

Abstract
Resurrection of non-processed pseudogenes may increase the efficacy of therapeutic gene editing, upon simultaneous targeting of a mutated gene and its highly homologous pseudogenes. To investigate the potency of this approach for clinical gene therapy of human diseases, we corrected a pseudogene-associated disorder, the immunodeficiency p47phox-deficient chronic granulomatous disease (p47phox CGD), using clustered regularly interspaced short palindromic repeats-associated nuclease Cas9 (CRISPR-Cas9) to target mutated neutrophil cytosolic factor 1 (NCF1). Being separated by less than two million base pairs, NCF1 and two pseudogenes are closely co-localized on chromosome 7. In healthy people, a two-nucleotide GT deletion (ΔGT) is present in the NCF1B and NCF1C pseudogenes only. In the majority of patients with p47phox CGD, the NCF1 gene is inactivated due to a ΔGT transfer from one of the two non-processed pseudogenes. Here we demonstrate that concurrent targeting and correction of mutated NCF1 and its pseudogenes results in therapeutic CGD phenotype correction, but also causes potentially harmful chromosomal deletions between the targeted loci in a p47phox-deficient CGD cell line model. Therefore, development of genome-editing-based treatment of pseudogene-related disorders mandates thorough safety examination, as well as technological advances, limiting concurrent induction of multiple double-strand breaks on a single chromosome.
Keywords: chronic granulomatous disease, CGD, gene editing, CRISPR, NCF1, pseudogene, gene therapy
Graphical Abstract
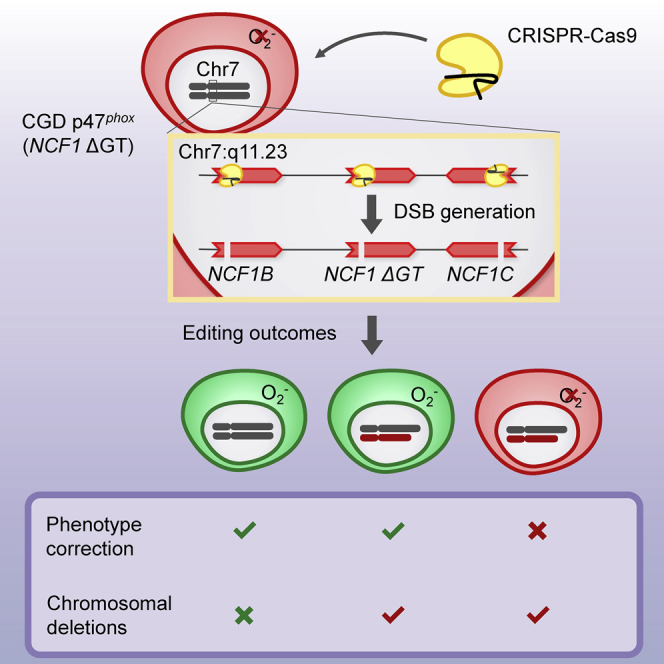
Efficacy of gene-editing may be improved by simultaneous targeting of a gene and its pseudogenes. Wrona et al. evaluate this approach for treatment of p47phox-deficient chronic granulomatous disease. They show that CRISPR-mediated phenotype correction is accompanied by chromosomal deletions, mandating technological improvements to limit induction of multiple DNA cuts.
Introduction
Chronic granulomatous disease (CGD) is characterized by defective respiratory burst,1 impaired microbicidal activity of phagocytes,2,3 and resulting life-threatening bacterial and fungal infections. This condition is caused by mutations of genes encoding gp91phox, p22phox, p67phox, p47phox, or p40phox subunits of the phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. In nearly all patients with p47phox CGD, the disease is caused by a two-nucleotide GT deletion (ΔGT) within exon 2 of the neutrophil cytosolic factor 1 (NCF1) gene.4,5 This ΔGT mutation causes a frameshift and an early translation termination. NCF1 is accompanied on chromosome 7 by two almost identical non-processed pseudogenes, NCF1B and NCF1C, which carry the ΔGT mutation also in healthy individuals.6,7 Although retroviral-based hematopoietic stem cell (HSC) gene therapy has been clinically successful in patients with the X-linked gp91phox-deficient form of CGD,8,9 autosomal recessive p47phox CGD has not been successfully addressed in gene therapy trials yet. Because more than 97% of patients with p47phox CGD share the same ΔGT mutation, genome-editing-based gene therapy may constitute an attractive alternative to lentiviral gene therapies for this subgroup of patients.
In general, genetic disorders such as p47phox CGD caused by mutation transfer from non-processed pseudogenes are particularly promising targets for genome editing, because parallel gene and pseudogene resurrection via the presence of highly homologous target sites may potentially increase the overall efficiency of the treatment. If the gene and pseudogene are located on the same chromosome, however, editing inducing double-strand breaks (DSBs) may cause chromosomal deletions as a side effect. At least 11 reported genetic disorders are associated with pseudogene-related gene conversion (Table S1),10,11 making them potentially attractive, but challenging targets for genome editing. In the case of p47phox CGD, the ΔGT mutation may be directly targeted and corrected, leading to conversion of the inactive NCF1 loci into p47phox-expressing genes. However, for other pseudogene-related disorders, various strategies may be considered, including exon replacement or minigene insertion.12,13
As a model representing pseudogene-related genetic disorders, here we study the efficacy and safety of genome editing of p47phox CGD by clustered regularly interspaced short palindromic repeats-associated nuclease Cas9 (CRISPR-Cas9). Diverse genome-editing systems (CRISPR-Cas9, zinc-finger nucleases [ZFNs], transcription activator-like effector nucleases [TALENs]) have been used preclinically for correction of CGD in cell line models14, 15, 16 and in human HSCs by cDNA delivery to a safe genomic harbor,17,18 by exon replacement,12 or by direct mutation targeting.19,20 Interestingly, ZFN-mediated correction of NCF1 pseudogenes in induced pluripotent stem cells (iPSCs) resulted in the expression of functional p47phox upon phagocytic differentiation.19 Whereas these studies focused primarily on the efficacy of CGD correction, we set out to evaluate the safety of ΔGT p47phox CGD correction in a cell line model of p47phox CGD.21
Results
Reconstitution of p47phox Expression and NADPH Oxidase Function upon CRISPR-Cas9-Mediated Correction of NCF1 Gene and Pseudogene Loci
First, the PLB-985 wild type (WT) and the corresponding isogenic p47phox CGD model cell line, PLB-985 NCF1 ΔGT,21 were nucleofected with a CRISPR-Cas9 and GFP co-expressing plasmid, along with a corrective single-stranded oligodeoxynucleotide (ssODN) template (Figure 1A). Single-guide RNA (sgRNA) sequences were designed to guide Cas9 to the ΔGT mutation site in mutated NCF1, which is also present in NCF1B and NCF1C pseudogenes (Figure 1B). The on-target correction efficiency in Cas9-expressing cells was determined by PCR-based restriction fragment length polymorphism (RFLP) method (PCR-RFLP) that detects restoration of the BsrGI restriction site upon correction, quantified as GTGT content in genomic DNA derived from edited cells (Figures 1B and 1C).
Figure 1.

CRISPR-Cas9 Correction of the ΔGT Mutation in PLB-985 WT and PLB-985 NCF1 ΔGT Cells
(A) Scheme depicting the correction strategy of NCF1 gene and pseudogene loci by CRISPR-Cas. (B) NCF1 locus: sequence of tested sgRNAs, cleavage sites for Cas9 (red arrowheads), position of the ΔGT mutation (filled red rectangle), protospacer adjacent motifs (PAMs) (blue rectangles), corrected NCF1 sequence (green rectangle), digestion sites for BsrG1 (orange arrowheads), and the BsrG1 restriction site (orange rectangle). (C) Polyacrylamide gel of PCR-RFLP analysis of bulk CRISPR-Cas9-treated PLB-985 WT and PLB-985 NCF1 ΔGT cell lines. Band intensities were analyzed by the displayed formula. The 161-bp band within the dashed rectangle resulted from digestion of corrected NCF1 (n = 4; bars: means with standard deviations; statistical analysis with unpaired t test with Welch’s correction, ∗∗p < 0.01, ∗∗∗p < 0.001). (D) TIDER analysis of cleavage and correction efficiencies for bulk CRISPR-Cas9-treated and untreated PLB-985 WT and PLB-985 NCF1 ΔGT cell lines (n = 4; bars: means with standard deviations; statistical analysis with unpaired t test with Welch’s correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (E) Western blot of control p67phox (another cytosolic NADPH oxidase subunit, which is complexed with p47phox and p40phox, p47phox, and GAPDH for differentiated bulk CRISPR-Cas9-treated PLB-985 WT and PLB-985 NCF1 ΔGT cell lines. (F) Light microscopy images of NBT test performed on differentiated bulk CRISPR-Cas9-treated PLB-985 WT and PLB-985 NCF1 ΔGT cell lines. ns, not significant.
This analysis demonstrated that the ΔGT mutation within the NCF1 gene and its pseudogenes can be corrected, and that two out of three tested sgRNAs (sgRNA #1 and #2; Figures 1B and 1C) successfully reconstituted the BsrG1 restriction site (Figure 1B), restoring the normal NCF1 gene sequence. The efficiencies of NCF1 gene and pseudogene loci correction corresponded to the GTGT content values observed for clinically healthy ΔGT p47phox CGD carriers.22 Estimated GTGT content values for CRISPR-Cas9-treated PLB-985 NCF1 ΔGT bulk cultures were 0.22 ± 0.02 and 0.17 ± 0.02 for sgRNA #1 and #2, respectively, suggesting that on average one NCF1 gene or one pseudogene locus was corrected in Cas9-expressing cells. Interestingly, CRISPR-Cas9 treatment of PLB-985 WT cells led to an increase of the GTGT content, from 0.39 ± 0.04 (untreated) to 0.70 ± 0.03, indicating the correction of NCF1B or NCF1C pseudogenes.
High efficacy of CRISPR-Cas9 editing was observed for all tested sgRNAs, as additionally assayed by TIDER (tracking of insertions, deletions, and recombination events by decomposition) method (Figure 1D).23 Due to the highly homologous NCF1 gene and pseudogene loci in humans, as well as a naturally present ΔGT in NCF1 pseudogenes, TIDER analysis of PLB-985 WT cells, performed on PCR co-amplified NCF1 gene and pseudogene loci, detects the correct NCF1 gene as “cleaved and corrected.” Therefore, the estimated reference cleavage and correction frequencies for the CRISPR-Cas9-untreated wild-type cells were 46.4% ± 1.3% and 41.8% ± 0.7%, respectively. The cleavage efficiency determined for CRISPR-Cas9-treated PLB-985 WT cells was 93.3% ± 1.7%, and the correction efficiency was 63.1% ± 5.6%.
The true cleavage and correction efficacies in PLB-985 WT cells, corresponding to the difference between values detected in CRISPR-Cas9-treated and untreated cells, confirmed the results observed with PCR-RFLP. In CRISPR-Cas9-treated PLB-985 NCF1 ΔGT bulk cultures, all tested sgRNAs exhibited high levels of cleavage efficiency: 89.8% ± 0.6%, 56.8% ± 4.4%, and 81.9% ± 6.5% for sgRNAs #1, #2, and #3, respectively. Correction of the ΔGT mutation was observed only with sgRNAs #1 and #2, reaching 29.3% ± 0.9% for sgRNA #1 and 18.9% ± 2.0% for sgRNA #2 (Figure 1D), confirming the results of the PCR-RFLP analysis (Figure 1C).
Analysis of p47phox protein expression in CRISPR-Cas9-treated cells (Figure 1E) showed that correction of the NCF1 pseudogenes in PLB-985 WT cells led to increased p47phox expression. Correction of PLB-985 NCF1 ΔGT (achieved with sgRNA #1 and #2) restored p47phox protein expression. In addition, the NADPH oxidase function was reconstituted in CRISPR-Cas9-treated PLB-985 NCF1 ΔGT cells, as measured by nitroblue tetrazolium (NBT) test (Figure 1F). Remarkably, CRISPR-Cas9 treatment of PLB-985 NCF1 ΔGT cells without corrective template also reconstituted NADPH oxidase function, although in fewer cells. This was not due to alternative splicing, but likely occurred because of the reading frame-restoring indel mutations in NCF1 (Figure S1).
Simultaneous Editing of Three NCF1 Gene and Pseudogene Loci Present on Chromosome 7 Leads to Complex Genomic Aberrations
To characterize potential adverse effects of concurrent CRISPR-Cas9 targeting of NCF1 gene and pseudogene loci, we generated single clones of CRISPR-Cas9-treated (sgRNA #1) PLB-985 NCF1 ΔGT cells (Figures S2–S6). The sgRNA #1 has been selected for subsequent experiments because it showed the highest specificity in cleaving the ΔGT carrying sequence among tested sgRNAs (Figure S2). The T7 Endonuclease I assay performed on the top predicted off-target sites showed high specificity of sgRNA #1 toward the mutated NCF1 and NCF1 pseudogenes (Figure S7A; Table S2). GUIDE sequencing (GUIDE-seq) analysis24 revealed three off-target sequences, of which one is represented within five distinct genomic locations in the proximity of members of a large multigene family, olfactory receptors (Figure S7B; Table S3).25 Analysis by single-molecule real-time (SMRT) sequencing26 of the NCF1/NCF1B/NCF1C PCR co-amplification products, surrounding the ΔGT mutation of CRIPSR-Cas9-treated PLB-985 NCF1 ΔGT individual clones, revealed high frequency of indel mutations at the NCF1 on-target site, exceeding 90% of reads from non-corrected NCF1 gene or pseudogene loci (Table S4). Sixty percent of Cas9-expressing clones exhibited corrected reads (Figure S3A). Furthermore, 5 out of 45 tested clones carried a 0.5-kb deletion at the CRISPR-Cas9 cut site that deleted NCF1 exon 2 (Figure 2A) and affected the results observed by PCR-RFLP (Figure S3B). Complementarity between the genomic sequences adjacent to the deleted region (Figure 2A) likely contributed to the induction of this deletion upon DSBs repair.
Figure 2.

Detection of Chromosomal Aberrations in CRISPR-Cas9-Treated PLB-985 NCF1 ΔGT Cells
(A) PCR product of NCF1 gene and pseudogene loci, with indicated primers. ΔGT, deletion-end adjacent partially complementary sequences, and detected deletion (red) between them are indicated. (B) Scheme of chromosome 7 q11.23 fragment: locations of FISH probes, CEP7, binding the centromere of chromosome 7, and RP11-100C23, binding the region between NCF1B and NCF1, as well as binding sites of four primer pairs used for CNV analysis are shown. A, B and C indicate the amplified chromosomal segments; c (centromeric), m (medial), and t (telomeric) sites. Location of NCF1, NCF1B, and NCF1C in blocks Bm, Bc, and Bt, respectively, is indicated by gray arrowheads. (C) Relative copy number of regions surrounding NCF1 gene and pseudogene loci in CRISPR-Cas9-treated PLB-985 NCF1 ΔGT cells determined by qPCR. Clones with deletion between NCF1B and NCF1C (red squares) and clones with deletion between NCF1 and NCF1C (red circles) are shown. (D) Number of CRISPR-Cas9-treated clones that exhibited no chromosomal aberrations within NCF1 loci (blue), number of clones with a 0.5-kb deletion at the CRISPR-Cas9 cleavage site (gray), and heterozygous deletions between NCF1 and NCF1C (yellow) and between NCF1B and NCF1C (red) confirmed by FISH and aCGH. (E) Immunofluorescence microscopy images of FISH analysis for a control clone with centromeric CEP7 Spectrum Aqua binding and with binding sites for RP11-100C23 Fluorescein probe on two chromosomes, as well as a clone with a heterozygous deletion of the region between NCF1B and NCF1 (binding of RP11-100C23 on one chromosome). (F) aCGH of chromosome 7 q11.22-23 region of untreated PLB-985 NCF1 ΔGT cells and one clone of CRISPR-Cas9-treated PLB-985 NCF1 ΔGT without (no deletion) and two clones with a heterozygous deletion are shown.
In addition to the observed indel formation, simultaneous CRISPR-Cas9 cleavage of two or three ΔGT-carrying NCF1 loci that are located on the same chromosome may also lead to large chromosomal rearrangements, which could span the regions between NCF1B and NCF1 (1.5 Mb), NCF1 and NCF1C (0.4 Mb), or NCF1B and NCF1C (1.9 Mb) (Figure 2B). We therefore quantified copy number variation (CNV) using quantitative PCR (qPCR) of non-repetitive genes, located between NCF1 loci (EIF4H between NCF1B and NCF1; WBSCR16 between NCF1 and NCF1C) and genes located outside of the NCF1 gene and pseudogene loci (CALN1 upstream of NCF1B; HIP1 downstream of NCF1C) (Figures 2B and 2C; Table S5). We identified 8 out of 49 clones (Figure 2C) that exhibited unaltered copy number of CALN1 and HIP1 genes located outside the NCF1 gene and pseudogene loci, but decreased copy number of EIF4H and WBSCR16, located between NCF1 gene and pseudogenes, suggesting the presence of heterozygous deletions between NCF1B and NCF1C. Interestingly, one clone exhibited decreased CNV only for WBSCR16, suggesting a heterozygous deletion between NCF1 and NCF1C. No clones were identified with a homozygous deletion of genes between NCF1 gene and pseudogene loci, which could result from reduced viability due to homozygous deletion of genes affecting cellular fitness.
Subsequently, all clones were tested for the presence of deletions between NCF1B and NCF1 by fluorescence in situ hybridization (FISH) (Figure 2B), and a genome-wide analysis of chromosomal aberrations was performed on selected clones using microarray-based comparative genomic hybridization (aCGH) (Figures 2C and 2F; Figure S6; Tables S6 and S7). FISH and aCGH analyses confirmed deletions between NCF1B and NCF1C detected by qPCR, and aCGH identified a second clone with a deletion between NCF1 and NCF1C (Figures 2C–2F). Duplications or chromosomal translocations within these regions were not observed, which suggests that deletions of chromosomal fragments are the predominant chromosomal aberration type after gene editing (see clones 21 and 27 in Figure S6 and Table S7).
Discussion
Although holding great promises to potentially cure monogenetic diseases at their origin, application of gene-editing technologies has also been linked to unpredictable and complex editing outcomes at the targeted site,27 which is a major hindrance to application of DSB-based gene editing in clinical settings. Our results suggest that CRISPR-Cas9-based gene therapy may indeed efficiently correct mutated NCF1 and its pseudogenes, and thus rescue the impaired NADPH oxidase activity in ΔGT p47phox CGD patients. However, simultaneous induction of two or three DSBs on a single chromosome can be associated with induction of large chromosomal aberrations that primarily affect the sequences between the targeted loci, as we and others have shown.28, 29, 30, 31, 32
The genotype of cells with CRISPR-Cas9-induced heterozygous deletions within NCF1 gene and pseudogene loci resembles the genotype present in patients with Williams syndrome.33 Haploinsufficiency of genes residing in the deleted region is primarily linked to cardiovascular and neurological manifestations of Williams syndrome. Hemizygosity of the elastin (ELN) gene has been linked to the connective-tissue abnormalities and hypertension, whereas hemizygosity of other genes has been associated with impaired visuospatial and motor abilities, as well as with mild-to-moderate intellectual disability. Although Williams syndrome is generally not considered as a cancer-predisposing condition, reports linking the reduced copy number of the BCL7B gene that is located between NCF1B and NCF1 to blood malignancies exist.34,35 A homozygous intra-chromosomal deletion of the region between NCF1 gene and pseudogene loci has not been observed in clones of CRISPR-Cas9-treated PLB-985 NCF1 ΔGT cells. Although after gene editing of the NCF1 loci of a ΔGT p47phox CGD patient, the deleted chromosomal fragments would be restricted to cells of the hematopoietic compartment, their long-term impact and potential adverse functional implications need to be evaluated carefully.
As we demonstrate for pseudogene-associated p47phox CGD, the risk for inducing chromosomal deletions is likely to apply to other diseases caused by mutations associated with highly homologous pseudogenes located on the same chromosome, such as autosomal dominant polycystic kidney disease,36 type 2 Gaucher disease,37 or neural tube defects.38 For later clinical application, future studies will thus require a substantial improvement of gene-editing enzyme delivery protocols, or application of different editing strategies, and development of rigorous post-editing diagnostic protocols. Measures to minimize adverse effects could include tightly controlled transient exposure of the genome to the gene-editing enzyme, application of other gene-editing strategies not generating DSBs, or modulation of repair pathway choice after DSB generation. Base editors39 inducing NCF1 exon 2 skipping by targeting the splice acceptor site adjacent to the ΔGT mutation may not lead to reconstitution of the phagocytic NADPH oxidase activity (Figures S1B–S1F); however, introduction of missing nucleotides at the ΔGT mutation without induction of DSBs by prime editors40 may constitute an appealing treatment alternative.
In conclusion, safety evaluation is of paramount importance for preclinical development of gene-editing-based approaches, especially for pseudogene-related diseases, where targeted genes and homologous pseudogenes are co-localized on the same chromosome. The extent of chromosomal aberrations, as well as their potential effects on the patient’s health, should be carefully addressed in preclinical studies on genome-editing gene therapy approaches, especially for pseudogene-related diseases (Table S1). As to the current state of the art, the application of gene-editing technology for treatment of pseudogene-related genetic disorders should therefore be limited to those cases in which the therapy provides immediate amelioration of life-threatening symptoms or the therapeutic benefit for the patient balances out the risk for potential adverse effects.
Materials and Methods
Plasmid Construction
The sgRNA sequences were designed using the Optimized CRISPR Design (F. Zhang laboratory, MIT, 2015; http://zlab.bio/guide-design-resources). Single-stranded DNA oligonucleotides were obtained from Microsynth (Balgach, Switzerland), cloned into pSpCas9(BB)-2A-GFP (PX458) (F. Zhang, Addgene plasmid #48138),41 and the plasmid sequence was confirmed by Sanger sequencing (Microsynth).
Cell Culture Conditions
PLB-985 WT and PLB-985 NCF1 ΔGT cell lines21 were cultured in RPMI 1640 medium (PAN-Biotech, Aidenbach, Germany), supplemented with 10% (v/v) fetal calf serum (FCS) (PAN-Biotech), 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific, Reinach, Switzerland). For granulocytic differentiation, cells were cultured for 7 days in RPMI 1640 medium supplemented with 5% (v/v) FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.5% (v/v) N,N-dimethylformamide (DMF) (Sigma Aldrich, Buchs, Switzerland). Throughout all experiments, cells were grown at 37°C in a humidified atmosphere containing 5% (v/v) CO2.
CRISPR-Cas9 Treatment of the PLB-985 NCF1 ΔGT Cell Line
PX458 plasmids (15–40 μg) expressing sgRNA, Cas9, and GFP proteins were delivered into 2 × 106 PLB-985 WT or PLB-985 NCF1 ΔGT cells by nucleofection, using the Amaxa Cell Nucleofector Kit V and Amaxa Nucleofector II, program C-023 (Lonza, Basel, Switzerland), along with a 100-nt ssODN (Microsynth) at a final concentration of 3 μM (Figure 1A). The sequence of the ssODN was: 5′-GCC TCT TTG GAG GCT GAA TGG GGT CCC CCG ACT CTG GCT TTC CCC CAG GTG TAC ATG TTC CTG GTG AAA TGG CAG GAC CTG TCG GAG AAG GTG GTC TAC C-3′. The locations of the binding sites for sgRNA are presented in Figure 1B. Nucleofected cells were supplemented with 500 μL growth medium and incubated at room temperature for 10 min, and cells were transferred to 10 mL growth medium thereafter. 1 μM SCR7 (BioVision, Milpitas, CA, USA) was added 3–4 h after nucleofection, and cells were cultured for 48 h. GFP-positive cells were sorted into a bulk culture (FACSAria III FCF; Becton Dickinson, Allschwil, Switzerland) in preconditioned, sterile filtered growth medium supplemented with 1 μM SCR7. Sorted cells were expanded for 1 month, and individual clones were generated by limiting dilution.
Assessment of Correction Efficacy by PCR-RFLP and TIDER
Genomic DNA of CRISPR-Cas9-treated cells was isolated using DNeasy Blood & Tissue Kit (QIAGEN, Hombrechtikon, Switzerland). Correction efficiency was assessed by PCR-RFLP method, as described previously.22 In brief, PCR co-amplification products of NCF1, NCF1B, and NCF1C were digested with BsrG1 and Pst1 restriction enzymes, and developed by electrophoresis in a 7.5% polyacrylamide gel. GTGT content was calculated based on size-normalized band intensities. The band intensity 169 bp was divided by the sum of band intensities 181 and 201 bp (Figure 1C).
TIDER analysis23 was performed using an online tool (https://tider.deskgen.com/) to assess CRISPR-Cas9 editing efficiency. Approximately 590 bp PCR products of NCF1 and its pseudogenes were amplified using the forward primer, 5′-CCA AGGT CTC AAG CAA TTC TCC-3′, and the reverse primer, 5′-CCA AAG GGT GGA GCT GGA AC-3′. TIDER analysis was performed on the Sanger sequencing chromatograms using the default parameters of the tool.
SMRT Sequencing
Barcoded PCR co-amplification products derived from the CRISPR-Cas9-treated PLB-985 NCF1 ΔGT clones (i.e., ΔGT carrying NCF1 and its pseudogenes) had a size of approximately 2.4 kb and were produced using combinations of the following barcoded forward (Fwd) and reverse (Rev) primers: Fwd1, 5′-TTA GGT CTA GGA TCC AGT CAA GGA T-3′; Fwd2, 5′-AAA GGA TCC AGT CAA GGA TCA ATG T-3′; Fwd3, 5′-TTT TCA GGT CTA GGA TCC AGT CAA-3′; Rev1, 5′-TTA GGT TCT GGG AGA TCC TGT CT-3′; Rev2, 5′-AAG TTC TGG GAG ATC CTG TCT GTT-3′; Rev3, 5′-CCA GCA GGT GCA TTT ATT TGG G-3′. Gel-purified amplification products were pooled and analyzed by the Functional Genomics Center Zurich, University of Zurich, and ETH Zurich, Zurich, Switzerland, as described before.21
Western Blot
Protein isolation from differentiated PLB-985 WT, PLB-985 NCF1 ΔGT, and CRISPR-Cas9-treated PLB-985 WT or PLB-985 NCF1 ΔGT cells was performed as described previously.42 In brief, cells were homogenized in a modified radioimmunoprecipitation assay (RIPA) lysis buffer and centrifuged for 10 min at 13,000 × g. Protein content in the supernatant was analyzed by Bradford assay (Sigma Aldrich). 10 μg total protein was denatured by boiling in a modified Laemmli loading buffer, separated by SDS-PAGE using 10% polyacrylamide gel, and wet-transferred onto Amersham Protran Premium NC nitrocellulose membrane (GE Healthcare Life Sciences, UK). The membrane was blocked with 3% skim milk and immune stained using the following primary antibodies: mouse anti-human p47phox monoclonal antibody clone 1 (Becton Dickinson, Allschwil, Switzerland), mouse anti-human p67phox monoclonal antibody clone D-6, or mouse anti-human GAPDH monoclonal antibody (0411) (both from Santa Cruz Biotechnology, Heidelberg, Germany). The membrane was incubated overnight with primary antibodies diluted 1:500 in an antibody buffer at 4°C, followed by 1-h incubation at room temperature with the mouse IgG kappa binding protein conjugated to horseradish peroxidase (Santa Cruz Biotechnology) diluted 1:3,000. The signal was developed by incubation of the membrane in SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific), visualized with ImageQuant LAS 4000 Biomolecular Imager (GE Healthcare Life Sciences, UK). Densitometric analysis of protein bands was performed using ImageJ.43
NBT Test
Differentiated PLB-985 WT, PLB-985 NCF1 ΔGT, and CRISPR-Cas9-treated clones of PLB-985 NCF1 ΔGT cells were incubated in 100 μg/mL phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) and 200 ng/mL NBT for 30 min at 37°C and 5% CO2 followed by cell fixation in 1% (w/v) formaldehyde. Fixed cells were analyzed visually for the presence of formazan precipitates using a Leica DM IL Fluo light microscope equipped with a DFC420 digital camera and LEICA application suite acquisition software (Leica Microsystems, Glattbrugg, Switzerland).
Chemiluminescence Assay
Chemiluminescence assay was performed on differentiated CRISPR-Cas9-treated PLB-985 cells in 96-well plates. A total of 1 × 105 cells/well were mixed with 200 μM luminol and 200 ng/mL PMA. Chemiluminescence signal was recorded with a Mithras LB 940 Multimode Microplate Reader (Berthold Technologies, Zug, Switzerland).
CNV Assessment by qPCR
The CNV of genomic locations surrounding the NCF1 gene and pseudogene loci in individual clones of CRISPR-Cas9-treated PLB-985 NCF1 ΔGT cells was assessed by qPCR. Primer sequences are listed in Table S5, and primer annealing locations are displayed in Figure 2B. qPCR was performed with the QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific) in 384-well plates, using 2.5 ng/μL genomic DNA in a total reaction volume of 10 μL, 500 nM forward and reverse primers, and SsoAdvanced Universal SYBR Green Supermix (Bio-Rad Laboratories, Cressier, Switzerland). PCR conditions included initial denaturation at 95°C for 3 min, 40 cycles of 30-s denaturation at 95°C, 30-s primer annealing at 65°C, and 15-s elongation at 72°C. Obtained mean cycle threshold (Ct) values of three measurements from individual plates were used for calculation of mean Ct values of at least three independent measurements. Relative copy number values of tested genomic regions were calculated using the 2−ΔΔCt method.44
FISH
Chromosome preparations of individual CRISPR-Cas9-treated PLB-985 NCF1 ΔGT clones were performed according to the manufacturers’ instructions. In brief, cells were incubated in hypotonic 0.075 M KCl solution for 30 min at 37°C, followed by dropwise fixation, and subsequent three rounds of washing with fixative solution (methanol:acetic acid; v/v 3:1 ratio). Fixed cells were dropped on a microscopic slide and hybridized with FISH probes. The FISH probes used for hybridization were a chromosome 7 centromere binding probe Vysis CEP7, labeled with Spectrum Aqua (Abbott, Abbott Molecular, Baar, Switzerland), and a BAC library probe RP11-100C23, conjugated to Green 5-Fluorescein dUTP (Empire Genomics, Buffalo, NY, USA). The slides were denatured at 75°C for 5 min, followed by overnight hybridization at 37°C and humid conditions using a Leica ThermoBrite System (Biosystems Switzerland, Muttenz, Switzerland). Then the slides were rinsed with a 0.4× saline sodium citrate (SSC)/0.3% (v/v) IGEPAL buffer (Sigma Aldrich) for 120 s at 72°C ± 2°C and 2× SSC/0.1% (v/v) IGEPAL (Sigma-Aldrich) for 60 s at room temperature. The slides were air-dried, and the nuclei of hybridized cells were visualized with Vectashield Mounting Medium containing DAPI (REACTOLAB, Servion, Switzerland). Microscopic images were acquired using the Axio Imager.Z2 microscope (Carl Zeiss, Feldbach, Switzerland) and analyzed using Isis software (MetaSystems Hard & Software, Altlussheim, Germany). For each tested clone, at least 200 interphase nuclei were analyzed.
aCGH
The PLB-985 NCF1 ΔGT cell line, as well as selected CRISPR-Cas9-treated PLB-985 NCF1 ΔGT clones, were subjected to aCGH using the CytoScan HD Array Kit (Affymetrix, Thermo Fisher Scientific, Schlieren, Switzerland) according to the manufacturer’s protocol. Results were analyzed with the Chromosome Analysis Suite (ChAS) software (version 3.1.1.27; Affymetrix, Thermo Fisher Scientific).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7.03 (GraphPad Software, La Jolla, CA, USA).
Author Contributions
D.W., U.S., and J.R. designed the experiments, designed figures, and wrote the manuscript; D.W. made the figures and performed the experiments with help of O.P., F.R., R.S.P., and J.T.; M.J. and J.T. gave advice for the experiments and revised the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by the CGD Society (grant no. CGDS16/01), Hochspezialisierte Medizin Schwerpunkt Immunologie (HSM-2-Immunologie), and the Gottfried und Julia Bangerter-Rhyner-Stiftung. D.W. received a research grant from the University of Zurich (Forschungskredit; FK-17-041). O.P. received a research grant from the University of Zurich (Forschungskredit; FK-17-053). J.R. is funded by the Uniscientia Foundation. J.R. and M.J. are supported by the Clinical Research Priority Program ImmuGene of University of Zurich. M.J. is an International Research Scholar of the Howard Hughes Medical Institute and Vallee Scholar of the Bert N & L Kuggie Vallee Foundation. We thank the Functional Genomics Center Zurich, University of Zurich, and ETH Zurich for performing SMRT sequencing, GUIDE-seq, data analysis, and technical support.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.015.
Supplemental Information
References
- 1.Segal B.H., Leto T.L., Gallin J.I., Malech H.L., Holland S.M. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi M., Niemiec M.J., Siler U., Urban C.F., Reichenbach J. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J. Allergy Clin. Immunol. 2011;127:1243–1252.e7. doi: 10.1016/j.jaci.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi M., Hakkim A., Brinkmann V., Siler U., Seger R.A., Zychlinsky A., Reichenbach J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–2622. doi: 10.1182/blood-2009-05-221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noack D., Rae J., Cross A.R., Ellis B.A., Newburger P.E., Curnutte J.T., Heyworth P.G. Autosomal recessive chronic granulomatous disease caused by defects in NCF-1, the gene encoding the phagocyte p47-phox: mutations not arising in the NCF-1 pseudogenes. Blood. 2001;97:305–311. doi: 10.1182/blood.v97.1.305. [DOI] [PubMed] [Google Scholar]
- 5.Roos D., de Boer M., Kuribayashi F., Meischl C., Weening R.S., Segal A.W., Ahlin A., Nemet K., Hossle J.P., Bernatowska-Matuszkiewicz E., Middleton-Price H. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood. 1996;87:1663–1681. [PubMed] [Google Scholar]
- 6.Görlach A., Lee P.L., Roesler J., Hopkins P.J., Christensen B., Green E.D., Chanock S.J., Curnutte J.T. A p47-phox pseudogene carries the most common mutation causing p47-phox- deficient chronic granulomatous disease. J. Clin. Invest. 1997;100:1907–1918. doi: 10.1172/JCI119721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hockenhull E.L., Carette M.J., Metcalfe K., Donnai D., Read A.P., Tassabehji M. A complete physical contig and partial transcript map of the Williams syndrome critical region. Genomics. 1999;58:138–145. doi: 10.1006/geno.1999.5815. [DOI] [PubMed] [Google Scholar]
- 8.van den Berg J.M., van Koppen E., Ahlin A., Belohradsky B.H., Bernatowska E., Corbeel L., Español T., Fischer A., Kurenko-Deptuch M., Mouy R. Chronic granulomatous disease: the European experience. PLoS ONE. 2009;4:e5234. doi: 10.1371/journal.pone.0005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siler U., Paruzynski A., Holtgreve-Grez H., Kuzmenko E., Koehl U., Renner E.D., Alhan C., de Loosdrecht A.A., Schwäble J., Pfluger T. Successful Combination of Sequential Gene Therapy and Rescue Allo-HSCT in Two Children with X-CGD - Importance of Timing. Curr. Gene Ther. 2015;15:416–427. doi: 10.2174/1566523215666150515145255. [DOI] [PubMed] [Google Scholar]
- 10.Sen K., Ghosh T.C. Pseudogenes and their composers: delving in the ‘debris’ of human genome. Brief. Funct. Genomics. 2013;12:536–547. doi: 10.1093/bfgp/elt026. [DOI] [PubMed] [Google Scholar]
- 11.Bischof J.M., Chiang A.P., Scheetz T.E., Stone E.M., Casavant T.L., Sheffield V.C., Braun T.A. Genome-wide identification of pseudogenes capable of disease-causing gene conversion. Hum. Mutat. 2006;27:545–552. doi: 10.1002/humu.20335. [DOI] [PubMed] [Google Scholar]
- 12.Sweeney C.L., Zou J., Choi U., Merling R.K., Liu A., Bodansky A., Burkett S., Kim J.W., De Ravin S.S., Malech H.L. Targeted Repair of CYBB in X-CGD iPSCs Requires Retention of Intronic Sequences for Expression and Functional Correction. Mol. Ther. 2017;25:321–330. doi: 10.1016/j.ymthe.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klatt D., Cheng E., Philipp F., Selich A., Dahlke J., Schmidt R.E., Schott J.W., Büning H., Hoffmann D., Thrasher A.J., Schambach A. Targeted Repair of p47-CGD in iPSCs by CRISPR/Cas9: Functional Correction without Cleavage in the Highly Homologous Pseudogenes. Stem Cell Reports. 2019;13:590–598. doi: 10.1016/j.stemcr.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou J., Sweeney C.L., Chou B.K., Choi U., Pan J., Wang H., Dowey S.N., Cheng L., Malech H.L. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117:5561–5572. doi: 10.1182/blood-2010-12-328161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreyer A.-K., Hoffmann D., Lachmann N., Ackermann M., Steinemann D., Timm B., Siler U., Reichenbach J., Grez M., Moritz T. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials. 2015;69:191–200. doi: 10.1016/j.biomaterials.2015.07.057. [DOI] [PubMed] [Google Scholar]
- 16.Merling R.K., Sweeney C.L., Chu J., Bodansky A., Choi U., Priel D.L., Kuhns D.B., Wang H., Vasilevsky S., De Ravin S.S. An AAVS1-targeted minigene platform for correction of iPSCs from all five types of chronic granulomatous disease. Mol. Ther. 2015;23:147–157. doi: 10.1038/mt.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Ravin S.S., Reik A., Liu P.Q., Li L., Wu X., Su L., Raley C., Theobald N., Choi U., Song A.H. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 2016;34:424–429. doi: 10.1038/nbt.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Ravin S.S., Li L., Wu X., Choi U., Allen C., Koontz S., Lee J., Theobald-Whiting N., Chu J., Garofalo M. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017;9:eaah3480. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- 19.Merling R.K., Kuhns D.B., Sweeney C.L., Wu X., Burkett S., Chu J., Lee J., Koontz S., Di Pasquale G., Afione S.A. Gene-edited pseudogene resurrection corrects p47phox-deficient chronic granulomatous disease. Blood Adv. 2016;1:270–278. doi: 10.1182/bloodadvances.2016001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sürün D., Schwäble J., Tomasovic A., Ehling R., Stein S., Kurrle N., von Melchner H., Schnütgen F. High Efficiency Gene Correction in Hematopoietic Cells by Donor-Template-Free CRISPR/Cas9 Genome Editing. Mol. Ther. Nucleic Acids. 2018;10:1–8. doi: 10.1016/j.omtn.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wrona D., Siler U., Reichenbach J. CRISPR/Cas9-generated p47phox-deficient cell line for Chronic Granulomatous Disease gene therapy vector development. Sci. Rep. 2017;7:44187. doi: 10.1038/srep44187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wrona D., Siler U., Reichenbach J. Novel Diagnostic Tool for p47 phox -Deficient Chronic Granulomatous Disease Patient and Carrier Detection. Mol. Ther. Methods Clin. Dev. 2019;13:274–278. doi: 10.1016/j.omtm.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brinkman E.K., Kousholt A.N., Harmsen T., Leemans C., Chen T., Jonkers J., van Steensel B. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018;46:e58. doi: 10.1093/nar/gky164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maßberg D., Hatt H. Human Olfactory Receptors: Novel Cellular Functions Outside of the Nose. Physiol. Rev. 2018;98:1739–1763. doi: 10.1152/physrev.00013.2017. [DOI] [PubMed] [Google Scholar]
- 26.Levene M.J., Korlach J., Turner S.W., Foquet M., Craighead H.G., Webb W.W. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 2003;299:682–686. doi: 10.1126/science.1079700. [DOI] [PubMed] [Google Scholar]
- 27.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traxler E.A., Yao Y., Wang Y.D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A., Li C., Weiss M.J. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blasco R.B., Karaca E., Ambrogio C., Cheong T.C., Karayol E., Minero V.G., Voena C., Chiarle R. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 2014;9:1219–1227. doi: 10.1016/j.celrep.2014.10.051. [DOI] [PubMed] [Google Scholar]
- 30.Maddalo D., Manchado E., Concepcion C.P., Bonetti C., Vidigal J.A., Han Y.C., Ogrodowski P., Crippa A., Rekhtman N., de Stanchina E. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long J., Hoban M.D., Cooper A.R., Kaufman M.L., Kuo C.Y., Campo-Fernandez B., Lumaquin D., Hollis R.P., Wang X., Kohn D.B., Romero Z. Characterization of Gene Alterations following Editing of the β-Globin Gene Locus in Hematopoietic Stem/Progenitor Cells. Mol. Ther. 2018;26:468–479. doi: 10.1016/j.ymthe.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunet E., Jasin M. Induction of Chromosomal Translocations with CRISPR-Cas9 and Other Nucleases: Understanding the Repair Mechanisms That Give Rise to Translocations. Adv. Exp. Med. Biol. 2018;1044:15–25. doi: 10.1007/978-981-13-0593-1_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pober B.R. Williams-Beuren syndrome. N. Engl. J. Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 34.Decimi V., Fazio G., Dell’Acqua F., Maitz S., Galbiati M., Rizzari C., Biondi A., Cazzaniga G., Selicorni A. Williams syndrome and mature B-Leukemia: A random association? Eur. J. Med. Genet. 2016;59:634–640. doi: 10.1016/j.ejmg.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Kimura R., Ishii Y., Tomiwa K., Awaya T., Nakata M., Kato T., Okazaki S., Heike T., Hagiwara M. Williams-Beuren Syndrome as a Potential Risk Factor for Burkitt Lymphoma. Front. Genet. 2018;9:368. doi: 10.3389/fgene.2018.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bogdanova N., Markoff A., Gerke V., McCluskey M., Horst J., Dworniczak B. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics. 2001;74:333–341. doi: 10.1006/geno.2001.6568. [DOI] [PubMed] [Google Scholar]
- 37.Horowitz M., Wilder S., Horowitz Z., Reiner O., Gelbart T., Beutler E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. 1989;4:87–96. doi: 10.1016/0888-7543(89)90319-4. [DOI] [PubMed] [Google Scholar]
- 38.Campbell I.G., Jones T.A., Foulkes W.D., Trowsdale J. Folate-binding protein is a marker for ovarian cancer. Cancer Res. 1991;51:5329–5338. [PubMed] [Google Scholar]
- 39.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huwiler A., Pfeilschifter J. Stimulation by extracellular ATP and UTP of the mitogen-activated protein kinase cascade and proliferation of rat renal mesangial cells. Br. J. Pharmacol. 1994;113:1455–1463. doi: 10.1111/j.1476-5381.1994.tb17160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.