

Graphical abstract
Abbreviations: ACE2, angiotensin-converting enzyme 2; AnxA5, annexinA5; APC, antigen-presenting cell; aPL, anti-phospholipid; APS, anti-phospholipid syndrome; ARDS, acute respiratory distress syndrome; BMP/SMAD, bone morphogenetic/small mother against decapentaplegic; β2GPI, β2-glicoprotein I; CME, clathrin-mediated endocytosis; COPD, chronic obstructive pulmonary disease; COVID-19, Sars-Cov-2 infection; CQ, Chloroquine; DC, dendritic cell; DMT1, divalent metal-ion transporter 1; EE, early endosome; EPO, erythropoietin; eNOS, endothelial nitric oxide synthetase; eNOX, endosomal NADPH oxidase; ER, endoplasmic reticulum; ERFE, erythroferrone; ERGIC, endoplasmic reticulum-Golgi intermediate compartment; FPN1, ferroportin 1; FT, ferritin; HAEC, human aortic endotelial cell; HAMP, hepcidin; HCQ, hydroxychloroquine; HCMV, Human Cytomegalovirus; HCV, hepatitis C virus; HFE, hemochromatosis gene; HH, hemochromatosis; HIV-1, Human Deficiency Virus 1; HP, haptoglobin; HPX, hemopexin; HUVEC, human umbilical vein endothelial cell; ICU, intensive care unit; ID, iron deficiency; IDA, iron deficiency anemia; IDU, infectious disease unit; IFN-γ, interferon-γ; IFN1, type interferon; IgG, immunoglobulin G; IL-1β, interleukin-1β; IL-4, interleukin-4; IL-6, interleukin-6; IL-12, interleukin-12; IL-13, interleukin-13; IL-17, interleukin-17; JAK/STAT3, Janus kinase/signal transducers and activators of transcription 3; LF, lactoferrin; LEL, late endosome-lysosome; LPS, lipopolysaccharide; M, viral envelope membrane protein; mAb, monoclonal antibody; MAPK, mitogen-activated protein kinase; MHC, Major Histocompatibility Complex; MHV-3, mouse hepatitis virus type 3; MMP, matrix metalloproteinase; MOI, multiplicity of infection; NET, neutrophil extracellular trap; NK, natural killer; NOX2, NADPH oxidase 2; NP, virus nucleoprotein; PICALM, phosphatidylinositol binding clathrin assembly protein; RA, rheumatoid arthritis; RBC, red blood cell; S, viral spike protein; SLE, systemic lupus erythematosus (SLE); STEAP3, metalloreductase six-transmembrane epithelial antigen of the prostate 3; TF, tissue factor; TFR1, transferrin receptor 1; TGN, trans-Golgi network; TGT, thrombin generating time; Th, T helper; TLR, Toll-like receptor; TMPRSS2, transmembrane serine protease 2; TNF-α, tumor necrosis factor-α; TRPML1/MCOLN1, mucolipin 1; VCAM-1, vascular cellular adhesion molecule 1
Keywords: Sars-Cov-2, COVID-19, Chloroquine/hydroxychloroquine, Iron, Inflammation, Thrombosis
Abstract
The anti-malarial drugs chloroquine (CQ) and primarily the less toxic hydroxychloroquine (HCQ) are currently used to treat autoimmune diseases for their immunomodulatory and anti-thrombotic properties. They have also been proposed for the treatment of several viral infections, due to their anti-viral effects in cell cultures and animal models, and, currently, for the treatment of coronavirus disease 2019 (COVID-19), the pandemic severe acute respiratory syndrome caused by coronavirus 2 (Sars-Cov-2) infection that is spreading all over the world. Although in some recent studies a clinical improvement in COVID-19 patients has been observed, the clinical efficacy of CQ and HCQ in COVID-19 has yet to be proven with randomized controlled studies, many of which are currently ongoing, also considering pharmacokinetics, optimal dosing regimen, therapeutic level and duration of treatment and taking into account patients with different severity degrees of disease. Here we review what is currently known on the mechanisms of action of CQ and HCQ as anti-viral, anti-inflammatory and anti-thrombotic drugs and discuss the up-to-date experimental evidence on the potential mechanisms of action of CQ/HCQ in Sars-Cov2 infection and the current clinical knowledge on their efficacy in the treatment of COVID-19 patients. Given the role of iron in several human viral infections, we also propose a different insight into a number of CQ and HCQ pharmacological effects, suggesting a potential involvement of iron homeostasis in Sars-Cov-2 infection and COVID-19 clinical course.
1. Introduction
Chloroquine (CQ) and its hydroxy-analogue hydroxychloroquine (HCQ), both 4-aminoquinolines, have been extensively used in the treatment of malaria in the last century, but the continuous emergence of drug-resistant strains of Plasmodium falciparum has accelerated the development of new antimalarial drugs. Nevertheless, these compounds, particularly the less toxic HCQ, are currently widely used to treat autoimmune diseases like rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and anti-phospholipid syndrome (APS), due to their immunomodulatory and anti-thrombotic properties. CQ and HCQ have also been proposed for the treatment of viral infections, since they have been demonstrated to directly inhibit viral entry and spread in several in vitro and in vivo models. Treatment with these drugs has then been proposed for several viruses in humans, including coronaviruses like Sars-Cov and Sars-Cov-2. Nonetheless, a clear evidence of their utility in human viral infectious diseases still lacks [1]. Translation from laboratory to clinic should be based on detailed analysis of results from randomized controlled studies and observational outcome registries focused on the efficacy, duration and toxicities of treatments with these drugs that could be useful to understand their real effectiveness.
Here we review the current knowledge on the mechanisms of action of CQ and HCQ as anti-viral, anti-inflammatory and anti-thrombotic drugs and discuss the current experimental evidence on the potential mechanisms of action of CQ/HCQ on Sars-Cov2. We also propose a different insight into some of CQ and HCQ effects, suggesting a potential role of iron homeostasis in Sars-Cov-2 disease (COVID-19), similarly to several other human viral infections [[2], [3], [4]]. Finally, we briefly review and discuss the current knowledge on their efficacy in the treatment of patients with COVID-19.
2. Methodology and literature search strategy
We conducted a literature search using different database (PubMed, Science Direct and Web of Science) up to April 20th 2020. The search strategy was to use different search terms alone and in any combination, such as “Sars-Cov-2 disease”, “COVID-19”, “Sars-Cov-2”, “coronavirus”, “clinical trial”, “treatment”, “drug”, “chloroquine”, “hydroxychloroquine”, “iron”, “virus”, “viral entry”, “viral spread”, “anti-viral activity”, “infection”, “inflammation”, “immunity”, “innate immunity”, “cytokine”, “IL-6”, “TNF-α”, “IL-1β”, “adaptive immunity”, “thrombosis”, “in vitro”. Only English articles with available data were included.
3. Mechanisms of CQ/HCQ as anti-viral drug
Several mechanisms have been proposed for CQ/HCQ mode of action as antiviral molecules, mostly based on their weak basic properties, widely known and classically employed in biomedical research to increase the pH of acidic intracellular organelles like endosomes, lysosomes, autophagic and Golgi vesicles in which these drugs concentrate, in order to study autophagic flux [5]
CQ/HCQ may inhibit entry, replication and spread of several viruses by different mechanisms [6,7], some of which have already been demonstrated [[8], [9], [10]] or proposed [11] for Sars-Cov-2 infection. See Table 1 for a schematic list of CQ/HCQ biological activities as anti-viral drugs.
Table 1.
Main biological activities of chloroquine (CQ) and hydroxychloroquine (HCQ) as anti-viral drugs.
| Biological activity | References |
|---|---|
| Inhibition of viral attachment and entry in the host cell | |
Inhibition of the biosynthesis of sialic acids
|
[12] [ 12,13] [14] |
| Inhibition of PICALM expression and CME | [15,16] |
| Endosomal alkalinization and inhibition of cellular endosomal protease (cathepsin and/or TMPRSS2) | [17] |
| Inhibition of new viral particle maturation and spread | |
| Endosomal alkalinization and inhibition of endosome-lysosome membrane fusion | [18] |
| ERGIC and TGN vesicle alkalinization and inhibition of post-translational modifications of viral proteins | [7,19,20] |
| ERGIC vesicle alkalinization and inhibition of viral budding | [21] |
| Inhibition of p38 MAPK activation | [23,24] |
| Inhibition of phospholipase A2 and membranous structures essential for replication and transcription | [35] |
3.1. Inhibition of viral attachment and entry in the host cell
Coronavirus infection begins with the interaction of viral particles with host cell membranes, through viral structural envelope proteins and specific cellular membrane receptors. CQ/HCQ could inhibit viral entry by acting as inhibitors of the biosynthesis of sialic acids, critical actors of virus-cell ligand recognition. The in vitro action of CQ against coronaviruses has been attributed to the inhibition of the N-glycosylation of the cell surface viral receptor, the angiotensin-converting enzyme 2 (ACE2) for both Sars-Cov and Sars-Cov-2, and/or possibly viral spike (S) proteins, in turn resulting in reduced binding affinity between cellular ACE2 and viral S protein, although glycosylation of Sars-Cov S protein seems to be unchanged by therapeutic doses of CQ [12]. S protein of Sars-Cov-2 is also glycosylated and its glycosylation pattern exhibits common sites with Sars-Cov, but also novel different potential positions [13]. By in silico analysis, Fantini and colleagues [14] have suggested that Sars-Cov-2, through its S protein, might use not only ACE2 receptor for entry but also sialic acids linked to host cell surface gangliosides, possibly improving the cellular attachment of the virus. In silico modelling suggests that CQ/HCQ could bind host sialic acids and gangliosides with high affinity, possibly inhibiting S protein interaction with the host plasma-membrane. Considering all these observations, CQ/HCQ could then act through two ways: decreasing viral entry and/or reducing infectivity of newly produced virions.
CQ has been shown to reduce the expression of phosphatidylinositol binding clathrin assembly protein (PICALM) [15], a cargo-selecting adaptor and one of the most abundant proteins in clathrin-coated pits that regulates the rate of cellular clathrin-mediated endocytosis (CME), implicated in Sars-Cov entry in human cells [16].
Following receptor binding, S protein of coronaviruses undergoes an acid-dependent proteolytic cleavage by cellular endosomal proteases like cathepsin or transmembrane serine protease 2 (TMPRSS2). The cleavage results in the fusion of viral and cellular endosomal membranes and may be inhibited by pH increase. Sars-Cov-2 S protein cleavage is obtained through the enzymatic activity of both cathepsin and TMPRSS2 [17].
Then, CQ/HCQ could have inhibitory effects on virus attachment and entry in the host cell, possibly resulting in blocking the viruses in endocytic vesicles.
3.2. Inhibition of new viral particle maturation and spread
CQ/HCQ have also been shown to display anti-viral activity even when administered after viral infection. This effect has been observed in vitro also in Sars-Cov and Sars-Cov 2 infections [8,9,12]. Further mechanisms could then be involved in antiviral drug action.
Through the alkalization of endosomes, CQ/HCQ might also act inhibiting or preventing endosome-lysosome membrane fusion that leads to membrane viral receptor recycling, viral uncoating and viral genome release into the cytosol, as observed for Sars-Cov [18].
CQ/HCQ may interfere with viral protein maturation processes, occurring in the endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC) and trans-Golgi network (TGN) vesicles and requiring a low pH. Elevation of pH may disrupt post-translational modifications like glycosylations and proteolytic processing of viral proteins. Like S protein, the envelope membrane proteins M of coronaviruses, the most abundant viral structural proteins, are glycosylated and their modifications also occur in vesicles of the ERGIC and TGN [7]. Cleavage sites other than those recognized by cathepsin and TMPRSS2 have been identified in the S protein of Sars-Cov-2 that may be cleaved by furin-like proteases. Furin is localized in the TGN, is highly expressed in the lung, its cleavage of S protein could be implicated in virus egress and spread [19] and might be inhibited by CQ, as observed in Chikungunya virus infection [20].
Due to their basic properties and consequent disruption of cellular vesicle compartments, CQ/HCQ may also inhibit virion budding, occurring when encapsidated viral genomes bud into the ERGIC membranes in which viral envelope proteins are inserted, forming mature virions [21].
Within infected airway epithelium, through their binding to Toll-like receptors (TLRs), several respiratory viruses (among them some coronaviruses) activate the mitogen-activated protein kinase (MAPK) pathways, particularly the p38 MAPK. Respiratory viruses harness MAPK activation to their own advantage, exploiting the triggering of their downstream targets for trafficking their own proteins and for viral assembly and spread. These effects are particularly important in people suffering for underlying airway diseases, like asthma or chronic obstructive pulmonary disease (COPD), who undergo a respiratory viral infection, since these pathways are already turned on and aberrantly activated [22]. CQ has been shown to hinder viral infections through the inhibition of p38 MAPK activation [23,24]. This inhibitory action could then be of particular importance in COVID-19 since people with COPD are among the worst affected by Sars-Cov-2 infection.
Therefore, besides viral attachment and entry, also viral uncoating, genome release, protein maturation process and assembly of new virions for budding and spread may be inhibited by the basic drugs, resulting in reduced infectivity.
3.3. Current evidence on the mechanism(s) of action of CQ/HCQ in Sars-Cov-2 infection
Little is currently known about the mechanism(s) of action of CQ/HCQ in the treatment of Sars-Cov-2 infection. The first published experimental evidence of the potential efficacy of CQ/HCQ in this disease comes from in vitro experiments. Wang and colleagues [8] found that CQ potently blocked Sars-Cov-2 infection (multiplicity of infection, MOI = 0,05) in Vero E6 cells at low-micromolar concentrations (EC50 = 1.13 μM, EC90 = 6.90 μM, 48 h), clinically achievable with 500 mg/day administration. Efficacy was evaluated by quantification of viral copies in the cell supernatant by RT-PCR, immunofluorescence microscopy and western blot analysis for virus nucleoprotein (NP). The authors found that CQ blocked infection at both entry and post-entry steps (drug added 2 h before viral treatment, western blot performed after 14 h). The same researchers [9] later compared HCQ and CQ efficacies in the same cell model and found that cytotoxicity was not significantly different between the two drugs. Measuring anti-viral efficacy at different MOIs (0.01, 0.02, 0.2, 0.8) by RT-PCR they observed lower E50 values for CQ at all MOIs (2.71, 3.81, 7.14, 7.36 for CQ and 4.51, 4.06, 17.31, 12.96 for HCQ), confirmed by immunofluorescence analysis for NP. These results suggested that CQ could be more efficacious than HCQ. The authors also confirmed that CQ acts at both entry and post-entry stages and this double action was also observed for HCQ. In order to shed light on potential mechanisms of action of the drugs, the authors studied the cellular localization of viral particles and found virions partly in early endosomes (EEs) and more in late endosome-lysosomes (LELs) in control conditions. When cells were treated with CQ and HCQ, more viral particles were observed in EEs, suggesting that the drugs block endocytotic vesicle maturation at intermediate stages and probably stall the virus transport from EEs to LELs, a crucial step for the release of viral genome. The authors also observed that both CQ and HCQ treatments resulted in abnormally enlarged EEs, but while HCQ increased LEL size and number, CQ treatment induced no changes in number and size of LELs but the vesicle structure was disrupted, suggesting partially distinct mechanisms of action of the two drugs. The anti-Sars-Cov-2 in vitro action of CQ and HCQ was confirmed in the same cellular model by [10]. These authors found EC50 values of 23.90 and 5.47 for CQ and 6.14 and 0.72 mM at 24 and 48 h for CQ and HCQ respectively, that, in contrast with the previous report, was a better performance for HCQ. They also confirmed that the drugs have anti-viral activity also when administered prior to viral infection, with EC50 values of >100, and 18.01 for CQ and 6.25 and 5.85 mM for HCQ respectively at 24 and 48 h.
4. Mechanisms of CQ/HCQ as anti-inflammatory drugs
CQ/HCQ are also used as anti-inflammatory and immunomodulatory drugs in autoimmune diseases, like RA and SLE. These properties derive from their multiple effects on the immune system cells and their modulation of crucial pro-inflammatory cytokines [25,26]. See Table 2 for a schematic list of CQ/HCQ biological activities as anti-inflammatory drugs.
Table 2.
Main biological activities of chloroquine (CQ) and hydroxychloroquine (HCQ) as anti-inflammatory drugs.
| Biological activity | References |
|---|---|
| Modulation of innate and adaptive immune cell activation, cytokine response and inflammation | |
Inhibition of antigen presentation by APCs
|
[15] [ 27,28] [27,28] |
| Inhibition of Ca2+ signaling and T and B cell activation | [29] |
| Inhibition of Th17 proliferation and differentiation | [30] |
| Vesicle alkalinization and inhibition of the TLR signaling and MMPs | [31,32] |
| Inhibition of phospholipase A2 and of the release of prostaglandins | [33] |
| Inhibition of p38 MAPK activation and of the release of cytokines | [22,23] |
| Inhibition of TNF-α release | [37,48,49,50] |
| Inhibition of vasodilation, infiltration and adhesion of leukocytes at the site of inflammation | |
| Inhibition of respiratory burst in polymorphonuclear leukocytes | |
| Inhibition of IL- β release | [38,51] |
| Inhibition of neutrophil recruitment and Th17 differentiation | |
| Inhibition of IL-6 release | [37,56] |
| Inhibition of Th17 differentiation | |
| Induction of cytotoxic activity of CD8 + T cells | |
| Activation of Treg cell functions | |
| Reduction of tissue injury | |
| Reduction of microorganism immune evasion strategy |
4.1. CQ/HCQ modulate innate and adaptive immune cell activation, cytokine response and inflammation
CQ/HCQ may impair the correct maturation and recognition of viral antigens by antigen-presenting cells (APCs) that require endosomal acidification for antigen processing. By phagocytosis and macropynocytosis, dendritic cells (DCs) capture pathogens, process and present their antigens to activate T cells. Through their antigen-specific B cell receptor and CME and clathrin-independent endocytosis, B cells recognize specific antigens and present their peptides to specific T cells. Through phagocytosis, CME, caveolin-mediated endocytosis and macropinocytosis, macrophages internalize pathogens, process their proteins and present antigens to T cells. T cells, through their antigen specific receptors, interact with APCs, recognizing antigenic peptides immobilized on their surface by Major Histocompatibility Complex (MHC) class I (for CD8+ T cytotoxic cells) and MHC class II (for CD4+ T helper cells) molecules. MHC class I molecules are loaded with those antigens mainly derived from proteasome degradation of cytosolic proteins (like viral proteins) in the ER, but also from phagocytosed/endocytosed material, processed through endosomes and lysosomes. MHC class II molecules bind antigens obtained by endosomal and lysosomal processing and, as MHC class I, require the low pH of these organelles to activate protease and other enzymes and form the complexed antigen [27,28]. CQ downregulates PICALM affecting CME and possibly antigen-processing [15]. CQ/HCQ also alkalinize vesicles of the endosome-lysosome-autophagy pathway, possibly further interfering with antigen presentation by APCs, both acting on antigen degradation and MHC molecules processing. The inhibition of antigen presentation may reduce T cell activation and differentiation and in turn decrease the production of pro-inflammatory cytokines.
HCQ also acts modulating T and B cell activation, independently of the above described inhibition of antigen presentation. The drug inhibits Ca2+ signaling and downstream events, involved in T and B cell receptor engagement with antigen, reducing their activation [29]. Further, HCQ hinders the differentiation and proliferation of T helper (Th) 17 cells, a subpopulation of CD4+ T cells characterized by the production of the pro-inflammatory cytokine interleukin-17 (IL-17) and associated with the pathogenesis of several inflammatory diseases, like RA and SLE [30].
TLRs are involved in innate immunity against bacteria and viruses. TLR7 and TLR9 are located in the ER and move to endosomes and lysosomes where they recognize bacterial and viral nucleic acids. By this binding, their activation results in the production of pro-inflammatory cytokines and chemokine like interleukin-6 (IL-6), interleukin-12 (IL-12), type I interferons (IFN1s), interferon-γ (IFN-γ) and cytokines promoting the stimulation of Th1 cells and disrupting the Th1/Th2 balance. The activation of TLR7 and TLR9 by their ligands requires the low pH of endosomes and lysosomes and an active autophagic pathway, then CQ/HCQ, through vesicle alkalinization, may inhibit this interaction and TLR signaling, in turn inhibiting the inflammatory response [31]. Matrix metalloproteinases (MMPs) are a family of zinc endopeptidases that regulate inflammation and tissue repair at several levels. MMPs regulate pro-inflammatory and anti-inflammatory cytokine and chemokine production by proteolytic processing and may in turn be induced by cytokines and chemokines. CQ has been shown to downregulate MMP-9 expression through the inhibition of TLR9 signaling [32].
CQ has also been demonstrated to inhibit phospholipase A2 and block the arachidonic acid cascade that leads to the production of prostaglandins and tromboxanes. In this context, CQ may have both anti-inflammatory and anti-thrombotic effects [33]. This is an important point, since Sars-Cov-2 can induce pulmonary microthrombi and coagulopathy, being characterized in its severe forms by D-dimer level elevation [34,35]. Interestingly, phospholipase A2 has recently been shown to be critically involved in coronavirus replication due to its role in the production of lipids required to form the membranous structures that are essential for virus replication and transcription [36]. Then, inhibiting phospholipase A2, CQ may be a triple arm against Sars-Cov-2, acting through the anti-inflammatory, anti-thrombotic and anti-viral mechanisms.
As stated above, respiratory viruses can activate the p38 MAPK pathway in infected airway epithelium and harness the trafficking machinery of infected cells to assist viral assembly and spread. p38 MAPK activation also leads to the release of cytokines like tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-17 and IL-6 that turn on the inflammatory response, but this reaction can be inhibited by the CQ [22,23].
4.2. CQ/HCQ inhibit IL-6, IL-1β and TNF-α release
As previously mentioned, COVID-19 is characterized by the release of high levels of IL-6, IL-1β and TNF-α that are key modulators of inflammation [37,38]. CQ/HCQ have been shown to inhibit their release by immune cells through several mechanisms like transcription and post-transcriptional regulation, p38 MAPK signaling block as stated above or disruption of cellular iron metabolism among others [6,[39], [40], [41], [42], [43], [44], [45], [46], [47]]. TNF-α is produced by macrophages, T and B cells, natural killer (NK) cells and several non-immune cells in response to a broad range of infections. Through several signaling pathways, TNF-α controls vasodilation, infiltration and adhesion of leukocytes at the site of inflammation, complement and coagulation cascade activation, respiratory burst in polymorphonuclear leukocytes and it is also a pyrogenic cytokine. TNF-α up-regulation has also been observed in coronavirus infections other than Sars-Cov-2 [48,49] and seems to play a key role in lung injury [50]. IL-1β is expressed in a tightly regulated way by myeloid cells and released by the inflammasome. This cytokine promotes neutrophil recruitment and Th17 differentiation [51]. Monoclonal antibodies (mAbs) blocking IL-1β pathway has been proposed to treat COVID-19 [52]. IL-6 plays a crucial role in COVID-19, particularly in the related acute respiratory distress syndrome (ARDS) and cytokine storm that can lead to death [34]. Preliminary clinical data with mAbs blocking IL-6 have demonstrated a certain degree of efficacy in severe COVID-19 patients [53] and in some single cases [54,55], but these pilot results needs further clinical data and well-planned clinical trial on the efficacy of this treatment, especially considering timing of treatment since, as suggested by Mc Gonagle and colleagues [34], this aspect could be crucial for the following proper tissue repair. IL-6 is produced in response to tissue damage and infections by several cell types, including B and T cells, monocytes, DCs, macrophages, fibroblasts and endothelial cells. The cytokine activates the Janus kinase/signal transducers and activators of transcription 3 (JAK/STAT3) signal transduction pathway that pleiotropically controls multiple downstream associated genes, having however both pro- and anti-inflammatory effects. IL-6 promotes Th2 cell response and inhibits Th1 polarization, induces cytolytic activity of CD8+ T cells, regulates acute phase protein production in the liver, promotes hematopoietic cell growth and differentiation, organizes migration and phagocytic activities of macrophages and accumulation of neutrophils at sites of infection or trauma, increases immunoglobulin G (IgG) production by B cells, suppresses some viral infection, orchestrates tissue remodeling and repair. On the other hand, cytotoxic activities of CD8+ T cells may also be negatively regulated by IL-6. IL-6 can induce Th17 cell differentiation that both acts in pathogen defense and viral persistence, while inhibiting Treg cell functions. Excessive IL-6 production may increase inflammation and exacerbate tissue injury. Some microorganisms can up-regulate IL-6 as an immune evasion strategy [56]. Then, the challenge for anti-IL-6 therapy in COVID-19 is to identify when IL-6 is detrimental or beneficial that is when clinicians would act on this pathway [34].
5. Mechanisms of CQ and HCQ as anti-thrombotic
A possible role of HCQ in the prevention of thrombosis in hip replacement surgery has been proposed since the late 1970s [[57], [58], [59], [60]]. In this pathology, a decrease of pulmonary embolism has been reported using HCQ doses between 600 and 1600 mg per day [61,62]. HCQ is now commonly used as therapeutic in SLE and APS, also due to its ability to decrease the incidence of thrombosis at the dose of 200 mg–400 mg per day [[63], [64], [65], [66], [67], [68]]. Some authors associated this anti-thrombotic effect to the decrease of the anti-phospholipid (aPL) antibody titers [[69], [70], [71]], while others to a more general and transversal contribute to immunomodulatory, anti-inflammatory, metabolic and anti-thrombotic effects [25,72].
Chinese cardiologists have reported diffuse micro-vascular thrombosis in various organs on autopsy examination in patients died for COVID-19, particularly in the lung. Taking into consideration this diffuse thrombosis, Chinese physicians have recommended anti-coagulant therapy in these patients, but no pharmacological protocols have currently been published [73].
The anti-thrombotic effect of CQ/HCQ may derive from different mechanisms. See Table 3 for a schematic list of CQ/HCQ biological activities as anti-thrombotic drugs.
Table 3.
Main biological actions of chloroquine (CQ) and hydroxychloroquine (HCQ) as anti-thrombotic drugs.
| Biological activity | References |
|---|---|
| Interference with platelet aggregation | |
| Decrease of collagen activation | [74] |
| Decrease of alpha granule discharge | [75] |
| Inhibition of phospholipase A2 and of the release of tromboxanes | [33,77] |
| Increase of fibrinogen with decrease of plasmatic and blood viscosity | [78] |
| Decrease of rheological properties of RBCs | [79] |
| Inhibition of NETs | [84] |
| Interference with membrane binding of blood clotting proteins | |
| Inhibition of the binding of aPL antibody- β2GPI complex to the phospholipid bilayer | [85] |
| Restoration of the AnxA5 anticoagulant shield | [86,87,88] |
| Improvement of biomarkers of endothelial dysfunction | |
| Amelioration of NO bioavailability and decrease of oxidative stress | [89] |
| Improvement of endothelial relaxation | [90] |
| Increase of p-eNOS/eNOS ratio, with improvement of NO production | [90] |
| Inhibition of eNOX and NOX2 | [91] |
| Improvement of lipid profile | [92,93,94] |
5.1. Interference with platelet aggregation
CQ/HCQ could act by interfering with platelet aggregation, decreasing the activation of collagen and alpha granule discharge [74,75]. Platelet activation induces degranulation with the release of adenine diphosphate (ADP) and other molecules in order to amplify the platelet activation process. HCQ has been shown to decrease ADP effects on platelet activation and in vitro platelet aggregation induced by the antibiotic ristocetin [74,76]. As mentioned above, CQ/HCQ could inhibit phospholipase A2 and decrease arachidonic acid, prostaglandin and tromboxane release [33,77]. When administered within 48 h before surgery, HCQ induces the increase of fibrinogen causing a decrease of plasmatic and blood viscosity [78]. A possible role in rheological property decrease of red blood cells (RBCs) has been proposed, but this effect is not perfectly clarified [79].
Neutrophil extracellular traps (NETs) are released by activated neutrophils that unleash intracellular molecules such as granules, proteins, DNA and histones in tissues or circulation to capture and kill pathogens during infections [80]. NETs have been shown to increase thrombosis in autoimmune diseases and in inflammation [81,82]. Golonka and colleagues [83] have recently hypothesized that neutrophil abundance in COVID-19 could be associated to the increase of NETs. In a murine model of pancreatic adenocarcinoma, NETs have been shown to stimulate the discharge of tissue factors and promote platelet activation and aggregation. In this model, CQ/HCQ showed, besides their proper anti-thrombotic characteristics, the ability to inhibit NETs, in turn further decreasing hypercoagulability [84].
5.2. Interference with membrane binding of blood clotting proteins
HCQ has been shown to inhibit the binding of the aPL antibody-β2-glicoprotein I (β2GPI) complex to the phospholipid bilayer, decreasing the pro-thrombotic effect [85]. AnnexinA5 (AnxA5) is an anticoagulant protein with great affinity for negatively charged phospholipids. This protein forms a 2-dimensional crystal over membrane phospholipids and exerts its potent anticoagulant activity shielding the phospholipid bilayer from the interaction with coagulation enzymes [86]. In vitro experiments on human umbilical vein endothelial cells (HUVEC) have evidenced the protective effect of HCQ, which acts restoring the AnxA5 anticoagulant shield and therefore prevents the beginning of coagulation cascade [87,88].
5.3. Improvement of biomarkers of endothelial dysfunction
HCQ is used in the treatment of APS. Its ability to reduce endothelial dysfunction has been studied in animal models. Vascular reactivity has been studied in mice injected for three weeks with aPL antibodies directed against β2GPI to induce APS, with or without HCQ co-treatment. HCQ improved endothelium-dependent dilatation by the amelioration of NO bioavailability and by the decrease of oxidative stress [89]. In another study, aPL antibodies were administered in mice and in human aortic endotelial cells (HAEC), in the presence or absence of HCQ, to evaluate endothelial function and endothelial nitric oxide synthetase (eNOS) modulation. In both models, thrombosis was evaluated by thrombin generating time (TGT) and tissue factor (TF) production. aPL antibodies increased vascular cellular adhesion molecule 1 (VCAM-1) expression and decreased endothelial relaxation induced by acetylcholine in HAEC cells. In mice, aPL antibodies increased thrombus size and shortened the time needed to generate arterial occlusion. TGT and TF were increased in both models. HCQ confirmed its anti-thrombotic activity decreasing clot formation, ameliorating TGT and improving endothelial relaxation. In addition, HCQ ameliorated eNOS activation, increasing the p-eNOS/eNOS ratio with consequent improvement of nitric oxide (NO) production [90]. Cytokines like TNF-α, IL-1β and aPL antibodies induce endosomal NADPH oxidase (eNOX) that is implicated in the pro-inflammatory signal transduction. HCQ inhibits these up-regulation in human monocytes, preventing the translocation of the catalytic subunit gp91phox in the endosome and so interfering with eNOX functions. Further, HCQ decreases the thrombotic effect induced by aPL antibodies by inhibiting NDAPH oxidase 2 (NOX2), the NADPH oxidase platelet isoform that is involved in platelet activation [91]. An additional indirect effect of HCQ on thrombosis is its ability to improve blood lipid profile. Studies in SLE and RA patients treated with steroids have evidenced lower LDL cholesterol and triglyceride levels [92]. These data were confirmed in further SLE cohorts treated with HCQ in association with steroids: the treatment decreased total cholesterol and VLDL level and increased HDL cholesterol [93,94].
6. CQ/HCQ and iron metabolism: is there a role for iron in Sar-Cov-2 infection?
Iron is an essential element for all organisms. This is due to its redox potential which makes it an essential cofactor for several proteins and enzymes involved in vital cellular functions like energy production, DNA replication and transcription. Even most viruses need iron, since they require the host metabolic apparatus to replicate their genome and produce mRNAs for their translation in functional viral proteins [2]. Therefore, while cellular iron repletion may boost viral replication and spread, iron deficiency may interfere with viral life cycle.
During infections and inflammation, anemia is frequently observed and caused by pro-inflammatory cytokines. Some of them directly affect iron homeostasis, like IL-1β, TNF-α and IL-6. The release of these cytokines, mainly IL-6, results in the upregulation of the iron-regulatory hormone hepcidin (HAMP), primarily produced by hepatocytes and released in the blood flow to regulate systemic iron homeostasis. Systemic HAMP blocks cellular iron export through ferroportin 1 (FPN1), resulting in reduced intestinal iron absorption, increased iron retention in hepatocytes and macrophages and ultimately anemia of infection/inflammation [4,95]. Several cells other than hepatocytes have been demonstrated to produce and release HAMP that can act as autocrine and paracrine molecule, modulating local iron homeostasis [95,96]. Not only cells of the immune system like lymphocytes, monocytes and macrophages (including alveolar macrophages) but also airway epithelial cells have been demonstrated to produce HAMP during infection and inflammation and potentially contribute to lung injury [[97], [98], [99]]. HAMP is also a peptide involved in innate antimicrobial immunity and an acute phase protein [100]. Further acute phase iron-related proteins like transferrin (Tf), lactoferrin (LF), ferritin (FT), haptoglobin (HP) and hemopexin (HPX) are modulated by viral infections, further underlining the crucial role of iron in anti-viral host defense. The role of iron metabolism has been thoroughly investigated in several human viral infections. For extensive reviews on this topic and for details on systemic and cellular iron metabolism, please refer to [[2], [3], [4],101].
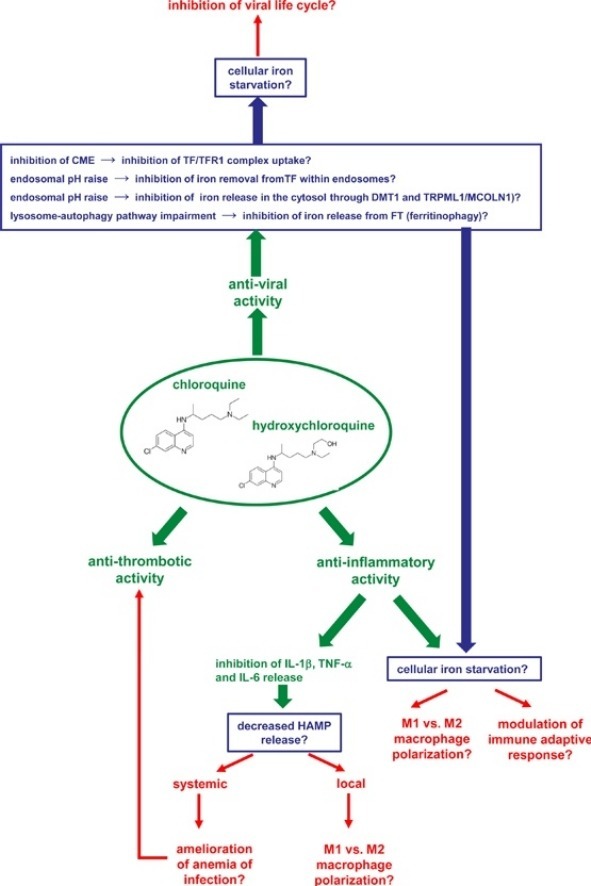
CQ/HCQ have been shown to modulate iron metabolism, impairing its homeostasis at different levels [43,102], and to decrease inflammatory cytokines like IL-6, IL-1β and TNF-α. Here we summarize the more striking evidences on CQ/HCQ action on cellular iron trafficking and their impact on cellular and systemic iron metabolism. We further propose alternative modes of action for these drugs, currently used in the treatment of COVID-19, that might also work through the interference with local and/or systemic iron metabolism, restricting this essential element in infected cells and/or immune cells involved in virus clearance and/or acting on virus cell cycle (Fig. 1 ).
Fig. 1.

Graphic representation of the possible pharmacological effects (in green) of chloroquine and hydroxychloroquine as anti-viral, anti-inflammatory and anti-thrombotic drugs and their possible links with systemic and cellular iron homeostasis. For each pharmacological effect, hypothetical activities of the drugs on iron homeostasis are in blue, while the possible consequences on the virus and the host are in red.
6.1. CQ/HCQ may induce cellular iron starvation: a potential inhibition of Sars-Cov-2 replication
In several experimental models, CQ has been shown to restrict iron entry into the cells. In the eukaryotic model Saccharomyces cerevisiae, CQ interferes with iron uptake, acting as a competitive inhibitor of iron entry and inducing iron starvation. Iron-deprived yeast, by means of knocking-out genes involved in iron uptake or by using iron chelators, shows increased sensitivity to CQ [103]. In mammalian cells, CQ has similar effects acting through the inhibition of the Tf/transferrin receptor 1 (TFR1) complex endocytosis. The first evidence of CQ inhibition of Tf uptake by cells was obtained in cultured rat embryo fibroblasts [104]. Tf is the main plasma iron carrier which, maintaining this cofactor in a redox inert state, distributes it to most cells of the human body. Tf tightly binds two Fe3+. TFR1, located on the plasma membrane of most types of cell, binds and internalizes Tf into endocytic vesicles through CME [105]. As stated above, CQ has been demonstrated to reduce PICALM expression in macrophages of treated mice [15]. This ubiquitously expressed protein is involved in CME and its deficiency has been demonstrated to result in anemia and abnormal iron metabolism in mice [106,107] and iron starvation, with increased surface TFR1 expression and decreased intracellular iron levels, in murine embryonic fibroblasts [108]. Then, CQ/HCQ treatment may result in the inhibition of Tf/TFR1 complex uptake and cellular iron starvation.
The release of iron from Tf after iron-loaded Tf/TFR1 complex CME and its translocation into the cytosol is a fundamental step for iron cellular acquisition and further usage. The acidic milieau of endosomes weakens the binding of Tf to Fe3+, enabling its release within these vesicles [105]. CQ/HCQ, by virtue of their basic properties, raise pH of endocytic vesicles, possibly inhibiting iron removal from Tf within endocytic vesicles.
Within endocytic vesicles, released Fe3+ is reduced to Fe2+ by the metalloreductase six-transmembrane epithelial antigen of the prostate 3 (STEAP3) and exported from the lysosomes to the cytosol through the divalent metal-ion transporter 1 (DMT1), mucolipin 1 (TRPML1/MCOLN1) and other transporters [105]. Iron transport through DMT1 is pH dependent, being stimulated at low pH. DMT1 is indeed a H+/Fe2+ symporter that needs a proton electrochemical potential gradient as driving force to transport iron from endosomes into the cytoplasm [109]. TRPML1/MCOLN1 is a non-selective channel permeable to various cations such as Ca2+, Na+ and K+ that can also transport Fe2+. It mainly localizes in late endosomes and lysosomes and the acidic environment of these vesicles activates this channel and the release of cations from the lumen into the cytosol [110]. Then CQ/HCQ could also inhibit iron release from endosomes into the cytosol.
The alkalizing properties of CQ/HCQ have been widely used to impair the endosome/lysosome fusion and inhibit autophagic flux [5]. FT is the main iron storage cellular protein that compartmentalizes iron in a non-reactive form within the cell, until use. Iron release from FT mainly occurs through protein degradation by a selective lysosome-autophagy pathway named ferritinophagy [101,111] that is inhibited by CQ [112]. Further, DMT1 and TRPML1 have been implicated in the release of iron derived from ferritinophagy and entrapped in autophagic vesicles and, as stated above, both require an acidic environment for their channel functions.
All the steps described above result in cellular iron starvation. This condition might possibly influence Sars-Cov-2 life cycle, although currently there is no experimental evidence in this new pandemic infection, but we suggest research in this direction. Sars-Cov-2 enter cells through ACE2 receptor that is expressed broadly in the human body, mainly in the vascular endothelia, gastrointestinal system, heart, kidney, muscle, skin and bronchial and lung alveolar epithelial cells [113]. In all human cells, then also Sars-Cov-2 target cells, iron is a cofactor of several crucial proteins, involved in bioenergetics, cellular growth and nucleic acid synthesis and all viruses need the cellular metabolic apparatus to replicate and synthesize their proteins. Several viral infections have been shown to increase cellular iron uptake and the inhibitory effect of iron starvation on viral cell cycle have indeed been thoroughly demonstrated in numerous human viruses like Human Deficiency Virus 1 (HIV-1), hepatitis C virus (HCV) and Human Cytomegalovirus (HCMV) [2,114]. Increased cellular iron uptake and FT synthesis has interestingly been demonstrated in the liver of mice infected with the mouse hepatitis virus type 3 (MHV-3), a member of the Coronaviridae [115].
6.2. CQ/HCQ may induce cellular iron starvation: a potential beneficial modulation of innate and adaptive immune response
A second important point raised by the iron starvation conditions induced by CQ/HCQ is their effect on immune cells, involved in the innate and adaptive immune responses against the virus. As all cells in the body, immune cells require iron for their proper functions and for their activation and proliferation. Iron excess often results in impaired immune response to infections, as observed in patients suffering from hemochromatosis (HH) or thalassemia [116]. Excessive or dysregulated immune response is of particular importance in the pathogenesis of COVID-19 [117] and diseases caused by other coronaviruses [[118], [119], [120]]. Direct infection of innate and adaptive immune cells has been described in some coronavirus infections [118,119], then iron starvation could also possibly inhibit these infections. Resident macrophages can polarize under the stimuli of cytokines in classically activated pro-inflammatory macrophages (M1), induced by interferon-γ (IFN-γ) and TNF-α or, under interleukin-4 (IL-4) and 13 (IL-13) stimuli, alternatively activated macrophages (M2), involved in pathogen clearance, tissue repair and inflammation reduction. While M2 macrophages have low iron levels, M1 macrophages are characterized by iron retention, secrete high levels of pro-inflammatory cytokines, produce high amounts of radicals to kill pathogens and produce HAMP that acts in an autocrine manner to minimize iron exit. Increased iron deposition in macrophages has been shown to induce M1 polarization and the persistence of a pro-inflammatory state due to an incomplete switch to M2 state [121]. Iron retention in macrophages could then favor intracellular virus life cycle in the case of their infection and further promote the process of inflammation, while iron starvation could result in the opposite effects. Further, iron excess in macrophages favors secondary infections with other microbes. Chronic administration of CQ has been shown to decrease iron content in a rat model. Legssyer and colleagues [122,123] showed that CQ decreased iron content in the liver of control, iron-loaded and iron-deficient rats, in the spleen of control and iron-loaded animals and, importantly, in alveolar macrophages of iron-loaded rats. They also interestingly observed that CQ treatment of primary cultures of alveolar macrophages obtained from all three groups of rats and treated with lipopolysaccharide (LPS) significatively reduced the oxidative response, measured by NO2 release, suggesting that CQ might prevent infections, particularly those associated with diseases characterized by iron overload, through limiting iron availability not only in infected cells but also in macrophages, in turn reducing inflammation. However, heterozygous and homozygous mutations in the hemochromatosis gene (HFE) reduce the CQ effect of iron removal in porphyria cutanea tarda [124]. Also in murine models, CQ has been shown to decrease macrophage activation syndrome (hemophagocytic syndrome) induced by pristane, by reducing macrophage infiltration, phagocytic functions, cytokine production and reducing FT, lactate dehydrogenase and triglyceride levels [125]. Although immune cells need iron for their proliferation and activation, excess iron has also been reported to impair antigen processing and presentation by APCs, suppress CD4+ cells and alter CD4+/CD8+ lymphocyte ratio, increase circulating immunoglobulin-producing B cells, reduce natural killer (NK) cell lysing efficiency and perturb complement activation [126], while iron deficiency has an immunosuppressive effect on T cells [127]. Then, CQ/HCQ treatment, through their action on iron homeostasis, could possibly have also a broad role in the modulation of the adaptive response to Sars-Cov-2.
6.3. CQ/HCQ may decrease IL-1β, TNF-α and IL-6 release: a potential reduction of local and systemic HAMP release
A third important point to highlight derives from the inhibitory action of CQ/HCQ on IL-1β, TNF-α and IL-6 release. First, TNF-α acts on iron homeostasis, reducing intestinal iron absorption and blocking iron recycling from macrophages, then inducing iron retention in these cells and their polarization to M1 pro-inflammatory phenotype [102]. Conversely, CQ inhibits TNF-α acting also through the induction of cellular iron starvation [40]. Then, the inhibition of the cytokine by CQ/HCQ, also through their effect on iron homeostasis, could alleviate inflammation, restoring a balanced systemic iron homeostasis and rescuing erythropoiesis. Secondly, IL-1β, TNF-α and IL-6 as stated above, induce the systemic and local release of HAMP. Iron is essential for life, but excess iron is toxic, due to its redox potential that can hamper oxidative stress damaging crucial cellular components. Iron availability is then tightly regulated both at the cellular and systemic levels [[2], [3], [4],95,101]. The main systemic regulator is HAMP. This hormone peptide is produced mainly in the liver and is regulated by circulating and tissue iron levels through the bone morphogenetic/small mother against decapentaplegic (BMP/SMAD) pathway in such a way that iron load induces while iron deficiency inhibits its expression, mainly acting on intestinal iron absorption and iron retention/release by macrophages and hepatocytes. HAMP expression is also downregulated by hypoxia and erythropoietin (EPO) through erythroferrone (ERFE) to allow iron mobilization for erythropoiesis, while it is upregulated by inflammation. IL-6 is the main signal that induce HAMP during inflammation, through the JAK/STAT3 pathway in association with the BMP/SMAD pathway, but also IL-1β and TNF-α have a direct role on HAMP regulation [95,128,129]. As stated above, CQ/HCQ not only interfere with cellular iron inducing its starvation in alveolar macrophages [122,123], then possibly resulting in a switch to M2 anti-inflammatory state, but also inhibit IL-6, IL-1β and TNF-α release, possibly reducing local HAMP release by macrophages. This reduction can result in further decreased iron retention in these cells allowing direction towards inflammation resolution. Further, cytokine decrease could result in systemic HAMP decrease that, through increased intestinal iron absorption, may ameliorate anemia of infection. Interestingly EPO treatment has recently been found to attenuate COVID-19 severe symptoms [130] while, through in silico modeling, Sars-Cov-2 has been found to possibly bind heme dissociating iron from porphyrin [131].
6.4. CQ/HCQ treatment, decreasing cytokine and HAMP release, may recover anemia of infection and thrombosis
Numerous studies correlated thrombocytosis induced by iron deficiency anemia (IDA) with thrombotic events [[132], [133], [134], [135]]. A recent study has evidenced that IDA patients manifesting thrombocytosis had 2-fold augmented risk of thrombosis when compared with IDA patient with normal platelet count [136]. Interestingly, COVID-19 patients with severe pneumonia seem to have high platelet counts compared with patients with severe non-COVID-19 pneumonia [137] and this increase is more evident among the non-survivor compared with survivor COVID-19 patients [138]. To clarify the effect of iron deficiency on thrombotic propensity in animal models, thrombosis was induced in Sprague-Dawley rat fed with iron-deficient diet or normal diet as control. Iron deficiency induced thrombocytosis and platelet number resulted proportional with thrombus size. In addition, platelet adhesion and aggregation were impaired. Taking into account data obtained in this model, the authors concluded that anemia of inflammation, caused by HAMP-mediated iron sequestration in the liver, spleen and macrophages, as possibly occurs in COVID-19 patients, may be considered a functional iron deficiency (ID) and patients affected by this condition should be treated as patients with high risk of thrombosis [139].
In conclusion, the multiple action of CQ and HCQ as anti-viral, anti-inflammatory and anti-thrombotic drug may be also strictly linked to their effects on iron homeostasis, both at the local and systemic level. Interestingly, another common drug, frequently used in the treatment of COVID-19 patients with markedly elevated D-dimer levels is heparin. Like CQ and HCQ, heparin is a versatile drug, as defined by Jecko Thachil [35], due to its possible actions as anti-coagulant, anti-inflammatory and anti-viral drug. It is interesting to note that, like CQ/HCQ, heparin too has been demonstrated to modulate iron metabolism: this anti-thrombotic drug has indeed been demonstrated to inhibit HAMP expression in human macrophages, to increase FPN1 plasma-membrane expression and promote iron export, resulting in cellular iron starvation [140].
All this evidence suggest a possible role of iron in Sars-Cov-2 infection that would be explored in future basic and clinical research, also considering it as a potential target for COVID-19 therapy as proposed for other human infectious diseases [[2], [3], [4]].
7. Current evidence on the efficacy of CQ/HCQ treatment in patients with Sars-Cov-2 infection
Molecular mechanism and theoretical mode of action of CQ and HCQ on multiple steps of the viral pathway and the demonstrated in vitro activity against COVID-19 [[8], [9], [10]] have prompted their off-label use in clinical setting during this pandemic emergency.
Several clinical trials have been launched to evaluate the effectiveness of these drugs [141,142]. Until now, published clinical data on CQ and HCQ are limited and regard only small, poorly controlled or uncontrolled clinical studies. We found only four published reports of these trials in PubMed till April 20th, 2020. The first clinical study to evaluate the efficacy and safety of CQ/HCQ in patients with COVID-19 has been reported by Huang and colleagues [143] from China. The authors screened 22 patients tested positive for Sars-Cov-2 by RT-PCR assay. Ten patients (3 with severe and 7 with moderate symptoms) were treated with CQ 500 mg twice a day for 10 days, while 12 patients (5 with severe and 7 with moderate symptoms) were treated with Lopinavir/Ritonavir 400/100 mg twice a day for 10 days. Based on RNA results, CQ was slightly better in its anti-viral activity with all patients tested negative by day 13. Lung improvement based on imaging was more than double at day 14 in CQ-treated compared with Lopinavir/Ritonavir-treated patients. CQ-treated patients were also discharged earlier. The authors did not observe serious adverse events. Gautret and colleagues [144] performed a single-arm protocol with 26 patients receiving 600 mg HCQ daily (200 mg for three times a day) and 16 control patients. Six HCQ-treated patients were lost during follow-up (3 of them were transferred in intensive care unit, ICU, 1 died, 1 left hospital, 1 stopped the treatment because of nausea). Among HCQ-treated patients, 6 received also azithromycin. None of the control group was lost. Twenty-two% of enrolled patients had pneumonia. At day 6 post-treatment, 100 % of HCQ/azithromycin-treated patients and 57 % of HCQ-treated patients tested negative for viral RNA versus 12,5% of controls. Treatment was more effective in patients with symptoms than in asymptomatic patients. The same researchers [145] further conducted an uncontrolled non-comparative observational study with 80 enrolled patients and the same treatment protocol for a maximum of 10 days. Six of these patients were from the previous study. Most patients (65, 81,3%) were discharged from the infectious disease unit (IDU) with a mean length stay of 4.6 days, while 13 were still hospitalized, 1 died and 1 stopped treatment for the potential interaction with other drugs. On the total, 15 % required oxygen therapy, 3 were transferred in ICU and two of them improved and returned to IDU, the third remained in ICU. Overall, for almost all enrolled patients the clinicians obtained a better clinical improvement with this treatment when compared with other treatments. Further, viral load rapidly decreased, with 83 % negative patients at day 7 and 93 % at day 8 of treatment. Only 2 patients had still detectable viral load at day 10. Molina and colleagues [146] studied 11 COVID-19 patients (10/11 with fever and nasal oxygen therapy, 8 with significant comorbidities) treated with HCQ plus azithromycin, using the same dosing regimen reported by Gautret and colleagues [144,145]. One patient died and 2 were transferred to ICU within 5 days after treatment initiation, one therapy was discontinued after 4 days for severe adverse event (prolongation of the QT interval), suggesting the poor clinical outcome of the combined treatment. In contrast with results obtained by [144,145], nasopharyngeal swabs were still positive for viral RNA in 8/10 patients at days 5–6. Researchers concluded by saying that, in their experience, they found no evidence of anti-viral activity and clinical benefit by using the combined therapy in severe COVID-19.
The currently available data failed to demonstrate or exclude a beneficial effect of CQ/HCQ on clinical progression of COVID-19 (as inferred by radiological findings; RR: 0.61; 95 % CI: 0.26, 1.43), or on viral clearance by RT-PCR tests (RR: 2.00; 95 % CI: 0.02, 20.00) although a somewhat higher proportion in the CQ/HCQ group experienced clinical improvement (RR: 1.47; 95 % CI 1.02, 2.11) [147].
Not only clinical efficacy but also optimal dosing regimen, therapeutic level, duration of treatment and pharmacokinetics in patients with different severity degrees of the disease are uncertain and currently there are no standard dosages or duration of treatment around the world for COVID-19 patient treatment [[148], [149], [150], [151], [152]]. Numerous studies are under way to evaluate their efficacy in treating and prevention of COVID-19 and to establish benefit versus harm of CQ/HQC treatment [153]. Notwithstanding, in the emergency phase of COVID-19 pandemic many old drugs have been off-label used for the treatment of the infection, based only on theoretical or in vitro efficacy and without enough clinical evidence based on randomized clinical trial.
8. Conclusions
As we have described, CQ/HCQ are likely to have: (1) a direct antiviral action stopping viral infection in several steps; (2) a hypothetical ability to attenuate the progression of COVID-19 to severe disease exploiting its inflammatory mechanisms, but also (3) through its potential anti-thrombotic effect.
Based on our review and because of their minimal toxicity profile and complex action, we suggest to use CQ/HCQ CQ beyond 5–10 days of treatment in patients with COVID-19 according to the hypothesis that their utility can extend also after ending Sars-Cov-2 high replication phase and considering also the possibility of a reactivation of the infection [154]. Randomized controlled studies and observational outcome registries focused on efficacy, duration and toxicities of treatment with these drugs could be useful to understand their real effectiveness while more specific anti-COVID-19 drugs are available.
Author contributions
E.Q.R. and I.Z. conceived and planned the manuscript; E.Q.R. performed the literature review of clinical data and wrote the related section of the manuscript; G.B. performed the literature review of data on anti-thrombotic effects and links with iron homeostasis and wrote the related section of the manuscript; I.Z. performed the literature review of data on anti-viral and anti-inflammatory effects and links with iron homeostasis and wrote the related sections of the manuscript; P.M. integrated the manuscript and produced tables and figure; all authors performed the critical revision of the manuscript, read and approved the final manuscript.
Funding
This study was partly supported by the University of Brescia (fondi ex 60 % to Eugenia Quiros-Roldan, Giorgio Biasiotto and Isabella Zanella).
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: E.Q.R. received travel grants from Bristol-Myers Squibb, Gilead Sciences, ViiV Healthcare, Janssen-Cilag, Merck Sharp & Dohme and consultancy fees from Janssen-Cilag, ViiV Healthcare and Merck Sharp & Dohme. The remaining authors declare that they have no conflicts of interest.
Acknowledgements
We would like to thank Andrea Zanella for helpful language editing. Isabella Zanella wishes to dedicate this work to her father, Antonio Zanella, who recently died for COVID-19.
Due to space constraints, we were unable to provide a comprehensive citation of all the relevant primary literature. We apologize to those whom we have even unintentionally omitted.
References
- 1.Touret F., de Lamballerie X. Of chloroquine and COVID-19. Antiviral Res. 2020;5(March (177)) doi: 10.1016/j.antiviral.2020.104762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drakesmith H., Prentice A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008;6(July (7)):541–552. doi: 10.1038/nrmicro1930. [DOI] [PubMed] [Google Scholar]
- 3.Drakesmith H., Prentice A.M. Hepcidin and the iron-infection axis. Science. 2012;338(November (6108)):768–772. doi: 10.1126/science.1224577. [DOI] [PubMed] [Google Scholar]
- 4.Ganz T. Iron and infection. Int. J. Hematol. 2018;107(January (1)):7–15. doi: 10.1007/s12185-017-2366-2. [DOI] [PubMed] [Google Scholar]
- 5.Klionsky D.J., Abdelmohsen K., Abe A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. doi: 10.1080/15548627.2015.1100356. (3rd edition) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savarino A., Boelaert J.R., Cassone A., Majori G., Cauda R. Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect. Dis. 2003;3(November (11)):722–727. doi: 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(March (3)):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J., Cao R., Xu M., Wang X., Zhang H., Hu H., Li Y., Hu Z., Zhong W., Wang M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;18(March (6)):16. doi: 10.1038/s41421-020-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao X., Ye F., Zhang M., Cui C., Huang B., Niu P., Liu X., Zhao L., Dong E., Song C., Zhan S., Lu R., Li H., Tan W., Liu D. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Clin. Infect. Dis. 2020;(March (9)):ciaa237. doi: 10.1093/cid/ciaa237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devaux C.A., Rolain J.M., Colson P., Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int. J. Antimicrob. Agents. 2020;11(March) doi: 10.1016/j.ijantimicag.2020.105938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincent M.J., Bergeron E., Benjannet S., Erickson B.R., Rollin P.E., Ksiazek T.G., Seidah N.G., Nichol S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005;22(August (22)):69. doi: 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S., Maurya V.K., Prasad A.K., Bhatt M.L.B., Saxena S.K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV) Virusdisease. 2020;31(March (1)):13–21. doi: 10.1007/s13337-020-00571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fantini J., Scala C.D., Chahinian H., Yahi N. Structural and molecular modeling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents. 2020;3(April) doi: 10.1016/j.ijantimicag.2020.105960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolfram J., Nizzero S., Liu H., Li F., Zhang G., Li Z., Shen H., Blanco E., Ferrari M. A chloroquine-induced macrophage-preconditioning strategy for improved nanodelivery. Sci. Rep. 2017;7(October (1)) doi: 10.1038/s41598-017-14221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue Y., Tanaka N., Tanaka Y., Inoue S., Morita K., Zhuang M., Hattori T., Sugamura K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007;81(August (16)):8722–8729. doi: 10.1128/JVI.00253-07. Epub 2007 May 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Müller M.A., Drosten C., Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;(March 4) doi: 10.1016/j.cell.2020.02.052. pii: S0092-8674(20)30229-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H., Yang P., Liu K., Guo F., Zhang Y., Zhang G., Jiang C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008;18(February (2)):290–301. doi: 10.1038/cr.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N.G., Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176(April) doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozden S., Lucas-Hourani M., Ceccaldi P.E., Basak A., Valentine M., Benjannet S., Hamelin J., Jacob Y., Mamchaoui K., Mouly V., Desprès P., Gessain A., Butler-Browne G., Chrétien M., Tangy F., Vidalain P.O., Seidah N.G. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 2008;283(August (32)):21899–21908. doi: 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- 21.Schoeman D., Fielding B.C. Coronavirus envelope protein: current knowledge. Virol. J. 2019;16(May (1)):69.. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manley G.C.A., Parker L.C., Zhang Y. Emerging regulatory roles of dual-specificity phosphatases in inflammatory airway disease. Int. J. Mol. Sci. 2019;20(February (3)):E678. doi: 10.3390/ijms20030678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kono M., Tatsumi K., Imai A.M., Saito K., Kuriyama T., Shirasawa H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: involvement of p38 MAPK and ERK. Antiviral Res. 2008;77(February (2)):150–152. doi: 10.1016/j.antiviral.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang M., Huang L., Li X., Kuang E. Chloroquine inhibits lytic replication of Kaposi’s sarcoma-associated herpesvirus by disrupting mTOR and p38-MAPK activation. Antiviral Res. 2016;133(September):223–233. doi: 10.1016/j.antiviral.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Ben-Zvi I., Kivity S., Langevitz P., Shoenfeld Y. Hydroxychloroquine: from malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012;42(April (2)):145–153. doi: 10.1007/s12016-010-8243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plantone D., Koudriavtseva T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin. Drug Investig. 2018;38(August (8)):653–671. doi: 10.1007/s40261-018-0656-y. [DOI] [PubMed] [Google Scholar]
- 27.Roche P.A., Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015;15(April (4)):203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerriero J.L. Macrophages: their untold story in t cell activation and function. Int. Rev. Cell Mol. Biol. 2019;342:73–93. doi: 10.1016/bs.ircmb.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Goldman F.D., Gilman A.L., Hollenback C., Kato R.M., Premack B.A., Rawlings D.J. Hydroxychloroquine inhibits calcium signals in T cells: a new mechanism to explain its immunomodulatory properties. Blood. 2000;95(June (11)):3460–3466. [PubMed] [Google Scholar]
- 30.Yang J., Yang X., Yang J., Li M. Hydroxychloroquine inhibits the differentiation of Th17 cells in systemic lupus erythematosus. J. Rheumatol. 2018;45(June (6)):818–826. doi: 10.3899/jrheum.170737. [DOI] [PubMed] [Google Scholar]
- 31.Saitoh S., Miyake K. Regulatory molecules required for nucleotide-sensing Toll-like receptors. Immunol. Rev. 2009;227(January (1)):32–43. doi: 10.1111/j.1600-065X.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- 32.Lim E.J., Lee S.H., Lee J.G., Chin B.R., Bae Y.S., Kim J.R., Lee C.H., Baek S.H. Activation of toll-like receptor-9 induces matrix metalloproteinase-9 expression through Akt and tumor necrosis factor-alpha signaling. FEBS Lett. 2006;580(August (18)):4533–4538. doi: 10.1016/j.febslet.2006.06.100. [DOI] [PubMed] [Google Scholar]
- 33.Nosál R., Jancinová V. Cationic amphiphilic drugs and platelet phospholipase A(2) (cPLA(2)) Thromb. Res. 2002;105(February (4)):339–345. doi: 10.1016/s0049-3848(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 34.McGonagle D., Sharif K., O’Regan A., Bridgewood C. The role of cytokines including Interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun. Rev. 2020;3(April) doi: 10.1016/j.autrev.2020.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thachil J. The versatile heparin in COVID-19. J. Thromb. Haemost. 2020;(April (2)) doi: 10.1111/jth.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller C., Hardt M., Schwudke D., Neuman B.W., Pleschka S., Ziebuhr J. Inhibition of cytosolic phospholipase a(2)α impairs an early step of coronavirus replication in cell culture. J. Virol. 2018;92(January (4)):e01463–17. doi: 10.1128/JVI.01463-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H., Zhang X., Zhang M., Wu S., Song J., Chen T., Han M., Li S., Luo X., Zhao J., Ning Q. Clinical and immunologic features in severe and moderate Coronavirus Disease 2019. J. Clin. Invest. 2020;(March (27)):137244. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conti P., Gallenga C.E., Tetè G., Caraffa A., Ronconi G., Younes A., Toniato E., Ross R., Kritas S.K. How to reduce the likelihood of coronavirus-19 (CoV-19 or SARS-CoV-2) infection and lung inflammation mediated by IL-1. J. Biol. Regul. Homeost. Agents. 2020;34(March (2)) doi: 10.23812/Editorial-Conti-2. [DOI] [PubMed] [Google Scholar]
- 39.Sperber K., Quraishi H., Kalb T.H., Panja A., Stecher V., Mayer L. Selective regulation of cytokine secretion by hydroxychloroquine: inhibition of interleukin 1 alpha (IL-1-alpha) and IL-6 in human monocytes and T cells. J. Rheumatol. 1993;20(May (5)):803–808. [PubMed] [Google Scholar]
- 40.Picot S., Peyron F., Donadille A., Vuillez J.P., Barbe G., Ambroise-Thomas P. Chloroquine-induced inhibition of the production of TNF, but not of IL-6, is affected by disruption of iron metabolism. Immunology. 1993;80(September (1)):127–133. [PMC free article] [PubMed] [Google Scholar]
- 41.Van den Borne B.E., Dijkmans B.A., de Rooij H.H., le Cessie S., Verweij C.L. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J. Rheumatol. 1997;24(January (1)):55–60. [PubMed] [Google Scholar]
- 42.Jeong J.Y., Jue D.M. Chloroquine inhibits processing of tumor necrosis factor in lipopolysaccharide-stimulated RAW 264.7 macrophages. J. Immunol. 1997;158(May (10)):4901–4907. [PubMed] [Google Scholar]
- 43.Boelaert J.R., Piette J., Sperber K. The potential place of chloroquine in the treatment of HIV-1-infected patients. J. Clin. Virol. 2001;20(February (3)):137–140. doi: 10.1016/s1386-6532(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 44.Seitz M., Valbracht J., Quach J., Lotz M. Gold sodium thiomalate and chloroquine inhibit cytokine production in monocytic THP-1 cells through distinct transcriptional and posttranslational mechanisms. J. Clin. Immunol. 2003;23(November (6)):477–484. doi: 10.1023/b:joci.0000010424.41475.17. [DOI] [PubMed] [Google Scholar]
- 45.Hong Z., Jiang Z., Liangxi W., Guofu D., Ping L., Yongling L., Wendong P., Minghai W. Chloroquine protects mice from challenge with CpG ODN and LPS by decreasing proinflammatory cytokine release. Int. Immunopharmacol. 2004;4(February (2)):223–234. doi: 10.1016/j.intimp.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 46.Jang C.H., Choi J.H., Byun M.S., Jue D.M. Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology (Oxford) 2006;45(June (6)):703–710. doi: 10.1093/rheumatology/kei282. Epub 2006 Jan 17. [DOI] [PubMed] [Google Scholar]
- 47.Farias K.J., Machado P.R., de Almeida Junior R.F., de Aquino A.A., da Fonseca B.A. Chloroquine interferes with dengue-2 virus replication in U937 cells. Microbiol. Immunol. 2014;58(June (6)):318–326. doi: 10.1111/1348-0421.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S.F., Tseng S.P., Yen C.H., Yang J.Y., Tsao C.H., Shen C.W., Chen K.H., Liu F.T., Liu W.T., Chen Y.M., Huang J.C. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem. Biophys. Res. Commun. 2014;451(August (2)):208–214. doi: 10.1016/j.bbrc.2014.07.090. Epub 2014 Jul 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahallawi W.H., Khabour O.F., Zhang Q., Makhdoum H.M., Suliman B.A. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. 2018;104(April):8–13. doi: 10.1016/j.cyto.2018.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malaviya R., Laskin J.D., Laskin J.D.L. Anti-TNFα therapy in inflammatory lung diseases. Pharmacol. Ther. 2017;180(December):90–98. doi: 10.1016/j.pharmthera.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keyel P.A. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine. 2014;69(September (1)):136–145. doi: 10.1016/j.cyto.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Ceribelli A., Motta F., De Santis M., Ansari A.A., Ridgway W.M., Gershwin M.E., Selmi C. Recommendations for coronavirus infection in rheumatic diseases treated with biologic therapy. J. Autoimmun. 2020;109(May) doi: 10.1016/j.jaut.2020.102442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang C., Wu Z., Li J.W., Zhao H., Wang G.Q. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int. J. Antimicrob. Agents. 2020;29(March) doi: 10.1016/j.ijantimicag.2020.105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Luna G., Habibi A., Deux J.F., Colard M., d’Alexandry d’Orengiani A.L.P.H., Schlemmer F., Joher N., Kassasseya C., Pawlotsky J.M., Ourghanlian C., Michel M., Mekontso-Dessap A., Bartolucci P. Rapid and severe Covid-19 pneumonia with severe acute chest syndrome in a sickle cell patient successfully treated with tocilizumab. Am. J. Hematol. 2020;(April (13)) doi: 10.1002/ajh.25833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cellina M., Orsi M., Bombaci F., Sala M., Marino P., Oliva G. Favorable changes of CT findings in a patient with COVID-19 pneumonia after treatment with tocilizumab. Diagn. Interv. Imaging. 2020;(March (31)) doi: 10.1016/j.diii.2020.03.010. pii: S2211-5684(20)30087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hunter C.A., Jones S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015;16(May (5)):448–457. doi: 10.1038/ni.3153. Review. Erratum in: Nat Immunol. 2017 Oct 18;18(11):1271. [DOI] [PubMed] [Google Scholar]
- 57.Carter A.E., Eban R. Prevention of postoperative deep venous thrombosis in legs by orally administered hydroxychloroquine sulphate. Br. Med. J. 1974;3:94–95. doi: 10.1136/bmj.3.5923.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pilcher D.B. Hydroxychloroquine sulfate in prevention of thromboembolic phenomena in surgical patients. Am. Surg. 1975;41:761–766. [PubMed] [Google Scholar]
- 59.Loudon J.R. Hydroxychloroquine and postoperative thromboembolism after total hip replacement. Am. J. Med. 1988;85:57–61. doi: 10.1016/0002-9343(88)90364-6. [DOI] [PubMed] [Google Scholar]
- 60.Snook G.A., Chrisman O.D., Wilson T.C. Thromboembolism after surgical treatment of hip fractures. Clin. Orthop. Relat. Res. 1981;155:21–24. [PubMed] [Google Scholar]
- 61.Johnson R., Green J.R., Charnley J. Pulmonary embolism and its prophylaxis following the Charnley total hip replacement. Clin. Orthop. Relat. Res. 1977;127:123–132. [PubMed] [Google Scholar]
- 62.Johnson R., Charnley J. Hydroxychloroquine in prophylaxis of pulmonary embolism following hip arthroplasty. Clin. Orthop. Relat. Res. 1979;144:174–177. [PubMed] [Google Scholar]
- 63.Belizna C. Hydroxychloroquine as an anti-thrombotic in antiphospholipid syndrome. Autoimmun. Rev. 2015;14:358–362. doi: 10.1016/j.autrev.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 64.Ruiz-Irastorza G., Egurbide M.V., Pijoan J.I., Garmendia M., Villar I., MartinezBerriotxoa A. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus. 2006;15:577–583. doi: 10.1177/0961203306071872. [DOI] [PubMed] [Google Scholar]
- 65.Kaiser R., Cleveland C.M., Criswell L.A. Risk and protective factors for thrombosis in systemic lupus erythematosus: results from a large, multi-ethnic cohort. Ann. Rheum. Dis. 2009;68:238–241. doi: 10.1136/ard.2008.093013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tektonidou M.G., Laskari K., Panagiotakos D.B., Moutsopoulos H.M. Risk factors for thrombosis and primary thrombosis prevention in patients with systemic lupus erythematosus with or without antiphospholipid antibodies. Arthritis Rheum. 2009;61:29–36. doi: 10.1002/art.24232. [DOI] [PubMed] [Google Scholar]
- 67.Petri M. Use of hydroxychloroquine to prevent thrombosis in systemic lupus erythematosus and in antiphospholipid antibody-positive patients. Curr. Rheumatol. Rep. 2011;13:77–80. doi: 10.1007/s11926-010-0141-y. [DOI] [PubMed] [Google Scholar]
- 68.Ho K.T., Ahn C.W., Alarco G.S., Baethge B.A., Tan F.K., Roseman J. Systemic lupus erythematosus in a multiethnic cohort (LUMINA): XXVIII. Factors predictive of thrombotic events. Rheumatology (Oxford) 2005;44:1303–1307. doi: 10.1093/rheumatology/kei014. [DOI] [PubMed] [Google Scholar]
- 69.Broder A., Putterman C. Hydroxychloroquine use is associated with lower odds of persistently positive antiphospholipid antibodies and/or lupus anticoagulant in systemic lupus erythematosus. J. Rheumatol. 2013;40:30–33. doi: 10.3899/jrheum.120157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nuri E., Taraborelli M., Andreoli L., Tonello M., Gerosa M., Calligaro A. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol. Res. 2017;65:17–24. doi: 10.1007/s12026-016-8812-z. [DOI] [PubMed] [Google Scholar]
- 71.Kravvariti E., Koutsogianni A., Samoli E., Sfikakis P.P., Tektonidou M.G. The effect of hydroxychloroquine on thrombosis prevention and antiphospholipid antibody levels in primary antiphospholipid syndrome: a pilot open label randomized prospective study. Autoimmun. Rev. 2020;19(April (4)) doi: 10.1016/j.autrev.2020.102491. [DOI] [PubMed] [Google Scholar]
- 72.Andrade D., Tektonidou M. Emerging therapies in antiphospholipid syndrome. Curr. Rheumatol. Rep. 2016;18:1–9. doi: 10.1007/s11926-016-0566-z. [DOI] [PubMed] [Google Scholar]
- 73.Campbell C.M., Kahwash R. Will Complement Inhibition be the New Target in Treating COVID-19 Related Systemic Thrombosis? Circulation. 2020;(April (9)) doi: 10.1161/CIRCULATIONAHA.120.047419. [DOI] [PubMed] [Google Scholar]
- 74.Bertrand E., Cloitre B., Ticolat R., Bile R.K., Gautier C., Abiyou G.O. Antiaggregation action of chloroquine. Med Trop. 1990;50:143–146. [PubMed] [Google Scholar]
- 75.Prowse C., Pepper D., Dawes J. Prevention of the platelet alpha-granule release reaction by membrane-active drugs. Thromb. Res. 1982;25:219–227. doi: 10.1016/0049-3848(82)90241-9. [DOI] [PubMed] [Google Scholar]
- 76.Jancinova V., Nosal R., Petrikova M. On the inhibitory effect of chloroquine on blood platelet aggregation. Thromb. Res. 1994;74:495–504. doi: 10.1016/0049-3848(94)90270-4. [DOI] [PubMed] [Google Scholar]
- 77.Espinola R.G., Pierangeli S.S., Gharavi A.E., Harris E.N. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb. Haemost. 2002;87:518–522. [PubMed] [Google Scholar]
- 78.Ernst E., Rose M., Lee R. Modification of transoperative changes in blood fluidity by hydroxychloroquine: a possible explanation for the drug’s antithrombotic effect. Pharmatherapeutica. 1984;4:48–52. [PubMed] [Google Scholar]
- 79.Bird H.A., Harkness J., Wright V. Some rheological properties of blood during antirheumatoid therapy. Pharmatherapeutica. 1981;3:36–39. [PubMed] [Google Scholar]
- 80.Brinkmann V., Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007;5(August (8)):577–582. doi: 10.1038/nrmicro1710. Review. [DOI] [PubMed] [Google Scholar]
- 81.Meng H., Yalavarthi S., Kanthi Y., Mazza L.F., Elfline M.A., Luke C.E. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheum. 2017;69(3):655–667. doi: 10.1002/art.39938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Doring Y., Soehnlein O., Weber C. Neutrophil extracellular traps in atherosclerosis and Atherothrombosis. Circ. Res. 2017;120(4):736–743. doi: 10.1161/CIRCRESAHA.116.309692. [DOI] [PubMed] [Google Scholar]
- 83.Golonka R.M., Saha P., Yeoh B.S., Chattopadhay S., Gewirtz A.T., Joe B., Vijay-Kumar M. Harnessing innate immunity to eliminate SARS-CoV-2 and ameliorate COVID-19 disease. Physiol. Genomics. 2020;(April (10)) doi: 10.1152/physiolgenomics.00033.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boone B.A., Murthy P., Miller-Ocuin J., Doerfler W.R., Ellis J.T., Liang X., Ross M.A., Wallace C.T., Sperry J.L., Lotze M.T., Neal M.D., Zeh H.J., 3rd Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. 2018;18(June (1)):678. doi: 10.1186/s12885-018-4584-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rand J.H., Wu X.X., Quinn A.S., Chen P.P., Hathcock J.J., Taatjes D.J. Hydroxychloroquine directly reduces the binding of antiphospholipid antibody—beta2-glycoprotein I complexes to phospholipid bilayers. Blood. 2008;112:1687–1695. doi: 10.1182/blood-2008-03-144204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andree H.A., Stuart M.C., Hermens W.T., Reutelingsperger C.P., Hemker H.C., Frederik P.M., Willems G.M. Clustering of lipid-bound annexin V may explain its anticoagulant effect. J. Biol. Chem. 1992;267(September (25)):17907–17912. [PubMed] [Google Scholar]
- 87.Rand J.H., Wu X.X., Quinn A.S., Ashton A.W., Chen P.P., Hathcock J.J. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug. Blood. 2010;115:2292–2299. doi: 10.1182/blood-2009-04-213520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rand J.H., Wu X.X., Quinn A.S., Taatjes D.J. The annexin A5-mediated pathogenic mechanism in the antiphospholipid syndrome: role in pregnancy losses and thrombosis. Lupus. 2010;19:460–469. doi: 10.1177/0961203310361485. [DOI] [PubMed] [Google Scholar]
- 89.Urbanski G., Caillon A., Poli C., Kauffenstein G., Begorre M.A., Loufrani L., Henrion D., Belizna C. Hydroxychloroquine partially prevents endothelial dysfunction induced by anti-beta-2-GPI antibodies in an in vivo mouse model of antiphospholipid syndrome. PLoS One. 2018;13(November (11)) doi: 10.1371/journal.pone.0206814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miranda S., Billoir P., Damian L., Thiebaut P.A., Schapman D., Le Besnerais M., Jouen F., Galas L., Levesque H., Le Cam-Duchez V., Joannides R., Richard V., Benhamou Y. Hydroxychloroquine reverses the prothrombotic state in a mouse model of antiphospholipid syndrome: role of reduced inflammation and endothelial dysfunction. PLoS One. 2019;14(March (3)) doi: 10.1371/journal.pone.0212614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Müller-Calleja N., Manukyan D., Canisius A., Strand D., Lackner K.J. Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann. Rheum. Dis. 2017;76(May (5)):891–897. doi: 10.1136/annrheumdis-2016-210012. [DOI] [PubMed] [Google Scholar]
- 92.Wallace D.J., Metzger A.L., Stecher V.J., Turnbull B.A., Kern P.A. Cholesterol-lowering effect of hydroxychloroquine in patients with rheumatic disease: reversal of deleterious effects of steroids on lipids. Am. J. Med. 1990;89:322–326. doi: 10.1016/0002-9343(90)90345-e. [DOI] [PubMed] [Google Scholar]
- 93.Petri M., Lakatta C., Magder L., Goldman D. Effect of prednisone and hydroxychloroquine on coronary artery disease risk factors in systemic lupus erythematosus: a longitudinal data analysis. Am. J. Med. 1994;96:254–259. doi: 10.1016/0002-9343(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 94.Borba E.F., Bonfá E. Longterm beneficial effect of chloroquine diphosphate on lipoprotein profile in lupus patients with and without steroid therapy. J. Rheumatol. 2001;28:780–785. [PubMed] [Google Scholar]
- 95.Camaschella C., Nai A., Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020;105(January (2)):260–272. doi: 10.3324/haematol.2019.232124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cocco E., Porrini V., Derosas M., Nardi V., Biasiotto G., Maccarinelli F., Zanella I. Protective effect of mitochondrial ferritin on cytosolic iron dysregulation induced by doxorubicin in HeLa cells. Mol. Biol. Rep. 2013;40(December (12)):6757–6764. doi: 10.1007/s11033-013-2792-z. [DOI] [PubMed] [Google Scholar]
- 97.Nguyen N.B., Callaghan K.D., Ghio A.J., Haile D.J., Yang F. Hepcidin expression and iron transport in alveolar macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;291(September (3)):L417–25. doi: 10.1152/ajplung.00484.2005. Epub 2006 Apr 28. [DOI] [PubMed] [Google Scholar]
- 98.Chen Q.X., Song S.W., Chen Q.X.H., Zeng C.L., Zheng X., Wang J.L., Fang X.M. Silencing airway epithelial cell-derived hepcidin exacerbates sepsis induced acute lung injury. Crit. Care. 2014;18(August (4)):470. doi: 10.1186/s13054-014-0470-8. PM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang Y., Zeng C., Yang S., Zhang Y., Song S., Liu S., Shu Q., Fang X., Chen Q. Airway epithelial hepcidin coordinates lung macrophages and immunity against bacterial pneumonia. Shock. 2019;(November (15)) doi: 10.1097/SHK.0000000000001471. [DOI] [PubMed] [Google Scholar]
- 100.Nairz M., Dichtl S., Schroll A., Haschka D., Tymoszuk P., Theurl I., Weiss G. Iron and innate antimicrobial immunity-Depriving the pathogen, defending the host. J. Trace Elem. Med. Biol. 2018;48(July):118–133. doi: 10.1016/j.jtemb.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 101.Biasiotto G., Di Lorenzo D., Archetti S., Zanella I. Iron and neurodegeneration: is ferritinophagy the link? Mol. Neurobiol. 2016;53(October (8)):5542–5574. doi: 10.1007/s12035-015-9473-y. [DOI] [PubMed] [Google Scholar]
- 102.Muriuki J.M., Atkinson S.H. How eliminating malaria may also prevent Iron deficiency in african children. Pharmaceuticals (Basel) 2018;11(October (4)):E96. doi: 10.3390/ph11040096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Emerson L.R., Nau M.E., Martin R.K., Kyle D.E., Vahey M., Wirth D.F. Relationship between chloroquine toxicity and iron acquisition in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 2002;46(March (3)):787–796. doi: 10.1128/AAC.46.3.787-796.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Octave J.N., Schneider Y.J., Hoffmann P., Trouet A., Crichton R.R. Transferrin uptake by cultured rat embryo fibroblasts. The influence of lysosomotropic agents, iron chelators and colchicine on the uptake of iron and transferrin. Eur. J. Biochem. 1982;123(April (2)):235–240. doi: 10.1111/j.1432-1033.1982.tb19758.x. [DOI] [PubMed] [Google Scholar]
- 105.Gammella E., Buratti P., Cairo G., Recalcati S. The transferrin receptor: the cellular iron gate. Metallomics. 2017;9(October (10)):1367–1375. doi: 10.1039/c7mt00143f. [DOI] [PubMed] [Google Scholar]
- 106.Potter M.D., Shinpock S.G., Popp R.A., Godfrey V., Carpenter D.A., Bernstein A., Johnson D.K., Rinchik E.M. Mutations in the murine fitness 1 gene result in defective hematopoiesis. Blood. 1997;90(September (5)):1850–1857. [PubMed] [Google Scholar]
- 107.Schultze A.E., Poppenga R.H., Johnson D.K. Alterations in serum and tissue iron profiles associated with mutations in thefitness14226sb locus of mice. Comp. Haematol. Int. 1998;8:72–76. doi: 10.1007/BF02642494. [DOI] [Google Scholar]
- 108.Scotland P.B., Heath J.L., Conway A.E., Porter N.B., Armstrong M.B., Walker J.A., Klebig M.L., Lavau C.P., Wechsler D.S. The PICALM protein plays a key role in iron homeostasis and cell proliferation. PLoS One. 2012;7(8) doi: 10.1371/journal.pone.0044252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shawki A., Knight P.B., Maliken B.D., Niespodzany E.J., Mackenzie B. H(+)-coupled divalent metal-ion transporter-1: functional properties, physiological roles and therapeutics. Curr. Top. Membr. 2012;70:169–214. doi: 10.1016/B978-0-12-394316-3.00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Di Paola S., Scotto-Rosato A., Medina D.L. TRPML1: the Ca(2+) retaker of the lysosome. Cell Calcium. 2018;69(January):112–121. doi: 10.1016/j.ceca.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 111.Mancias J.D., Wang X., Gygi S.P., Harper J.W., Kimmelman A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(May (7498)):105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kong Z., Liu R., Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2019;109(January):2043–2053. doi: 10.1016/j.biopha.2018.11.030. [DOI] [PubMed] [Google Scholar]
- 113.Hamming I., Timens W., Bulthuis M.L., Lely A.T., Navis G., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203(June (2)):631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vogel J.U., Schmidt S., Schmidt D., Rothweiler F., Koch B., Baer P., Rabenau H., Michel D., Stamminger T., Michaelis M., Cinatl J. The thrombopoietin receptor agonist eltrombopag inhibits human cytomegalovirus replication via Iron chelation. Cells. 2019;9(December (1)):E31. doi: 10.3390/cells9010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tiensiwakul P., Husain S.S. Effect of mouse hepatitis virus infection on iron retention in the mouse liver. Br. J. Exp. Pathol. 1979;60(April (2)):161–166. [PMC free article] [PubMed] [Google Scholar]
- 116.Porto G., Cruz E., Teles M.J., de Sousa M. HFE related hemochromatosis: uncovering the inextricable link between Iron homeostasis and the immunological system. Pharmaceuticals (Basel) 2019;12(August (3)):E122. doi: 10.3390/ph12030122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cao X. COVID-19: immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020;(April (9)) doi: 10.1038/s41577-020-0308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Perlman S., Dandekar A.A. Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 2005;5(December (12)):917–927. doi: 10.1038/nri1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Frieman M., Heise M., Baric R. SARS coronavirus and innate immunity. Virus Res. 2008;133(April (1)):101–112. doi: 10.1016/j.virusres.2007.03.015. Epub 2007 Apr 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Channappanavar R., Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017;39(July (5)):529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Recalcati S., Locati M., Gammella E., Invernizzi P., Cairo G. Iron levels in polarized macrophages: regulation of immunity and autoimmunity. Autoimmun. Rev. 2012;11(October (12)):883–889. doi: 10.1016/j.autrev.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 122.Legssyer R., Ward R.J., Crichton R.R., Boelaert J.R. Effect of chronic chloroquine administration on iron loading in the liver and reticuloendothelial system and on oxidative responses by the alveolar macrophages. Biochem. Pharmacol. 1999;57(April (8)):907–911. doi: 10.1016/s0006-2952(98)00368-2. [DOI] [PubMed] [Google Scholar]
- 123.Legssyer R., Josse C., Piette J., Ward R.J., Crichton R.R. Changes in function of iron-loaded alveolar macrophages after in vivo administration of desferrioxamine and/or chloroquine. J. Inorg. Biochem. 2003;94(February (1-2)):36–42. doi: 10.1016/s0162-0134(02)00633-5. [DOI] [PubMed] [Google Scholar]
- 124.Stölzel U., Köstler E., Schuppan D., Richter M., Wollina U., Doss M.O., Wittekind C., Tannapfel A. Hemochromatosis (HFE) gene mutations and response to chloroquine in porphyria cutanea tarda. Arch. Dermatol. 2003;139(March (3)):309–313. doi: 10.1001/archderm.139.3.309. [DOI] [PubMed] [Google Scholar]
- 125.Ouyang Q., Huang Z., Wang Z., Chen X., Ni J., Lin L. Effects of pristane alone or combined with chloroquine on macrophage activation, oxidative stress, and TH1/TH2 skewness. J. Immunol. Res. 2014;2014 doi: 10.1155/2014/613136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martins A.C., Almeida J.I., Lima I.S., Kapitão A.S., Gozzelino R. Iron Metabolism and the Inflammatory Response. IUBMB Life. 2017;69(June (6)):442–450. doi: 10.1002/iub.1635. [DOI] [PubMed] [Google Scholar]
- 127.Kemp J.D. The role of iron and iron binding proteins in lymphocyte physiology and pathology. J. Clin. Immunol. 1993;13(March (2)):81–92. doi: 10.1007/BF00919264. [DOI] [PubMed] [Google Scholar]
- 128.Kanamori Y., Murakami M., Sugiyama M., Hashimoto O., Matsui T., Funaba M. Hepcidin and IL-1β. Vitam. Horm. 2019;110:143–156. doi: 10.1016/bs.vh.2019.01.007. [DOI] [PubMed] [Google Scholar]
- 129.Shu W., Pang Z., Xu C., Lin J., Li G., Wu W., Sun S., Li J., Li X., Liu Z. Anti- TNF-α monoclonal antibody therapy improves anemia through downregulating hepatocyte hepcidin expression in inflammatory bowel disease. Mediators Inflamm. 2019;13(November (2019)) doi: 10.1155/2019/4038619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hadadi A., Mortezazadeh M., Kolahdouzan K., Alavian G. Does recombinant human Erythropoietin administration in critically ill COVID-19 patients have miraculous therapeutic effects? J. Med. Virol. 2020;(April (8)) doi: 10.1002/jmv.25839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wenzhong liu, hualan Li. COVID-19: attacks the 1-beta chain of hemoglobin and captures the porphyrin to inhibit human heme metabolism. ChemRxiv. 2020 doi: 10.26434/chemrxiv.11938173.v7. Preprint. [DOI] [Google Scholar]
- 132.Keung Y.K., Owen J. Iron deficiency and thrombosis: literature review. Clin. Appl. Thromb. Hemost. 2004;10(4):387–391. doi: 10.1177/107602960401000412. [DOI] [PubMed] [Google Scholar]
- 133.Azab S.F., Abdelsalam S.M., Saleh S.H. Iron deficiency anemia as a risk factor for cerebrovascular events in early childhood: a case-control study. Ann. Hematol. 2014;93(4):571–576. doi: 10.1007/s00277-013-1922-y. [DOI] [PubMed] [Google Scholar]
- 134.Chang Y.L., Hung S.H., Ling W., Lin H.C., Li H.C., Chung S.H.D. Association between ischemic stroke and iron-deficiency anemia: a population-based study. PLoS One. 2013;8(12) doi: 10.1371/journal.pone.0082952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hung S.H., Lin H.C., Chung S.H.D. Association between venous thromboembolism and iron-deficiency anemia: a population-based study. Blood Coagul. Fibrinolysis. 2015;26(4):368–372. doi: 10.1097/MBC.0000000000000249. [DOI] [PubMed] [Google Scholar]
- 136.Song A.B., Kuter D.J., Al-Samkari H. Characterization of the rate, predictors, and thrombotic complications of thrombocytosis in iron deficiency anemia. Blood. 2019;134(Suppl_1):959. doi: 10.1002/ajh.25925. [DOI] [PubMed] [Google Scholar]
- 137.Yin S., Huang M., Li D., Tang N. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis. 2020;(April (3)) doi: 10.1007/s11239-020-02105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tang N., Bai H., Chen X., Gong J., Li D., Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020;(March (27)) doi: 10.1111/jth.14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jimenez K., Leitner F., Leitner A., Scharbert G., Schwabl P., Kramer A.M., Krnjic A., Friske J., Helbich T., Evstatiev R., Khare V., Gasche C. Iron deficiency induced thrombocytosis increases thrombotic tendency in rats. Haematologica. 2020;(February (20)) doi: 10.3324/haematol.2019.245092. pii: haematol.2019.245092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Abreu R., Essler L., Loy A., Quinn F., Giri P. Heparin inhibits intracellular Mycobacterium tuberculosis bacterial replication by reducing iron levels in human macrophages. Sci. Rep. 2018;8(May (1)):7296. doi: 10.1038/s41598-018-25480-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harrison C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020;(February (27)) doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- 142.Zhai P., Ding Y., Wu X., Long J., Zhong Y., Li Y. The epidemiology, diagnosis and treatment of COVID-19. Int. J. Antimicrob. Agents. 2020;(March (28)) doi: 10.1016/j.ijantimicag.2020.105955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Huang M., Tang T., Pang P., Li M., Ma R., Lu J., Shu J., You Y., Chen B., Liang J., Hong Z., Chen H., Kong L., Qin D., Pei D., Xia J., Jiang S., Shan H. Treating COVID-19 with chloroquine. J. Mol. Cell Biol. 2020;(April (1)):mjaa014. doi: 10.1093/jmcb/mjaa014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gautret P., Lagier J.C., Parola P., Hoang V.T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V.E., Dupont H.T., Honoré S., Colson P., Chabrière E., La Scola B., Rolain J.M., Brouqui P., Raoult D. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020;(March (20)) doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gautret P., Lagier J.C., Parola P., Hoang V.T., Meddeb L., Sevestre J., Mailhe M., Doudier B., Aubry C., Amrane S., Seng P., Hocquart M., Eldin C., Finance J., Vieira V.E., Dupont H.T., Honoré S., Stein A., Million M., Colson P., La Scola B., Veit V., Jacquier A., Deharo J.C., Drancourt M., Fournier P.E., Rolain J.M., Brouqui P., Raoult D. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: a pilot observational study. Travel Med. Infect. Dis. 2020;(April (11)) doi: 10.1016/j.tmaid.2020.101663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Molina J.M., Delaugerre C., Goff J.L., Mela-Lima B., Ponscarme D., Goldwirt L., de Castro N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med. Mal. Infect. 2020;(March (30) doi: 10.1016/j.medmal.2020.03.006. pii: S0399-077X(20)30085-30088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Available at: https://www.idsociety.org/globalassets/idsa/practice-guidelines/covid-19/treatment/supplementary-information.pdf. Accessed 20 April 2020.
- 148.Perinel S., Launay M., Botelho-Nevers É, Diconne É, Louf-Durier A., Lachand R., Murgier M., Page D., Vermesch R., Thierry G., Delavenne X. Towards optimization of hydroxychloroquine dosing in intensive care unit COVID-19 patients. Clin. Infect. Dis. 2020;(April (7)):ciaa394. doi: 10.1093/cid/ciaa394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Available at: https://www.fda.gov/media/136534/download. Accessed 20 April 2020.
- 150.Available at: https://www.cdc.gov/coronavirus/2019-ncov/hcp/therapeutic-options.html. Accessed 20 April 2020.
- 151.Available at: http://www.simit.org/IT/simit/sezioni-regionali.xhtml/sezione/112-lombardia/comunicazioni/1. Accessed 20 April 2020.
- 152.Available at: https://www.aemps.gob.es/la-aemps/ultima-informacion-de-la-aemps-acerca-del-covid%e2%80%9119/tratamientos-disponibles-para-el-manejo-de-la-infeccion-respiratoria-por-sars-cov-2/?lang=en. Accessed 20 April 2020.
- 153.Available at: https://clinicaltrials.gov/ct2/results?cond=%22Coronavirus+Infections%22. Accessed 20 April 2020.
- 154.Ye G., Pan Z., Pan Y., Deng Q., Chen L., Li J., Li Y., Wang X. Clinical characteristics of severe acute respiratory syndrome coronavirus 2 reactivation. J. Infect. 2020;(March (20)) doi: 10.1016/j.jinf.2020.03.001. pii: S0163-4453(20)30114-30116. [DOI] [PMC free article] [PubMed] [Google Scholar]