

Abstract
Friedreich’s Ataxia (FRDA) is an incurable genetic disease caused by an expanded trinucleotide AAG repeat within intronic RNA of the frataxin (FXN) gene. We have previously demonstrated that synthetic antisense oligonucleotides or duplex RNAs that are complementary to the expanded repeat can activate expression of FXN and return levels of FXN protein to near normal. The potency of these compounds, however, was too low to encourage vigorous pre-clinical development. We now report testing of “gapmer” oligonucleotides consisting of a central DNA portion flanked by chemically modified RNA that increases binding affinity. We find that gapmer antisense oligonucleotides are several fold more potent activators of FXN expression relative to previously tested compounds. The potency of FXN activation is similar to a potent benchmark gapmer targeting the nuclear noncoding RNA MALAT-1, suggesting that our approach has potential for developing more effective compounds to regulate FXN expression in vivo.
Keywords: Antisense oligonucleotide, Friedreich’s Ataxia, Frataxin, RNA, Gene activation
Graphical Abstract
1. Introduction
After many years of slow progress, synthetic oligonucleotides are beginning to have a major impact on clinical practice.1,2 In 2016, the antisense oligonucleotide Spinraza was approved for treatment of spinal muscular atrophy (SMA).3,4 The most severe form of SMA was invariably fatal and Spinraza has had a transformative impact on the lives of patients and their treatment. Spinraza also offered a convincing demonstration that relatively large, negatively charged oligonucleotides could be administered into the central nervous system, enter affected cells, modulate gene expression, and produce the desired physiologic effect without severe side effects.
Since the approval of Spinraza, several other antisense oligonucleotides and duplex RNAs have either been approved or had remarkable effects in clinical trials.5–9 Treatment groups ranges from thousands of patients to just one.5,10,11 The drug inclisiran has shown strikingly favorable efficacy and safety profiles in large scale clinical trials designed to lower cholesterol in patients who show an inadequate response to statins5. It is possible that inclisiran will be the first synthetic oligonucleotide prescribed to hundreds of thousands of individuals. At the other end of the spectrum, milasen is a synthetic antisense oligonucleotide that went from design to compassionate use clinical application in just a few months to treat a single patient with a devastating rare genetic disease.10
These successes have demonstrated the potential of synthetic nucleic acids as a therapeutic approach – a potential greater than even the most optimistic proponents would have imagined a decade previously. That success has set a high bar for the initiation of new clinical programs. Compounds must be carefully chosen to both fill a major unmet need and be potent enough in vivo to rival the favorable efficacy/safety profiles of the successful approved drugs and the promising candidates that are advancing in various company’s pipelines.
In this paper, we report progress towards improving the potency of antisense oligonucleotides designed to activate expression of human frataxin (FXN) as a potential treatment for Friedreich’s ataxia (FRDA).
FRDA is an inherited recessive genetic disorder caused by an expansion of the trinucleotide AAG within intron 1 of the FXN gene.12 This mutation does not affect the structure or sequence of FXN protein. Instead, it causes a decrease in FXN gene expression. Biochemical evidence suggests that the expanded AAG repeat binds to the complementary region of chromosomal DNA to form a DNA-RNA R-Loop that acts as a break on gene transcription.13 FXN protein expression is decreased by two thirds or more, causing the slowly progressing degenerative disease.
Our laboratory had been developing ASOs and dsRNAs to target other trinucleotide or hexanucleotide repeat expansions. We tested the hypothesis that synthetic nucleic acids designed to bind the expanded AAG repeat could block the mutant RNA, prevent R-loop formation, relieve the brake on gene expression, and promote increased expression of FXN RNA and FXN protein. We demonstrated that synthetic dsRNAs and ASOs complementary to the AAG repeat could activate expression of FXN protein to levels that occur in normal, non-mutant cells.14–17 We also observed activation by ss-siRNAs, compounds that are single-stranded RNAs yet act through the RNAi pathway.16
These compounds activated gene expression in both patient-derived fibroblast cells and in induced pluripotent stem cell (iPSC)-derived neuronal progenitor cells (Table 1).17 Activation was achieved in several patient-derived cell lines regardless of repeat length. This outcome suggest thats a single compound might be used to treat a broad spectrum FRDA patients.
Table 1.
Fibroblast and neural progenitor cell models.
| FA models | Age of Onset | # GAA Repeats | Type | |
|---|---|---|---|---|
| Allele 1 | Allele 2 | |||
| GM03816 | 36 | 330 | 380 | Fibroblasts |
| GM02153 | - | - | - | |
| F4259 | 15 | 340 | 690 | Neural Progenitor Cells |
Moving from a laboratory demonstration of activity to clinical testing requires that a development program have a favorable profile necessary to compete with development programs for other diseases. For example, it is beneficial for compounds to be tested for efficacy in animals. For anti-AAG ASOs, that testing is ongoing and results will be reported in due course. It is also important that compounds be as potent as possible. EC50 values for the compounds that block the AAG repeat showed good, but possibly inadequate potencies.17
In previous testing, we had used a potent benchmark ASO targeting the nuclear non-coding RNA MALAT-1. MALAT-1 is a good benchmark target because its expression can be reduced without detrimental effects. In contrast to our anti-AAG ASOs, that act as “steric blockers” to obstruct the AAG repeat, the anti-MALAT-1 compound is a gapmer consisting of a central DNA region flanked by chemically modified RNA bases that enhance binding affinity. The steric block ASOs were several-fold less potent than the anti-MALAT-1 gapmer.17
We hypothesized that anti-AAG gapmer oligonucleotides might possess improved potencies relative to steric block AAG ASOs because of the potential to induce cleavage of the RNA target. Here we report enhanced potencies that are comparable to anti-MALAT-1 ASOs. Cells grow normally with no evidence of toxicity. These data suggest that anti-AAG gapmers have favorable efficacies and may be a more promising route to compounds to treat FRDA.
2. Results
2.1. Design of gapmers
We had previously evaluated ASOs that possessed 2’- nucleotide modifications spread throughout the oligonucleotide. These ASOs were designed to block the expanded AAG repeat and prevent it from binding to chromosomal DNA and slowing transcription of the FXN gene.
We designed a new generation of oligonucleotide gapmers that consisted of a central DNA portion flanked by chemically modified RNA bases. The 2’-methoxyethyl (2’-O-MOE) or 2’,4’- linked constrained ethyl ((S)-cEt) modifications increase binding affinity, while the central DNA gap creates and RNA-DNA hybrid upon binding that can recruit RNase H and lead to cleavage of the target transcription (Fig 1). Both 2’-O-MOE and (S)-cET modifications act to reduce the conformational flexibility of the ribose, decrease the entropic penalty of hybridization, and increase the thermal stability of binding.1
Figure 1.
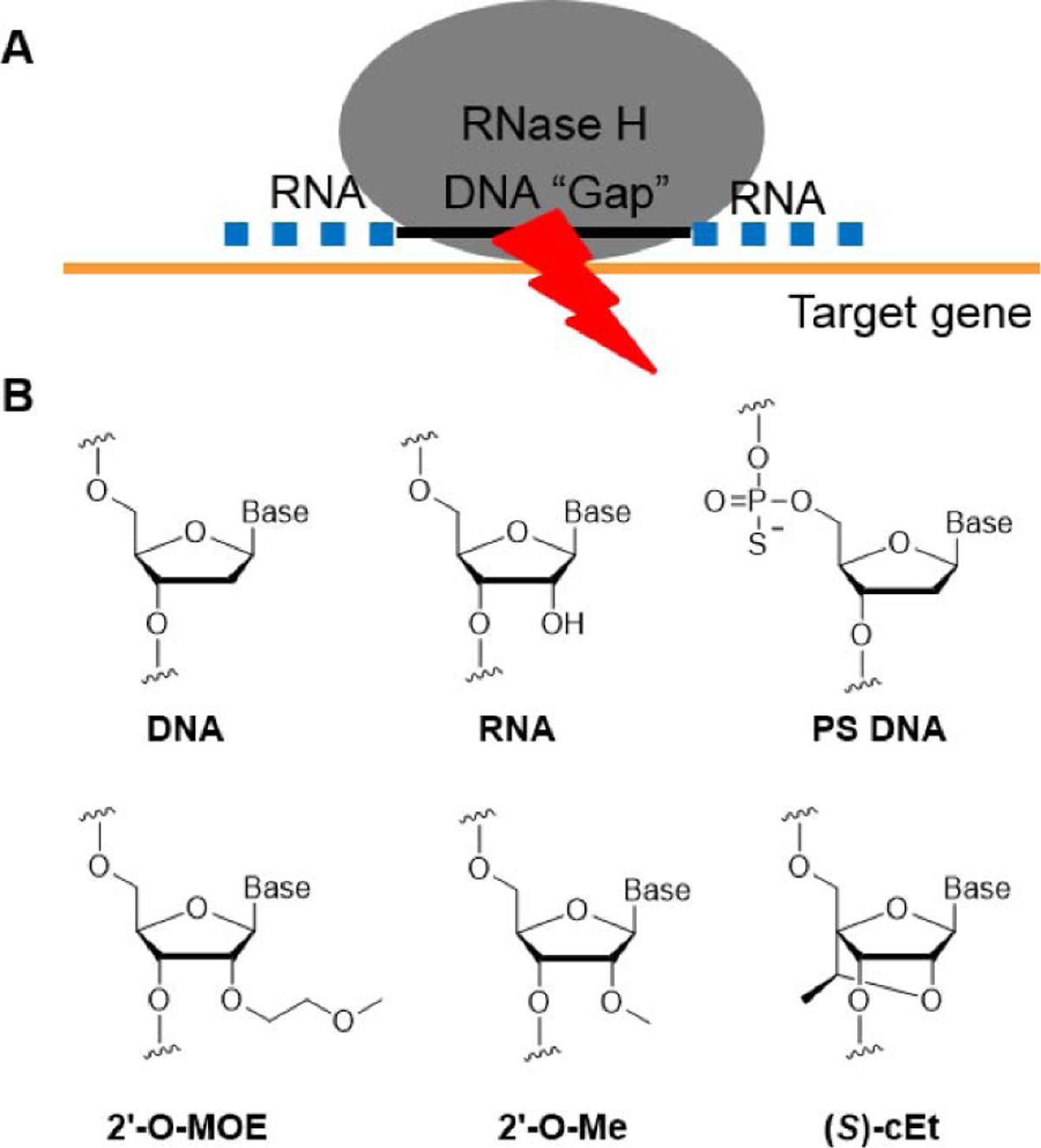
(A) Schematic of gapmer design and RNAse H recruitment; (B) Chemical modifications of gapmers in this study.
Nucleotides modified with (S)-cEt increase binding affinity more than 2’-methoxyethyl nucleotides. Therefore, we designed the flanking regions to contain a ten base central DNA region and either three nucleotide (S)-cEt (3-10-3) or five base 2’-O-MOE (5-10-5) flanking regions (gap 12–15, gap18–20, Table 2). In addition, we designed gapmers with phosphorothioate and phosphodiester mixed backbones to increase stability against endonucleases (gap 15–17, gap 21, 22 and 54).18 The introduction of a single 2′-O-methyl (2′-O-Me) modification at gap position 2 can reduce protein-binding, thus reducing hepatotoxicity and improving the therapeutic index, so we designed three gapmers (gap 55–57) with 2′-O-Me at gap position 2.19 Melting temperature determinations revealed similar values for all compounds, varying from 69°C to 79°C.
Table 2.
Sequences of 5-10-5 2’-O-MOE gapmers and 3-10-3 (S)-cEt gapmers that target the AAG repeat.
| No. | Chemistry | Sequence (5′−3′) | Tm (°C) |
|---|---|---|---|
| gap12 | MOE, DNA, PS | TTmCTTmCTTmCTTmCTTmCTTmCTT | 69.36 |
| gap13 | MOE, DNA, PS | mCTTmCTTmCTTmCTTmCTTmCTTmCT | 71.20 |
| gap14 | MOE, DNA, PS | TmCTTmCTTmCTTmCTTmCTTmCTTmC | 69.47 |
| gap 15 | MOE, DNA, PS, PO | TTmCTTmCTTmCTTmCTTmCTTmCT | 72.13 |
| gap16 | MOE, DNA, PS, PO | mCTTmCTTmCTTmCTTmCTTmCTTmCT | 74.80 |
| gap17 | MOE, DNA, PS, PO | TmCTTmCTTmCTTmCTTmCTTmCTTmC | 72.93 |
| gap18 | cET, DNA, PS | mCTTmCTTmCTTmCTTmCTTmC | 74.43 |
| gap19 | cET, DNA, PS | TmCTTmCTTmCTTmCTTmCTT | 75.95 |
| gap20 | cET, DNA, PS | TTmCTTmCTTmCTTmCTTmCT | 77.48 |
| gap21 | cET, DNA, PS, PO | mCTTmCTTmCTTmCTTmCTTmC | 79.06 |
| gap22 | cET, DNA, PS, PO | TmCTTmCTTmCTTmCTTmCTT | 77.21 |
| gap54 | cET, DNA, PS, PO | TTmCTTmCTTmCTTmCTTmCT | 79.13 |
| gap55 | cET, Me, DNA, PS | mCTTmCUTmCTTmCTTmCTTmC | 78.89 |
| gap56 | cET, Me, DNA, PS | TmCTTCTTmCTTmCTTmCTT | 75.08 |
| gap57 | cET, Me, DNA, PS | TTmCTUmCTTmCTTmCTTmCT | 78.86 |
2’-O-MOE, (S)-cET BNA, 2’-O-Me, DNA, mC: 5-methyl C, all PS backbone, Under line PO backbone
2.2. Evaluation of potency in FRDA patient-derived fibroblasts and neuronal progenitor cells
We began testing the gapmer ASOs using FRDA patient-derived fibroblast cell line GM03816 (330/380 repeats). ASOs were delivered to cells in complex with cationic lipid. Negative controls include siCM and CM-PO, a non-complementary duplex RNA and a non-complementary ASO, respectively. Positive controls include siGAA, an anti-AAG duplex RNA known to activate expression of FXN, and siExon-2, a duplex RNA that targets the coding region of FXN mRNA and represses FXN expression, according to our previous studies.14–17
Every repeat-targeted gapmer activated FXN protein expression (Fig 2) and RNA expression (Fig 3) 2–4 fold. This activation approximates the level found in the wild-type fibroblast lines. qPCR requires measurement of stably expressed gene for standardization, and it is useful to crosscheck results using more than one reference gene. Additional endogenous reference genes validated activation of FXN mRNA as assayed by qPCR (Supplemental Fig. 1).
Figure 2.
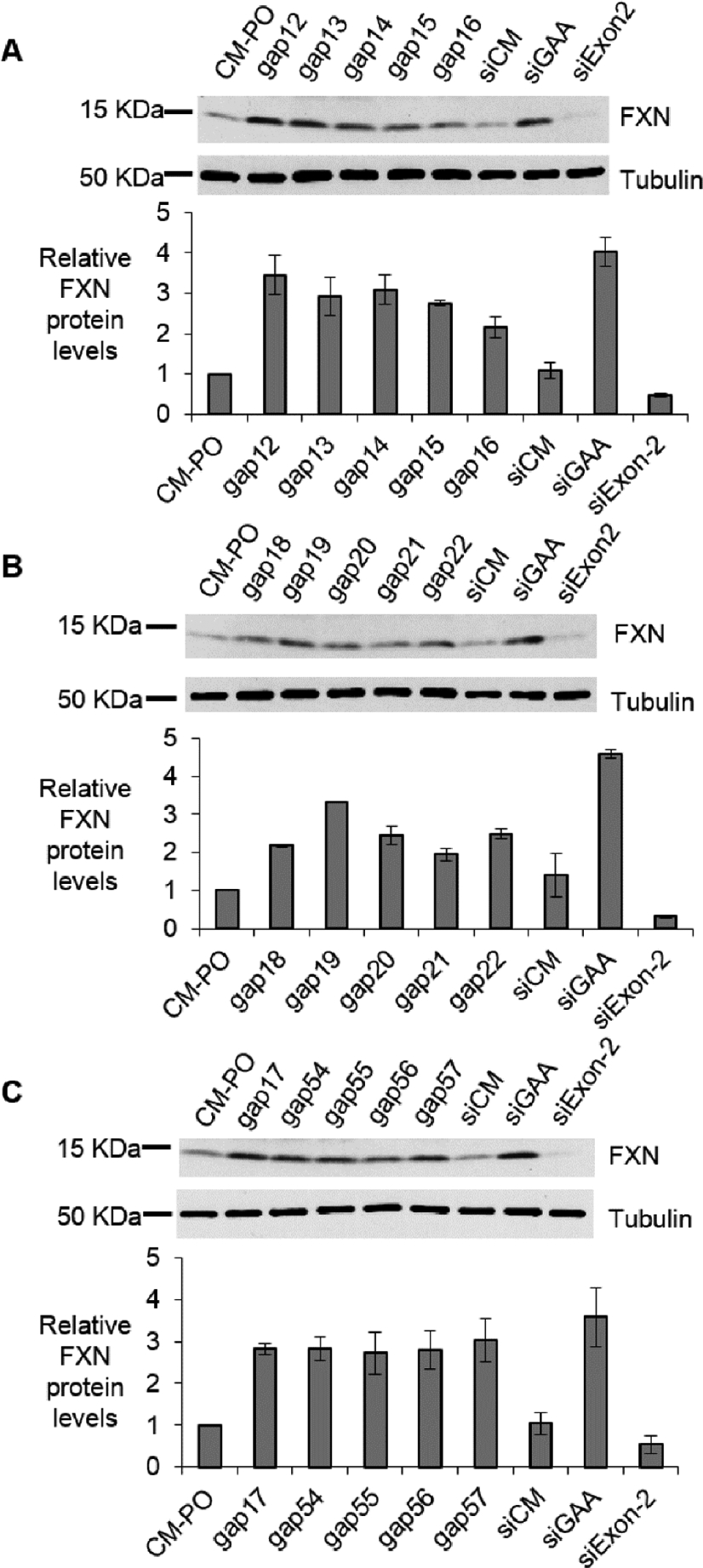
Activation of FXN protein expression in fibroblast line GM03816 by (A, C) 5-10-5 2’-O-MOE gapmers (12 nM), (B, C) 3-10-3 (S)-cEt gapmers (12 nM). Positive control duplex RNAs are used in 25 nM. All data are presented as ±STDEV (n=3).
Figure 3.
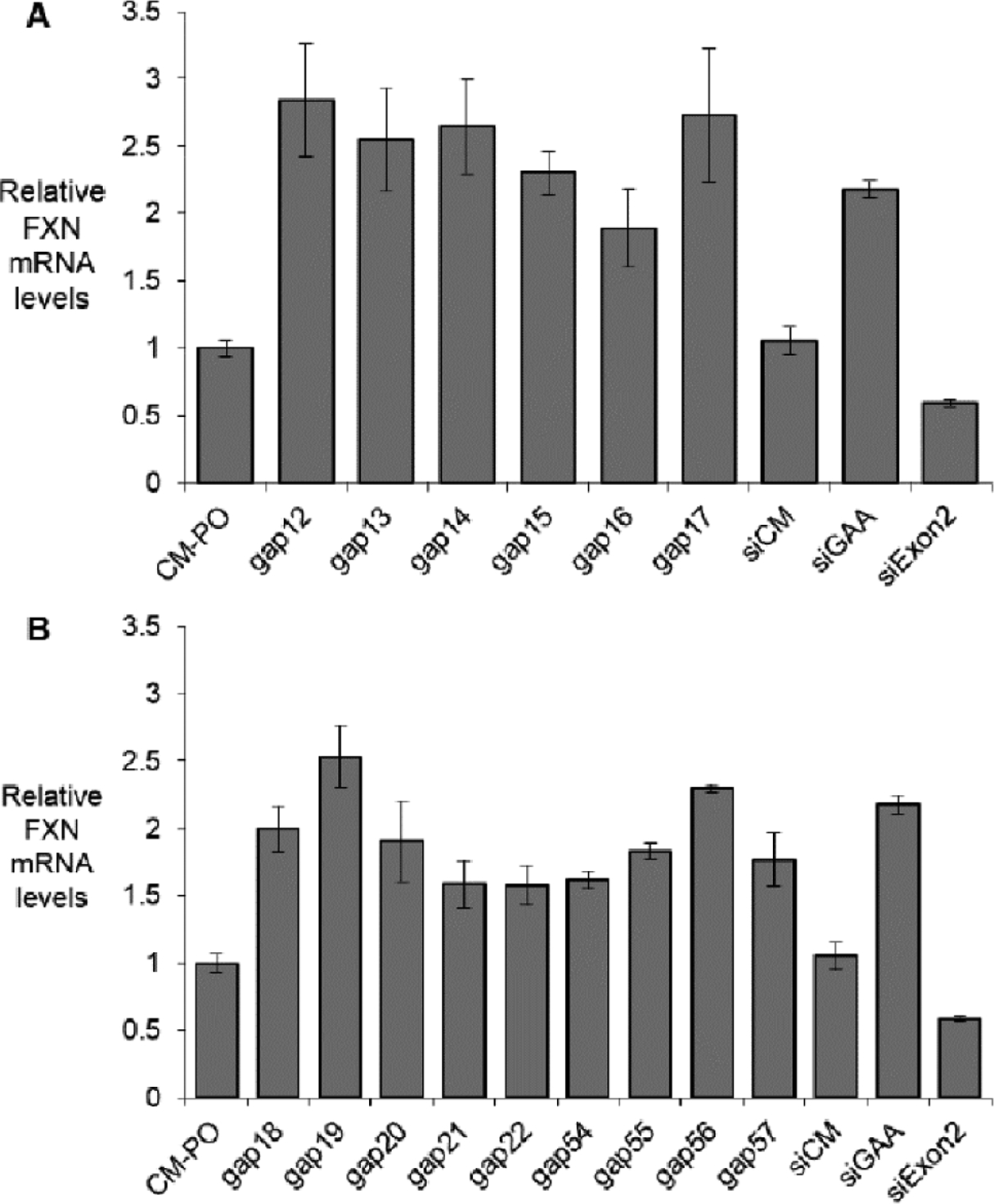
Activation of FXN mRNA expression in fibroblast line GM03816 by (A) 5-10-5 2’-O-MOE gapmers (12 nM), (B) 3-10-3 (S)-cEt gapmers (12 nM). Duplex RNAs are used in 25 nM. All data are presented as ±STDEV (n=3).
To better rank the candidate compounds we evaluated the potencies of three 3-10-3 (S)-cEt and three 5-10-5 2’-O-MOE gapmers at varied concentrations (Fig 4) in FRDA patient-derived fibroblast line GM03816. All potencies were determined in triplicate. For all six compounds, maximal activation was achieved at concentrations lower than 3 nM. Potencies were similar, ranging from 0.17 to 0.48 nM.
Figure 4.
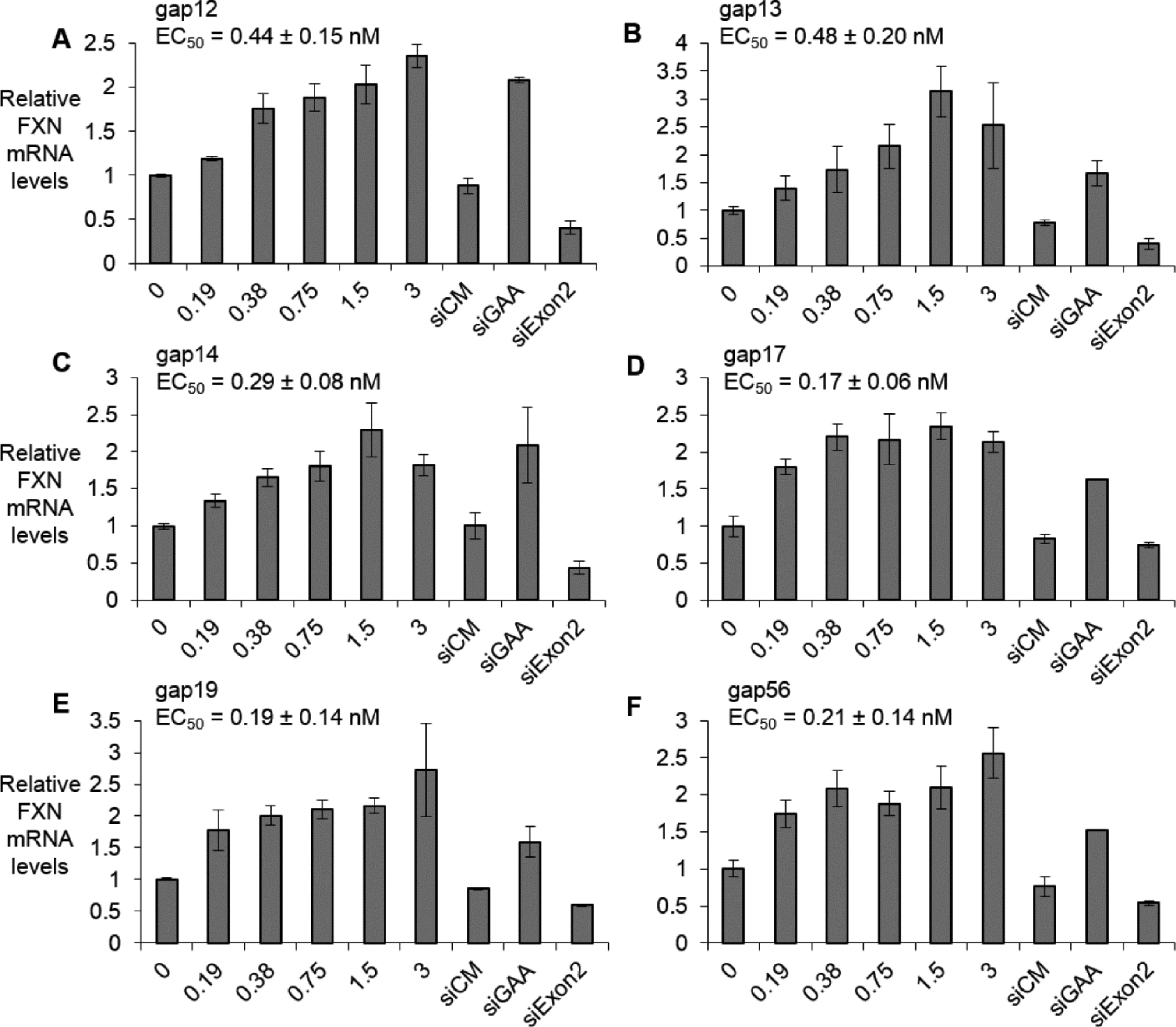
Dose dependent activation of FXN mRNA expression (lipofectamine RNAiMAX mediated transfection) in FRDA fibroblast line GM03816 by (A) gap12 (n=3), (B) gap13 (n=4), (C) gap14 (n=4), (D) gap17 (n=3), (E) gap19 (n=3), and (F) gap56 (n=3) at 0–3 nM range. Duplex RNAs are used in 25 nM. All data are presented as ±STDEV.
To evaluate the potency of RNA-mediated gene activation in a physiologically more relevant cell type we tested compounds in a patient-derived induced pluripotent stem cell derived neuronal progenitor cell line F4259 (iPSC-NPCs, 340/690 repeats). iPSC-NPC’s cannot be efficiently transfected with ASOs using cationic lipid but ASOs can be effectively delivered by electroporation.17 Electroporation requires high concentrations of ASO, so potencies cannot be directly compared with potencies obtained from assays using lipid-mediated transfection of fibroblast cells.
We observed dose dependent activation of gene expression in iPSC-NPC’s. The six compounds showed similar potencies of gene activation, ranging from 80 nM to 200 nM after triplicate or quadruplicate determinations. Maximal levels of activation ranged from 2 to 2.5 fold (Fig 5, Supplemental Fig. 2).
Figure 5.
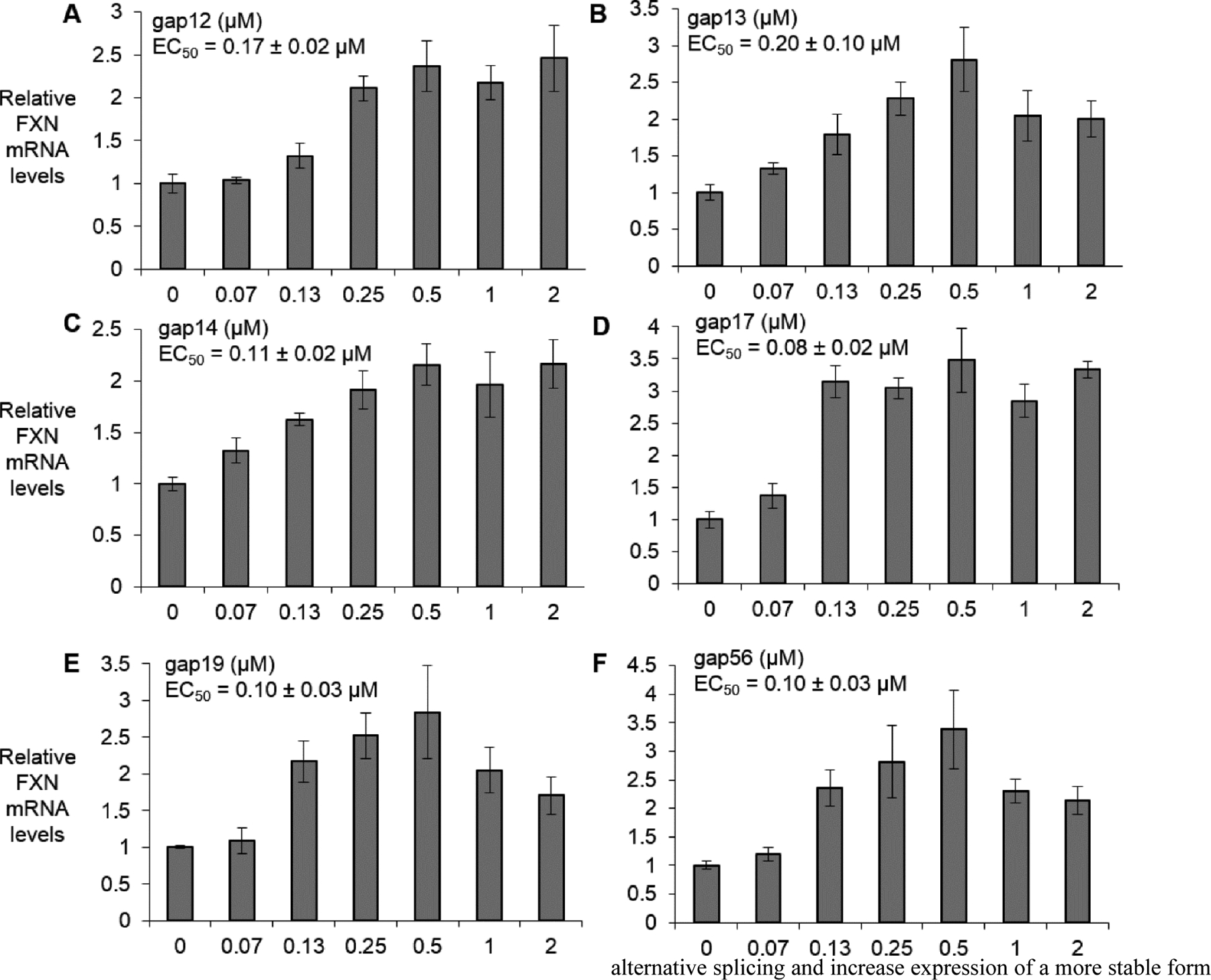
Dose dependent activation of FXN mRNA expression (electroporation) in FRDA NPC line F4259 by (A) gap12 (n=4), (B) gap13 (n=4), (C) gap14 (n=4), (D) gap17 (n=3), (E) gap19 (n=4), and (F) gap56 (n=3) at 0–2 μM range. All data are presented as ±STDEV.
2.3. Gapmers increase FXN pre-mRNA expression
To elucidate whether repeat-targeted gapmers increased expression of FXN pre-mRNA, we transfected gap 14 in FRDA patient-derived fibroblast line GM03816 and used qRT-PCR to assay FXN intron 1 and intron 3 pre-mRNA levels. Transfection of gap14 increased expression of all five regions of FXN introns to the levels comparable to the positive control siGAA (Fig 6A), consistent with the conclusion that activation of FXN protein expression by gapmers is at the level of transcription.
Figure 6.
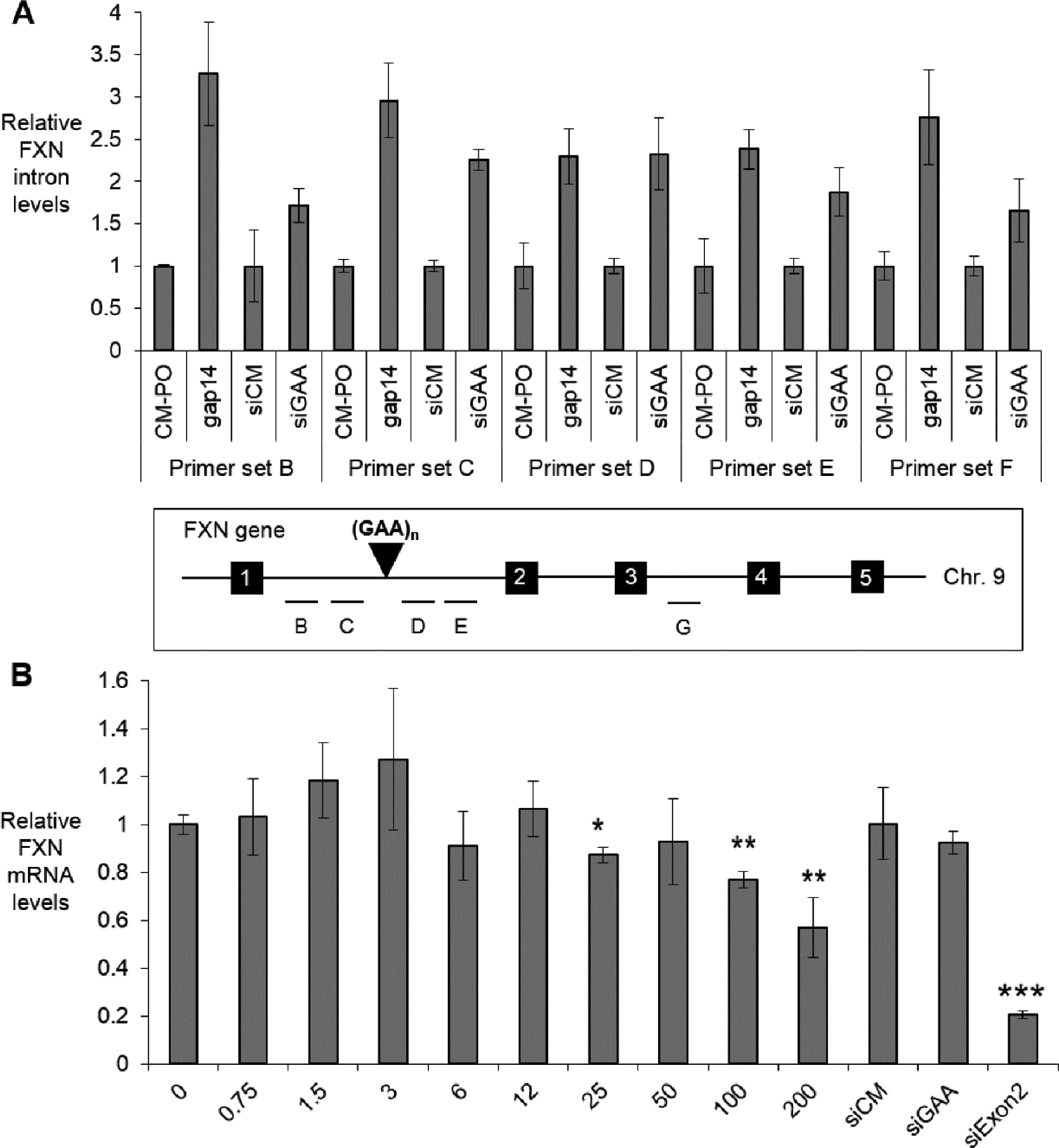
(A) Repeat-targeted gapmer gap 14 (12 nM) and duplex RNA siGAA (25 nM) activate FXN intron 1 and intron 3 pre-mRNA levels (n=2). Primer sets B-C refer to intron 1 upstream of GAA repeats, primer sets D-E refer to intron 1 downstream of GAA repeats, and prime set G refers to regions in intron 3. (B) Dose response curves of gap 14 (0–200 nM, n =3) in wild-type fibroblast line GM02153 (<50 GAA repeats on both alleles) with control duplex RNAs (25 nM). All data are presented as ±STDEV. *P < 0.05, **P < 0.01, ***P < 0.001, relative to EP (−) by Student t-test.
2.4. Activation is not achieved in healthy wild-type fibroblasts
As a control, we tested the possibility of FXN activation by gap14 in a healthy wild-type cell line GM02153 that does not have a GAA repeat expansion (<50 repeats) in either FXN allele. FXN mRNA levels were not affected significantly at 0–25 nM, the concentrations often used in cationic lipid mediated transfection in fibroblasts (Fig 6B). Even at high concentrations (100, 200 nM), which were rarely used in cationic lipid mediated transfection, there is no up-regulation of FXN mRNA. Instead, there is a slight decrease in FXN mRNA, consistent with general disruption of cellular pathways when cells are overloaded with excess oligonucleotide. Wild-type FXN levels are not impeded by R-loop formation and the result is consistent with the conclusion that anti-AAG oligonucleotides have no opportunity to change FXN expression in wild-type cells.
3. Discussion
Relative to traditional small molecule drug development candidates, oligonucleotides are large and negatively charged. Such properties would normally suggest poor intracellular uptake and pharmacological properties. Surprisingly, however, the opposite has been observed.
For the central nervous system, the uptake of oligonucleotides is sufficiently efficient for successful drug development. Spinraza, a single stranded antisense oligonucleotide designed to alter of survival motor neuron protein, has shown activity in humans. While oligonucleotides targeting the central nervous system have not yet demonstrated an ability to efficiently cross the blood brain barrier, Spinraza did show that an oligonucleotide could be dissolved in saline, administered into the spine by intrathecal injection, and be highly active.
Spinraza’s success sets the stage for the development of other oligonucleotide drug candidates designed to affect the expression of target genes in the central nervous system.
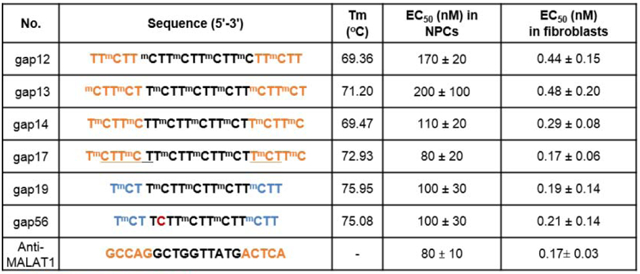
We had previously observed that a benchmark gapmer targeting MALAT-1 expression could achieve IC50 values for reducing MALAT-1 levels of 80 nM for iPSC-NPCs and 0.17 nM for fibroblast cells (Table 3). For comparison, the steric blocking anti-AAG ASO was significantly less potent when assayed for FXN activation, 500 nM in iPSC-NPCs (6.2 fold less) and 1.6 nM in fibroblast cells (9.4-fold less) (Supplemental Fig. 3).17
Table 3.
Activity of selected gapmers in FRDA patient-derived fibroblasts and neural progenitor cells in comparison with anti-MALAT1 gapmer (Shen, X., et al. RNA, 2019).
| No. | Sequence (5′−3′) | Tm (°C) | EC50 (nM) in NPCs | EC50 (nM) in fibroblasts |
|---|---|---|---|---|
| gap12 | TTmCTT mCTTmCTTmCTTmCTTmCTT | 69.36 | 170 ± 20 | 0.44 ± 0.15 |
| gap13 | mCTTmCT TmCTTmCTTmCTTmCTTmCT | 71.20 | 200 ± 100 | 0.48 ± 0.20 |
| gap14 | TmCTTmCTTmCTTmCTTmCTTmCTTmC | 69.47 | 110 ± 20 | 0.29 ± 0.08 |
| gap17 | TmCTTmCTTmTTmCTTmCTTmCTTmC | 72.93 | 80 ± 20 | 0.17 ± 0.06 |
| gap19 | TmCT TmCTTmCTTmCTTmCTT | 75.95 | 100 ± 30 | 019 ± 0.14 |
| gap56 | TmCT TCTTmCTTmCTTmCTT | 75.08 | 100 ± 30 | 0.21 ± 0.14 |
| Anti-MALAT1 | GCCAGGCTGGTTATGACTCA | - | 80 ± 10 | 0.17 ± 0.03 |
2’-O-MOE, (S)-cEt BNA, 2’-O-Me, DNA, mC: 5-methyl C, all PS backbone, Under line PO backbone
We now observe that anti-AAG gapmers can achieve potencies as low as 80 nM in iPSC-NPC’s and 0.17 nM in fibroblast cells. These potencies are the same as those achieved by the anti-MALAT gapmer and represent a several-fold improvement relative to steric block ASOs.
These data for anti-AAG gapmers suggest they have an advantage for drug discovery relative to steric blocking ASOs. One potential drawback of anti-AAG gapmers is that recruitment of RNaseH may lead to cleavage of “off-target” RNA sequences and toxic effects. We did not observe changes in cell growth or morphology, but effects might be more apparent in animals. Based on blast search, there are 11 human transcripts with more than 6 GAA repeat sequence (Supplemental Fig. 6). RNA-seq analysis and animal studies are ongoing, and will possibly reveal potential off-target effects. The amount of gapmer ASO dosed in animals will likely need to be carefully calibrated to ensure identification of an optimal therapeutic window.
4. Conclusions
Gapmer oligonucleotides complementary to the expanded AAG repeat within the FXN gene can activate expression of FXN RNA and protein. The potency for some gapmers is the same as a benchmark anti-MALAT-1 gapmer and superior to previously described anti-AAG steric blocking ASOs. Increased potency makes clinical application more feasible but additional studies of off-target effects and efficacy in FRDA model mice will be necessary to fully evaluate the potential of this class of anti-AAG compounds.
5. Experimental
5.1. Fibroblast cell culture and transfection
Fibroblast cells, GM03816 (Coriell Institute, Friedreich’s Ataxia patient cell line), GM02153 (Coriell Institute, healthy wild-type cell line) were cultured as described previously.15 Briefly, all cells were grown in minimum essential medium supplemented with 10% fetal bovine serum and 1% non-essential amino acids at 37°C in 5% CO2. Lipofectamine RNAiMAX (Invitrogen) was used to transfect oligonucleotides following the manufacturer’s recommended protocol in OptiMEM reduced serum medium (Invitrogen). OptiMEM was changed to complete medium after 24 h. Transfected cells were harvested 72 h and 96 h after transfection for qRT-PCR and western blot analyses, respectively. The timing is based on a time-course and are the earliest times at which maximal activation at mRNA and protein level is observed. Cells were dissociated with 1X trypsin, mixed together with equal volume of trypan blue (Sigma) and counted using cell counter (TC20 Automated Cell Counter; Bio-Rad).
5.2. Neural progenitor cell culture and transfection
A primary fibroblast line derived from FRDA patient (F4259) was reprogrammed to induced pluripotent stem cells (iPSCs) using integration-free Sendai virus transgene delivery (CytoTune 2.0 kit, ThermoFisher Scientific) per the manufacturer’s instructions. The iPSC lines were tested for pluripotency and differentiation capabilities.20 The iPSC lines were differentiated into neural progenitor cells (NPCs) via inhibiting TGF-β/SMAD signaling as described previously.21 NPCs were maintained in STEMdiff™ neural progenitor medium (Stemcell Technologies). Cells were dissociated with StemPro™ Accutase™ Cell Dissociation Reagent (Gibco) + 10 μM Y27632 (Selleck Chemicals).
MaxCyte system used pre-set protocols (Optimization 1 to 10, ranging from low energy to high energy) for most cell types. Transfection was performed by the MaxCyte STX® scalable transfection system using Optimization 4 electroporation protocols with OC-100 cuvettes (MaxCyte, Inc.). Prior to electroporation, oligonucleotides or duplex RNAs were added to OC-100. Cells were thawed and added to the corresponding maintenance medium (10 mL), washed one time and resuspended in HyClone™ electroporation buffer (MaxCyte, Inc.). Cells were counted using trypan blue staining (TC20™ Automated Cell Counter, Bio-Rad), 500,000 cells in the volume of 50 μL were added to OC-100 and electroporation was performed. Immediately after transfection, 50 μL of warm maintenance medium was added to the cuvettes, and the cuvettes were closed and rested in incubator (37°C and 5% CO2) for 15 min. Then, cells were plated (two wells per cuvette for RNA as two biological replicates) to 12-well plates pre-coated with Corning™ Matrigel™ membrane matrix (Fisher Scientific, CB-40234). FXN expression was assayed after 72 hours by qRT-PCR.
5.3. Quantitative real-time PCR
Total RNA was harvested and treated with DNase (removing genomic DNA contamination) at 72 hours post transfection with TRIzol™ reagent (Invitrogen, for fibroblasts) and NucleoSpin™ RNA XS kit (MACHEREY-NAGEL, for neural progenitor cells) following the manufacturer’s recommended protocol. Equal amount of treated RNA (representing approximately the same number of cells and ranging from 0.2–2 μg of RNA) were reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and diluted to 60–200 μL final volume after reaction. Q RT-PCR was performed with two technical replicates per sample using iTaq™ Universal SYBR® Green Supermix (BIO-RAD) with 5μ1 of cDNA as template and gene specific primer pairs (Supplemental Fig. 4).
5.4. Western blot analysis
Cell extracts were prepared using lysis buffer supplemented with 1% Protease Inhibitor Cocktail Set I (Calbiochem) as described previously.22 Protein were separated on 4–20% gradient Mini-PROTEAN® TGX™ precast gels (Bio-Rad). After gel electrophoresis, proteins were wet transferred to nitrocellulose membrane (0.45 μm, GE Healthcare Life Sciences) at 100 V for 45 min. Membranes were blocked for 2 hours at room temperature with 5% milk in 1x PBS containing 0.1% TWEEN-20 (PBST 0.1%). Blocked membranes were incubated with the primary antibodies at 4 °C in PBST 0.1% with 1% milk on rocking platform overnight: anti-FXN at 1:20,000 (4F9, from Dr. Hélène Puccio at IGBMC, France) and anti-β-Tubulin at 1:5,000 (Sigma-Aldrich, T5201). After primary antibody incubation, membranes were washed 4 × 10 min at room temperature with PBST 0.2% (1× PBS, 0.2% TWEEN-20) and then incubated for one hour at room temperature with HRP-conjugated anti-Mouse IgG secondary antibody (Jackson ImmunoResearch, 715-035-150, FXN 1:20,000, β-Tubulin 1:10,000) in PBST 0.1%. Membranes were washed again 4 × 10 min in PBST 0.1% and 4 × 10 min in 1x PBS at room temperature. Washed membranes were soaked with HRP substrate per the manufacturer’s recommendations (SuperSignal™ West Pico Plus Chemiluminescent substrate, Thermo Scientific) and exposed to films. The films were scanned and bands were quantified using ImageJ software. More biological replicates of Fig. 2 imaged were reported in Supplemental Fig. 5.
5.5. EC50 calculations
The program GraphPad Prism 7.03 was used to calculate EC50/IC50. The Hill equations was used for fitting curves in the following form: Y = Y0+(Ymax-Y0)Xn/(Kn+Xn), where Y is the normalized fold activation/inhibition, X is the oligo concentration, Y0 is baseline response (activation/inhibition at a oligo concentration 0), Ymax is the maximum fold activation/inhibition, K is the EC50 value and n is the Hill coefficient.23 Data sets from at least four replicates were used for curve fitting. The error of EC50 is standard error of the mean (SEM), which is calculated from combining the data of each individual dose curve.
5.6. Melting temperature determination
Thermal denaturation analysis of oligonucleotides to determine melting temperature, Tm, values was carried out using a CARY Varian 100 Bio UV-Vis spectrophotometer. 20 μl of single strand compound with equal volume of target sequence or 40 μl of double strand compound were added to 360μl buffer (0.25 M NaCl, 0.2 mM EDTA, 20 mM Sodium Phosphate, pH 7.0) and monitored at 260 nm in a 1 cm quartz cuvette (temperature range: 15 – 95 °C; ramp: 1 °C). The melting temperature was calculated and averaged from at least 7 technical replicates.
Supplementary Material
Acknowledgments
This study was supported R35GM118103 (DRC) from the National Institutes of Health, the Robert A. Welch Foundation I-1244 (DRC), the Friedreich’s Ataxia Research Alliance, and the Paul D. Wellstone MDCRC Trainee Fellowship Award (XS) from UT Southwestern Medical Center. DRC is the Rusty Kelley Professor of Biomedical Science. Work in the Napierala laboratory was supported by National Institutes of Health (R01NS081366) and Muscular Dystrophy Association (MDA418838). The research was also supported by a generous gift from the Doremus family. We thank MaxCyte for providing the MaxCyte STX system and related supplies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
DRC has filed a patent application related to early work on this topic.
Supplementary data
Supplementary data associated with this article can be found in the online version.
References and notes
- 1.Shen X, Corey DR Nucl. Acids Res 2018, 46, 1548–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levin AA N. Engl. J. Med 2019, 380, 57–70. [DOI] [PubMed] [Google Scholar]
- 3.Corey DR Nat. Neurosci 2017, 20, 497–499. [DOI] [PubMed] [Google Scholar]
- 4.Bennett CF, Krainer AR, Cleveland DW Annu. Rev. Neurosci 2019, 42, 385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macchi C, Sirtori CR, Corsini A, Santos RD, Watts GF, Ruscica M Pharmacol. Res 2019, 150, 104413. [DOI] [PubMed] [Google Scholar]
- 6.Sardh E, Harper P, Balwani M, Stein P, Bissell DM, Desnick R, Parker C, Phillips J, Bonkovsky HL, Vassiliou D, Penz C, Chan-Daniels A, He Q, Querbes W, Fitzgerald K, Kim JB, Garg P, Vainshaw A, Simon AR, Anderson KE N. Engl. J. Med 2019, 380, 549–558. [DOI] [PubMed] [Google Scholar]
- 7.Akinc A, Maier MA, Manoharan M, Fitzgerald K, Jayaraman M, Barros S, Ansell S, Du X, Hope MJ, Madden TD, Mui BL, Semple SC, Tam YK, Ciufolini M, Witzigmann D, Kulkarni JA, van der Meel R, Cullis PR Nat. Nanotech 2019, 14, 1084–1087. [DOI] [PubMed] [Google Scholar]
- 8.Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J, Rickards H, Posser A, Kordasiewicz HB, Czech C, Swayze EE, Norris DA, Baumann T, Gerlach I, Schobel SA, Paz E, Smith AV, Bennett CF, Lane RM N. Engl. J. Med 2019, 380, 2307–2316. [DOI] [PubMed] [Google Scholar]
- 9.Witzum JL, Gaudet D, Freedman SD, Alexander VJ, Williams KR, Yang Q, Hughes SG, Geary RS, Arca M, Stroes ESG, Bergeron J, Soran H, Civeira F, Hemphill L, Tsimikas S, Blom DJ, O’Dea L, Bruckert EN Engl. J. Med 2019, 381, 531–542. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Pendergast MK, Goldkind SF, Lee EA, Kuniholm A, Soucy A, Vaze J, Belur NR, Fredriksen K, Stojkovska I, Tsytsykova A, Armant M, DiDonato RL, Choi J, Cornelissen L, Rereira LM, Augustine EF, Genetti CA, Dies K, Barton B, Williams L, Goodlett BD, Riley BL, Pasternak A, Berry ER, Pflock KA, Chu S, Reed C, Tyndall K, Agrawal PB, Beggs AH, Grant PE, Urion DK, Snyder RO, Waisbren SE, Poduri A, Pakr PJ, Patterson A, Biffi A, Mazzulli JR, Bodamer O, Berde CB, and Yu TW N. Engl. J. Med 2019, 381, 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aartsma-Rus A and Watts JK Nucl. Acid. Ther 2019, 29(6), 302–304. [DOI] [PubMed] [Google Scholar]
- 12.Delatycki MB, Bidichandani SI Neurobiol. Dis 2019, 132, 104606. [DOI] [PubMed] [Google Scholar]
- 13.Groh M, Lufino MM, Wade-Martins R, Gromak N PLoS Genet. 2014,10, e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Matsui M, Corey DR Nat. Comm 2016, 7, 10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Shen X, Liu Z, Norrrborm M, Prakash TP, O’Rielly D, Sharma VK, Dahma MJ, Watts JL, Rigo F, Corey DR Nucl. Acid Ther 2018, 28, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen X, Kilikevcius A, O’Reilly D, Prakash TP, Damha MJ, Rigo F, Corey DR Bioorg. Med. Chem. Lett 2018, 28, 2850–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen X, Beasley S, Putnam JN, Li Y, Prakash TP, Rigo F, Napierala M, Corey DR RNA. 2019, 25, 1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt K, Prakash TP, Donner AJ, Kinberger GA, Gaus HJ, Low A, 0stergaard ME, Bell M, Swayze EE and Seth PP, 2017. Nucl. Acids. Res 2017, 45(5), 2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen W, De Hoyos CL, Migawa MT, Vickers TA, Sun H, Low A, Bell TA, Rahdar M, Mukhopadhyay S, Hart CE and Bell M, 2019. Nat. Biotechnol 2019, 37(6), 640–650. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Polak U, Bhalla AD, Rozwadowska N, Butler JS, Lynch DR, Dent SY and Napierala M,Mol. Ther 2015, 23(6), 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M and Studer L Nat. Biotechnol 2009, 27(3), 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watts JK, Yu D, Charisse K, Montaillier C, Potier P, Manoharan M and Corey DR Nucl. Acids Res, 2010, 38(15), 5242–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goutelle S, Maurin M, Rougier F, Barbaut X, Bourguignon L, Ducher M and Maire P Fundam. Clin. Pharmacol 2008, 22(6), 633–648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.