

Abstract
Rap1 is a small GTPase that has been implicated in dendritic development and plasticity. In this study, we investigated the role of Rap1 in axonal growth and its activation in response to neurotrophins and myelin-associated inhibitors. We report that Rap1 is activated by brain-derived neurotrophic factor (BDNF), and that this activation can be blocked by myelin-associated glycoprotein (MAG) or CNS myelin, which also induced increases in Rap1GAP1 levels. In addition, we demonstrate that adenoviral overexpression of Rap1 enhances neurite outgrowth in the presence of MAG and myelin, while inhibition of Rap1 activity through overexpression of Rap1GAP1 blocks neurite outgrowth. These findings suggest that Rap1GAP1 negatively regulates neurite outgrowth, making it a potential therapeutic target to promote axonal regeneration.
Introduction
The seminal work of David and Aguayo (1) demonstrated that neurons in the adult mammalian central nervous system (CNS) do not irreversibly lose regenerative capacity upon differentiation and establishment of connectivity. When provided with a permissive environment, the severed axons of adult neurons were able to regenerate over long distances (1). However, in the milieu of the injured CNS, attempts at regeneration fail, largely because of numerous inhibitory factors such as myelin-associated inhibitors and components of the astroglial scar (2, 3, 4).
While inhibitory molecules feature prominently in the injured CNS, potent promoters of axonal growth, such as neurotrophins (5) and the cell adhesion molecules L1 and laminin (6), are also upregulated. Regenerating axons therefore receive input from both growth-inhibiting and growth-promoting signals, with the resulting output skewed towards inhibition. Consequently, it is crucial to identify convergence points in the signaling mechanisms for pro-regenerative and inhibitory factors – with the goal of manipulating these pathways so that the outcome can be shifted towards regeneration.
Small GTPases are logical candidates for this role, as they serve as natural integration points for the positive and negative cues that influence cytoskeletal dynamics within the growth cone. Proteins such as Rac1, Cdc42, and RhoA have direct effects on actin polymerization, and RhoA has been identified as a key factor in mediating inhibition by MAG (7, 8). Like all small GTPases, these proteins are activated by guanine nucleotide exchange factors (GEFs) and inactivated by GTPase activating proteins (GAPs). The equilibrium between GEF and GAP activity determines whether the protein will assume an active, GTP-bound or inactive, GDP-bound conformation. While members of the Rho subfamily have well-established roles in neural plasticity, less is known about Rap1, a member of the Ras subfamily, but it has been shown to be an important regulator of diverse neuronal functions such as migration, dendrite formation, and neurotrophin signaling (9, 10, 11, 12). To date, nine GEFs and five GAPs have been identified for Rap1, with Rap1GAP1 being the most widely studied of the GAPs (13). The majority of the GEFs are activated by receptor tyrosine kinases and second messengers such as Ca2+ and diacylglycerol (13, 14), but Rap1 is also regulated by a unique class of GEFs known as exchange proteins directly activated by cyclic AMP (EPACs; 14, 15). In light of previous studies from our laboratory and others demonstrating that elevation of intracellular cyclic AMP (cAMP) levels allows neurons to overcome inhibition by MAG and myelin (16, 17, 18, 19), we hypothesized that Rap1 may regulate axonal growth by integrating signals from myelin-associated inhibitors and cAMP. We therefore investigated whether MAG and myelin block activation of Rap1, and if Rap1 activation has an impact on neurite outgrowth.
Materials and Methods
All animal experiments were approved by the Institutional Animal Care and Use Committee at Hunter College, City University of New York. Hunter College has AAALAC accreditation. The experiments were carried out in accordance with NIH guidelines for use and care for laboratory animals, and the United States Public Health Service’s Policy on Humane Care and Use of Laboratory Animals. These regulations are consistent with the EU Directive 2010/63/EU for animal experiments.
Isolation of primary neurons
Rat cerebellar granule neurons (CGN), hippocampal neurons, and cortical neurons were prepared as described previously (20). The cerebella from postnatal day 5-6 (P5-6) Long Evans rats were dissected, collected in Neurobasal-A media (Invitrogen), and treated with papain (0.5 mg/ml, Sigma) and DNAse (100 μg/ml, Worthington) for 30 minutes. Enzymatic activity was stopped with soybean trypsin inhibitor (Sigma). After washing with Neurobasal-A media and trituration of the tissue, neurons were collected by centrifugation and resuspended in modified SATO media for plating (20). Cerebral cortices and hippocampi were isolated from P1-3 Long Evan rats and treated with papain, followed by purification of neurons using OptiPrep density gradients (Sigma). Neurons were plated in modified SATO media. Rat dorsal root ganglion (DRG) neurons were prepared as described previously (21). The ganglia were removed from P5-7 Long Evans rats and incubated for 30 minutes at 37°C in L15 media with trypsin (0.025%, Gibco) and collagenase (0.3%, Worthington). DMEM with 10% fetal bovine serum (FBS) was added to inactivate the trypsin. Ganglia were triturated and DRG neurons were collected by centrifugation and resuspended in modified SATO media.
Treatments
Experiments with 8-(4-Chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-CPT)
For priming experiments, CGN were treated with 1 mM dbcAMP (Calbiochem; positive control), or with 8-CPT (Tocris) at concentrations of 0.25, 0.5, and 1 mM. Neurons were plated in poly-L-lysine (PLL)-coated 24-well culture dishes at a density of 1×106 neurons per well and incubated for 16 hours. Neurons were then trypsinized, and used in neurite outgrowth assays. Additional CGN were treated with 0.5 or 1 mM 8-CPT, or 1 mM dbcAMP and used immediately in neurite outgrowth assays.
Recombinant adenoviruses
Plasmids expressing wild type Rap1 or Rap1F64A were kindly provided by Dr. O. Daumke (22). Rap1GAP1-expressing plasmids were kindly provided by Dr. P.J. Casey (23). cDNAs were subcloned into pTRACK CMV and then inserted by homologous recombination into pAdeasy-1 (Agilent Technologies). The viral preparation and purification was carried out as previously described (24). For the experiments, DRG or hippocampal neurons were plated overnight at 37°C in PLL-coated 24-well plates. Neurons were then transduced with adenoviruses at a final concentration of 1010 plaque-forming units (PFU)/ml. Infected neurons were incubated overnight at 37°C before being trypsinized and used in neurite outgrowth assays.
siRNA experiments
Rap1GAP-specific short interfering RNA (siRNA) and negative control siRNA were obtained from Qiagen (FlexiTube siRNA, catalogue number SI01737043, target sequence 5′-CACAGCCAGAATCTACCGGAA-3′ and AllStars negative control siRNA, SI03650318) and transfected into neurons using the Neon Transfection system (Life Technologies). All procedures were carried according to the manufacturer’s protocol using two pulses of 1150 V and 10 milliseconds. Freshly isolated hippocampal or cortical neurons were washed once in PBS, collected by centrifugation, and resuspended in transfection buffer. For each transfection, 0.3 nmol of siRNA was added to the neurons and they were immediately plated in 24-well tissue culture dishes at a density of 0.5×106 neurons per well with Neurobasal-A media supplemented with B27 (Gibco). After an overnight incubation at 37°C, neurons were trypsinized and used in neurite outgrowth assays, or lysed for Western blotting to confirm Rap1GAP knockdown.
Neurite outgrowth assays
Monolayers of control or MAG-expressing CHO cells were prepared as described previously (21). 8-well tissue culture slides (Lab-Tek) were treated for 30 minutes at room temperature with poly-L-lysine (20 μg/ml, Sigma), followed by incubation with fibronectin (10 μg/ml, Sigma) for 2 hours at 37°C. CHO cells were plated in the wells a concentration of 250,000 cells/ml and grown overnight at 37°C to form near-confluent monolayers. As an alternative substrate, neurons were cultured on CNS myelin (25, 26). 8-well tissue culture glass slides (Lab-Tek) were coated with 20 μg/ml PLL at room temperature for 30 minutes. Rat CNS myelin was then added to the PLL-covered slides at 2 μg of total protein per well and desiccated overnight.
Neurons were plated at a density of 1.5×104 cells per well in modified SATO media (300 μl of media per well, 2×104 neurons per cm2). Neurons plated on the CHO cell monolayers were incubated for 18-22 hours, while neurons plated on myelin substrates were allowed to grow for 22-24 hours. Neurons were then fixed with 4% paraformaldehyde, permeabilized with ice-cold methanol, and immunostained with anti-β-III-tubulin antibody (1:1000, BioLegend, #801213) followed by Alexa Fluor 568-conjugated anti-mouse IgG (1:2000, Invitrogen, #10037). Images were acquired and the length of longest neurite for each neuron was measured using MetaMorph software (Molecular Devices). A minimum of 200 neurons per condition were measured in each experiment. For neurons infected with adenoviruses, neurite length was measured only for neurons that were positive for both GFP and βIII tubulin, which ensured that only infected neurons were analyzed.
Western blotting experiments examining Rap1GAP1 levels
For experiments examining the effects of MAG and myelin on Rap1GAP levels, CGN or cortical neurons were plated in PLL-coated dishes (20 million cells per plate) for 24 hours and then treated with either 20 μg/ml MAG-Fc or 20 μg/ml CNS myelin for 20 minutes at 37°C. In a subsequent series of experiments examining Rap1GAP expression, cortical neurons received one of the following treatments: 1 mM dbcAMP for 10 or 20 minutes, 0.5 mM 8-CPT for 20 minutes, or 2 ng/ml PTX (Sigma) for 30 or 60 minutes. Untreated neurons were used as controls in all experiments.
Cells were lysed in 1X RIPA buffer and samples containing equal amounts of protein were combined with Laemmli sample buffer. SDS-PAGE was performed using 6% polyacrylamide gels and proteins were transferred to PVDF membranes at 70V for 1 hour. Membranes were probed overnight with anti-Rap1GAP monoclonal antibody (1:1000, Millipore Sigma, #04-413), followed by HRP-conjugated anti-rabbit IgG (1:2000; Cell Signaling Technology, #7074). Signals were detected with Pierce ECL Western Blotting Substrate or SuperSignal West Femto Maximum Sensitivity Substrate. Membranes were stripped and reprobed using anti-actin polyclonal antibody (1:1000; Sigma, #A2066), followed by HRP-conjugated anti-rabbit IgG (1:2000; Cell Signaling Technology, #7074). Densitometric measurements of the resulting bands were made using ImageJ software.
Rap1 activation assays
CGN or cortical neurons were cultured on PLL-coated 10 cm plates (20 million neurons per plate) overnight in modified SATO media and 4 hours prior to treatment the media was changed to unsupplemented DMEM. Cells received one of the following treatments: 200 ng/ml BDNF for 20 minutes, 20 μg/ml MAG-Fc or CNS myelin for 20 minutes followed by 200 ng/ml BDNF for 20 minutes, 0.5 mM 8-CPT for 20 minutes, 20 μg/ml MAG-Fc for 20 minutes followed by 0.5 mM 8-CPT for 20 minutes, 20 μg/ml MAG-Fc or CNS myelin for 20 minutes, 1 mM dbcAMP for 20 minutes, or 20 μg/ml MAG-Fc for 30 minutes followed by 1 mM dbcAMP for 20 minutes. Rap1 activation assays were then performed using Rap1 Activation Assay Kits (Millipore Sigma, 17-321). Cells were lysed in lysis buffer supplemented with PMSF (0.1M), protease and phosphatase inhibitor cocktails (Calbiochem). The lysates were cleared by centrifugation and incubated with Ral GDS-RBD agarose slurry for 45 minutes at 4°C to pull down activated Rap1. Beads were precipitated by centrifugation, washed, and GTP-bound Rap1 was eluted from the beads using Laemmli sample buffer. Proteins were resolved by SDS-PAGE using 15% polyacrylamide gels, and transferred to PVDF membranes at 60V for 1 hour. Membranes were incubated overnight with rabbit anti-Rap1 (1:500, Millipore Sigma, #07-916) antibody, followed by incubation with HRP-conjugated anti-rabbit IgG (1:2000; Cell Signaling Technology, #7074), and detected with Pierce ECL Western Blotting Substrate or SuperSignal West Femto Maximum Sensitivity Substrate. Total Rap1 levels were assessed using input samples obtained prior to the Rap1 pulldown. The conditions for SDS-PAGE, Western blotting, and visualization of total Rap1 were identical to those used for analysis of GTP-bound Rap1. Densitometric measurements of the resulting bands were made using ImageJ software.
Statistical analysis
Statistical analyses were done with GraphPad Prism software (GraphPad). Data are presented as mean ± SEM from at least 3 independent experiments. Paired one-tailed Student’s t test was used to compare two groups and one-way ANOVA followed by Bonferroni’s multiple comparisons test was used to assess significance between three or more groups.
Results
MAG and myelin block activation of Rap1 by BDNF and EPAC.
We first examined whether neurotrophins and myelin-associated inhibitors have opposing effects on Rap1 activation. There is substantial evidence that neurotrophins act through extracellular signal-regulated kinase 1/2 (Erk1/2) to activate Rap1 in a variety of neuronal populations, including cortical and dorsal root ganglion neurons (11, 27). In addition, our laboratory has previously demonstrated that pre-treatment (priming) with brain-derived neurotrophic factor (BDNF) renders cerebellar granule neurons (CGN) insensitive to the inhibitory effects of MAG and myelin (16). We subsequently reported that BDNF elicits this response through activation of Erk1/2 and suppression of phosphodiesterase-4 activity, which in turn leads to increased cAMP levels, activation of cAMP-responsive element binding protein (CREB), and reversal of myelin-mediated inhibition (16, 28).
To determine if Rap1 is activated in response to BDNF, P5-7 CGN were treated with 200 ng/ml BDNF for twenty minutes and pull-down assays for active Rap1 were performed. Levels of GTP-bound Rap1 were significantly increased after treatment with BDNF (Fig. 1A–C); however, when neurons were treated with 20 μg/ml MAG-Fc or CNS myelin prior to the addition of BDNF, this increase in Rap1 activation was eliminated (Fig. 1A–C). In the case of the treatment with CNS myelin, a reduction in total Rap1 levels may have contributed to this effect (Fig. 1B). Since BDNF treatment leads to elevation of intracellular cAMP (16), it is probable that the activation of Rap1 was mediated through EPAC, and so, we next determined if MAG and myelin could inhibit the downstream effects of EPAC. CGN were treated with 0.5 mM 8-CPT-2Me-cAMP (8-CPT), a specific pharmacological activator of EPAC (29), and as expected, this induced a substantial increase in Rap1 activation (Fig. 2A). However, when neurons were pre-treated with MAG-Fc, activation of Rap1 by 8-CPT was inhibited (Fig. 2A). Several studies have shown that 8-CPT can enhance neurite outgrowth and growth cone turning responses in a permissive environment (30, 31, 32), and so, we tested whether 8-CPT could also enhance neurite growth in the presence of MAG. We found that, similar to the effects of BDNF (16) and dbcAMP (Fig. 2B), priming with 8-CPT significantly increased neurite outgrowth on MAG-expressing Chinese hamster ovary (CHO) cells (Fig. 2B). However, when the neurons were treated with 8-CPT immediately prior to plating, neurite outgrowth was still strongly inhibited by MAG when compared to neurons treated with dbcAMP (Fig. 2C). Together, these results indicate that induction of EPAC and Rap1 activity contribute to overcoming inhibition by MAG, while acute exposure to myelin-associated inhibitors blocks BDNF- and EPAC-mediated activation of Rap1. The latter effect also supports our previous findings showing that myelin-associated inhibitors interfere with neurotrophin signaling, thus preventing accumulation of cAMP (16) and activation of CREB (33), and leading to inhibition of neurite outgrowth.
Figure 1. MAG and myelin block activation of Rap1 by BDNF.
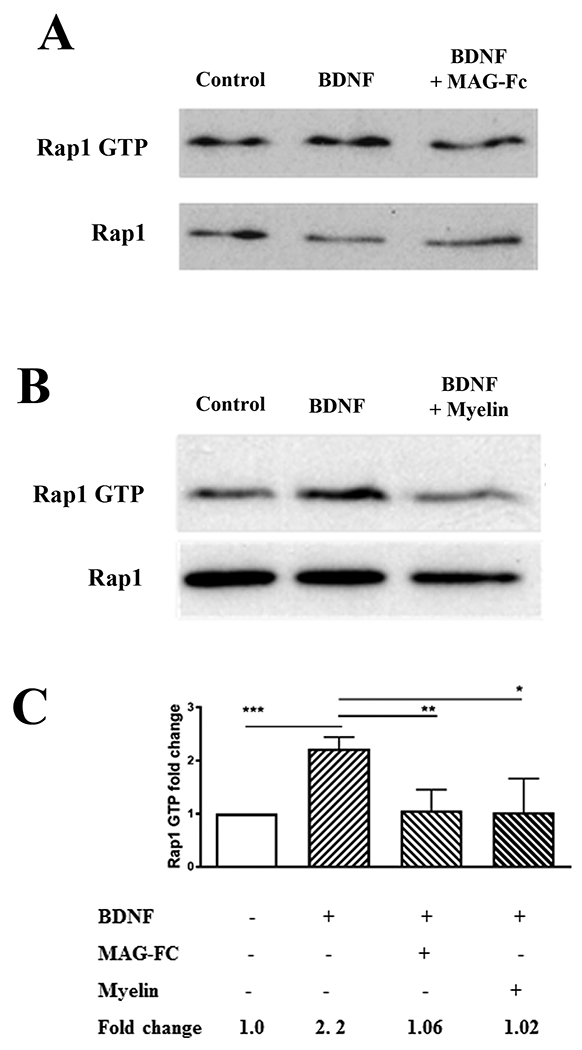
(A, B) Western blots of P5-6 CGN treated with BDNF (200 ng/ml) for 20 minutes, or with MAG-Fc (20 μg/ml) or CNS myelin (20 μg/ml) for 20 minutes prior to the addition of BDNF (n=4 for experiments with MAG-Fc, n=3 for experiments with myelin). Lysates were used for Rap1 activation assays detecting GTP-bound Rap1, and total Rap1 was assessed using input samples. (C) Quantification of Rap1 activation, where Rap1 GTP levels were normalized to the total Rap1 levels in the input lysate. Graphs depict average fold changes ± SEM (***p < 0.001, **p < 0.01, *p < 0.05 one-way ANOVA with Bonferroni’s multiple comparisons test).
Figure 2. The EPAC-specific cAMP analogue 8-CPT activates Rap1 and overcomes inhibition by MAG when used for priming.
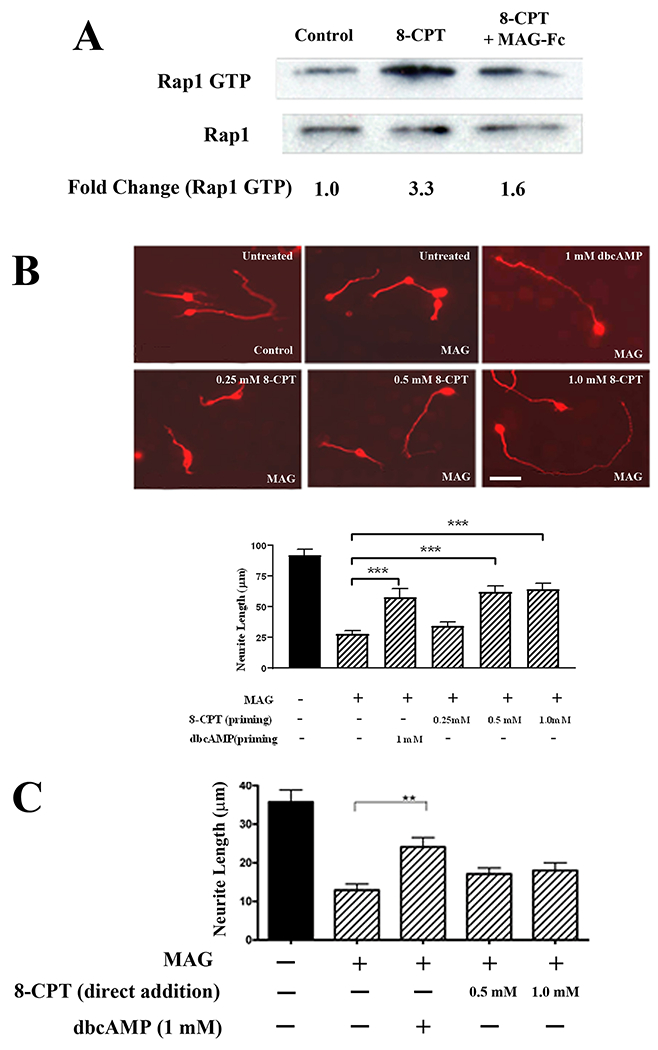
(A) Western blots of P5-6 CGN treated with 8-CPT (0.5 mM) for 20 minutes, or with MAG-Fc (20 μg/ml) for 20 minutes prior to the addition of 8-CPT (n=3). Lysates were used for Rap1 activation assays detecting GTP-bound Rap1, and total Rap1 was assessed using input samples. (B) Representative images of P5-6 CGN that were incubated overnight with 1 mM dbcAMP, 0.25, 0.5 or 1 mM 8-CPT, and transferred to monolayers of either MAG-expressing CHO cells or control CHO cells (scale bar=10 μm). Graph depicts average length of the longest neurite per neuron ±SEM for approximately 150-200 neurons per treatment (3 independent experiments, ***p < 0.001, one-way ANOVA with Bonferroni’s multiple comparisons test). (C). Quantification of neurite outgrowth for P5-6 CGN that were treated with 1 mM dbcAMP, 0.5 or 1 mM 8-CPT, and plated directly on monolayers of MAG-expressing CHO cells or control CHO cells. Graph depicts average length of the longest neurite per neuron ±SEM for approximately 150-200 neurons per treatment (3 independent experiments, **p < 0.01, one-way ANOVA with Bonferroni’s multiple comparisons test).
MAG and myelin increase Rap1GAP1 levels, leading to inactivation of Rap1.
We next addressed the question of whether MAG and myelin can influence Rap1 activation through inhibition of Rap1 GEFs, or stimulation of Rap1 GAP activity. Rap1 possesses low levels of intrinsic GTPase activity (34), and consequently, it has been proposed that inactivation of Rap1 is regulated primarily by Rap GAPs (35). Furthermore, Rap1GAP1 is widely expressed in CNS structures such as the striatum, hippocampus, mesencephalon, and cerebellum (10, 36), which places it in the proper spatial context to influence Rap1 activity in neurons. This prompted us to examine the effects that MAG and myelin exert on Rap1GAP1. We determined that levels of Rap1GAP1 were significantly higher following treatment with 20 μg/ml MAG-Fc or CNS myelin (Fig. 3A, B). Moreover, these MAG- and myelin-mediated increases in Rap1GAP1 levels resulted in a corresponding decrease in levels of GTP-bound Rap1 (Fig. 3C, D), which indicates that there is a direct correlation between levels of Rap1GAP1 and Rap1 activity. There are two possible explanations for the effects of myelin-associated inhibitors on Rap1GAP1 levels. The first is that MAG and myelin increase Rap1GAP1 expression, and the second is that proteosomal degradation of Rap1GAP1 is reduced in the presence of these inhibitors, leading to elevated levels of protein within the neuron. Our observations also place Rap1 among the other small GTPases, such as RhoA (8) and Rac (37), that are modulated by myelin-associated inhibitors.
Figure 3. MAG and CNS myelin increase Rap1GAP1 protein levels and suppress activation of Rap1.
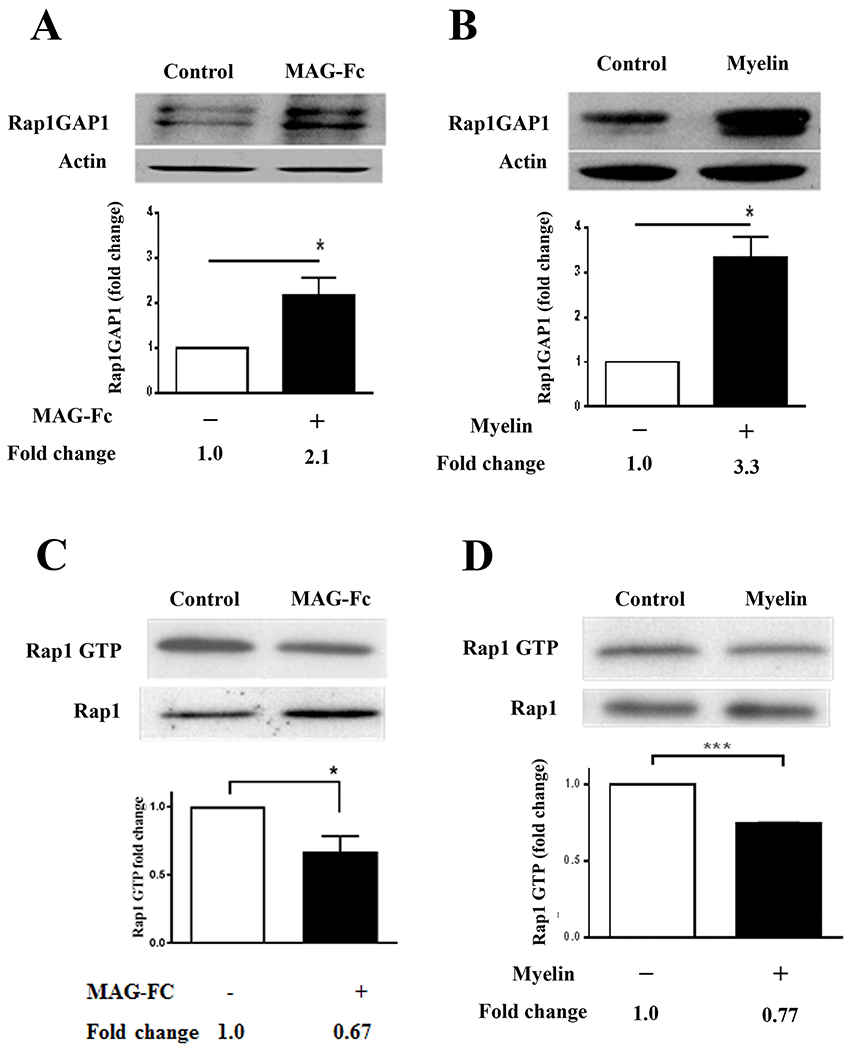
(A, B) Western blots of P5-6 CGN treated with MAG-Fc (20 μg/ml) or CNS myelin (20 μg/ml) for 20 minutes (n=8 for MAG-Fc, n=3 for myelin). Rap1GAP1 was quantified by normalizing Rap1GAP levels to their respective loading controls. Graphs depict average fold changes ± SEM. (*p < 0.05, paired one-tailed Student’s t-test). (C, D) Western blots of P5-6 CGN treated with MAG-Fc (20 μg/ml) or CNS myelin (20 μg/ml) for 20 minutes (n=3 for MAG-Fc, n=3 for myelin). Lysates were used for Rap1 activation assays detecting GTP-bound Rap1, and total Rap1 was assessed using input samples. For quantification, Rap1 GTP levels were normalized to the total Rap1 levels in the input lysate. Graphs depict average fold changes ± SEM (*p < 0.05, ***p < 0.001, paired one-tailed Student’s t-test).
Neurite outgrowth is influenced by Rap1 activity.
To test whether Rap1 activity has effects on axonal growth, we assessed neurite outgrowth on permissive and inhibitory substrates following either overexpression or suppression of Rap1. To examine the impact of Rap1 overexpression, we constructed GFP-tagged adenoviruses expressing either wild-type Rap1, or a constitutively active mutant form of Rap1 (F64A) that is insensitive to regulation by Rap1GAPs (22). Hippocampal and DRG neurons were infected with virus and after 1 day in culture, the neurons were transferred to monolayers of control or MAG-expressing CHO cells or CNS myelin substrates, where they were incubated for an additional 24 hours. For the measurements of neurite length, only cells that were positive for both the neuronal marker βIII-tubulin and GFP were included in the analysis. For neurons infected with a control adenovirus expressing GFP, neurite outgrowth on both MAG and CNS myelin was strongly inhibited (Fig. 4A, B), but neurons overexpressing wild-type Rap1 displayed significant increases in neurite length on both MAG and myelin (Fig 4A, B). Similarly, overexpression of Rap1F64A also significantly increased neurite outgrowth on inhibitory substrates compared to GFP controls (Fig. 4A, B). These results indicate that sustained induction of Rap1 activity can overcome inhibition by CNS myelin and thereby directly influence the rate of neurite extension.
Figure 4. Overexpression of wild type Rap1 or the RapGAP-insensitive Rap1 mutant Rap1F64A blocks inhibition by MAG and myelin.
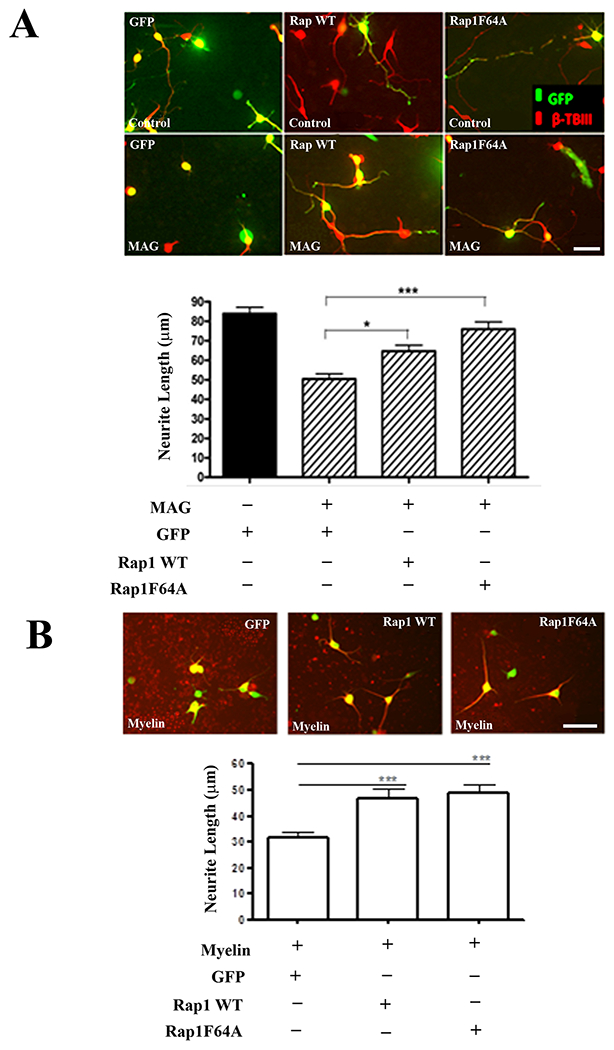
(A) Representative images of P1-3 hippocampal neurons infected with adenoviruses expressing GFP, GFP and wild type Rap1, or GFP and Rap1F64A. Neurons were incubated for 24 hours and transferred to control or MAG-expressing CHO cell monolayers (scale bar=20 μm). Graph depicts average length of the longest neurite per neuron ±SEM for approximately 50 neurons per treatment that were positive for both GFP and βIII tubulin (3 independent experiments, *p < 0.05, ***p < 0.001, one-way ANOVA with Bonferroni’s multiple comparisons test). (B) Representative images of P5-7 DRG neurons infected with adenoviruses expressing GFP, GFP and wild type Rap1, or GFP and Rap1F64A. Neurons were incubated for 24 hours and transferred to CNS myelin substrates (scale bar=40 μm). Graph depicts average length of the longest neurite per neuron ±SEM for approximately 50 neurons per treatment that were positive for both GFP and βIII tubulin (3 independent experiments, ***p < 0.001, one-way ANOVA with Bonferroni’s multiple comparisons test).
To demonstrate that neurite outgrowth can be adversely affected by reductions in Rap1 activity, neurons were infected with adenoviruses expressing Rap1GAP1. We observed that overexpression of RapGAP1 severely limited the ability of neurons to extend neurites on both control and MAG-expressing CHO cells, as neurite length was significantly reduced on both substrates (Fig. 5A). To confirm that this effect is mediated through Rap1GAP1, we then conducted additional experiments in which Rap1GAP1 expression was knocked down with siRNA (Fig. 5B). Neurons were transfected with non-targeting or Rap1GAP1 siRNA for 24 hours and then transferred to CHO cell monolayers or CNS myelin. Neurite outgrowth was strongly inhibited by MAG and myelin for neurons transfected with non-targeting siRNA (Fig. 5C), but siRNA knockdown of Rap1GAP1 reversed the inhibitory effects of MAG and myelin and enabled neurons to extend significantly longer neurites on both substrates (Fig. 5C). These observations reinforce our prior conclusion that Rap1 activity promotes axonal growth and in addition, they demonstrate that Rap1GAP1 plays a prominent role in facilitating myelin-mediated inhibition.
Figure 5. Overexpression of Rap1GAP1 inhibits neurite outgrowth while Rap1GAP1 knockdown blocks MAG-mediated inhibition of neurite outgrowth.
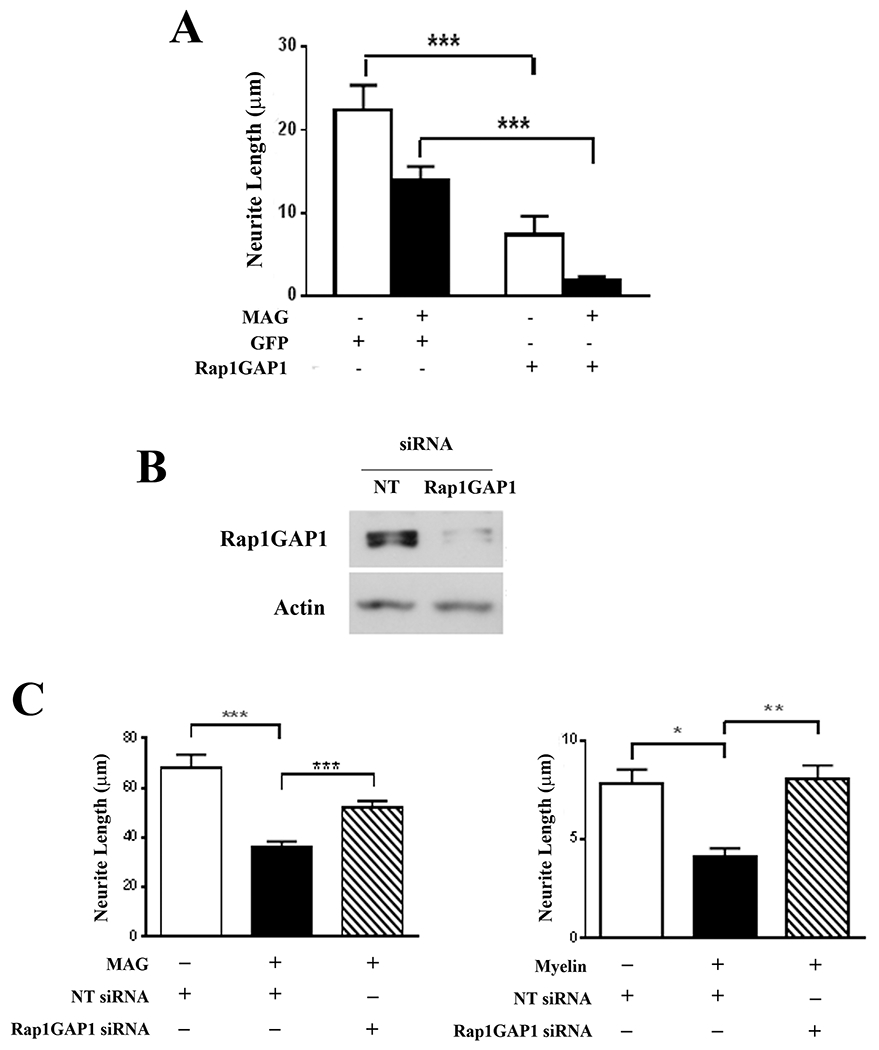
(A) Quantification of neurite outgrowth for P1-3 hippocampal neurons that were infected with adenoviruses expressing GFP, or GFP and Rap1GAPI. Neurons were incubated for 24 hours and transferred to control or MAG-expressing CHO cell monolayers. Graph depicts average length of the longest neurite per neuron ±SEM for approximately 50 neurons per treatment that were positive for both GFP and βIII tubulin (3 independent experiments, ***p < 0.001, one-way ANOVA with Bonferroni’s multiple comparisons test). (B) Western blots of P1-3 cortical neurons that were transfected with either non-targeting (NT) or Rap1GAP1 siRNA and incubated for 24 hours. (C) Quantification of neurite outgrowth for P1-3 cortical neurons that were transfected with either non-targeting (NT) or Rap1GAP1 siRNA and transferred to CHO cell monolayers or CNS myelin substrates. Graphs depict average length of the longest neurite per neuron ±SEM for approximately 100 neurons per treatment (3 independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with Bonferroni’s multiple comparisons test).
Dibutyryl cAMP decreases Rap1GAP1 protein levels.
In our preceding experiments, we demonstrated that the EPAC-specific cAMP analogue 8-CPT induces Rap1 activity, and that priming with 8-CPT overcomes inhibition by MAG. This is notably different from the effects of another cyclic AMP analogue, dibutyryl cAMP (dbcAMP), which stimulates downstream activation of both PKA and EPAC (28, 38) and blocks inhibition when added directly to neurons growing on MAG or myelin, with no need for priming (16). To investigate the reasons for this dichotomy, we examined whether these two cAMP analogues have differing effects on Rap1 activation and Rap1GAP1 expression. When neurons were treated with 1 mM dbcAMP for 20 minutes, we observed a significant increase in GTP-bound Rap1 (Fig. 6A), and this was reminiscent of the increased Rap1 activation that was observed following 8-CPT treatment (Fig. 2A). Interestingly, when the neurons were treated with MAG-Fc prior to the addition of dbcAMP, the activation of Rap1 was unaffected (Fig. 6A). This stands in sharp contrast to the results obtained with 8-CPT, which showed that activation of Rap1 by 8-CPT was blocked by MAG (Fig. 2A). Given the importance of Rap1GAP1 levels regulating Rap1 activity, we then tested if dbcAMP reduces Rap1GAP1 levels, which would counteract the increase in Rap1GAP1 induced by MAG (Fig. 3) and prevent inactivation of Rap1. Treatment with dbcAMP for 20 minutes significantly decreased levels of Rap1GAP1 (Fig. 6B), while 8-CPT had no effect on Rap1GAP1 levels (Fig. 6C). This suggests that direct reversal of myelin-mediated inhibition requires suppression of RapGAP1, and that this suppression does not occur with agents that require priming to enhance neurite outgrowth. To test this, we revisited our previous findings showing that priming with BDNF overcomes inhibition by MAG and myelin, and that treatment with pertussis toxin (PTX) eliminated the need for priming, allowing BDNF to directly increase neurite outgrowth in the presence of myelin-associated inhibitors (16). When neurons were treated with PTX, we observed a significant, time-dependent decrease in Rap1GAP1 levels (Fig. 6D), and this likely accounts for the ability of PTX to enhance neurite outgrowth on inhibitory substrates. It also provides further support for our hypothesis that reducing Rap1GAP1 levels is essential for directly overcoming inhibition by MAG and myelin.
Figure 6. dbcAMP activates Rap1 and decreases Rap1GAP1 protein levels.
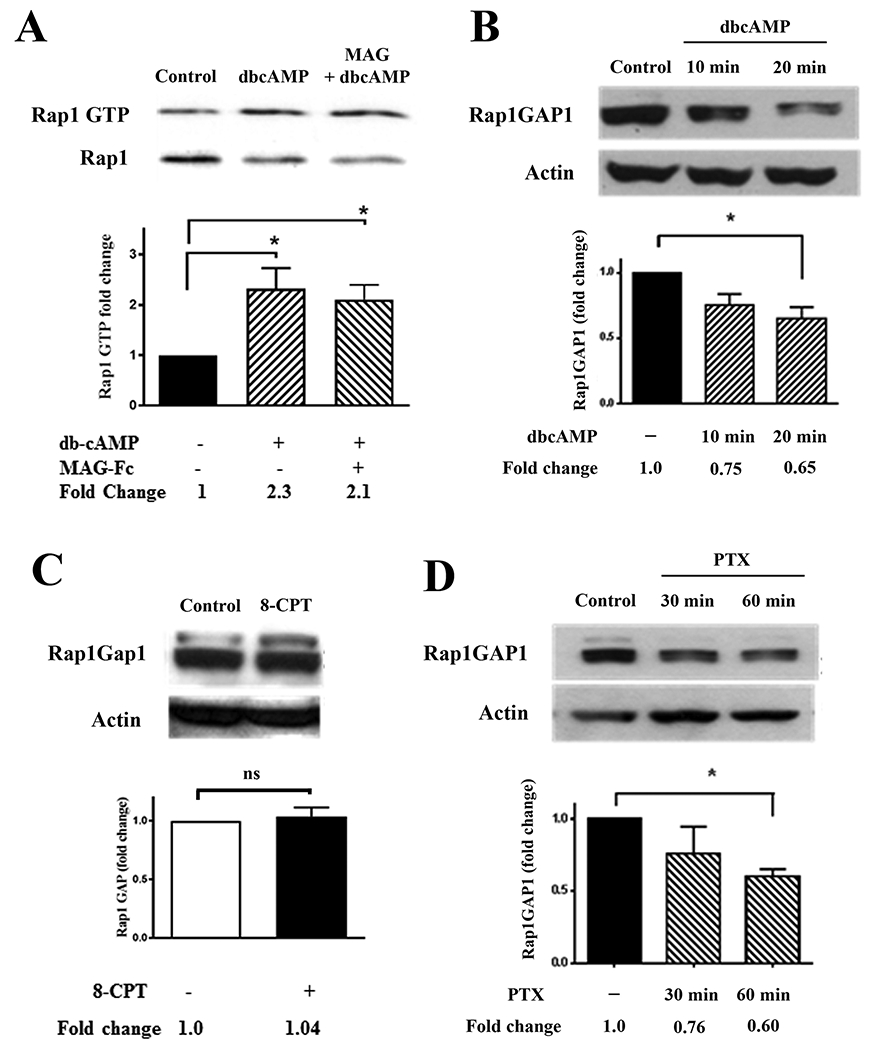
(A) Western blots of P1-3 cortical neurons treated with dbcAMP (1 mM) for 20 min or MAG-Fc (20 μg/ml) for 30 min prior to the addition of dbcAMP (n=3). Lysates were used for Rap1 activation assays detecting GTP-bound Rap1, and total Rap1 was assessed using input samples. Rap1 activation was quantified by normalizing Rap1 GTP levels to the total Rap1 levels in the input lysates. Graph depicts average fold changes ± SEM. (*p < 0.05, one-way ANOVA with Bonferroni’s multiple comparisons test). (B) Western blots of P1-3 cortical neurons treated with dbcAMP (1 mM) for 10 or 20 minutes (n=3). Rap1GAP1 was quantified by normalizing Rap1GAP levels to their respective loading controls. Graph depicts average fold changes ± SEM (*p < 0.05, one-way ANOVA with Bonferroni’s multiple comparisons test). (C) Western blots of P1-3 cortical neurons treated with 8-CPT (0.5 mM) for 20 minutes (n=3). Graph depicts average fold changes ± SEM (ns=not significant, paired one-tailed Student’s t-test). (D) Western blots of P1-3 cortical neurons treated with PTX (2 ng/ml) for 30 or 60 minutes (n=3). Rap1GAP1 was quantified by normalizing Rap1GAP levels to their respective loading controls. Graph depicts average fold changes ± SEM (*p < 0.05, one-way ANOVA with Bonferroni’s multiple comparisons test).
Discussion
Based on our observations, we have concluded that Rap1 serves as an important regulatory node for neurite outgrowth, integrating growth-promoting signals from neurotrophins and cAMP, as well as growth-inhibiting signals from CNS myelin. In addition, our data suggest that these factors influence Rap1 activity primarily through modulation of Rap1GAP1 levels. We therefore propose the following mechanism for Rap1’s ability to reverse inhibition by myelin-associated inhibitors (Fig. 7). Upon exposure to CNS myelin, Rap1GAP1 protein levels are increased and this facilitates the hydrolysis of GTP-bound Rap1 to its GDP-bound form, reducing its activity and contributing to the inhibition of neurite outgrowth. Conversely, elevation of intracellular cAMP, as can occur through administration of dbcAMP, reduces Rap1GAP1 levels and thereby maintains Rap1 activity at higher levels, which enables neurites to grow in the presence of myelin.
Figure 7. Schematic representation of the effects of MAG and cAMP on Rap1 activation and neurite outgrowth.
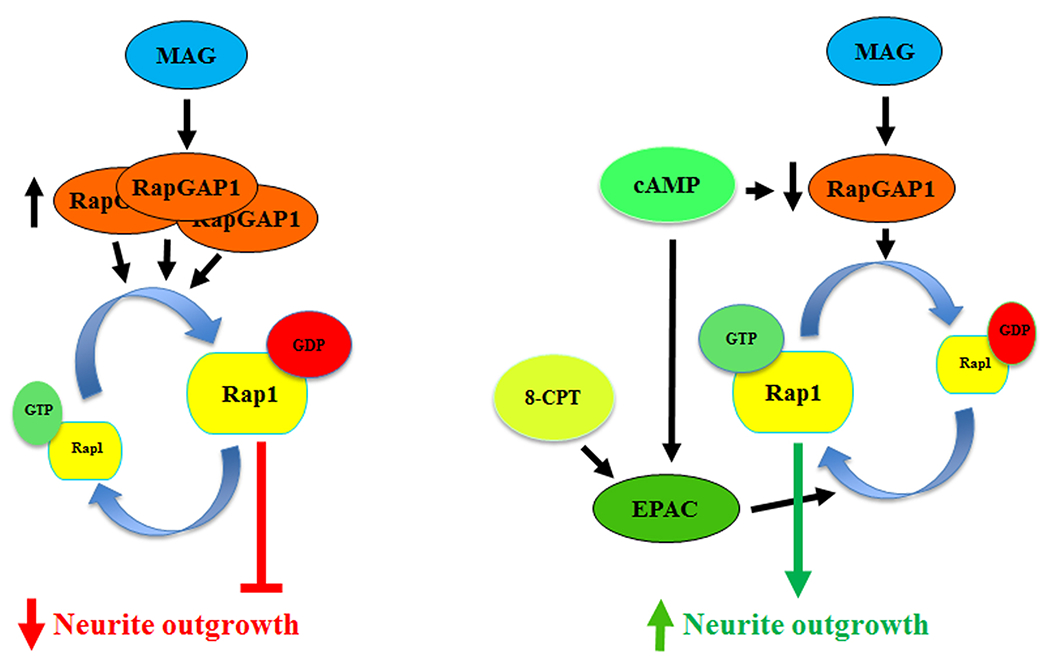
In the presence of MAG, Rap1GAP1 levels are increased, which leads to inactivation of Rap1 and inhibition of neurite outgrowth. Conversely, when intracellular cAMP is elevated, Rap1GAP1 levels are reduced and this leads to an increase in Rap1 activity, which facilitates neurite outgrowth. Activation of EPAC can also increase neurite outgrowth, but only in response to priming with agents such as 8-CPT.
This putative mechanism allows us to view the data obtained in earlier studies of myelin-mediated inhibition in a new and intriguing context. For example, we have previously reported that the ability of cAMP to overcome inhibition by MAG and myelin is dependent on the downstream activation of protein kinase A (PKA), as pharmacological inhibition of PKA with KT5720 or H89 blocked cAMP-induced increases in neurite outgrowth in vitro and in vivo (16, 17, 19). In a study of synaptic plasticity in striatal neurons, it was shown that PKA phosphorylates Rap1GAP1, leading to a decrease in its activity and a corresponding increase in Rap1 activation (10). PKA-mediated inhibition of Rap1GAP1 also significantly increased the head size of dendritic spines of cultured hippocampal neurons, which indicates that altering Rap1GAP1 function can elicit morphological changes as well (10). It is therefore plausible that the reductions in Rap1GAP1 levels and the increased neurite outgrowth observed in response to dbcAMP were mediated through PKA. This assertion is somewhat tempered by the fact that PKA also directly phosphorylates Rap1 and causes it to translocate from the cell membrane to the cytoplasm, where it encounters the cytoplasmic pool of Rap1GAP1 and is rendered inactive (39), but the concomitant reduction in Rap1GAP1 protein levels likely offsets this effect and allows Rap1 activity to be maintained at a level that promotes axonal growth.
The exact mechanism by which cAMP reduces Rap1GAP1 levels is not known, but one possibility is increased proteosomal degradation of the protein. In the human Neuro2A cell line, neurite outgrowth can be mediated through the CB1 cannabinoid receptor, which is coupled to the G protein Gαo/i (40). Treatment with the CB1 receptor agonist HU-210 activated the Gαo/i pathway, which in turn led to increased ubiquitination of Rap1GAP2 and its degradation by the proteasome (40). This resulted in increased Rap1 activity and neurite outgrowth, and importantly, the induction of Rap1 activity was dependent on the levels of Rap1GAP2 protein within the cell, as treatment with lactacystin, a proteasome inhibitor, blocked both the degradation of Rap1GAP2 and Rap1 activation (40). The findings of this study strongly parallel our current observations and therefore suggest that agents such as dbcAMP may enhance neurite outgrowth and Rap1 activation by promoting ubiquitination and degradation of Rap1GAP1.
Similarly, we must consider how myelin-associated inhibitors increase Rap1GAP1 levels and reduce activation of Rap1. MAG mediates inhibition by binding to a variety of receptors. The most well-defined of these is a receptor complex consisting of Nogo receptor 1 (NgR1), p75 neurotrophin receptor or TAJ/TROY, and LINGO-1 (41, 42, 43, 44, 45), but it can also act through paired immunoglobulin receptor B (PirB; 46) and low density lipoprotein receptor-related protein (LRP1; 47). Binding of MAG to these receptors leads to activation of RhoA (8), but there is no evidence that they enhance the expression of Rap1GAP1. As noted previously, Rap1GAP is found predominantly in the cytoplasm (39), but it has also been shown that activation of muscarinic receptors can induce translocation of Rap1GAP2 to the cell membrane, where it inactivates Rap1 (48). We conducted cellular fractionation experiments to examine the subcellular localization of Rap1GAP1 following MAG treatment, and our preliminary results indicated that Rap1GAP1 was predominantly localized to the membrane under these conditions (data not shown). This would place Rap1GAP1 in the appropriate position to hydrolyze GTP-bound Rap1 and we believe that this accounts for the reductions in Rap1 activity that we observed in response to MAG and myelin.
One of the more surprising findings of this study was our observation that while both dbcAMP and 8-CPT activate Rap1, 8-CPT’s effects on Rap1 can be blocked by MAG and it also did not reduce Rap1GAP1 levels. Unlike dbcAMP, it also did not directly overcome inhibition by MAG in neurite outgrowth assays, but did have an effect when used for priming. Given EPAC’s ability to activate Rap1 and the fact that it is activated by cAMP (14, 15), this raises questions about how dbcAMP and EPAC differentially promote axonal growth within an inhibitory environment. As we have noted above, it is likely that dbcAMP reduces Rap1GAP1 levels and enhances neurite outgrowth through downstream activation of PKA. Activation of EPAC occurs independently of PKA, which means that an alternative mechanism must be responsible for its effects on neurite outgrowth. Since EPAC is a GEF for Rap1, a direct effect on Rap1 activity is the most logical explanation. A previous study reported that 8-CPT significantly increased neurite outgrowth when adult DRG neurons were grown on explants of adult rat spinal cord, but the duration of these assays was not noted (49). They also proposed that EPAC acts during the initiation phase of neurite outgrowth (49). Acute activation of Rap1 by 8-CPT may therefore be sufficient to initiate a growth response, but as our data show, 8-CPT has no effect on Rap1GAP1 and this may limit the extent of growth over a longer period of time. Prolonged exposure to MAG (as occurs in our neurite outgrowth assays) may further dampen the effects of 8-CPT by promoting localization of Rap1GAP1 to the cell membrane and Rap1 inactivation. With priming, however, the extended treatment with 8-CPT prior to exposure to MAG may allow Rap1 activity and Rap1GAP1 suppression to reach levels that allow the neurons to extend neurites in an inhibitory environment.
Like 8-CPT, neurotrophins are only able to overcome inhibition by MAG with priming. In a prior study we have shown that priming with BDNF results in prolonged activation of Erk, which leads to elevation of cAMP and the activation of PKA and CREB (28). In neurons, Rap1 is a positive regulator of Erk (50, 51), and it has been shown to be critical for neurotrophin-mediated activation of Erk and cAMP pathways in a PC12 cell differentiation model (11). It is therefore possible that BDNF-mediated activation of Rap1 is responsible for the downstream activation of Erk and elevation of cAMP that we have observed. Cyclic AMP would then activate Rap1 through two parallel mechanisms: EPAC and PKA-dependent inactivation of Rap1GAP1. Conversely, the negative regulation of Rap1 by MAG may uncouple Erk and cAMP signaling from Rap1, thus preventing Erk and PKA-dependent activation of CREB by BDNF. Thus, it appears that Rap1 is a critical component of the Erk and cAMP signaling that occurs in response to BDNF, resulting in sustained activation of both pathways and the morphological effects that are seen with priming.
When we consider how Rap1 affects axonal growth, there is ample evidence to suggest that active Rap1 might exert a direct effect on the molecular machinery governing the extension of neurites. It was shown in non-neuronal cells that Rap1 promotes cell spreading by activating and recruiting Tiam1 and VAV2, which are GEFs for the small GTPase Rac1 (52). Interestingly, Tiam1 is known to regulate axonal growth in hippocampal and cortical neurons (53, 54), which suggests that activation of Rac1 by Rap1 may contribute to neurite outgrowth. It is well-established that signaling by myelin-associated inhibitors converges on RhoA activation and that this facilitates inhibition of axonal regeneration by inducing stress fiber formation (7, 8). In our model, Rap1 may counter MAG-mediated activation of RhoA through its ability to activate two distinct RhoGAPs: ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 3 (ARAP3) and RA-RhoGAP, which have both been shown to promote neurite outgrowth by inhibiting RhoA (55, 56). This positions Rap1 as an important factor in the integration and regulation of small GTPase activity and cytoskeletal dynamics within growing axons. Based on our data and the studies cited here, the net effect of Rap1 activation would be to shift the cytoskeleton towards growth-promoting events such as actin nucleation and filopodial extension, and away from inhibitory events such as actin polymerization and myosin II contraction.
Effective treatments for spinal cord injuries and other forms of CNS trauma remain elusive, due in large part to the highly complex nature of these injuries at both the pathophysiological and molecular levels. Promoting effective regeneration of axons is one facet that must be addressed to achieve functional recovery, and Rap1 could serve as a promising target to develop regenerative therapies. Agents that promote activation of Rap1 or stimulate Rap1GAP1 activity could prove to be effective components of combinatorial treatments that aim to limit cell death, reduce the expression of inhibitory molecules such as chondroitin sulfate proteoglycans, and ultimately, restore function by enhancing axonal regeneration and reestablishing neural circuits.
Acknowledgments
This work was supported by the Specialized Neuroscience Research Program (3U54NS041073; NINDS-NIH), RO1NS037060 (NINDS-NIH), and an infrastructure grant from the Research Centers in Minority Institutions Program (RR003037; NCRR-NIH) at Hunter College. The funders had no involvement with the design and execution of the study or the preparation of the manuscript. The authors declare no conflicts of interest. The manuscript has been prepared in accordance with the Uniform Requirements for Manuscripts Submitted to Biomedical Journals, International Committee of Medical Journal Editors.
References
- 1.David S, Aguayo AJ (1981) Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science 214:931–933. [DOI] [PubMed] [Google Scholar]
- 2.Filbin MT (2003) Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci 4:703–713. [DOI] [PubMed] [Google Scholar]
- 3.Fitch MT, Silver J (2008) CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol 209:294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu BP, Cafferty WB, Budel SO, Strittmatter SM (2006) Extracellular regulators of axonal growth in the adult central nervous system. Philos Trans R Soc Lond B Biol Sci 361:1593–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dougherty KD, Dreyfus CF, Black IB (2000) Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol Dis 7:574–585. [DOI] [PubMed] [Google Scholar]
- 6.Jones LL, Sajed D, Tuszynski MH (2003) Axonal regeneration through regions of chondroitin sulfate proteoglycan deposition after spinal cord injury: a balance of permissiveness and inhibition. J Neurosci 23:9276–9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N, Tigyi G, McKerracher L (1999) Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci 19:7537–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamashita T, Higuchi H, Tohyama M (2002) The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol 157:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah B, Lutter D, Tsytsyura Y, Glyvuk N, Sakakibara A, Klingauf J, Püschel AW (2017) Rap1 GTPases Are Master Regulators of Neural Cell Polarity in the Developing Neocortex. Cereb Cortex 27:1253–1269. [DOI] [PubMed] [Google Scholar]
- 10.McAvoy T, Zhou MM, Greengard P, Nairn AC (2009) Phosphorylation of Rap1GAP1, a striatally enriched protein, by protein kinase A controls Rap1 activity and dendritic spine morphology. Proc Natl Acad Sci USA 106:3531–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJ (1998) Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392:622–626. [DOI] [PubMed] [Google Scholar]
- 12.York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ (2000) Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol 20:8069–8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gloerich M, Bos JL (2011) Regulating Rap small G-proteins in time and space. Trends Cell Biol 21:615–623. [DOI] [PubMed] [Google Scholar]
- 14.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282:2275–2279. [DOI] [PubMed] [Google Scholar]
- 15.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396:474–477. [DOI] [PubMed] [Google Scholar]
- 16.Cai D, Shen Y, De Bellard M, Tang S, Filbin MT (1999) Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron 22:89–101. [DOI] [PubMed] [Google Scholar]
- 17.Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT (2001) Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci 21:4731–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34:885–893. [DOI] [PubMed] [Google Scholar]
- 19.Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34:895–903. [DOI] [PubMed] [Google Scholar]
- 20.Martinez J, Stessin AM, Campana A, Hou J, Nikulina E, Buck J, Levin LR, Filbin MT (2014) Soluble adenylyl cyclase is necessary and sufficient to overcome the block of axonal growth by myelin-associated factors. J Neurosci 34:9281–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeBellard ME, Tang S, Mukhopadhyay G, Shen YJ, Filbin MT (1996) Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci 7:89–101. [DOI] [PubMed] [Google Scholar]
- 22.Brinkmann T, Daumke O, Herbrand U, Kühlmann D, Stege P, Ahmadian MR, Wittinghofer A (2002) Rap-specific GTPase activating protein follows an alternative mechanism. J Biol Chem 277:12525–12531. [DOI] [PubMed] [Google Scholar]
- 23.Meng J, Glick JL, Polakis P, Casey PJ (1999) Functional interaction between Galpha(z) and Rap1GAP suggests a novel form of cellular cross-talk. J Biol Chem 274:36663–36669. [DOI] [PubMed] [Google Scholar]
- 24.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13:757–767. [DOI] [PubMed] [Google Scholar]
- 26.Shen YJ, DeBellard ME, Salzer JL, Roder J, Filbin MT (1998) Myelin-associated glycoprotein in myelin and expressed by Schwann cells inhibits axonal regeneration and branching. Mol Cell Neurosci 12:79–91. [DOI] [PubMed] [Google Scholar]
- 27.Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC (2003) NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron 39:69–84. [DOI] [PubMed] [Google Scholar]
- 28.Gao Y, Nikulina E, Mellado W, Filbin MT (2003) Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J Neurosci 23:11770–11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4:901–906. [DOI] [PubMed] [Google Scholar]
- 30.Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M (2007) Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol 179:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray AJ, Tucker SJ, Shewan DA (2009) cAMP-dependent axon guidance is distinctly regulated by Epac and protein kinase A. J Neurosci 29:15434–15444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inda C, Bonfiglio JJ, Dos Santos Claro PA, Senin SA, Armando NG, Deussing JM, Silberstein S (2017) cAMP-dependent cell differentiation triggered by activated CRHR1 in hippocampal neuronal cells. Sci Rep 7:1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT (2004) Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44:609–621. [DOI] [PubMed] [Google Scholar]
- 34.Noda M (1993) Structures and functions of the K rev-1 transformation suppressor gene and its relatives. Biochim Biophys Acta 1155:97–109. [DOI] [PubMed] [Google Scholar]
- 35.Bos JL (1998) All in the family? New insights and questions regarding interconnectivity of Ras, Rap1 and Ral. EMBO J 17:6776–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiao L, Zhang Y, Hu C, Wang YG, Huang A, He C (2011) Rap1GAP interacts with RET and suppresses GDNF-induced neurite outgrowth. Cell Res 21:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niederost B, Oertle T, Fritsche J, McKinney RA, Bandtlow CE (2002) Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J Neurosci 22:10368–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dasgupta B, Dugan LL, Gutmann DH (2003) The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci 23:8949–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi M, Dillon TJ, Liu C, Kariya Y, Wang Z, Stork PJ (2013) Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J Biol Chem 288:27712–27723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, Bivona TG, Philips MR, Devi LA, Iyengar R (2005) Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through G(alpha)o/i-triggered proteasomal degradation of Rap1GAP1II. J Biol Chem 280:11413–11421. [DOI] [PubMed] [Google Scholar]
- 41.Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB (2004) LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 7:221–228. [DOI] [PubMed] [Google Scholar]
- 42.Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, Garcia KC, He Z (2005) A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45:345–351. [DOI] [PubMed] [Google Scholar]
- 43.Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, Murray B, Jung V, Pepinsky RB, Mi S (2005) TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron 45:353–359. [DOI] [PubMed] [Google Scholar]
- 44.Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420:74–78. [DOI] [PubMed] [Google Scholar]
- 45.Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM (2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5:1302–1308. [DOI] [PubMed] [Google Scholar]
- 46.Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322:967–970. [DOI] [PubMed] [Google Scholar]
- 47.Stiles TL, Dickendesher TL, Gaultier A, Fernandez-Castaneda A, Mantuano E, Giger RJ, Gonias SL (2013) LDL receptor-related protein-1 is a sialic-acid-independent receptor for myelin-associated glycoprotein that functions in neurite outgrowth inhibition by MAG and CNS myelin. J Cell Sci 126:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mochizuki N, Ohba Y, Kiyokawa E, Kurata T, Murakami T, Ozaki T, Kitabatake A, Nagashima K, Matsuda M (1999) Activation of the ERK/MAPK pathway by an isoform of Rap1GAP1 associated with G alpha(i). Nature 400:891–894. [DOI] [PubMed] [Google Scholar]
- 49.Murray AJ, Shewan DA (2008) Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol Cell Neurosci 38:578–588. [DOI] [PubMed] [Google Scholar]
- 50.Grewal SS, York RD, Stork PJ (1999) Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol 9:544–553. [DOI] [PubMed] [Google Scholar]
- 51.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89:73–82. [DOI] [PubMed] [Google Scholar]
- 52.Arthur WT, Quilliam LA, Cooper JA (2004) Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J Cell Biol 167:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kunda P, Paglini G, Quiroga S, Kosik K, Caceres A (2001) Evidence for the involvement of Tiam1 in axon formation. J Neurosci 21:2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyamoto Y, Yamauchi J, Tanoue A, Wu C, Mobley WC (2006) TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc Natl Acad Sci USA 103:10444–10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeon CY, Kim HJ, Morii H, Mori N, Settleman J, Lee JY, Kim J, Kim SC, Park JB (2010) Neurite outgrowth from PC12 cells by basic fibroblast growth factor (bFGF) is mediated by RhoA inactivation through p190RhoGAP and ARAP3. J Cell Physiol 224:786–794. [DOI] [PubMed] [Google Scholar]
- 56.Yamada T, Sakisaka T, Hisata S, Baba T, Takai Y (2005) RA-RhoGAP, Rap-activated Rho GTPase-activating protein implicated in neurite outgrowth through Rho. J Biol Chem 280:33026–33034. [DOI] [PubMed] [Google Scholar]