other type of GPCRs. CB1 receptors are mostly located at the terminal parts of central and peripheral neurons where they are involved in inhibition of neurotransmission [17]. Many other tissues also have CB1 receptors albeit at much lower numbers. CB1 receptors can regulate various functions, including cardiovascular, reproductive, and respiratory functions as well as the neuronal development and neuromodulatory processes.
Signaling through the CB1 receptor negatively regulates neurotransmitter release by inhibiting the phosphorylation of the A-type potassium channels. In its unphosphorylated form, the A-type potassium channels sustain a continuous outward flow of potassium [18]. The activation of CB1 is also known to negatively regulate the inward flow of potassium through the D-type receptors. CB1 activation also results in the inhibition of the N-type calcium channels via a direct interaction with Gαi and Gαo proteins. Altogether, this results in the restriction of neurotransmission leading to cognitive impairment and sedative-like effects after the consumption of marijuana [19].
CB1 receptors in humans are more abundant in most regions of brain of healthy females as compared to males, as demonstrated by an in vivo positron emission tomography (PET) [20]. In addition, CB1 receptors abundance increases with age in females but not in males [21].
CB2 receptor activation
CB2 is expressed mostly in cells and tissues of the immune system, with the highest number being found in the spleen, lymph nodes, and blood, namely in the T and B lymphocytes [22,23], although some evidence exists that CB2 receptors are also found in the peripheral nervous system and even in the microglia population [24]. The expression of CB2 in the CNS is triggered by inflammation and is mainly localized to resident macrophages of microglia [25,26]. This expression of CB2 has been localized primarily to microglia, the resident macrophages of the CNS.
Anandamide is a principal endogenous ligand of the CB2 receptor. The CB2, in part, exerts its effects by initiating phospholipase C (PLC) and the inositol 1, 4, 5-triphosphate (IP3) signaling pathways. 2-AG was found to initiate ER Ca2+ ion depletion followed by the activation of capacitative Ca2+ entry (CCE) and a transient increase of Ca2+ ions in mitochondria [27].
The activation of the CB2 receptor by 2-AG modulates immune responses, including the proliferation, survival, and migration of immune cells [28]. The data indicate that CB2 regulates B cell immunity by promoting an appropriate localization and retention of marginal zone B cells such that they are able to respond to foreign antigens, resulting in the early production of IgM, an essential immune component of protective immunity against multivalent microorganisms [29]. However, phytocannabinoids, such as Δ9-THC, have been shown to suppress B and T lymphocyte proliferation in response to cell-specific mitogens [30,31] by suppressing the cytolytic activity and proliferation and maturation of cytotoxic T lymphocytes. However, some evidence exists that THC exerts such effect independently of CB1 and CB2 receptors [32]. Collectively, the data suggest that the exogenous application of Δ9-THC inhibits functional activities of various immunocytes, thus leading to a decreased resistance to infection.
The activation of other receptors
There are other type of receptors cannabinoids were shown to interact with, including transient receptor potential cation channel vanilloid (TRPV), TRP ankyrin (TRPA), TRP melastatin (TRPM), peroxisome proliferator-activated receptor gamma (PPARγ), N-arachidonoyl glycine (NAGly, AEA metabolite) receptor GPR18, the orphan receptor GPR55, GPR19 and many others [33,34]. According to human protein atlas (www.proteinatlas.org), TRPV1 is expressed in most organs, including brain, liver, gastrointestinal tract and skin, with the highest expression found in the latter two. PPARγ is also expressed in most tissues, with the highest expression found in bone marrow/immune system, kidneys, gastrointestinal tract, uterus and testes. In contrast, GPR18 is mainly expressed in bone marrow/immune system and testes, GPR19 – in the brain, testes and a bit in bone marrow, and GPR55 – in bone marrow and testes.
The analysis of the pharmacokinetics of Δ9-THC and anandamide in single- (CB1) and double- (CB1/CB2) knockout mice showed the activation of GPR55 which is another GPCR. Unlike CB1 and CB2, GPR55 is coupled to the Gα13 protein instead of Gαi and Gαo proteins [35]. Gα13 activation leads to the activation of RhoA and cell migration [36], and cannabidiol has been shown to be an antagonist of GPR55.
TRPV1-TRPV4, TRPA1, and TRPM8 receptors are termed the ionotropic cannabinoid receptors. The TRVP1 receptor is known to be activated by compounds like capsaicin and vanilloids with their structures similar to that of anandamide [37]. Some data exist that demonstrate the ability of anandamide to bind TRVP1, making this receptor an actual novel cannabinoid receptor [37,38]. AEA was the first endocannabinoid functioning as endogenous antagonist of TRPM8 [39]. THC, in contrast, does not modulate TRPV1 but rather works against TRPV2 (most potently), TRPV3, TRPV4, TRPA1, and TRPM8 [40]. Most fascinatingly, CBD is more active at binding TRPV1 and TRPM8 rather than CB1 and CB2 [40]. Details on the signaling through TRPV1 can be found in the review by Muller et al. [34].
Interaction of cannabinoids with receptors
Cannabinoids may function as both agonists and antagonists of cannabinoid receptors. Δ9-THC is considered a partial agonist of CB1 and CB2 receptors. The affinity of THC to these receptors is lower than that of endocannabinoids but higher than that of the phytocannabinoids Δ8-THC, Δ9-THCV, CBD, cannabigerol, and cannabinol [41]. However, THC may also act as a partial antagonist of CB receptors, inhibiting the effect of endogenous cannabinoids. The density of receptors may play a role; the low level of CB receptor expression may result in THC functioning as an antagonist [41,42], and an increase in the density may reverse its function to an agonist [41]. For example, Patel and Hillard [43] found that Δ9-THC exhibits the antagonist activity of CB1 similar to that of the specific synthetic inhibitor SR141716A [43] or R-(þ)-WIN55212 [44,45].
CBD affinity for CB1 and CB2 does not appear to be high; the displacement of [3 H]CP55940 from the cannabinoid CB1 and CB2 receptors has shown that CBD functions at micromolar concentrations, which are at least 1,000-fold higher than those observed while using THC [41]. More recently, CBD has been suggested to be a CB1 and CB2 receptor antagonist and has been shown to inhibit anandamide uptake and metabolism [46]. It has been suggested that at low concentrations, CBD acts as an inverse agonist binding to the same receptors as other agonists but causing a different physiological effect [47].
As far as other minor cannabinoids are concerned, the available information is rather limited. It has been demonstrated that CBG activates the α2-adrenoceptors and blocks the CB1 and 5-HT1A receptors [48]. Testing the activity of THCV has demonstrated that it functions as a CB1 and CB2 receptor antagonist at low concentrations [49] and as a CB2 agonist at higher concentrations [50].
Sex-specific differences in cannabis response
Considering the differences in the expression pattern of CB1 receptor and other receptors to which THC binds [51], it can be hypothesized that males and females respond to cannabis differently. Women seem to be more prone to addiction to cannabis as compared to men. Significantly greater withdrawal intensity and negative impact of withdrawal are observed in women [52]. Women are also more likely to have lifetime panic disorder, agoraphobia, reported more days of poor physical health and cannabis-related medical problems after cannabis cessation [52].
Smoking cannabis is known to increases heart rate greater in males than in females [53]. Also, greater sedative effects [53] and dizziness [54] is observed in women as compared to men. In addition, women also responded to cannabis extract with significantly greater fatigue, drowsiness, and psychomotor suppression compared to women treated with placebo [55]. Sex-specific differences in response to cannabis can be due to the different amounts of endocannabinoids produced, different rate of metabolism of these cannabinoids and different level of expression of receptors.
Metabolism of endo- and phytocannabinoids and its significance
Both AEA and 2-AG can be degraded by either hydrolysis or oxygenation. Hydrolysis is accomplished via two distinct routes: AEA is degraded by fatty-acid amide hydrolase (FAAH) and 2-AG is degraded by monoacylglycerol lipases (MAGs), although FAAH is also able to metabolize 2-AG (very minor pathway). In addition, in some conditions, and/or certain cells, both AEA and 2-AG can be metabolized by carboxylesterases 1 and 2 (CES1 and CES2); these enzymes metabolize 2-AG in human leukocytes equally well with MAGs [56]. Oxygenation can occur through several enzymes, including cyclooxygenase-2, lipoxygenases, and cytochrome P450, and these enzymes have different activity in different tissues [57].
Δ9-THC is metabolized in the liver by microsomal hydroxylation and oxidation catalyzed by enzymes of cytochrome P450 (CYP) complex leading to the production of various acidic metabolites, including 11-OH-THC and THCCOOH [58]. Similarly, CBD is also a substrate of CYP450 enzymes which converts CBD through extensive hydroxylation followed by further oxidations to many different metabolites, with 7-COOH-CBD and its derivatives being the most abundant [59].
Metabolism of phytocannabinoids is greatly influenced by mode of administration and natural variations in the activity of all metabolic enzymes [60]. There are also sex-specific differences in metabolism of cannabis. THC metabolism in male rats resulted in multiple metabolites, whereas only a single metabolite 11-OH-THC was produced in female rats [61]. Brain levels of metabolite 11-OH-THC were higher in THC-exposed female rats compared to male rats [62]. Similarly, in human, exposure to THC results in high plasma THC levels in females [63]. This information must be taken into consideration when cannabis is administered.
Cannabinoids as anticancer agents
The balanced expression of CB1 and CB2 receptors as well as other associated receptors is extremely important for the overall health of an organism. Alterations in the ECS occur in many disease states. For example, the upregulation of CB1 and CB2 receptors and an increase in the endogenous levels of endocannabinoids have been observed in many pathological states, including neurodegenerative and cardiovascular diseases as well as in cancer [64].
Changes in endocannabinoid system in cancer
Changes in the endocannabinoid system have been demonstrated in various cancers. These changes include the levels of produced endocannabinoids, the expression of their receptor targets and even oligomerization of such receptors. It is still not well understood whether the changes in ECS may contribute to malignization or are a consequence of it. One of the main functions of ECS is to maintain body homeostasis, and thus ECS responds to environmental stimuli in transient and sometime even in persisting manner [65], and thus may contribute to the process of malignization. On the other hand, ECS is involved in regulation of cell division, differentiation and fate [66] and therefore could change in parallel with the process of carcinogenesis or could change in response to it. Another possibility is that ECS in tumors changes in response to microenvironment/niche signals.
For example, changes in the CB1 and CB2 expression have been demonstrated to correlate with cancer cell motility, invasion, proliferation, adhesion, and apoptosis [17]. The level of anandamide and AEG is 2–3 times higher in adenomas and colorectal cancers than in normal mucosa [67]. The number of CB2-positive receptors is much higher in colon cancer cells than in the normal epithelial cells. The CB2 receptor agonist N-cyclopentyl-7-methyl-1-(2-morpholin-4-ylethyl)-1,8-naphthyridin-4(1 H)-on-3-carboxamide (CB13) increases receptor expression and leads to apoptosis in the tested cancer cell line [68].
Similarly, the CB1 and CB2 receptor expression is upregulated in human hepatocellular carcinoma compared to non-tumorous tissues [69]; the authors however suggest that a higher expression of these receptors may improve survival [69]. A similar scenario has been observed in prostate cancer; it has been suggested that the increased densities of these receptors may correlate with a better prognosis [70].
A higher expression of CB receptors may predict tumor responsiveness to treatment. For example, prostate cancer cells expressing high levels of CB receptors respond favorably to receptor agonists [70], whereas breast cancer cells with low expression levels respond to cannabinoids with an increased proliferation [71].
Other reports, in contrast, demonstrate that endocannabinoid‐degrading enzymes are upregulated in various cancer cell lines as well as in human tumors [72]. Silencing CB1 receptor promotes growth of intestinal adenoma, whereas overexpression of CB1 inhibits cancer [73]. Decrease in the expression of endocannabinoid‐metabolizing enzymes also resulted in the decrease in the tumor growth. Increase in endocannabinoids levels leads to reduction in formation of precancerous lesions in mouse colon [74] and inhibition of prostate cancer cell growth [75].
Although the expression level of CB1 and CB2 receptors was found to be higher in various cancers [76], in many cancers, the CB1/CB2 receptors are downregulated [77]. Wang et al. [73] found a downregulation of CB1 expression in human colorectal cancer due to methylation of the CB1 promoter and a lower expression of CB1 correlated with the enhanced cancer proliferation in mice xenografted with a colorectal tumor [73]. In the regulation of other receptors, many cancers have been shown to overexpress GPR55 [78]. In fact, high levels of GPR55 are strongly correlated with tumor aggressiveness.
In addition to changes in the expression of cannabinoid receptors in different cancers, there are changes in the receptor oligomerization. CB1 receptors were shown to interact with many different other GPCRs potentially forming heterodimers and tetramers, including adenosine A2A receptors [79], dopamine D2 receptors (changing coupling from Gi to Gs) [80], D2 and adenosine A2A receptors [81]; opioid µ and δ receptors (resulting in a negative cross-talk) [82], CB2 receptors (resulting in a negative cross-talk and cross-antagonism in neuronal cells) [83], adrenergic β2 receptor (inducing internalization of CB1 receptors) [84], and 5HT2A serotonin receptor [85], cannabinoid-related orphan receptor GPR55 [86] and several others (discussed in [87]). Information about the interaction of CB2 receptor with other receptors is scarce; CB2 forms heterodimers with CB1 (see above) as well as with GPR55 [88] and CXCR4 [89].
Above-mentioned interactions typically result in changes in the function of receptors or produce negative cross-talks; therefore, if such changes occur in cancer cells, they can significantly alter response of cancer cells to endocannabinoids. Indeed, CB1/CB2, CB2/CXCR4, CB2/GPR55 and CB2/HER2 heteromers have been found in breast and prostate cancer cells [89–91]. Increased levels of endocannabinoids (anandamide in colon cancer) and increased level of receptors and their dimerization in cancer are paralleled by elevated levels of degradation enzymes, FAAH and MAGL [92,93].
The contribution of cannabinoid receptors to cancer development as well as their relationship to the prognosis and therapy response is still not entirely clear. The lack of strong correlation between the expression of cannabinoid receptors and cancer progression paralleled by inhibition of cancer growth in response to treatment with cannabinoids may suggest that cannabinoids’ effect on cancer may also be triggered through cytoplasmic effects rather than only through the direct involvement of transmembrane CB1/CB2 receptors. The CB1/CB2 receptor activation in response to cannabinoids observed in cancers could either be part of the feedback mechanism or completely unrelated. Regardless of the mechanism, research data have demonstrated positive effects of cannabinoids on cancer progression, including proliferation, viability, vascularization and invasiveness (reviewed in [94]). Below, we will describe the impact of cannabinoids on various cancers and summarize the details in Table 2.
Table 2.
The effect of various cannabinoids on cancer cells in vitro and in vivo.
| Cancer type/Compound | Experiment details | Dose | Exposure time | Method/effect | Potential mechanism | Reference |
|---|---|---|---|---|---|---|
| Glioblastoma | ||||||
| CBD | U87, U373 | IC50 25 µM | 24 h | cell viability by MTT, apoptosis | ROS activation | Massi et al. [103] |
| U87 xenograft of athymic nude mice | 0.5 mg/mouse | 18 d | 70% reduction of tumor growth | Massi et al. [103] | ||
| CBD | U87 | IC50 5 µM | 6 h | cell migration by Boyden chamber | likely independent of CB1, CB2, TRPV1 | Vaccani et al. [95] |
| CBD | U251 | IC50 0.126 µM | 3 d | cell migration by Boyden chamber | Marcu et al. [145] | |
| CBD | U87 | 25 µM | 14 h | caspase activity, CaspACE Assay | activation of caspases 3, 8 and 9; glial cells not affected (concentrations up to 50 µM) – no ROS production, no increased GSH | Massi et al. [119] |
| 25 µM | 10 h | ELISA assay for cytochrome C | cytochrome c activation | Massi et al. [119] | ||
| 25 µM | 5 h | DCFH-DA, apoptosis | ROS activation | Massi et al. [119] | ||
| CBD | U87 | tumor growth, 5-LOX measurement, luekotriene B4 measurement | 5-LOX activation | Massi et al. [104] | ||
| U87 xenograft of athymic nude mice | 0.5 mg/mouse | 23 d | AEA and FAAH measurement | AEA degradation through FAAH; ↑ FAAH activity in vivo | Massi et al. [104] | |
| CBD | U251 | 1 µM | 3 d | invasion assay, western blot | Id1 downregulation ↑ mTOR, reduced PLCG1 | Soroceanu et al. [96] |
| primary tumor-derived cultures | 1-1.5 µM | 48 h | neurosphere formation | ¯Sox2 | Soroceanu et al. [96] | |
| U251 xenograft of athymic nude mice | 15 mg/kg | 4 w | tumor growth/95% reduction | downregulated Id-1 | Soroceanu et al. [96] | |
| CBD | glioma stem cell (GSC) line 3832 | IC50 3.5 µM | cell viability | ROS activation | Singer et al. [99] | |
| glioma stem cell (GSC) line 387 | IC50 2.6 µM | cell viability | ROS activation | Singer et al. [99] | ||
| GSC3832 and 387 xenografts | 15 mg/kg | 3-4 w | tumor growth, induced apoptosis | inhibited pAKT and Ki67, stimulated caspase-3, inhibits Id1 and Sox2 | Singer et al. [99] | |
| CBD | SF126 | IC50: 1.2 µM | 3 d | cell viability | Reduced Id1 | Marcu et al. [145] |
| U251 | IC50: 0.6 µM | 3 d | cell viability | Reduced Id1 | Marcu et al. [145] | |
| U87 | IC50: 0.6 µM | 3 d | cell viability | Reduced Id1 | Marcu et al. [145] | |
| CBD | T98 G, U87 MG, GL261 | 5.2–11.0 µM | 3 d | decreased cell viability | autophagy | Scott et al. [101] |
| CBD-rich extract | T98 G, U87 MG, GL261 | 10.0–11.0 µM | 3 d | decreased cell viability | autophagy | Scott et al. [101] |
| Δ9-THC | U87 MG | IC50: 1.5 µM | 3 d | cell viability (MTT) | autophagy and apoptosis | Torres et al. [128] |
| Δ9-THC | C6.9 glioma xenograft in mice | 500 µg/d THC | 8 d | Inhibition of tumor growth | Downregulation of MMP-2 | Blasquez et al. [130] |
| Δ9-THC | 2 glioblastoma multiforme patients | 1.29 or 1.46 mg/day | 30 and 26 d | Inhibition of tumor growth | Downregulation of MMP-2 | Blasquez et al. [130] |
| Δ9-THC | T98 G, U87 MG, GL261 | 12.0–14.0 µM | 3 d | decreased cell viability | autophagy | Scott et al. [101] |
| Δ9-THC | U87 MG | 6.0 µM | 8 h | Increased autophagy | ER stress pathway: upregulation of p8 and TRB3; inhibition of Akt and mTORC via TRB3 (this triggers autophagy) | Salazar et al. [124] |
| Δ9-THC | U87 MG xenografts | 15 mg/kg/d | increases autophagy | increases TRB3 expression, decreases S6 phosphorylation, active caspase-3 increased | Salazar et al. [124] | |
| Δ9-THC | T98 G, U87 MG, GL261 | 2–4 µM | 72 h | Cell viability | Increased Mdk and amphiregulin | Lorente et al. 2009 [189], Lorente et al. 2011 [190] |
| Δ9-THC | SF126 | IC50: 2.5 µM | 3 d | cell viability | Reduced Id1 | Marcu et al. [145] |
| U251 | IC50: 3.3 µM | 3 d | cell viability | Reduced Id1 | Marcu et al. [145] | |
| U87 | IC50: 3.3 µM | 3 d | cell viability | Reduced Id1 | Marcu et al. [145] | |
| Δ9-THC | U251 | IC50 85 nM | 3 d | cell migration by Boyden chamber | Marcu et al. [145] | |
| THC-rich extract | T98 G, U87 MG, GL261 | 11.0–13.0 µM | 3 d | decreased cell viability | autophagy | Scott et al. [101] |
| Δ9-THC:CBD | U251 | 0.1:0.1 µM | cell migration by Boyden chamber | No increase over THC or CBD alone | Marcu et al. [145] | |
| Δ9-THC:CBD | U251 | 0.4 µM: 1.7 µM | 3 d | apoptosis, downregulation of pERK | PARP, increased ROS and caspases 3, 7, 9 activity | Marcu et al. [145] |
| Δ9-THC:CBD | U87 MG | 0.7:0.7 µM | 72 h | decreased cell viability | enhanced apoptosis and autophagy | Torres et al. [128] |
| T98 G | 1.2:1.2 µM | 72 h | decreased cell viability | enhanced apoptosis and autophagy | Torres et al. [128] | |
| HG19 | 2.4:2.4 µM | 72 h | decreased cell viability | enhanced apoptosis and autophagy | Torres et al. [128] | |
| Δ9-THC:CBD | U87 MG xenograft | 7.5:7. 5 mg/kg/d | 6d | Reduction of tumor growth | Combo effect was stronger | Torres et al. [128] |
| Δ9-THC:CBD | T98 G | 10.0:10.0 µM | 4 h | pERK | Increase in pERK | Torres et al. [128] |
| Δ9-THC+TMZ | U87 MG | IC50: THC (1.8 µM):TMZ (100 µM) | 3 d | cell viability (MTT) | autophagy and apoptosis | Torres et al. [128] |
| Δ9-THC+CBD+TMZ | U87 MG | 0.9:0.9:75.0 µM | 72 h | decreased cell viability | enhanced apoptosis and autophagy | Torres et al. [128] |
| T98 G | 1.1:1.1:200.0 µM | 72 h | decreased cell viability | enhanced apoptosis and autophagy | Torres et al. [128] | |
| Δ9-THC+CBD+TMZ | U87 MG xenograft | 3.7:3.7:5.0 mg/kg/d | reduction in tumor growth | enhanced apoptosis and autophagy | Torres et al. [128] | |
| Δ9-THC+TMZ | T98 G xenograft | 15.0: 5.0 mg/kg/d | reduced tumor growth | caspase 3 activation | Torres et al. [128] | |
| THC:CBD:Radiation | T98 G, U87 MG, GL261 | 10.0:10.0 µM: 5 Gy | 4 h + 1 h | decreased cell viability | Decrease in pAKT2, pERK, Increased gamma-H2AX | Scott et al. [101] |
| THC:CBD:Radiation | GL261 xenograft | 4 mg/kg; 100.0:100.0 µM: 5 Gy | Decreased tumor growth, increased apoptosis and angiogenesis | Caspase 3 activation | Scott et al. [101] | |
| WIN-55,212-2 | C6.9 glioma | EC50: 14.7 µM | 48 h | decreased cell viability | Down-regulation of ERK1/2 and inhibition of AKT, activation of caspase 9 | Ellert-Miklaszewska et al. [131] |
| JWH-133 | C6.9 glioma xenograft in mice | 50 µg/day | 8 d | Inhibition of tumor growth | Downregulation of MMP-2 | Blasquez et al. [130] |
| KM-233 | U87 MG | 12 mg/kg/d | 20 d | 80% reduction of tumor | Change in phosphorylation profiles of MEK, ERK1/2, Akt, BAD, STAT3, and p70S6 K in U87 MG human GBM cells | Gurley et al. [168] |
| Human Leukemia | ||||||
| CBD | EL-4 cells (mouse lymphoma) | 2.5–10.0 µM | 24 h | decreased cell viability | Enhanced apoptosis, CB2 mediated | McKallip et al. [105] |
| Jurkat cells | 5.0 µM | 24 h | decreased cell viability | Decreased PARP, CB2 mediated | McKallip et al. [105] | |
| MOLT-4 cells | 2.5 µM | 24 h | decreased cell viability | Increased caspases, decreased PARP, increased cytochrome c, increased ROS, increased Nax4, p22 | McKallip et al. [105] | |
| EL-4 xenograft | 12.5 or 25 mg/kg | Decreased tumor growth | Apoptosis | McKallip et al. [105] | ||
| CBD | HL60 | IC50: 8.0 µM | 48 h | decreased cell viability | Enhanced apoptosis | Scott, Dalgleish, & Liu [147] |
| Δ9-THC | HL60 | IC50: 13.0 µM | 48 h | decreased cell viability | Enhanced apoptosis | Scott, Dalgleish, & Liu [147] |
| Δ9-THC | CEM, HL60, MOLT4 | IC50: CEM – 18 µM; HL60 – 14.0 µM; MOLT4 – 33.0 µM | 24 h | increased apoptosis | Reduced pERK | Liu et al. [143] |
| Δ9-THC | CEM | 1 µM | ↑ sensitivity to chemotherapeutic agents (increased effect of cytarabine, doxorubicin, and vincristine) | Reduced pERK | Holland et al. [106] | |
| Δ9-THC or CBD | CCRF-CEM (leukemia), CEM/VLB100 (multidrug resistant) | 10 µM | 72 h | ↑ cytotoxic effects of vinblastine (reduced IC50 of vinblastine by 3 fold in resistant cells) | Holland et al. [106] | |
| Δ9-THC or CBD | CEM/VLB100 cells | 10 µM | 72 h | ↑ P-glycoprotein substrate accumulation | ↓ P-glycoprotein expression | Holland et al. [106] |
| Δ9-THC:CBD | HL60 | IC50: 4.0 + 4.0 µM | 48 h | decreased cell viability | Enhanced apoptosis | Scott, Dalgleish, & Liu [147] |
| Human Lung Cancer | ||||||
| CBD | A549 | 0.1–10 µM | 24 – 72 h | ↓invasion | increased TIMP1 expression | Ramer et al. [108] |
| A549 xenograft | 5 mg/kg | 28 d | ↓metastasis | 84% inhibition of metastasis | Ramer et al. [108] | |
| CBD | A549, H460, H358 | from 0.1 µM | 48 – 72 h | ↓invasion | ↓PAI-1 secretion | Ramer et al. [109] |
| A549 xenograft | 5 mg/kg | 42 d | ↓tumor growth | ↓PAI-1 expression | Ramer et al. [109] | |
| CBD | A549, H460 | IC50: A549 – 3.47 µM; H460 – 2.80 µM | 2-48 h | decreased cell viability | ↑ PPARgamma, COX-2, PGE2, PGD2, 15d-PGJ2 | Ramer et al. [107] |
| primary tumor-derived cells | IC50 0.124 µM | increased PPARgamma, COX-2 | Ramer et al. [107] | |||
| A549 xenograft | 5 mg/kg | 72 d | reduced tumor volume | increased PPARgamma, COX-2 mRNA | Ramer et al. [107] | |
| CBD | MDA-MB231 and MDA-MB436 | IC50: 1.3 and 1.6 µM | 3 d | decreased cell viability, decreased cell migration (Boyden chamber) | Decreased Id1 expression | McAllister et al. [116] |
| Δ9-THC | MDA-MB231 and MDA-MB436 | IC50: 1.2 and 2.5 µM | 3 d | decreased cell viability, decreased cell migration (Boyden chamber) | Decreased Id1 expression | McAllister et al. [116] |
| Δ9-THC | A549, SW-1573 | up to 20 µM | 24 h | no effect on cell viability | Preet et al. [141] | |
| Δ9-THC | A549, SW-1573 | up to 20 µM | 72 h | induced apoptosis and inhibited proliferation | inhibiting the EGF-induced phosphorylation of ERK1/2, JNK1/2 and AKT | Preet et al. [141] |
| Δ9-THC | A549, SW-1573 | 10 µM | 72 h | inhibited migration | inhibiting the EGF-induced phosphorylation of ERK1/2, JNK1/2 and AKT | Preet et al. [141] |
| Δ9-THC | A549 xenograft | 5 mg/kg | 28 d | 50-60% reduction in tumor growth | Preet et al. [141] | |
| CBN | Lewis lung adenocarcinoma | 50 mg/kg | 20 d | increased animal survival | Manson et al. [149] | |
| Human breast cancer | ||||||
| CBD | MDA-MB231 | 1.5 µM | 3 d | ↓ invasion, ↓ metastasis | ↑ ROS, ↓ Id1, ↑ pERK | McAllister et al. [97] |
| 4T1 | IC50: 1.5 µM | McAllister et al. [97] | ||||
| 4T1 xenograft | 1 mg/kg | 15 d | decreased cell proliferation | ↓ G1/S transition, Id1 | McAllister et al. [97] | |
| 4T1 xenograft | 1 or 5 mg/kg | 15 d | reduced primary tumor growth and metastasis | McAllister et al. [97] | ||
| CBD | MCF-7 and ZR-75-1 (estrogen receptor +); SK-BR-3 (ER -) | 0–10 µM | 24 h | ↓ cell viability | Increased apoptosis, cytotoxicity seems to be independent of CB1, CB2, and TRPV1 receptors | Shrivastava et al. [98] |
| MDA-MB-231 | 5–7.5 µM | 16 h | ↓ cell viability | ↓ pAKT, ↓mTOR, ↓4EBP1, ↑PARP, ↑caspases, ↑t-Bid translocation (internal stimuli) | Shrivastava et al. [98] | |
| CBD | MDA-MB-231, 4T1 | IC50: 1.8–1.9 µM | 3 d | decreases cell proliferation | ↑ ROS, ↑ apoptosis, ↓ Id1 | Murase et al. [113] |
| CBD | 4T1 xenograft | 0.3–1.0 mg/kg | 6 w | ↓ cell viability, ↓ advanced stage metastasis | Murase et al. [113] | |
| CBD | SUM159, SCP2 | 9 µM | 48 h | ↓ cell proliferation, ↓ colony formation, ↓ migration | ↓ Nf-kB, ↓pEGFR, ↓pAKT | Elbaz et al. [112] |
| 4T1 and MVT-1 xenografts | 10 mg/kg | 3 w | inhibited tumor growth, angiogenesis and metastasis | decreased proliferative activity, vessel formation and p‐EGFR expression, lower activation of AKT, and ERK proteins | Elbaz et al. [112] | |
| CBD | MCF-7 | IC50: 8.2 µM | 4 d | decreases cell proliferation | Activation of CB and TRPV1 receptors | Ligresti et al. [153] |
| CBD-rich extract | MCF-7 | IC50: 6.0 µM | 4 d | decreases cell proliferation | Takeda et al. [151], Watanabe et al. [150] | |
| CBC | MCF-7 | IC50: 14.2 µM | 4 d | decreases cell proliferation | Ligresti et al. [153] | |
| CBG | MCF-7 | IC50: 9.8 µM | 4 d | decreases cell proliferation | Ligresti et al. [153] | |
| CBG | MDA-MB231 and MDA-MB436 | IC50: 2.3 and 2.1 µM | 3 d | decreased cell viability, decreased cell migration (Boyden chamber) | Decreased Id1 expression | McAllister et al. [116] |
| CBN | MDA-MB231 and MDA-MB436 | IC50: 1.2 and 2.6 µM | 3 d | decreased cell viability, decreased cell migration (Boyden chamber) | Decreased Id1 expression | McAllister et al. [116] |
| Δ9-THC | MCF-7 | EC50: 26.0 µM | inhibited 17beta-estradiol-induced proliferation | von Bueren et al. [136] | ||
| Δ9-THC | MCF-7 | 5 µM | 72 h | decreases cell proliferation | blocks of the G2-M transition; upregulates JunD | Caffarel et al. [137], Caffarel et al. [138] |
| Δ9-THC | MMTV-neu – Erb overexpressing mice | 0.5 mg/animal/day | 3 m | Decreased tumor growth and metastases | Downregulation of Act1 and Erb2 | Caffarel et al. [125] |
| Δ9-THC | MCF-7 | IC50: 14.2 µM | 4 d | decreases cell proliferation | Takeda et al. [151], Watanabe et al. [150] | |
| THC-rich extract | MCF-7 | IC50: 21.0 µM | 4 d | decreases cell proliferation | Takeda et al. [151], Watanabe et al. [150] | |
| CBN | MCF-7 | stimulated proliferation | HER2 upregulated | Takeda et al. [151], Watanabe et al. [150] | ||
| JWH-015 | MCF-7, MDA-MB-231 | 20 µM | 12 h | ↓ metastasis (hormone-sensitive breast cancer) | Inhibits CXCL12/CXCR4 signaling | Nasser et al. [163] |
| AEA | MCF-7 | IC50: 1.4 µM | 4 d | decreases cell proliferation | Via activation of CB1 receptor | Melck et al. [133]; Melck et al. [135] |
| AEA | MCF-7 | IC50: 1.0 µM | 3 d | decreases cell proliferation | Blocks transition from G1 to S | De Petrocellis et al. [134] |
| O-1663 | MDA-MB-231, 4T1 | IC50: 0.83–0.85 µM | decreases cell proliferation | More efficient than CBD in upregulating Id2 and survival, ↑autophagy | Murase et al. [113] | |
| O-1663 | 4T1 xenograft | 0.3–1 mg/kg | 6 w | ↑ survival | More efficient than CBD in upregulating Id2 and survival, ↑autophagy | Murase et al. [113] |
| JWH-018, JWH-073, JWH-122, JWH-210 | MCF-7 | 2–23 µM | 3 d | Anti-estrogenic properties in MCF-7 cells | Koller et al. [165] | |
| WIN55, 212–2 and JWH-133 | MDA-MB-231 | 10 µM | 24 h | decreases cell proliferation | Block G1 to S phase transition through COX-2/PGE-2 signaling pathway | Qamri et al. [157] |
| Human cervical cancer | ||||||
| CBD | HeLa, C33A | 10 µM | 24 h | ↓ invasion | induces TIMP1 | Ramer et al. [108] |
| CBD | HeLa | 10 µM | 24 h | decreases cell proliferation, increases apoptosis | Apoptosis: increased subG0/G1 and annexin V | Lukhele & Motadi [117] |
| Human prostate cancer | ||||||
| CBD, THC, CBG, CBC, CBN | LNCaP and DU-145 | IC50: 15–25 µM | 16 h | ↓ cell viability | ↓ IL-8, G1/S transition ↑ calcium, ROS, p53, p21, PUMA, tunel, CHOP, calcium | De Petrocellis et al. [118] |
| CBD, THC, CBG, CBC, CBN enriched cannabis extracts | LNCaP and DU-145 | IC50: 6–15 µM | 16 h | ↓ cell viability | De Petrocellis et al. [118] | |
| CBD + bicalutamide | LNCaP and DU-145 xenografts | 25 mg/kg | 4-5 w | ↓ tumor growth | De Petrocellis et al. [118] | |
| CBD, CBG, THC, CBC | DU-145 | IC50: 20–50 µM | 4 d | decreases cell proliferation | Activation of CB and TRPV1 receptors | Ligresti et al. [153] |
| anandamide, HU210, 2-AG | DU-145 | IC50: 0.1–0.3 µM | 3 d | Decreased proliferation | inhibits NGF, Trk, PRLr receptor expression | Melck et al. [133] |
| Human melanoma | ||||||
| Δ9-THC | CHL-1, A375 | 5 µM | 48 h | ↓ cell viability | ↑ autophagy | Armstrong et al. [146] |
| Δ9-THC | CHL-1 xenograft | 15 mg/kg | 20 d | ↓ tumor growth | ↑ apoptosis | Armstrong et al. [146] |
| Δ9-THC+CBD | CHL-1, A375 | 1 µM + 1 µM | 48 h | ↓ cell viability | ↑ apoptosis | Armstrong et al. [146] |
| Δ9-THC rich extract | CHL-1 xenograft | 7.5 mg/kg | 20 d | ↓ tumor growth | Armstrong et al. [146] | |
| WIN-55, 212–2 or JWH-133 | B16 xenograft | 50–120 mg/kg | 8-12 d | reduced malignant tumors | G1 cell cycle arrest via p-Akt inhibition and hypophosphorylation of the pRb retinoblastoma protein tumor suppressor | Blazquez et al. [158] |
| WIN-55, 212–2 or JWH-133 | B16 | 0.1–1 µM | 48 h | Anti-proliferative effect in epidermal cell lines (PDV.C57 and HaCa4) | G1 cell cycle arrest via p-Akt inhibition and hypophosphorylation of the pRb retinoblastoma protein tumor suppressor | Blazquez et al. [158] |
| Human Pancreatic Cancer | ||||||
| Δ9-THC | MIA PaCa2 | 2 µM | 66 h | decreases cell proliferation | CB2 receptor and ceramide-dependent upregulation of p8 and ATF-4 and TRB3 | Carracedo et al. [132] |
| Δ9-THC | MIA PaCa2 xenograft | 15 mg/kg | 15 d | ↓ tumor growth | ↑ apoptosis, increased TRB3 expression | Carracedo et al. [132] |
| JWH | MIA PaCa2 xenograft | 1.5 mg/kg | 15 d | ↓ tumor growth | ↑ apoptosis, increased TRB3 expression | Carracedo et al. [132] |
| ACEA, AM251, JWH-015, AM630 | MiaPaCa2 | 8.6–19.2 µM | 72 h | decreases cell proliferation | Receptor-independent; induced apoptosis; activation of JAC-STAT pathway | Fogli et al. [171] |
| Oral cancer | ||||||
| Δ9-THC, smoking cannabis | OSCC | Induced apoptosis in oral squamous cell carcinoma | Lopes et al. [140] | |||
| Thyroid carcinoma | ||||||
| JWH-133, WIN-55,212-2 | ARO | 1-2 µM | 24 h | Increased apoptosis | Induced apoptosis in ARO (anaplastic thyroid cancer cell line) and ARO-IL2 cells | Shi et al. [159] |
| JWH-133, WIN-55,212-2 | ARO xenograft | 50 µg/ml in 50 µl | 60 d | ↓ tumor growth | Shi et al. [159] | |
| Met-F-AEA | ARO, NPA | 5 µM | 72 h | Cell growth inhibition | Activation of p53 and p21 mediated pathway | Cozzolino et al. [155] |
| Bone cancer | ||||||
| WIN55,212-2 | NCTC-2472 xenograft | 0.5 mg/kg | 18 d | Increased apoptosis | Hald et al. [160] | |
| AM1241 | NCTC-2472 xenograft | 3 mg/kg | 10-14 d | Reduction in bone loss; reduced pain | Lozano-Ondoua et al. 2010 | |
| JWH-015 | 66.1 cells xenograft | 6 mg/kg | 18 d | Increased survival, decreased tumor-associated pain | Cytokine/chemokine suppression (of a mammary cell line implanted into the femur intermedullary space) | Lozano-Ondoua et al. [166] |
| Lymphoma | ||||||
| Win-55,212-2 | MCL | 5 µM | 24 h | Decreases cell viability | Flygare et al. [142] | |
| AEA | MCL | 5 µM | 24 h | Decreases cell viability | Flygare et al. [142] | |
| Δ9-THC | CEM, HL60, MOLT4 | 1 µM | 72 h | Induced apoptosis | MAPK/ERK pathway; reduces phosphorylated ERK expression; sensitizes to cytotoxic agents | Liu et al. [143] |
| R(+)-methanandamide and WIN-55,212–2 | MCL | 10 µM | 4 h | Induced apoptosis | Associated with ceramide accumulation and p38, depolarization of the mitochondrial membrane, and caspase activation | Gustafsson et al. [162] |
| Win-55,212-2 | MCL | 1.5–5.0 µM | 48 h | Paraptosis-like cell death | Wasik et al. [161] | |
| Colorectal Cancer | ||||||
| CBG | HCT 116 xenograft | 3-10 mg/kg | 10 d | Inhibits tumor growth | Likely via TRPM8 (CBG is an antagonist) (CB2 antagonists enhance the effect of CBG on cell viability) | Borrelli et al. [152] |
| Caco-2 and HCT 116 | 10 µM | 6-24 h | Decreases cell viability | Increased ROS | Borrelli et al. [152] |
The effects of CBD on cancer
CBD has been shown to have effects on glioblastoma, leukemia, lung cancer, breast cancer, cervical cancer, prostate cancer, and melanoma (see Table 2). CBD inhibits cell growth and migration, causes apoptosis of cancer cells and inhibits their invasion [95–98]. It also downregulates GSC self-renewal [99] and angiogenesis [96,100,101] in glioblastoma. Inhibition of angiogenesis was demonstrated on the level of cell proliferation and on the level of whole organisms in induced tumor models [102]. Multiple mechanisms of inhibition of angiogenesis were demonstrated, including cytostasis of endothelial cells, inhibition of endothelial cell migration, inhibition of invasion and sprouting in vitro and down-regulation of pro-angiogenic factors. All these effects are likely associated with a down-regulation of MMP2 and MMP9, urokinase-type plasminogen activator (uPA), endothelin-1 (ET-1), platelet-derived growth factor-AA (PDGF-AA) and chemokine (c-x-c motif) ligand 16 (CXCL16), molecules associated with angiogenesis [102].
The effect on CBD on cancer is most well studied in glioblastoma models. The following glioblastoma lines have been used for treatment with CBD: U87, U373, U251, GSC 3832, GSC 387, SF126, T98 G, U87 MG, and GL261. The IC50 varied from 0.125 to 5.0 µM. Similarly, CBD was used to treat athymic nude mice xenografted with U87, U251, GSC3832, or GSC387. The amounts of 15 mg/kg to 20 mg/kg (0.5 mg/mouse) administered over 18–28 days were shown to be effective in tumor reduction and prolonging the overall survival of treated animals [96,99,103,104] (Table 2).
CBD has been shown to downregulate leukemia cell viability and upregulate apoptosis, and these effects have also been observed in vivo where CBD downregulates tumor growth and upregulates apoptosis [105]. Treatment of leukemia with CBD upregulates the accumulation of P-glycoprotein substrate and increases cytotoxic effects of vinblastine [106]. CBD also inhibits the growth of EL-4 (mouse lymphoma), Jurkat, and MOLT-4 cells at concentrations of 2.5–10.0 µM [105]. In lung cancer, CBD has been shown to downregulate tumor invasion, metastasis and growth [107–109]. Invasion is inhibited in vitro and in vivo during CBD treatment of A549 lung cancer cells [110,111]. CBD downregulates colony formation and migration of cancer cells [112] and increases survival in a mouse model of human breast cancer [113]. In MDA-MB-231 breast cancer cells, CBD blocks the lisophosphatidylinositol-stimulated cell migration and invasion [114,115]. In mice xenografted with 4T1, a mammary metastatic cell line, CBD reduces a primary tumor size and lung metastasis [97,116]. In cervical cancer, invasion and metastasis are downregulated by CBD [108], and CBD halts cell proliferation and increases apoptosis (Figure 3) [117]. CBD downregulates cell viability and tumor growth in prostate cancer and affects the antitumor activity of first-line agents [118] (Table 2).
Figure 3.
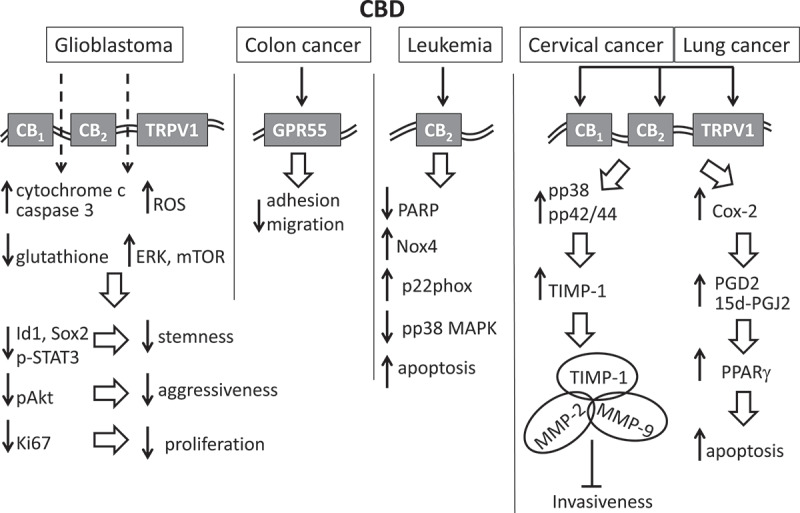
Molecular aspects of anti-cancer effects of CBD. The effect of CBD on glioblastoma is in part mediated through CB1, CB2 and TRPV1 receptors. This leads to the increased levels of cytochrome c, caspase 3 and ROS as well as decreased levels of glutathione and increase expression of ERK and mTOR. More downstream effects include: decrease in stemness due to the decrease in Id1, Sox2 and p-STAT3; decrease in cancer aggressiveness due to the decrease in pAkt; decrease in proliferation due to the decrease in Ki67. The effect on colon cancer is in part mediated through GPR55 which leads to the decrease in adhesion and migration. The effect on leukemia is mediated through CB2 receptor which leads to inhibition of PARP, increased levels of Nox4 and p22phox and reduced levels of pp38MAPK, resulting in increased frequency of apoptosis. In cervical cancer and lung cancer, CBD activates CB1, CB2 and TRPV1 receptors leading in part to upregulation of COX-2. The proapoptotic activity of CBD in lung cancer is associated with the upregulation of COX-2 and activation of PPARγ by the COX-2-derived products PGD2 and 15d-PGJ2. CBD exposure also leads to upregulation of pp38 and pp42/44 which in turn likely upregulates the TIMP-1 expression. TIMP-1 forms inhibitory complexes with MMP-2 and MMP-9 leading to inhibition of cancer invasiveness.
Massi et al. [119] suggest that CBD effects on glioblastoma are likely mediated through a CB1-, CB2-, and TRPV1-independent pathway involving the upregulation of caspase 3, cytochrome c, and ROS as well as a decrease in glutathione [119] (Figure 3). Many cancers have also been shown to overexpress GPR55 [78], and exposure of the metastatic colon cancer cell line HCT116 to cannabidiol reveals a substantial reduction in adhesion and migration, which is inhibited when GPR55 is knocked down by siRNA [120]. However, studies on CBD’s effects on leukemia, namely on EL-4 cells and Jurkat cells, have shown that the enhanced apoptosis and decreased PARP expression is CB2 receptor-dependent since the CB2-selective antagonist inhibits the reduction of cell viability [105]. It has been shown that CBD increases the levels of Nox4 and p22phox and decreases the amount of phosphorylated p38 MAPK (pp38). Similarly, Ramer et al. (2010a) have demonstrated that the CBD-induced antitumor effect is reversed by antagonists to CB1, CB2, and TRPV1 receptors in cervical and lung cancer [108]. CBD has also appeared to upregulate the tissue inhibitor of matrix metalloproteinases-1 (TIMP-1), and the siRNA-mediated knockdown of TIMP-1 reverses the effect of CBD on tumor cells invasiveness (Figure 3) [108]. In contrast to the report of McKallip et al. [105], CBD exposure resulted in the upregulation of pp38; both pp38 and pp42/44 appeared to be the upstream targets that likely regulate the TIMP-1 expression [108]. TIMP-1 inhibits cancer cell invasiveness by forming inhibitory complexes with MMP-2 and MMP-9 [111]. The proapoptotic activity of CBD is associated with the upregulation of COX-2 and activation of PPARγ by the COX-2-derived products PGD2 and 15d-PGJ2 [107]. Pretreating cancer cells with the PPARγ inhibitor GW9662 reduces cancer cell apoptosis. CBD also affects cell metabolism as indicated by changes in reactive oxygen species (ROS) produced in the mitochondria [17]. High levels of ROS production have been associated with triggering apoptosis [121], and CBD modulates ROS and ERK pathways to down-regulate Id-1 [97,116]. Furthermore, pretreatment with antioxidants N-acetyl cysteine and a-tocopherol diminishes the pro-apoptotic effect of CBD.
Moreover, CBD inhibits the self-renewal and stemness. Stem cell key regulators such as Id1, Sox2, and p-STAT3 are inhibited by CBD in a ROS-dependent manner. The transcriptional regulator Id-1 plays a critical role in modulating the invasiveness of glioblastoma; the overexpression of Id1 promotes a stem-like phenotype [96]. By silencing the marker of stemness, Sox2 has been shown to stop the proliferation and lead to the loss of tumorigenicity in vivo [122]. CBD also upregulates mTOR, reduces the expression of PLCG1 [96] and inhibits pAKT (a marker of glioblastoma aggressiveness) and Ki67 (a marker of proliferation) [99].
In vivo effects were shown to be mediated through the lipoxygenase (LOX) pathway, specifically through the lipoxygenase-catalyzed arachidonic acid metabolism [103,104,119]. Massi et al. (2008) have demonstrated the importance of LOX and COX pathways and the endocannabinoid system in controlling tumor growth. In vivo in the CBD-treated tumor tissues, CBD augments NRF2, an activator of the antioxidant response element (ARE) present in the promoters of ROS-detoxifying enzymes, including NQO1, GST, HMOX-1, and xCT [99]. xCT, a critical determinant of growth and invasion of cancer cells, regulates cysteine metabolism.
Finally, it is important to stress out that different processes of cancer growth and stages of cancer can be affected by cannabinoids in different manner. Proliferation, migration, invasiveness and metastases are affected differently. For example, recent work using CBD and synthetic CB ligands demonstrated that glioblastoma cell invasion can be inhibited in a receptor and cell type specific manner that is independent of proliferation and apoptosis [123].
The effects of THC treatment
Like CBD, THC has been shown to have effects on glioblastoma, breast cancer, oral cancer, lung cancer, lymphoma, and leukemia. THC inhibits proliferation, upregulates apoptosis, downregulates angiogenesis, induces autophagy, inhibits cell migration and metastasis. Notably, the THC-induced cell growth inhibitory effects and suppression of tumor growth in xenografted tumors have been achieved at higher concentrations when compared to the effects of CBD (Table 2). THC upregulates apoptosis, downregulates angiogenesis [101] and increases autophagy in glioblastoma [124]. It also shows pro-apoptotic, anti-proliferative, anti-angiogenic and anti-invasive effects in vitro and in vivo in breast cancer cells ErbB2 [125–127]. In glioblastoma, THC reduces cell viability and tumor growth; THC combined with the chemotherapeutic agent temozolomide (TMZ) enhances the antitumor activity of TMZ in tumor xenografts [128]. When combined with synthetic cannabinoids, THC has various effects on glioblastoma growth [129–132]. In breast cancer, Δ9-THC inhibits the proliferation of MCF7 and MCF7-AR1 cell lines [133–136], as well as the proliferation of estrogen receptor-negative/progesterone receptor-positive breast cancer cells [134,137–139]. In oral squamous cell carcinoma, Δ9-THC induces apoptosis [140]. In A549 and SW-1573 non-small cell lung cancer cells, THC inhibits the EGF-induced invasion, tumor growth, and lung metastasis in an in vivo mouse model injected with A549 cells [140,141]. Δ9-THC inhibits cell viability, increases apoptosis in lymphoma [142] and induces apoptosis when combined with other cytotoxic agents [143]. THC also increases the sensitivity of leukemia cells to chemotherapy [143].
THC’s anticancer effects appear to be directly related to its ability to bind to CB1, CB2, and other G protein-coupled receptors. In contrast to CBD, THC causes tumor cell death by directly engaging these receptors [119,144]. The activation of CB1/CB2 receptors triggers the accumulation of ceramide, activating autophagy and apoptosis [124]. The link between the THC-dependent activation of CB receptors and the activation of apoptosis through the ceramide pathway has been supported by the following experiments: pretreatment with CB1 and CB2 specific receptor antagonists SR141716 (SR1) and SR144528 (SR2), respectively, prevents apoptosis; the inhibition of serine palmitoyl-transferase which is an enzyme that catalyzes the first step of ceramide biosynthesis by ISP-1 also prevents apoptosis; and the pharmacologic or genetic inhibition of autophagy prevents the efficient induction of glioma cell death by THC [128]. Curiously, none of these events prevents the CBD-induced apoptosis. In contrast to the effect of CBD, the manipulation of the antioxidant machinery does have a significant effect on apoptosis in treatments with THC, further indicating that THC and CBD trigger glioma cell death through different pathways [128].
Regarding the molecular mechanisms of action, THC, like CBD, results in the downregulation of Id1 [145], the inhibition of the AKT and ERK [106,143] pathways, and an increase in caspase-3 levels [141]. Furthermore, THC, in some of the studies, down-regulates MMP-2, matrix metalloproteinase which promotes cell migration in glioblastoma [130], upregulates the components of the ER stress pathway, namely p8 and TRB3, decreases S6 phosphorylation, inhibits Akt and mTORC via TRB3, and reduces p70S6 kinase phosphorylation (mTORC1 substrate) by triggering autophagy in glioblastoma [124] (Figure 4). The activation of the ER stress pathway and autophagy appears to be a critical component of the inhibition of THC-induced tumor growth as autophagy-deficient tumors do not respond to the growth-inhibiting activity of THC. Additionally, exposure to ISP-1 prevents the THC-induced ER dilation, eIF2α phosphorylation, the upregulation of p8, ATF4, CHOP, and TRB3, and autophagy, thus further supporting the importance of the de novo synthesis and ceramide accumulation for the THC-induced action. Support for the role of eIF2α phosphorylation at Ser51 and the regulation of p8 expression and its downstream targets in response to THC has come from studies using eIF2α S51A knock-in mice; cells from these mice lack the upregulation of p8, ATF4, CHOP and TRB3 as well as autophagy in response to THC. Similarly, siRNAs against p8 are also effective in abrogating autophagy in response to THC. THC also induces caspase-3 in an ATG1-, ATG5-, and AMBRA1-dependent manner because the selective knockdown of these genes prevents caspase-3 activation and THC-induced apoptosis (Figure 4).
Figure 4.
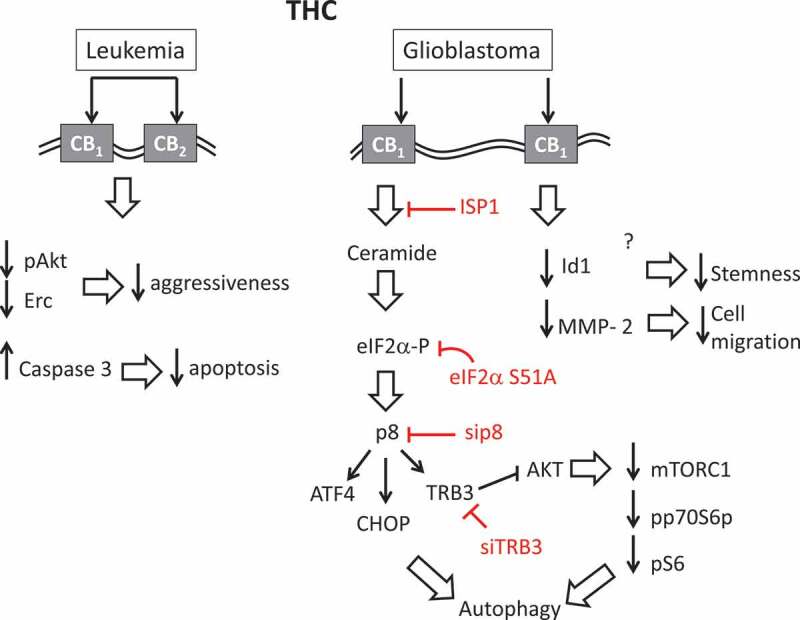
Molecular aspects of anti-cancer effects of THC. The effect of THC on leukemia is in part mediated through CB1, CB2 receptors. Binding of THC to these receptors leads to downregulation of pAct and Erc and decreases in cancer aggressiveness. The effect on glioblastoma is mediated through CB1 receptor. The activation of CB1 receptors triggers the accumulation of ceramide, leading to eIF2α phosphorylation, the upregulation of p8, ATF4, CHOP, and TRB3, and resulting in autophagy. TRB3 can also inhibit Akt, which in turn leads to the decreased in the expression of mTORC and reduction in the phosphorylated form of 70S6 kinase, also leading to autophagy. THC, in some of the studies, down-regulates Id1 leading to decrease in stemness and MMP-2 (matrix metalloproteinase) leading to the change in cell migration. ISP1, eIF2α S51A, sip8 and siTRB3 inhibit this process at multiple steps.
The effects of combining CBD and THC treatment
THC and CBD are definitely not similar in their influence on various cancers and cancer stages. Very little is known about it, however. Due to different levels of affinity of CBD and THC for cannabinoid receptors and effects on other receptors, combining CBD and THC in one treatment may be beneficial for cancer therapy. The effects of combination therapy with CBD and THC have been reported in glioblastoma and melanoma. Treating a U87 MG mouse xenografted with 7.5 mg/kg of THC and CBD has been demonstrated to be more effective in reducing tumor growth than THC or CBD alone [128]. The combination of CBD and THC enhances the antitumor effect of oral chemotherapy drug temozolomide (TMZ) on glioblastoma [128]. Using T98 G, U87 MG and GL261 glioblastoma cells, Scott et al. (2014) have demonstrated that adding a THC/CBD mixture potentiates the effect of gamma radiation on apoptosis [101]. However, in glioblastoma, combining 0.1 µM of THC and CBD downregulates cell viability and invasion and upregulates apoptosis, but this effect is not larger than the effect observed with either cannabinoid alone [145].
In melanoma, CBD combined with THC downregulates cell viability and tumor growth; 1.0 µM THC and CBD has a similar effect on apoptosis as 5.0 µM of CBD or THC alone [146]. More recently, treating HL60 leukemia cells with CBD and THC has revealed that they have a superior effect when used together compared to being used alone, and they are synergistic with the anti-leukemia drugs cytarabine and vincristine [147] (Table 2). The greatest induction of apoptosis occurs when chemotherapy is followed by cannabinoid administration. Combining CBD, THC and cytotoxic drugs sensitize leukemia to cytotoxic effects and reduce the therapeutic dose of anti-leukemia drugs.
The effects of CBN, CBG and CBC treatments
Cannabinol (CBN) is a weak agonist at CB1 and CB2 receptors and is a metabolite of Δ9-THC [148]. CBN has been demonstrated to increase the animal survival rate in mice xenografted with Lewis lung adenocarcinoma by 27% [149]. CBN inhibits the proliferation of MDA-MB231 and MDA-MB436 breast cancer cells by decreasing Id1 expression [116], but it increases the proliferation of human MCF-7 breast cancer cells through Her2 upregulation [150,151]. Cannabigerol (CBG), a less common phytocannabinoid than CBD and THC, has been shown to have an effect on colorectal cancer. It increases apoptosis in Caco-2 and HCT 116 cells through ROS production and selectively inhibits the growth of colorectal cancer in an HCT 116 xenograft likely via TRPM8 [152]. CBG has also shown its activity in breast and prostate cancer cells [153].
Cannabichromene (CBC) is another relatively rare cannabinoid that also exhibits the anticancer activity; the relatively high potency has been shown in breast cancer and prostate cancer. The IC50 for MCF-7 and MDA-MB-231 breast cancer cells and DU-145 prostate cancer cells were 14.2 and 20.4 µM [153]. CBC has been shown to have a stronger anticancer activity than THC but lower than CBG [153].
The effects of treatment with THC-A
THCA is a precursor of THC; it is abundant in cannabis but is converted to THC through heat-induced decarboxylation, a process that occurs during the combustion of cannabis flowers (smoking) or during the oil extraction and cooking processes to obtain cannabis-infused oils and edibles. Some data related to the effect of THCA on cancer are scarce, but THCA has been shown to decrease cell proliferation in MCF-7 and MDA-MB-231 breast cancer cells and DU-145 prostate cancer cells, with IC50 9.8 µM, 18.2 µM and approximately 25 µM, respectively [153].
The effects of endocannabinoids and cannabinoid analogues
Endocannabinoids demonstrate a higher efficacy in binding to cannabinoid receptors than phytocannabinoids. Both AEA and 2-AG have been shown to be more active than Δ9-THC at CB1 and CB2 receptors (reviewed in [41]). Among endocannabinoids, the effect of AEA on cancer cells is better studied than the effect of 2-AG. AEA has been shown to be active against the human breast cancer line MCF7 [133–135] as well as the human prostate cancer line DU-145 [133], with an IC50 of 1.0–1.4 µM. Specifically, AEA inhibits the proliferation of HBCC breast cancer cells and prostate cancer DU-145 cells by down-regulation of prolactin receptor PRL [133]. AEA inhibited the proliferation of MCF-7 and EFM-19 cells with IC50 values between 0.5 and 1.5 μM; AEA did not result in cell toxicity or apoptosis but rather due to the cell cycle arrest – reduction of cells in the S phase of the cell cycle [133–135]. Similarly, 2-AG has also been able to inhibit the proliferation of human breast cancer MCF7 cells and also through the long-form PRL receptor [133]. In addition, 2-AG inhibited proliferation of pancreatic ductal adenocarcinoma cells and promoted an immunosuppressive microenvironment via increasing the suppressive immune cell population of myeloid-derived suppressor cells [154]. AEA at a concentration of 5.0 µM reduces the growth of MCL lymphoma cells [142].
A metabolically stable analogue of anandamide, Met-F-AEA, has been shown to be active against NPA and ARO thyroid carcinoma cell lines where it causes a 50% decrease in cell growth at a concentration of 5.0 µM [155].
Several synthetic cannabinoids have been developed, and both their affinity for cannabinoid receptors and their activity in cancer cell lines have been tested. For example, HU-210, CP55940 and R-(+)-WIN55212 synthetic cannabinoids have been shown to have a higher CB1 and CB2 agonist efficacy than Δ9-THC. WIN-55,212–2 is an aminoalkylindole derivative with a chemical formula completely different from endocannabinoids. WIN-55,212–2 is a highly potent CB1, CB2 and TRPV1 receptor agonist [156] that has demonstrated its activity against C6.9 glioma cells [131], MDA-MB-231 breast cancer cells at 10.0 µM [157], human melanoma B16 cells [158], ARO cells of thyroid cancer [159], an NCTC-2472 bone cancer cell xenograft [160], and MCL lymphoma cells [142,161], with IC50 s ranging from 1.0 to 15.0 µM. WIN-55,212–2 and R(+)-methanandamide have been shown to induce apoptosis in MCL lymphoma cells [162], and WIN-55,212–2 also causes paraptosis in MCL lymphoma cells [161].
JWH-015 [163] and JWH-133 [164] are CB2 selective agonists (over 200-fold over CB1). JWH-133 has been shown to inhibit C6.9 glioma xenograft growth [130] and ARO cell thyroid carcinoma in mice [159]. Other chemicals in the JWH series, JWH-015, JWH-018, JWH-073, JWH-122 and JWH-210 also exhibit antiestrogenic and anticancer properties, including inhibiting growth in MCF-7 and MDA-MB-231 breast cancer [163,165], MIA PaCa2 pancreatic cell xenografts [132] and 66.1 bone cancer xenografts [166]. Like THC, JWN-133 appears to regulate tumor growth through the ceramide biosynthetic pathway and downregulate MMP-2 because the application of fumonisin B1 appears to abrogate tumor growth and regulate the aforementioned pathways.
KM-233 is a structural analog of Δ8-THC with an affinity for CB2 and CB1 receptors; the affinity to the former is greater by approximately13-fold [167]. KM-233 reduces the growth of U87 MG glioma xenografts by 80% over the 20-day application period [168]. Its action appeared to be similar to the activity of THC; treating U87 MG glioma cells with KM-233 causes a time-dependent change in the phosphorylation profiles of MEK, ERK1/2, Akt, BAD, STAT3 and p70S6 K, as well as an increase in cleaved caspase 3 [168].
Other analogues include O-1663, Hu-210 and arachidonyl-2ʹ-chloroethylamide (ACEA). O-1663 is a non-classical cannabinoid and a derivative of CBD. Its activity was found to be higher than that of CBD in inhibiting the growth of MDA-MB-231 and 4T1 breast cancer cells, reducing tumor growth and preventing metastases in a 4T1 xenograft model [113]. Hu-210 is a synthetic cannabinoid with an activity up to 800-fold higher than that of THC [169]. Hu-210 functions as a CB1/CB2 agonist and inhibits DU-145 prostate cancer cell growth at lower concentrations than anandamide [133]. Arachidonyl-2ʹ-chloroethylamide (ACEA) is a synthetic preferential agonist of the CB1 receptor [170]. ACEA decreases the proliferation of MIA PaCa2 pancreatic cancer cells [171].
Comparing the effects of pure cannabinoids and cannabis extracts
A great majority of the data on the effects cannabinoids on cancer are obtained from experiments that use phytocannabinoids, endocannabinoids and synthetic cannabinoids in their pure form. Very few reports have demonstrated the effect of actual cannabis extracts on cancer cells. Armstrong et al. (2015) have demonstrated that THC-enriched extracts are nearly twice as efficient in treating CHL-1 xenografts in mice than pure THC; a comparable reduction in tumor growth has been achieved with 7.5 mg/kg of THC-enriched extracts and 15 mg/kg of THC only [146]. Similarly, the effect of cannabis extract enriched with ~70% of CBD is more potent in inhibiting cell proliferation than a pure CBD compound [153]. Finally, in a reasonably comprehensive study, the effect of CBD, THC, CBN, CBG, CBC, CBDV, THCV, CBGV, THCA, THCVA and CBGA cannabinoids has been compared to the effect of cannabis extracts enriched with the corresponding cannabinoids; it has been observed that the enriched extracts have a significantly stronger effect on the growth of DU-145 and LNCaP human prostate cancer cells [118]. More recently, the effect of 12 different extracts was tested on 12 different cancer cell lines; there was a positive correlation observed between the level of THC in extracts and the anti-cancer properties of these extracts on A549 adenocarcinomic human alveolar basal epithelial cells [172]. Comparison to the effect of pure THC demonstrated that most of the extracts had much stronger anti-cancer effect [172]. Thus, it appears that the anticancer properties of extracts are more potent than those of pure cannabinoids.
The effect of full extracts in comparison to purified cannabinoids can be explained by the presence of other molecules, such as terpenoids, flavonoids, amino acids, sugars and other molecules. The additional effect of molecules besides cannabinoids is often called “the entourage effect.” It is believed that this effect is mainly caused by terpenoids, but this is not well documented. Terpenoids alone exhibit anti-cancer properties. Limonene inhibits the development of chemically induced rodent cancers, including skin, liver and mammary gland [173], induces autophagy [174] and activates immune system by inducing NO production (reviewed in [173]). Little to no information exists on potential synergism between cannabinoids and terpenoids in terms of their anti-cancer properties. One report demonstrated that CBN capacity to inhibit breast cancer cell growth by the inhibition of breast cancer resistance protein (BCRP, an ABC transporter) was potentiated by limonene [175]. On the other hand, botanical extract prepared from dry flowers was more potent for the inhibition of triple-negative breast cancer cells in vitro and in vivo as compared to pure THC, and reconstitution experiment using pure THC supplemented with top five most abundant terpenoids (β-caryophyllene, linalool, α-humulene, nerolidol 1 and β-pinene), present in the extract did not demonstrate an additive effect [176]. Therefore, much more effort is needed to demonstrate whether the entourage effect is due to the presence of terpenoids or other molecules.
Clinical trials
Clinical trials have begun to determine the effects of cannabinoid formulations in humans. The results of very few clinical trials are published in order to conclude whether cannabis or/and cannabinoids are effective for treatment of cancer. The route of administration is important and can include direct tumor injection, ingestion, inhaling, transdermal and others. Bioavailability of cannabinoids and terpenoids directly depend on the mode of administration; ingestion results in as a little as 6–20% of all active compounds reaching bloodstream [177], whereas upon inhaling 10–60% becomes bioavailable [178,179]. Onset time and duration of action also differ; whereas inhalation leads to the onset within minutes and lasts only couple hours, oral administration results in onset at 60–90 min with the effects lasting 6–8 h [180]. Therefore, direct comparison of the effectiveness of specific formulation is only possible if the administration route is similar.
An early Phase 1 study was conducted in which recurrent glioblastoma patients who had failed standard therapy were administered THC intracranially; under these conditions, THC administration was safe and no significant adverse effects were reported [181]. Since the analysis of the safety of intracranial THC administration was the primary goal of the study, no comparison group between THC treatment and controls were established to demonstrate the efficacy. Some patients responded to THC treatment, however, overall, no significant clinical benefit was observed; samples obtained pre- and post-treatment indicated that the mechanisms previously defined in mouse models were activated in these patients [181].
A Phase 2 safety study of Sativex in combination with TMZ (NCT01812603) was conducted in patients with grade 4 glioblastoma multiforme exposed to the maximum tolerated dose of Sativex, a spray that contained a 1:1 ratio of Δ9-THC and CBD. The study has confirmed the feasibility and safety of individualized dosing and provided preliminary evidence that Sativex combined with temozolomide offers some efficacy in patients with recurrent glioblastoma multiforme because the one-year survival rate has been higher in the treatment group than placebo [182]. Since many glioblastoma patients display a resistance to TMZ [183], this is likely clinically relevant.
Other studies have investigated the safety of synthetic cannabinoid formulations in patients with solid tumors (NCT01489826, NCT02423239), but these studies have not yet been completed. A double-blind, placebo-controlled clinical trial has been set to begin the investigation of the effects of cannabis for pain and inflammation in patients receiving radiation therapy for lung cancer (NCT02675842). The results of upcoming clinical trials will help guide future research in the field of cannabinoids and cancer treatment in humans.
Conclusions
Cannabinoids have great promise. Although the exact mechanism is not understood, in some models, cannabinoids have been shown to decrease cancer cell growth and invasion, and similar effects have been observed in mouse models. More research is necessary to investigate whether there is a role for cannabinoids in treatment of cancer in humans. For example, T98 G has much higher MGMT expression and methylation levels and is much more resistant to TMZ than U87 MG; nevertheless, combining THC with TMZ greatly reduces tumor growth in T98 G xenografts, while no effect has been observed in either therapy alone [128].
It appears that an increase in the CB1 and CB2 receptor expression may be beneficial for cancer cell response to phytocannabinoids. Therefore, chemicals that increase the density of these receptors in cancer cells may function synergistically with cannabinoids, such as THC and CBD. However, caution needs to be exercised because both THC and CBD have the activity of partial antagonists, and in some cancers, an overall increase in the CB1/CB2 expression may increase an antagonistic effect of THC/CBD, leaving the agonistic effect intact. It should also be noted that repeated exposure to THC may lower the density of cannabinoid receptors in neurons, which is especially noticeable for the CB1 receptor, resulting in tolerance and a reduced efficiency of THC in triggering a therapeutic effect [184]. It remains to be shown whether a similar effect is observed in cancer cells.
In addition, caution must be exercised as to the potential negative effect of cannabinoids on healthy tissues surrounding tumor or on entire organism. Due to the versatility of cannabinoids in binding multiple receptors and activating/inhibiting multiple signaling pathways, their effect on healthy tissues may be unpredictable and more studies are needed, using in vitro and in vivo models, to ensure that we are aware of all potential negative effects of these molecules when they are used in cancer therapy.
Immune system plays a critical role in prevention of malignization and immunotherapy nowadays is one of the therapies of choice for many cancers. Due to the modulating effect of CBD on immune system [185], it can potentially interfere with immunotherapy. Very few reports suggest that [186] and therefore studies are needed to demonstrate what immunotherapy drug if any can be used in combination with cannabinoids in general, and with CBD in particular, for treatment of cancer.
The use of cannabinoids to alleviate side-effects, such as lack of appetite, nausea and pain is also an important direction of research and potential clinical trials. Similarly, the use of cannabinoids for potentiation of the effects of chemo- or radiotherapy is also understudied; in this case, cannabinoids may allow to decrease the amount of chemotherapy drug or dose of radiation to achieve the same anti-cancer effect and at the same time to decrease potential side-effects.
Finally, it remains to be shown whether cancers in males and females respond to cannabis in a different manner and whether there are specific combinations of cannabinoids and terpenoids that are better suitable for females or males.
Funding Statement
We acknowledge NSERC CRD and MITACS grants to IK and OK.
Highlights
Endocannabinoids and phytocannabinoids can be used for cancer therapy
Cannabis extracts have stronger anti-tumor capacity than single cannabinoids
Combination of several cannabinoids may have more potent effect on cancer.
Author contributions
Authors contributed equally to data collection, analysis and manuscript preparation.
Disclosure statement
Authors are co-founders of Pathway Rx Inc, a research and development start-up company in the field of medical cannabis.
References
- [1].Fonseca BM, Teixeira NA, Correia-da-Silva G.. Cannabinoids as modulators of cell death: clinical applications and future directions. Rev Physiol Biochem Pharmacol. 2017;173:63–88. [DOI] [PubMed] [Google Scholar]
- [2].Fellermeier M, Eisenreich W, Bacher A, et al. Biosynthesis of cannabinoids. Incorporation experiments with (13)C-labeled glucoses. Eur J Biochem. 2001;268(6):1596–1604. [DOI] [PubMed] [Google Scholar]
- [3].Huang WJ, Chen WW, Zhang X. Endocannabinoid system: role in depression, reward and pain control (Review). Mol Med Rep. 2016;14(4):2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reggio PH. Endocannabinoid binding to the cannabinoid receptors: what is known and what remains unknown. Curr Med Chem. 2010;17(14):1468–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Le Boisselier R, Alexandre J, Lelong-Boulouard V, et al. Focus on cannabinoids and synthetic cannabinoids. Clin Pharmacol Ther. 2017;101(2):220–229. [DOI] [PubMed] [Google Scholar]
- [6].Aizpurua-Olaizola O, Soydaner U, Ozturk E, et al. Evolution of the cannabinoid and terpene content during the growth of cannabis sativa plants from different chemotypes. J Nat Prod. 2016;79(2):324–331. [DOI] [PubMed] [Google Scholar]
- [7].Russo EB, Taming THC.. Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258(5090):1946–1949. [DOI] [PubMed] [Google Scholar]
- [9].Mackie K. Cannabinoid receptor homo- and heterodimerization. Life Sci. 2005;77(14):1667–1673. [DOI] [PubMed] [Google Scholar]
- [10].Pacher P, Mechoulam R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res. 2011;50(2):193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mackie K. Cannabinoid receptors: where they are and what they do. J Neuroendocrinol. 2008;20(Suppl 1):10–14. [DOI] [PubMed] [Google Scholar]
- [12].Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459(7245):356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nogueras-Ortiz C, Yudowski GA. The multiple waves of cannabinoid 1 receptor signaling. Mol Pharmacol. 2016;90(5):620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Flores-Otero J, Ahn KH, Delgado-Peraza F, et al. Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat Commun. 2014;5:4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ahn KH, Mahmoud MM, Shim JY, et al. Distinct roles of beta-arrestin 1 and beta-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J Biol Chem. 2013;288(14):9790–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rozenfeld R, Devi LA. Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. Faseb J. 2008;22(7):2311–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chakravarti B, Ravi J, GR K. 2014 cannabinoid targets in cancer.pdf. Oncotarget. 2014;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Howlett AC, Mukhopadhyay S. Cellular signal transduction by anandamide and 2-arachidonoylglycerol. Chem Phys Lipids. 2000;108(1–2):53–70. [DOI] [PubMed] [Google Scholar]
- [19].Gifford AN, Makriyannis A, Volkow ND, et al. In vivo imaging of the brain cannabinoid receptor. Chem Phys Lipids. 2002;121(1–2):65–72. [DOI] [PubMed] [Google Scholar]
- [20].Normandin MD, Zheng MQ, Lin KS, et al. Imaging the cannabinoid CB1 receptor in humans with [11C]OMAR: assessment of kinetic analysis methods, test-retest reproducibility, and gender differences. J Cereb Blood Flow Metab. 2015;35(8):1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Van Laere K, Goffin K, Casteels C, et al. Gender-dependent increases with healthy aging of the human cerebral cannabinoid-type 1 receptor binding using [(18)F]MK-9470 PET. Neuroimage. 2008;39(4):1533–1541. [DOI] [PubMed] [Google Scholar]
- [22].Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. [DOI] [PubMed] [Google Scholar]
- [23].Felder CC, Glass M. Cannabinoid receptors and their endogenous agonists. Annu Rev Pharmacol Toxicol. 1998;38:179–200. [DOI] [PubMed] [Google Scholar]
- [24].Nunez E, Benito C, Pazos MR, et al. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse. 2004;53(4):208–213. [DOI] [PubMed] [Google Scholar]
- [25].Ramirez BG, Blazquez C, Gomez del Pulgar T, et al. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25(8):1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cabral GA, Marciano-Cabral F. Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J Leukoc Biol. 2005;78(6):1192–1197. [DOI] [PubMed] [Google Scholar]
- [27].Zoratti C, Kipmen-Korgun D, Osibow K, et al. Anandamide initiates Ca(2+) signaling via CB2 receptor linked to phospholipase C in calf pulmonary endothelial cells. Br J Pharmacol. 2003;140(8):1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Basu S, Dittel BN. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res. 2011;51(1):26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Basu S, Ray A, Dittel BN. Cannabinoid receptor 2 is critical for the homing and retention of marginal zone B lineage cells and for efficient T-independent immune responses. J Immunol. 2011;187(11):5720–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Burnette-Curley D, Cabral GA. Differential inhibition of RAW264.7 macrophage tumoricidal activity by delta 9tetrahydrocannabinol. Proc Soc Exp Biol Med. 1995;210(1):64–76. [DOI] [PubMed] [Google Scholar]
- [31].Robinson RH, Meissler JJ, Breslow-Deckman JM, et al. Cannabinoids inhibit T-cells via cannabinoid receptor 2 in an in vitro assay for graft rejection, the mixed lymphocyte reaction. J Neuroimmune Pharmacol. 2013;8(5):1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Karmaus PW, Chen W, Kaplan BL, et al. Delta9-tetrahydrocannabinol suppresses cytotoxic T lymphocyte function independent of CB1 and CB 2, disrupting early activation events. J Neuroimmune Pharmacol. 2012;7(4):843–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Console-Bram L, Marcu J, Abood ME. Cannabinoid receptors: nomenclature and pharmacological principles. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38(1):4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Muller C, Morales P, Reggio PH. Cannabinoid ligands targeting TRP channels. Front Mol Neurosci. 2018;11:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ryberg E, Larsson N, Sjogren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152(7):1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shen B, Estevez B, Xu Z, et al. The interaction of Galpha13 with integrin beta1 mediates cell migration by dynamic regulation of RhoA. Mol Biol Cell. 2015;26(20):3658–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Szallasi A. Vanilloid (capsaicin) receptors in health and disease. Am J Clin Pathol. 2002;118(1):110–121. [DOI] [PubMed] [Google Scholar]
- [38].Smart D, Gunthorpe MJ, Jerman JC, et al. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br J Pharmacol. 2000;129(2):227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].De Petrocellis L, Starowicz K, Moriello AS, et al. Regulation of transient receptor potential channels of melastatin type 8 (TRPM8): effect of cAMP, cannabinoid CB(1) receptors and endovanilloids. Exp Cell Res. 2007;313(9):1911–1920. [DOI] [PubMed] [Google Scholar]
- [40].De Petrocellis L, Ligresti A, Moriello AS, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163(7):1479–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Iwamura H, Suzuki H, Ueda Y, et al. In vitro and in vivo pharmacological characterization of JTE-907, a novel selective ligand for cannabinoid CB2 receptor. J Pharmacol Exp Ther. 2001;296(2):420–425. [PubMed] [Google Scholar]
- [43].Patel S, Hillard CJ. Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: further evidence for an anxiolytic role for endogenous cannabinoid signaling. J Pharmacol Exp Ther. 2006;318(1):304–311. [DOI] [PubMed] [Google Scholar]
- [44].Sim LJ, Hampson RE, Deadwyler SA, et al. Effects of chronic treatment with delta9-tetrahydrocannabinol on cannabinoid-stimulated [35S]GTPgammaS autoradiography in rat brain. J Neurosci. 1996;16(24):8057–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569(Pt 2):501–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kluger B, Triolo P, Jones W, et al. The therapeutic potential of cannabinoids for movement disorders. Mov Disord. 2015;30(3):313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cascio MG, Gauson LA, Stevenson LA, et al. Evidence that the plant cannabinoid cannabigerol is a highly potent alpha2-adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br J Pharmacol. 2010;159(1):129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Thomas A, Stevenson LA, Wease KN, et al. Evidence that the plant cannabinoid Delta9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146(7):917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bolognini D, Costa B, Maione S, et al. The plant cannabinoid Delta9-tetrahydrocannabivarin can decrease signs of inflammation and inflammatory pain in mice. Br J Pharmacol. 2010;160(3):677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Calakos KC, Bhatt S, Foster DW, et al. Mechanisms underlying sex differences in cannabis use. Curr Addict Rep. 2017;4(4):439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sherman BJ, McRae-Clark AL, Baker NL, et al. Gender differences among treatment-seeking adults with cannabis use disorder: clinical profiles of women and men enrolled in the achieving cannabis cessation-evaluating N-acetylcysteine treatment (ACCENT) study. Am J Addict. 2017;26(2):136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Penetar DM, Kouri EM, Gross MM, et al. Transdermal nicotine alters some of marihuana’s effects in male and female volunteers. Drug Alcohol Depend. 2005;79(2):211–223. [DOI] [PubMed] [Google Scholar]
- [54].Mathew RJ, Wilson WH, Davis R. Postural syncope after marijuana: a transcranial Doppler study of the hemodynamics. Pharmacol Biochem Behav. 2003;75(2):309–318. [DOI] [PubMed] [Google Scholar]
- [55].Kaufmann RM, Kraft B, Frey R, et al. Acute psychotropic effects of oral cannabis extract with a defined content of Delta9-tetrahydrocannabinol (THC) in healthy volunteers. Pharmacopsychiatry. 2010;43(1):24–32. [DOI] [PubMed] [Google Scholar]
- [56].Xie S, Borazjani A, Hatfield MJ, et al. Inactivation of lipid glyceryl ester metabolism in human THP1 monocytes/macrophages by activated organophosphorus insecticides: role of carboxylesterases 1 and 2. Chem Res Toxicol. 2010;23(12):1890–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tsuboi K, Uyama T, Okamoto Y, et al. Endocannabinoids and related N-acylethanolamines: biological activities and metabolism. Inflamm Regen. 2018;38:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sharma P, Murthy P, Bharath MM. Chemistry, metabolism, and toxicology of cannabis: clinical implications. Iran J Psychiatry. 2012;7(4):149–156. [PMC free article] [PubMed] [Google Scholar]
- [59].Harvey DJ, Mechoulam R. Metabolites of cannabidiol identified in human urine. Xenobiotica. 1990;20(3):303–320. [DOI] [PubMed] [Google Scholar]
- [60].Hryhorowicz S, Walczak M, Zakerska-Banaszak O, et al. Pharmacogenetics of cannabinoids. Eur J Drug Metab Pharmacokinet. 2018;43(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Narimatsu S, Watanabe K, Yamamoto I, et al. Sex difference in the oxidative metabolism of delta 9-tetrahydrocannabinol in the rat. Biochem Pharmacol. 1991;41(8):1187–1194. [DOI] [PubMed] [Google Scholar]
- [62].Tseng AH, Harding JW, Craft RM. Pharmacokinetic factors in sex differences in Delta 9-tetrahydrocannabinol-induced behavioral effects in rats. Behav Brain Res. 2004;154(1):77–83. [DOI] [PubMed] [Google Scholar]
- [63].Klumpers LE, Cole DM, Khalili-Mahani N, et al. Manipulating brain connectivity with delta(9)-tetrahydrocannabinol: a pharmacological resting state FMRI study. Neuroimage. 2012;63(3):1701–1711. [DOI] [PubMed] [Google Scholar]
- [64].Miller LK, Devi LA. The highs and lows of cannabinoid receptor expression in disease: mechanisms and their therapeutic implications. Pharmacol Rev. 2011;63(3):461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].De Laurentiis A, Araujo HA, Rettori V. Role of the endocannabinoid system in the neuroendocrine responses to inflammation. Curr Pharm Des. 2014;20(29):4697–4706. [DOI] [PubMed] [Google Scholar]
- [66].Galve-Roperh I, Chiurchiu V, Diaz-Alonso J, et al. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res. 2013;52(4):633–650. [DOI] [PubMed] [Google Scholar]
- [67].Ligresti A, Bisogno T, Matias I, et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology. 2003;125(3):677–687. [DOI] [PubMed] [Google Scholar]
- [68].Cianchi F, Papucci L, Schiavone N, et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor alpha-mediated ceramide de novo synthesis in colon cancer cells. Clin Cancer Res. 2008;14(23):7691–7700. [DOI] [PubMed] [Google Scholar]
- [69].Xu X, Liu Y, Huang S, et al. Overexpression of cannabinoid receptors CB1 and CB2 correlates with improved prognosis of patients with hepatocellular carcinoma. Cancer Genet Cytogenet. 2006;171(1):31–38. [DOI] [PubMed] [Google Scholar]
- [70].Sarfaraz S, Afaq F, Adhami VM, et al. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005;65(5):1635–1641. [DOI] [PubMed] [Google Scholar]
- [71].McKallip RJ, Nagarkatti M, Nagarkatti PS. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J Immunol. 2005;174(6):3281–3289. [DOI] [PubMed] [Google Scholar]
- [72].Nomura DK, Long JZ, Niessen S, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140(1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wang D, Wang H, Ning W, et al. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008;68(15):6468–6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Izzo AA, Aviello G, Petrosino S, et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J Mol Med (Berl). 2008;86(1):89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Orellana-Serradell O, Poblete CE, Sanchez C, et al. Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol Rep. 2015;33(4):1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sarfaraz S, Adhami VM, Syed DN, et al. Cannabinoids for cancer treatment: progress and promise. Cancer Res. 2008;68(2):339–342. [DOI] [PubMed] [Google Scholar]
- [77].Bifulco M, Laezza C, Portella G, et al. Control by the endogenous cannabinoid system of ras oncogene-dependent tumor growth. Faseb J. 2001;15(14):2745–2747. [DOI] [PubMed] [Google Scholar]
- [78].Andradas C, Caffarel MM, Perez-Gomez E, et al. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene. 2011;30(2):245–252. [DOI] [PubMed] [Google Scholar]
- [79].Carriba P, Ortiz O, Patkar K, et al. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology. 2007;32(11):2249–2259. [DOI] [PubMed] [Google Scholar]
- [80].Kearn CS, Blake-Palmer K, Daniel E, et al. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol Pharmacol. 2005;67(5):1697–1704. [DOI] [PubMed] [Google Scholar]
- [81].Navarro G, Carriba P, Gandia J, et al. Detection of heteromers formed by cannabinoid CB1, dopamine D2, and adenosine A2A G-protein-coupled receptors by combining bimolecular fluorescence complementation and bioluminescence energy transfer. ScientificWorldJournal. 2008;8:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rios C, Gomes I, Devi LA. mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148(4):387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Callen L, Moreno E, Barroso-Chinea P, et al. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J Biol Chem. 2012;287(25):20851–20865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hudson BD, Hebert TE, Kelly ME. Physical and functional interaction between CB1 cannabinoid receptors and beta2-adrenoceptors. Br J Pharmacol. 2010;160(3):627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Vinals X, Moreno E, Lanfumey L, et al. Cognitive impairment induced by Delta9-tetrahydrocannabinol occurs through heteromers between cannabinoid CB1 and serotonin 5-HT2A receptors. PLoS Biol. 2015;13(7):e1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Martinez-Pinilla E, Reyes-Resina I, Onatibia-Astibia A, et al. CB1 and GPR55 receptors are co-expressed and form heteromers in rat and monkey striatum. Exp Neurol. 2014;261:44–52. [DOI] [PubMed] [Google Scholar]
- [87].Moreno E, Cavic M, Krivokuca A, et al. The endocannabinoid system as a target in cancer diseases: are we there yet? Front Pharmacol. 2019;10:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Balenga NA, Martinez-Pinilla E, Kargl J, et al. Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. Br J Pharmacol. 2014;171(23):5387–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Coke CJ, Scarlett KA, Chetram MA, et al. Simultaneous activation of induced heterodimerization between CXCR4 chemokine receptor and cannabinoid receptor 2 (CB2) reveals a mechanism for regulation of tumor progression. J Biol Chem. 2016;291(19):9991–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Scarlett KA, White EZ, Coke CJ, et al. Agonist-induced CXCR4 and CB2 heterodimerization inhibits Galpha13/RhoA-mediated migration. Mol Cancer Res. 2018;16(4):728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Blasco-Benito S, Moreno E, Seijo-Vila M, et al. Therapeutic targeting of HER2-CB2R heteromers in HER2-positive breast cancer. Proc Natl Acad Sci U S A. 2019;116(9):3863–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Thors L, Bergh A, Persson E, et al. Fatty acid amide hydrolase in prostate cancer: association with disease severity and outcome, CB1 receptor expression and regulation by IL-4. PLoS One. 2010;5(8):e12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pagano E, Borrelli F, Orlando P, et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol Res. 2017;119:227–236. [DOI] [PubMed] [Google Scholar]
- [94].Pyszniak M, Tabarkiewicz J, Luszczki JJ. Endocannabinoid system as a regulator of tumor cell malignancy - biological pathways and clinical significance. Onco Targets Ther. 2016;9:4323–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Vaccani A, Massi P, Colombo A, et al. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br J Pharmacol. 2005;144(8):1032–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Soroceanu L, Murase R, Limbad C, et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res. 2013;73(5):1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].McAllister SD, Murase R, Christian RT, et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res Treat. 2011;129(1):37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Shrivastava A, Kuzontkoski PM, Groopman JE, et al. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol Cancer Ther. 2011;10(7):1161–1172. [DOI] [PubMed] [Google Scholar]
- [99].Singer E, Judkins J, Salomonis N, et al. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015;6:e1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Freimuth N, Ramer R, Hinz B. Antitumorigenic effects of cannabinoids beyond apoptosis. J Pharmacol Exp Ther. 2010;332(2):336–344. [DOI] [PubMed] [Google Scholar]
- [101].Scott KA, Dalgleish AG, Liu WM. The combination of cannabidiol and Delta9-tetrahydrocannabinol enhances the anticancer effects of radiation in an orthotopic murine glioma model. Mol Cancer Ther. 2014;13(12):2955–2967. [DOI] [PubMed] [Google Scholar]
- [102].Solinas M, Massi P, Cantelmo AR, et al. Cannabidiol inhibits angiogenesis by multiple mechanisms. Br J Pharmacol. 2012;167(6):1218–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Massi P, Vaccani A, Ceruti S, et al. Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J Pharmacol Exp Ther. 2004;308(3):838–845. [DOI] [PubMed] [Google Scholar]
- [104].Massi P, Valenti M, Vaccani A, et al. 5-Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor activity of cannabidiol, a non-psychoactive cannabinoid. J Neurochem. 2008;104(4):1091–1100. [DOI] [PubMed] [Google Scholar]
- [105].McKallip RJ, Jia W, Schlomer J, et al. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol Pharmacol. 2006;70(3):897–908. [DOI] [PubMed] [Google Scholar]
- [106].Holland ML, Panetta JA, Hoskins JM, et al. The effects of cannabinoids on P-glycoprotein transport and expression in multidrug resistant cells. Biochem Pharmacol. 2006;71(8):1146–1154. [DOI] [PubMed] [Google Scholar]
- [107].Ramer R, Heinemann K, Merkord J, et al. COX-2 and PPAR-gamma confer cannabidiol-induced apoptosis of human lung cancer cells. Mol Cancer Ther. 2013;12(1):69–82. [DOI] [PubMed] [Google Scholar]
- [108].Ramer R, Merkord J, Rohde H, et al. Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases-1. Biochem Pharmacol. 2010;79(7):955–966. [DOI] [PubMed] [Google Scholar]
- [109].Ramer R, Rohde A, Merkord J, et al. Decrease of plasminogen activator inhibitor-1 may contribute to the anti-invasive action of cannabidiol on human lung cancer cells. Pharm Res. 2010;27(10):2162–2174. [DOI] [PubMed] [Google Scholar]
- [110].Massi P, Solinas M, Cinquina V, et al. Cannabidiol as potential anticancer drug. Br J Clin Pharmacol. 2013;75(2):303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ramer R, Hinz B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J Natl Cancer Inst. 2008;100(1):59–69. [DOI] [PubMed] [Google Scholar]
- [112].Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol Oncol. 2015;9(4):906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Murase R, Kawamura R, Singer E, et al. Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. Br J Pharmacol. 2014;171(19):4464–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ford LA, Roelofs AJ, Anavi-Goffer S, et al. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br J Pharmacol. 2010;160(3):762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Nevalainen T, Irving AJ. GPR55, a lysophosphatidylinositol receptor with cannabinoid sensitivity? Curr Top Med Chem. 2010;10(8):799–813. [DOI] [PubMed] [Google Scholar]
- [116].McAllister SD, Christian RT, Horowitz MP, et al. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol Cancer Ther. 2007;6(11):2921–2927. [DOI] [PubMed] [Google Scholar]
- [117].Lukhele ST, Motadi LR. Cannabidiol rather than Cannabis sativa extracts inhibit cell growth and induce apoptosis in cervical cancer cells. BMC Complement Altern Med. 2016;16(1):335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].De Petrocellis L, Ligresti A, Schiano Moriello A, et al. Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: pro-apoptotic effects and underlying mechanisms. Br J Pharmacol. 2013;168(1):79–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Massi P, Vaccani A, Bianchessi S, et al. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell Mol Life Sci. 2006;63(17):2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kargl J, Andersen L, Hasenohrl C, et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br J Pharmacol. 2016;173(1):142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Gangemi RM, Griffero F, Marubbi D, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27(1):40–48. [DOI] [PubMed] [Google Scholar]
- [123].Hohmann T, Feese K, Greither T, et al. Synthetic cannabinoids influence the invasion of glioblastoma cell lines in a cell- and receptor-dependent manner. Cancers (Basel). 2019;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Salazar M, Carracedo A, Salanueva IJ, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119(5):1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Caffarel MM, Andradas C, Mira E, et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol Cancer. 2010;9:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhang H, Berezov A, Wang Q, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Ursini-Siegel J, Schade B, Cardiff RD, et al. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7(5):389–397. [DOI] [PubMed] [Google Scholar]
- [128].Torres S, Lorente M, Rodriguez-Fornes F, et al. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther. 2011;10(1):90–103. [DOI] [PubMed] [Google Scholar]
- [129].Blazquez C, Casanova ML, Planas A, et al. Inhibition of tumor angiogenesis by cannabinoids. Faseb J. 2003;17(3):529–531. [DOI] [PubMed] [Google Scholar]
- [130].Blazquez C, Salazar M, Carracedo A, et al. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008;68(6):1945–1952. [DOI] [PubMed] [Google Scholar]
- [131].Ellert-Miklaszewska A, Kaminska B, Konarska L. Cannabinoids down-regulate PI3K/Akt and Erk signalling pathways and activate proapoptotic function of bad protein. Cell Signal. 2005;17(1):25–37. [DOI] [PubMed] [Google Scholar]
- [132].Carracedo A, Gironella M, Lorente M, et al. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006;66(13):6748–6755. [DOI] [PubMed] [Google Scholar]
- [133].Melck D, De Petrocellis L, Orlando P, et al. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology. 2000;141(1):118–126. [DOI] [PubMed] [Google Scholar]
- [134].De Petrocellis L, Melck D, Palmisano A, et al. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci U S A. 1998;95(14):8375–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Melck D, Rueda D, Galve-Roperh I, et al. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999;463(3):235–240. [DOI] [PubMed] [Google Scholar]
- [136].von Bueren AO, Schlumpf M, Lichtensteiger W. Delta(9)-tetrahydrocannabinol inhibits 17beta-estradiol-induced proliferation and fails to activate androgen and estrogen receptors in MCF7 human breast cancer cells. Anticancer Res. 2008;28(1A):85–89. [PubMed] [Google Scholar]
- [137].Caffarel MM, Sarrio D, Palacios J, et al. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006;66(13):6615–6621. [DOI] [PubMed] [Google Scholar]
- [138].Caffarel MM, Moreno-Bueno G, Cerutti C, et al. JunD is involved in the antiproliferative effect of Delta9-tetrahydrocannabinol on human breast cancer cells. Oncogene. 2008;27(37):5033–5044. [DOI] [PubMed] [Google Scholar]
- [139].Laezza C, Pisanti S, Crescenzi E, et al. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006;580(26):6076–6082. [DOI] [PubMed] [Google Scholar]
- [140].Lopes CF, de Angelis BB, Prudente HM, et al. de Azambuja Ribeiro RI: concomitant consumption of marijuana, alcohol and tobacco in oral squamous cell carcinoma development and progression: recent advances and challenges. Arch Oral Biol. 2012;57(8):1026–1033. [DOI] [PubMed] [Google Scholar]
- [141].Preet A, Ganju RK, Groopman JE. Delta9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene. 2008;27(3):339–346. [DOI] [PubMed] [Google Scholar]
- [142].Flygare J, Gustafsson K, Kimby E, et al. Cannabinoid receptor ligands mediate growth inhibition and cell death in mantle cell lymphoma. FEBS Lett. 2005;579(30):6885–6889. [DOI] [PubMed] [Google Scholar]
- [143].Liu WM, Scott KA, Shamash J, et al. Enhancing the in vitro cytotoxic activity of Delta9-tetrahydrocannabinol in leukemic cells through a combinatorial approach. Leuk Lymphoma. 2008;49(9):1800–1809. [DOI] [PubMed] [Google Scholar]
- [144].Velasco G, Carracedo A, Blazquez C, et al. Cannabinoids and gliomas. Mol Neurobiol. 2007;36(1):60–67. [DOI] [PubMed] [Google Scholar]
- [145].Marcu JP, Christian RT, Lau D, et al. Cannabidiol enhances the inhibitory effects of delta9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol Cancer Ther. 2010;9(1):180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Armstrong JL, Hill DS, McKee CS, et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J Invest Dermatol. 2015;135(6):1629–1637. [DOI] [PubMed] [Google Scholar]
- [147].Scott KA, Dalgleish AG, Liu WM. Anticancer effects of phytocannbinoids used with chemotherapy in leukaemia cells can be improved by altering the sequence of their administration. Int J Oncol. 2017;51(1):369–377. [DOI] [PubMed] [Google Scholar]
- [148].Guindon J, Hohmann AG. The endocannabinoid system and cancer: therapeutic implication. Br J Pharmacol. 2011;163(7):1447–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Munson AE, Harris LS, Friedman MA, et al. Antineoplastic activity of cannabinoids. J Natl Cancer Inst. 1975;55(3):597–602. [DOI] [PubMed] [Google Scholar]
- [150].Watanabe K, Motoya E, Matsuzawa N, et al. Marijuana extracts possess the effects like the endocrine disrupting chemicals. Toxicology. 2005;206(3):471–478. [DOI] [PubMed] [Google Scholar]
- [151].Takeda S, Yamaorib SB, Motoyab E, et al. Delta(9)-tetrahydrocannabinol enhances MCF-7 cell proliferation via cannabinoid receptor-independent signaling. Toxicology. 2008;245(1–2):141–146. [DOI] [PubMed] [Google Scholar]
- [152].Borrelli F, Pagano E, Romano B, et al. Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a Cannabis-derived non-psychotropic cannabinoid. Carcinogenesis. 2014;35(12):2787–2797. [DOI] [PubMed] [Google Scholar]
- [153].Ligresti A, Moriello AS, Starowicz K, et al. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther. 2006;318(3):1375–1387. [DOI] [PubMed] [Google Scholar]
- [154].Qiu C, Yang L, Wang B, et al. The role of 2-arachidonoylglycerol in the regulation of the tumor-immune microenvironment in murine models of pancreatic cancer. Biomed Pharmacother. 2019;115:108952. [DOI] [PubMed] [Google Scholar]
- [155].Cozzolino R, Cali G, Bifulco M, et al. A metabolically stable analogue of anandamide, Met-F-AEA, inhibits human thyroid carcinoma cell lines by activation of apoptosis. Invest New Drugs. 2010;28(2):115–123. [DOI] [PubMed] [Google Scholar]
- [156].Patwardhan AM, Jeske NA, Price TJ, et al. The cannabinoid WIN 55,212-2 inhibits transient receptor potential vanilloid 1 (TRPV1) and evokes peripheral antihyperalgesia via calcineurin. Proc Natl Acad Sci U S A. 2006;103(30):11393–11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Qamri Z, Preet A, Nasser MW, et al. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol Cancer Ther. 2009;8(11):3117–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Blazquez C, Carracedo A, Barrado L, et al. Cannabinoid receptors as novel targets for the treatment of melanoma. Faseb J. 2006;20(14):2633–2635. [DOI] [PubMed] [Google Scholar]
- [159].Shi Y, Zou M, Baitei EY, et al. Cannabinoid 2 receptor induction by IL-12 and its potential as a therapeutic target for the treatment of anaplastic thyroid carcinoma. Cancer Gene Ther. 2008;15(2):101–107. [DOI] [PubMed] [Google Scholar]
- [160].Hald A, Ding M, Egerod K, et al. Differential effects of repeated low dose treatment with the cannabinoid agonist WIN 55,212-2 in experimental models of bone cancer pain and neuropathic pain. Pharmacol Biochem Behav. 2008;91(1):38–46. [DOI] [PubMed] [Google Scholar]
- [161].Wasik AM, Almestrand S, Wang X, et al. WIN55,212-2 induces cytoplasmic vacuolation in apoptosis-resistant MCL cells. Cell Death Dis. 2011;2:e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Gustafsson K, Christensson B, Sander B, et al. Cannabinoid receptor-mediated apoptosis induced by R(+)-methanandamide and Win55,212-2 is associated with ceramide accumulation and p38 activation in mantle cell lymphoma. Mol Pharmacol. 2006;70(5):1612–1620. [DOI] [PubMed] [Google Scholar]
- [163].Nasser MW, Qamri Z, Deol YS, et al. Crosstalk between chemokine receptor CXCR4 and cannabinoid receptor CB2 in modulating breast cancer growth and invasion. PLoS One. 2011;6(9):e23901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Marriott KS, Huffman JW. Recent advances in the development of selective ligands for the cannabinoid CB(2) receptor. Curr Top Med Chem. 2008;8(3):187–204. [DOI] [PubMed] [Google Scholar]
- [165].Koller VJ, Zlabinger GJ, Auwarter V, et al. Toxicological profiles of selected synthetic cannabinoids showing high binding affinities to the cannabinoid receptor subtype CB(1). Arch Toxicol. 2013;87(7):1287–1297. [DOI] [PubMed] [Google Scholar]
- [166].Lozano-Ondoua AN, Hanlon KE, Symons-Liguori AM, et al. Disease modification of breast cancer-induced bone remodeling by cannabinoid 2 receptor agonists. J Bone Miner Res. 2013;28(1):92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Krishnamurthy M, Ferreira AM, Moore BM 2nd:. Synthesis and testing of novel phenyl substituted side-chain analogues of classical cannabinoids. Bioorg Med Chem Lett. 2003;13(20):3487–3490. [DOI] [PubMed] [Google Scholar]
- [168].Gurley SN, Abidi AH, Allison P, et al. Mechanism of anti-glioma activity and in vivo efficacy of the cannabinoid ligand KM-233. J Neurooncol. 2012;110(2):163–177. [DOI] [PubMed] [Google Scholar]
- [169].Devane WA, Breuer A, Sheskin T, et al. A novel probe for the cannabinoid receptor. J Med Chem. 1992;35(11):2065–2069. [DOI] [PubMed] [Google Scholar]
- [170].Hillard CJ, Manna S, Greenberg MJ, et al. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther. 1999;289(3):1427–1433. [PubMed] [Google Scholar]
- [171].Fogli S, Nieri P, Chicca A, et al. Cannabinoid derivatives induce cell death in pancreatic MIA PaCa-2 cells via a receptor-independent mechanism. FEBS Lett. 2006;580(7):1733–1739. [DOI] [PubMed] [Google Scholar]
- [172].Baram L, Peled E, Berman P, et al. The heterogeneity and complexity of Cannabis extracts as antitumor agents. Oncotarget. 2019;10(41):4091–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Kuttan G, Pratheeshkumar P, Manu KA, et al. Inhibition of tumor progression by naturally occurring terpenoids. Pharm Biol. 2011;49(10):995–1007. [DOI] [PubMed] [Google Scholar]
- [174].Yu X, Lin H, Wang Y, et al. d-limonene exhibits antitumor activity by inducing autophagy and apoptosis in lung cancer. Onco Targets Ther. 2018;2018(11):1833–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Holland ML, Allen JD, Arnold JC.. Interaction of plant cannabinoids with the multidrug transporter ABCC1 (MRP1). Eur J Pharmacol. 2008;591(1–3):128–131. [DOI] [PubMed] [Google Scholar]
- [176].Blasco-Benito S, Seijo-Vila M, Caro-Villalobos M et al. Appraising the. “entourage effect”: antitumor action of a pure cannabinoid versus a botanical drug preparation in preclinical models of breast cancer. Biochem Pharmacol. 2018;157:285–293. [DOI] [PubMed] [Google Scholar]
- [177].Adams IB, Martin BR. Cannabis: pharmacology and toxicology in animals and humans. Addiction. 1996;91(11):1585–1614. [PubMed] [Google Scholar]
- [178].Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet. 2003;42(4):327–360. [DOI] [PubMed] [Google Scholar]
- [179].Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].MacCallum CA, Russo EB. Practical considerations in medical cannabis administration and dosing. Eur J Intern Med. 2018;49:12–19. [DOI] [PubMed] [Google Scholar]
- [181].Guzman M, Duarte MJ, Blazquez C, et al. A pilot clinical study of Delta9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95(2):197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Twelves C, Short S, Wright S. A two-part safety and exploratory efficacy randomized double-blind, placebo-controlled study of a 1: 1 ratio of the cannabinoids cannabidiol and delta-9-tetrahydrocannabinol (CBD: THC) plus dose-intense temozolomide in patients with recurrent glioblastoma multiforme (GBM). J clin oncol. 2017;35:2046. [Google Scholar]
- [183].Mrugala MM. Advances and challenges in the treatment of glioblastoma: a clinician’s perspective. Discov Med. 2013;15(83):221–230. [PubMed] [Google Scholar]
- [184].Sim-Selley LJ, Martin BR. Effect of chronic administration of R-(+)-[2,3-Dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxaz inyl]-(1-naphthalenyl)methanone mesylate (WIN55,212-2) or delta(9)-tetrahydrocannabinol on cannabinoid receptor adaptation in mice. J Pharmacol Exp Ther. 2002;303(1):36–44. [DOI] [PubMed] [Google Scholar]
- [185].Kaplan BL, Springs AE, Kaminski NE. The profile of immune modulation by cannabidiol (CBD) involves deregulation of nuclear factor of activated T cells (NFAT). Biochem Pharmacol. 2008;76(6):726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Taha T, Meiri D, Talhamy S, et al. Cannabis impacts tumor response rate to nivolumab in patients with advanced malignancies. Oncologist. 2019;24(4):549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Soderstrom K, Soliman E, Van Dross R. Cannabinoids modulate neuronal activity and cancer by CB1 and CB2 receptor-independent mechanisms. Front Pharmacol. 2017;8:720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Turcotte C, Blanchet MR, Laviolette M, et al. The CB2 receptor and its role as a regulator of inflammation. Cell Mol Life Sci. 2016;73(23):4449–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Lorente M, Carracedo A, Torres S, et al. Amphiregulin is a factor for resistance of glioma cells to cannabinoid-induced apoptosis. Glia. 2009 Oct;57(13):1374–1385. doi: 10.1002/glia.20856. PubMed PMID: 19229996. [DOI] [PubMed] [Google Scholar]
- [190].Lorente M, Torres S, Salazar M, et al. Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell Death Differ. 2011 Jun;18(6):959–73. doi: 10.1038/cdd.2010.170. Epub 2011 Jan 14. PubMed PMID: 21233844; PubMed Central PMCID: PMC3131933. [DOI] [PMC free article] [PubMed] [Google Scholar]