

Abstract
Cell migration is essential for proper development and the defense against pathogens. Our previous work detailed a pathway of REversion-inducing-Cysteine-rich protein with Kazal motifs (RECK) isoform-mediated invasion in which a shorter RECK protein competes with MMP9 for interaction with the canonical RECK protein on the cell surface. Here we demonstrate that the mechanism through which RECK isoforms affect cell migration is mediated through changes in the levels of post-translational modifications (PTM) of α-tubulin. We show that both the canonical and short RECK isoforms modulate levels of tubulin acetylation and detyrosination. We demonstrate that these changes are sufficient to modulate the rate of fibroblast migration. If these tubulin PTMs are not altered, the canonical RECK isoform no longer affects cell migration. In defining the molecular pathway linking RECK and tubulin PTMs, we found that MMP9 and integrin activity both act as upstream regulators of tubulin acetylation and detyrosination. Overall, we propose a mechanism in which RECK isoforms on the cell surface have opposing effects on cell migration through MMP9-modulated changes to integrin-extracellular matrix (ECM) interactions that, in turn, affect microtubule PTMs.
Keywords: RECK isoforms, Fibroblast migration, Tubulin PTM, MMP9, Integrin
1. Introduction
The process of cell migration is multifaceted and driven through an array of both intracellular and extracellular rearrangements. Cytoskeletal components are important for the formation of protrusions emanating from the leading edges of migrating cells [1]. In addition to microfilaments, microtubules also affect cell migration. Stabilization of microtubule plus ends is essential for proper fibroblast migration [2]. In response to the generation of a wound in monolayer cultures of fibroblasts or treatment with lysophosphatidic acid (LPA), cells generate an asymmetric microtubule array with stable microtubules mostly oriented toward the wound or LPA treatment [3,4,5,6]. Stable microtubules can accumulate PTMs of α-tubulin including detyrosination (removal of the C-terminal tyrosine to reveal a glutamine) or acetylation on lysine 40, which faces the lumen [7]. Tyrosination is catalyzed by tubulin-tyrosine ligase (TTL), while detyrosination [8] was recently discovered to be performed by vasohibins [9]. Acetylation is performed by αTAT1 [10] and is removed by histone deacetylase 6 (HDAC6) [11] or sirtuin 2 [12].
RECK is a membrane-bound, GPI-anchored glycoprotein that has been extensively studied as a direct regulator of matrix metalloproteinases (MMPs) that control cell migration [13]. RECK can directly bind MMPs to reduce MMP activity, change levels of ECM proteins, and alter cell motility. Morioka and colleagues found that RECK-null mouse embryonic fibroblasts (MEFs) have lower levels of detyrosinated tubulin [14]; however, this finding has not been pursued further.
In our previous study, we showed that RECK isoforms can have opposing roles in fibroblast invasion, with the longer RECK isoform (long RECK) inhibiting cell invasion and the shorter RECK isoform (short RECK) enhancing cell invasion [15]. We discovered that short RECK interacts with long RECK on the cell surface and interferes with MMP9–long RECK interaction, thus liberating MMP9. In our previous study, invasion was modeled by coating cells with Matrigel to simulate the complex ECM found in vivo. Here we address how RECK isoforms modulate cell migration and report our findings that RECK mediates these effects through tubulin PTMs.
2. Materials and methods
2.1. Cell culture
Primary human fibroblast cell lines were established using samples of human foreskin from the National Disease Research Interchange as previously described [16]. BJ5ta (immortalized fibroblasts) and 293T cells were purchased from America Type Culture Collection (ATCC). All cell lines were stored in a 5% CO2-controlled humid incubator at 37°C and were cultured on tissue culture plates in 10% FBS in DMEM. The above protocols were subject to approval by the University of California, Los Angeles IRB (protocol number 14–000145).
2.2. Migration assay
Eighteen hours prior to experimentation, cells were seeded into individual wells of an IncuCyte® ImageLock 96-well microplate (Essen Bioscience) at a concentration of 25,000 cells/well and were stored in a 37°C incubator. The following day, the IncuCyte® WoundMaker™ (Essen Bioscience) was used to generate a 700–800 μm width scratch in the center of each well. The wells were then filled with 100 microliters of complete medium. Repopulation of the denuded area was tracked in 2 hour intervals for up to 96 hours using the IncuCyte ZOOM® live imaging software. Relative would density was calculated as cell density in the wound area relative to cell density outside of the wound area over time. More details can be found on the manufacturer’s website (https://www.essenbioscience.com/en/applications/live-cell-assays/scratch-wound-cell-migration-invasion/).
2.3. Real-time RT-PCR
Fifty nanograms of RNA were reverse transcribed and amplified in a total volume of 25 μl per reaction using SuperScript® III Platinum® SYBR® Green One-Step qRT-PCR Kit (Invitrogen). The CFX96 Real-time system (Bio-Rad) was used for real-time RT PCR, and ubiquitin-C (UBC) levels were referenced for normalization of gene expression. We calculated gene expression using the 2−ΔΔCt method. Primer sequences are as follows: αTAT1, 5’-AACTCGACACTCTCGTGCTG-3’ and 5’-CTGTTTAGGGGCCAAGGAGG-3’; TTL 5’-GGAGAGGGCAACGTTTGGAT-3’ and 5’-CAAGACCCAGCTTCGGATGT-3’.
2.4. Western blot
Prior to the western blot, proteins were extracted from harvested fibroblasts using radioimmunoprecipitation assay (RIPA) lysis buffer (10mM Tris-Cl pH8.0, 0.1% sodium deoxycholate, 0.5mM EGTA, 1mM EDTA, 0.1% SDS, 1% Triton X-100, 140mM NaCl) that also contained a single tablet of protease inhibitor (Roche Applied Science). Cell lysates containing all proteins were analyzed using Thermo Scientific™ Pierce™ BCA™ Protein Assay kit (ThermoFischer). Twenty μgs of protein was loaded per lane for western blots. Each western blot was repeated two or three times.
2.5. Antibodies
To detect alpha-tubulin and Flag, antibodies were purchased from Sigma-Aldrich. For detection of MMP9 and long RECK, antibodies were obtained from Thermo Fisher Scientific. Antibodies against acetyl-tubulin and Glu-tubulin were purchased from Cell Signaling Technology and Millipore, respectively. All primary antibodies were diluted in 5% skim milk in 0.1% Tween-TBS and the dilution rates were as follows. Alpha-tubulin: 1:5,000; Flag: 1:2,500; MMP9: 1:1,000; long RECK: 1:1,000; acetyl-tubulin: 1:1,000; Glu-tubulin: 1:1,000.
2.6. shRNA cell line engineering
shRNAs were designed through https://rnaidesigner.thermofisher.com/rnaiexpress. Knockdown shRNAs were cloned into the lentiviral plasmid pGreenPuro (Systems Biosciences) at BamHI and EcoRI sites, and the following sequences were expressed under the control of the H1 promoter. Sequences are provided as below:
shControl.1: 5’-GATCCCCTAAGGTTAAGTCGCCCTCGTTCAAGAGACGAGGGCGACTTAACCTTAGGTTTTTG-3’ and 5’-AATTCAAAAACCTAAGGTTAAGTCGCCCTCGTCTCTTGAACGAGGGCGACTTAACCTTAGGG-3’;
shControl.2: 5’-GATCCCAACAAGATGAAGAGCACCAACTTCCTGTCAGATTGGTGCTCTTCATCTTGTTGTTTTTG-3’ and 5’-AATTCAAAAACAACAAGATGAAGAGCACCAATCTGACAGGAAGTTGGTGCTCTTCATCTTGTTGG-3’;
shShortRECK.1: 5’-GATCCGCTTTGTACATCCTGGAATCCCTTCCTGTCAGAGGATTCCAGGATGTACAAAGCTTTTTG-3’ and 5’-AATTCAAAAAGCTTTGTACATCCTGGAATCCTCTGACAGGAAGGGATTCCAGGATGTACAAAGCG-3’;
shShortRECK.2: 5’-GATCCGATCTGTGCTCTCTGACATTTCTTCCTGTCAGAAAATGTCAGAGAGCACAGATCTTTTTG-3’ and 5’-AATTCAAAAAGATCTGTGCTCTCTGACATTTTCTGACAGGAAGAAATGTCAGAGAGCACAGATCG-3’;
shLongRECK.1: 5’-GATCCGCGTGGCAGTCGATTACTATGTTCAAGAGACATAGTAATCGACTGCCACGCTTTTTG-3’ and 5’-AATTCAAAAAGCGTGGCAGTCGATTACTATGTCTCTTGAACATAGTAATCGACTGCCACGCG-3’;
shLongRECK.2: 5’-GATCCGCATCAAAGCATCTTGCATTACTTCCTGTCAGATAATGCAAGATGCTTTGATGCTTTTTG-3’ and 5’-AATTCAAAAAGCATCAAAGCATCTTGCATTATCTGACAGGAAGTAATGCAAGATGCTTTGATGCG-3’;
shαTAT1.1: 5’-GATCCGGATGATCGTGAGGCTCATAACTTCCTGTCAGATTATGAGCCTCACGATCATCCTTTTTG-3’ and 5’-AATTCAAAAAGGATGATCGTGAGGCTCATAATCTGACAGGAAGTTATGAGCCTCACGATCATCCG-3’;
shαTAT1.2: 5’-GATCCGCGAGAACTCTTCCAGTATATCTTCCTGTCAGAATATACTGGAAGAGTTCTCGCTTTTTG-3’ and 5’-AATTCAAAAAGCGAGAACTCTTCCAGTATATTCTGACAGGAAGATATACTGGAAGAGTTCTCGCG-3’;
shTTL: 5’-GATCCGCTTCAGAGCTTCTCGATTTCCTTCCTGTCAGAGAAATCGAGAAGCTCTGAAGCTTTTTG-3’ and 5’-AATTCAAAAAGCTTCAGAGCTTCTCGATTTCTCTGACAGGAAGGAAATCGAGAAGCTCTGAAGCG-3’.
Lentivirus concentrations were tested by flow cytometry analysis using 293T cells. Virus titrated to a multiplicity of infection (M.O.I.) of 1 was used to treat fibroblasts with polybrene (final concentration: 10 μg/ml) and infected cells were selected using 2μg/ml puromycin (Invivogen) in 10% FBS DMEM for 3 days.
2.7. Inhibitor treatment
All inhibitors were incubated with cultured cells at 37°C for 24 hours. The manufacturers and final concentrations are as follows: SB-3CT (iMMP2/iMMP9) (Santa Cruz Biotechnology) 13.9nM; MMP-9 Inhibitor I (iMMP9) (Santa Cruz Biotechnology) 5nM; Cilengitide (ApexBio) 37nM; RGD (Arg-Gly-Asp) peptides (ApexBio) 30μM; Cyclo(-RGDfK) (ApexBio) 50μM; Firategrast (ApexBio) 50μl.
2.8. Statistical and image analysis
Excel 2013 and GraphPad Prism 5 were used to analyze all results. Data are shown as mean ± S.D. or S.E. Significance was determined by repeated measures two-way ANOVA with Dunnett’s multiple comparison test or unpaired two-sided t-test. P<0.05 was considered significant. Image J was used to quantify western blots.
3. Results & Discussion
3.1. Long RECK & short RECK isoforms modulate fibroblast migration.
We wondered whether short RECK could regulate cell migration beyond ECM digestion. To test this idea, we monitored the dynamics of fibroblast migration using the IncuCyte® WoundMaker™ in the absence of Matrigel (Fig. 1A). We investigated fibroblasts stably expressing shRNAs that target either long RECK or short RECK. Isoform-specific knockdown of either short or long RECK in these fibroblasts was demonstrated in our previous report [15]. Long RECK knockdown increased cell migration, while short RECK knockdown decreased cell migration (Fig. 1B and C). These results were similar to those observed with the invasion assay [15]. Taken together, we have found that long RECK promotes both fibroblast invasion and migration, while short RECK inhibits both invasion and migration. Without Matrigel, the fibroblasts in all states migrated more quickly than when Matrigel was present (compare Fig. 1A and B with Lee et al, 2018, Fig. 2C and D), consistent with reduced ECM without Matrigel treatment. Because we found that RECK can affect cell migration even without Matrigel present, the results suggested to us that RECK may have additional effects on cell migration besides ECM degradation.
Fig. 1. RECK isoforms differentially regulate cell migration and tubulin PTM levels.
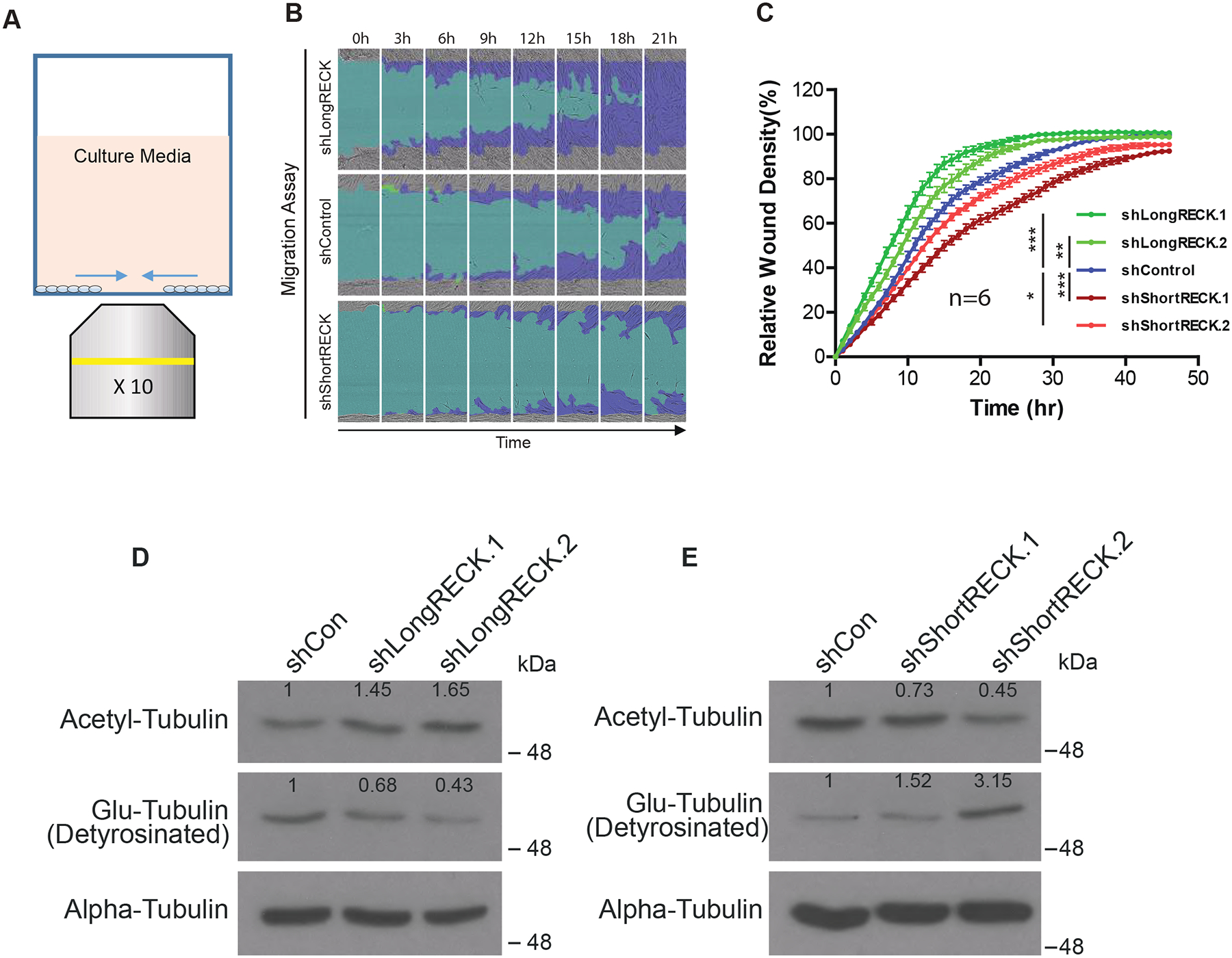
(A) Schematic representing in vitro cell migration assay. (B) Representative time-lapse images of fibroblast migration as a result of RECK isoform-specific shRNA knockdown. The light blue region depicts the initial wound created by IncuCyte WoundMaker, while the dark blue region depicts cell movement over a span of 24 hours. shLongRECK-expressing cells migrated more rapidly, while fibroblasts expressing shShortRECK migrated more slowly. (C) Quantification of the migration assay as relative wound density (100 * density in wound/density outside wound) over time. Data is shown as mean ± S.E. with 6 replicates. Significance was calculated using a repeated measures two-way ANOVA with Dunnett’s multiple comparison test (simplified as ANOVA). One asterisk indicates p < 0.05, two asterisks indicate p < 0.01, three asterisks indicates p < 0.001. Three independent experiments were performed with similar results, and data from one experiment is presented. (D and E) RECK isoforms can modulate levels of tubulin PTMs. Western blotting reveals long RECK knockdown increases acetyl-tubulin levels and decreases Glu-tubulin levels; conversely, short RECK knockdown decreases levels of acetylated tubulin and increases tubulin detyrosination. Two independent experiments showed similar results, and data from one experiment is shown. Two types of shRNAs that target different regions of the same RECK isoform showed similar trends. The stable fibroblast cell lines with RECK isoform knockdown used here are the same as those that we characterized in our previous report [15]. Numbers on the western blots indicate relative protein expression level compared to control.
Fig 2. Tubulin PTMs can regulate cell migration.
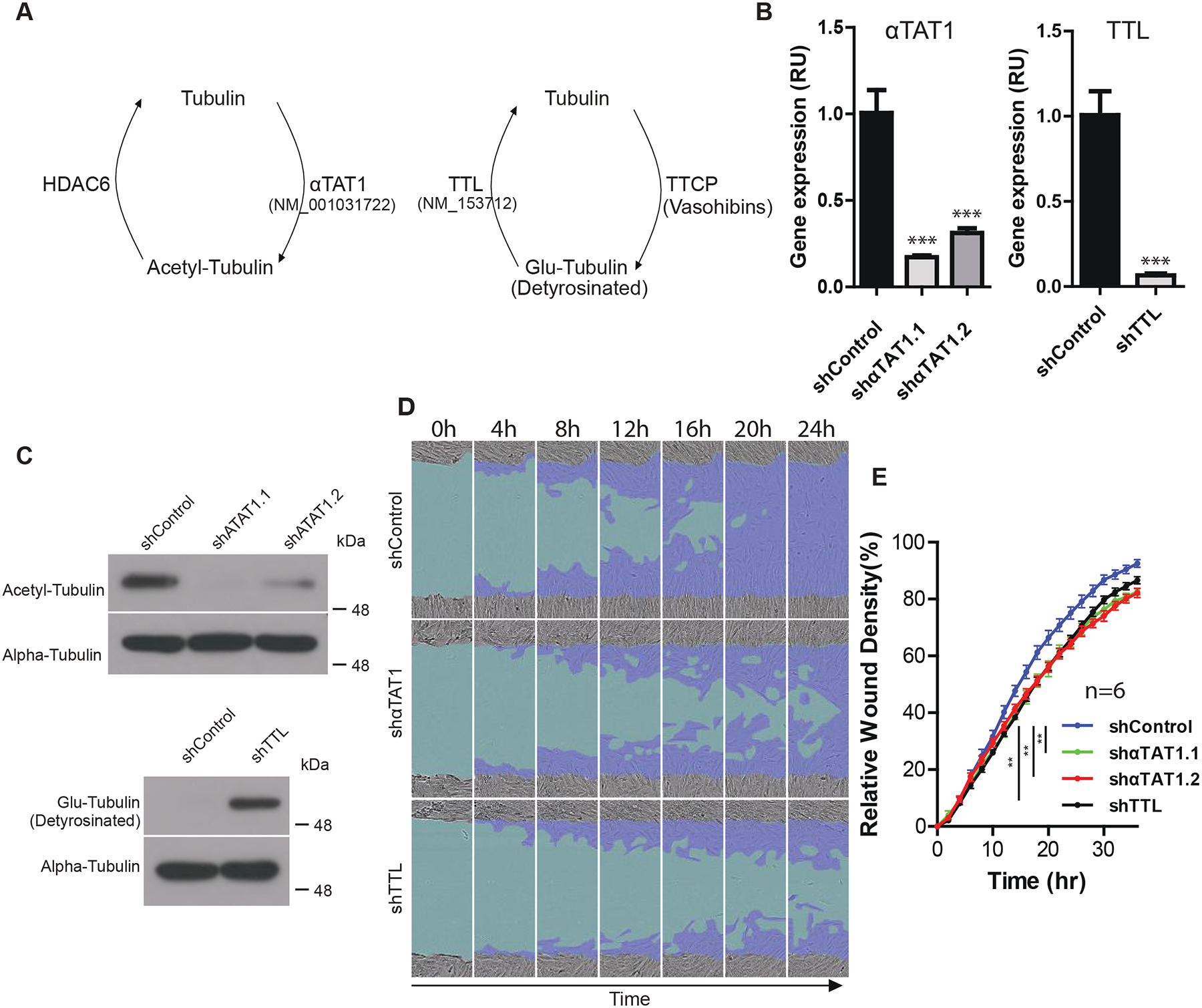
(A) Diagram depicting cycles of tubulin PTMs. (B) Gene expression following αTAT1 and TTL shRNA knockdown in immortalized fibroblasts measured with real-time RT-PCR. Gene expression was normalized to UBC and then the relative fold change compared to the control shRNA was calculated. Data shown as mean ± S.D. Three asterisks indicates p < 0.001 (unpaired two-sided t-test). (C) Levels of acetylated tubulin and Glu-tubulin as a result of αTAT1 and TTL knockdown, respectively. (D) Representative images of decreased fibroblast migration over a timespan of 24 hours as a result of reduction in tubulin acetylation and increased Glu-tubulin. Light blue region depicts the initial wound created by IncuCyte WoundMaker, while dark blue region depicts cell movement over a span of 24 hours. (E) Quantification of migration assay. Data are shown as mean ± S.E. with 6 replicates. Two asterisks indicate p < 0.01 based on ANOVA analysis. Three independent experiments showed similar results. Data from one experiment is shown.
3.2. RECK isoforms influence levels of tubulin PTMs.
We sought to determine whether RECK affects intracellular factors that mediate cell migration. Previous studies showed that RECK knockout cell lines have lower levels of detyrosinated (Glu) tubulin [14]. In addition, metastatic and aggressive breast cancers contain high levels of acetylated α-tubulin [17]. We thus sought to elucidate the role of RECK isoforms in regulating tubulin PTMs as a possible mechanism for RECK’s effects on cell migration. To begin our analysis, we knocked down either long RECK or short RECK with shRNAs and monitored the levels of acetylated tubulin or Glu-tubulin. Knockdown of long RECK resulted in increased levels of acetyl-tubulin and decreased levels of Glu-tubulin, while knocking down short RECK resulted in decreased levels of acetyl-tubulin and increased levels of Glu-tubulin (Fig. 1D and E). These results show that short and long RECK isoforms have opposing effects on tubulin PTMs that could potentially contribute to cytoskeletal organization through tubulin flexibility and stability. The findings are also somewhat surprising because in previous studies, both acetylated tubulin and Glu-tubulin were associated with stable microtubules [9,18], while in our experiments, the acetylated tubulin and Glu-tubulin levels moved in opposite directions with either short or long RECK knockdown.
3.3. Tubulin acetylation and detyrosination decrease fibroblast migration.
Having demonstrated that both short and long RECK affect the levels of the acetylated and Glu-forms of tubulin, we next investigated the effect of these tubulin modifications on fibroblast motility. We modulated the levels of acetylated or Glu-tubulin forms by designing shRNAs against αTAT1 or TTL to decrease levels of acetylation and increase levels of Glu-tubulin, respectively (Fig. 2A). Knockdown of αTAT1 and TTL enzymes was confirmed through real-time RT-PCR (Fig. 2B). As expected, αTAT1 knockdown decreased levels of acetyl-tubulin, while TTL knockdown increased levels of Glu-tubulin (Fig. 2C). Neither αTAT1 nor TTL knockdown affected total tubulin levels (Fig. 2C). Fibroblasts transduced with either αTAT1-targeted or TTL-targeted shRNAs exhibited decreased cell migration (Fig. 2D and E). The findings show that altering tubulin PTMs can affect fibroblast cell migration. Furthermore, the directionality of the effects of these tubulin PTMs is consistent with tubulin PTMs mediating the effects of RECK isoforms on cell migration. Because long RECK knockdown results in faster migration, increased acetyl-tubulin and decreased Glu-tubulin due to long RECK knockdown would also be predicted to increase migration. Likewise, because short RECK knockdown resulted in slower migration, the decreased acetyl-tubulin and increased Glu-tubulin that follow short RECK knockdown would be predicted to decrease fibroblasts migration.
3.4. Knockdown of tubulin-modifying enzymes prevents increased fibroblast migration following long RECK knockdown.
We monitored the effect of shRNA-mediated knockdown of long RECK and tubulin-modifying enzymes on fibroblast migration. We characterized levels of acetyl-tubulin and Glu-tubulin after shRNA-mediated knockdown of long RECK, αTAT1, and TTL. When compared to fibroblasts only expressing an shRNA against long RECK, fibroblasts expressing shRNAs to long RECK, αTAT1, and TTL saw a reversal in tubulin acetylation and detyrosination levels (Fig. 3A). We then tested whether reversing tubulin PTMs while knocking down long RECK would revert the migration-promoting effects of long RECK knockdown. Indeed, when long RECK was knocked down along with αTAT1 and TTL, the cells migrated similarly to the rate in cells without RECK knockdown (Fig. 3B and C). These results are consistent with tubulin PTMs playing a role in the change in migration rate mediated by RECK. To further explore this possibility, we monitored the migration of fibroblasts with knockdown of RECK or a control shRNA and knockdown of αTAT1 and TTL or control shRNAs. While fibroblasts without manipulation of tubulin PTMs migrated more rapidly when long RECK was knocked down, fibroblasts with a reversal of tubulin PTMs did not migrate more rapidly when long RECK was knocked down (Fig. 3D and E). We concluded that modulation of tubulin PTMs is important for the effects of long RECK on fibroblast migration.
Fig. 3. Knockdown of αTAT1 and TTL reverts faster migration with long RECK knockdown.
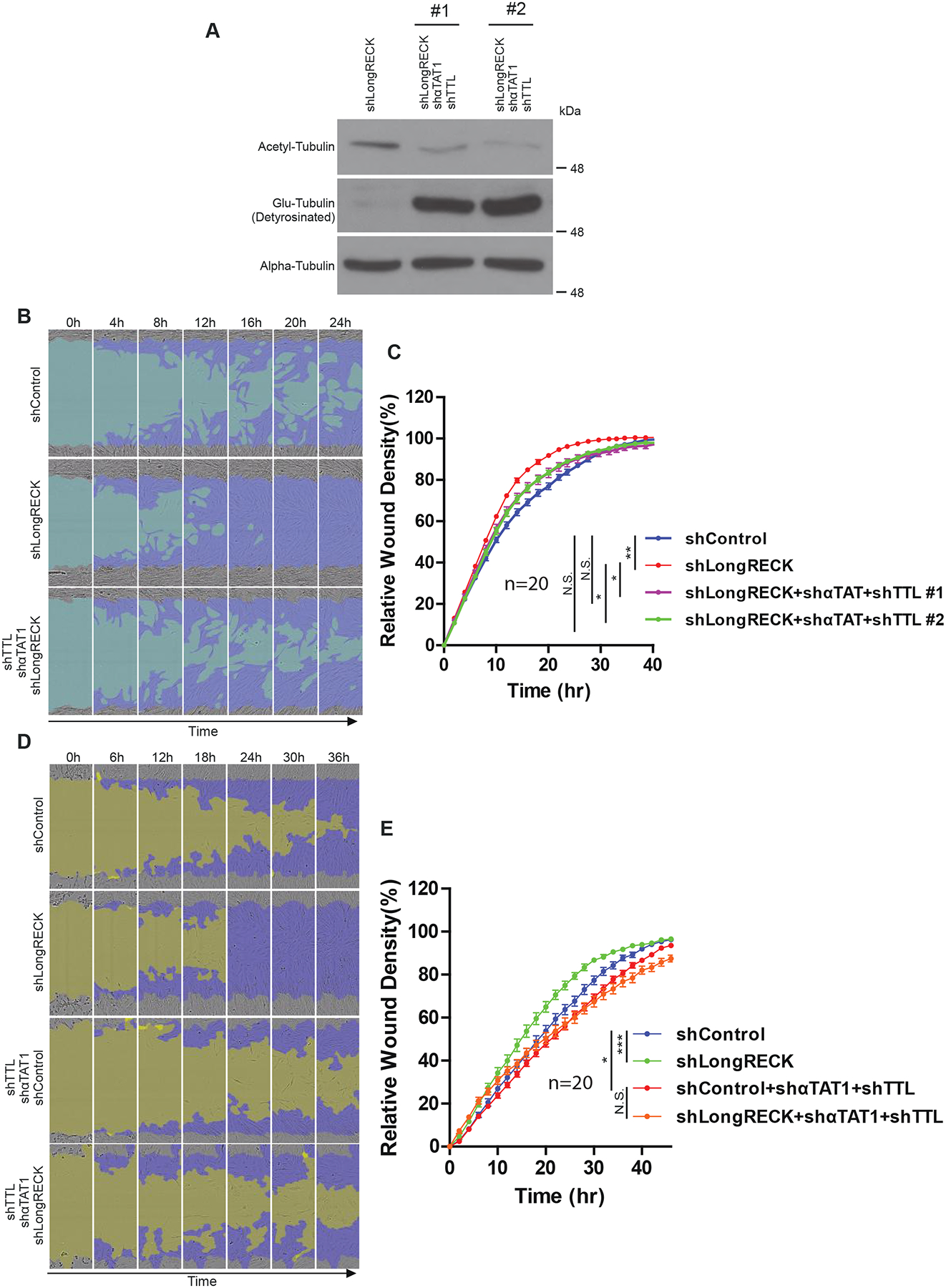
(A) Western blot of acetyl-tubulin and Glu-tubulin levels following shRNA knockdown of either the long RECK isoform alone or both long RECK and tubulin-modifying enzymes in immortalized fibroblasts. (B) Representative images of fibroblast migration after transfection with shRNAs targeting either long RECK or long RECK, αTAT1, and TTL. The light blue region depicts the initial wound created by IncuCyte WoundMaker, while the dark blue region depicts cell movement over a span of 24 hours. (C) Quantification of migration assay. Data are shown as mean ± S.E. with 20 replicates. Cells only targeted for long RECK knockdown migrate further than control cells, as expected; however, knockdown of tubulin-modifying enzymes reverts this phenotype. Two independent experiments showed similar results. Data from one experiment is shown. (D) Representative images of fibroblast migration. Cells expressing the indicated combinations of shRNAs were monitored for migration rate. The dark yellow region depicts the initial wound created by IncuCyte WoundMaker, while the dark blue region resembles cell movement over a span of 36 hours. (E) Quantification of migration assay. Data are shown as mean ± S.E. with 20 replicates. Two independent experiments were performed with similar results. Data from one experiment is presented. One asterisk indicates p < 0.05, two asterisks indicate p < 0.01, three asterisks indicates p < 0.001 based on ANOVA analysis.
3.5. MMP9 and integrin activation affect acetyl-tubulin but not Glu-tubulin levels.
Knockdown of long RECK promotes ECM degradation by MMP9, which would be expected to clear a path for cells to move [15,19]. We hypothesized that RECK’s effects on MMP9 might also contribute to changes in migration through tubulin PTMs. To test this hypothesis, we asked whether MMP9 activity can affect tubulin acetylation or detyrosination. 293T cells or fibroblasts were treated with either an MMP9 inhibitor or an inhibitor of MMP2/9, and the levels of tubulin PTMs were monitored. Treatment with any of the inhibitors resulted in decreased levels of acetyl-tubulin, with no effect on the levels of Glu-tubulin (Fig. 4A and B). The results show that MMP2/9 activity can affect tubulin acetylation but not detyrosination.
Fig. 4: MMP9 and the integrin signaling pathway regulate levels of acetyl-tubulin.
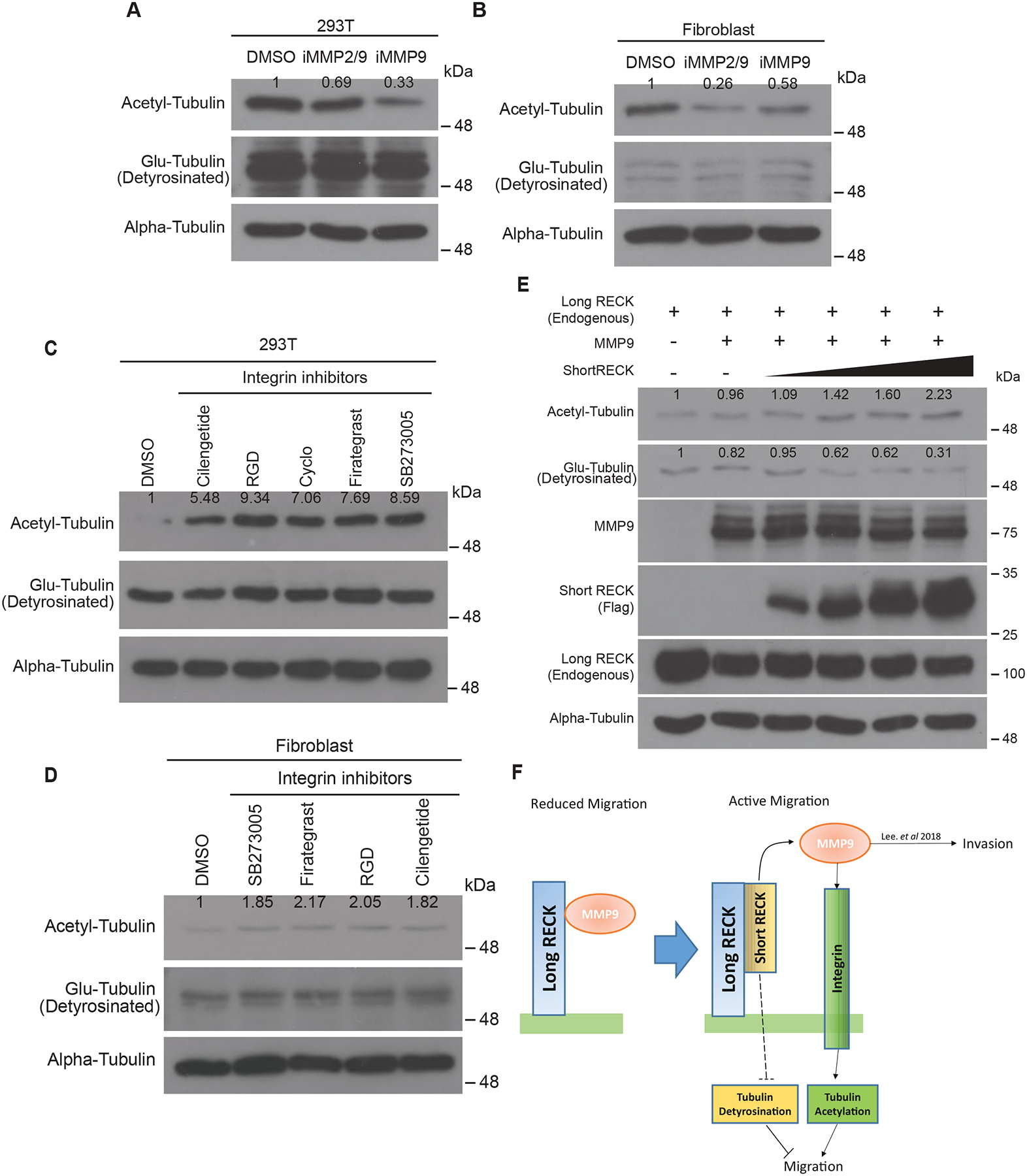
(A and B) Western blots demonstrating acetylated tubulin levels, but not Glu-tubulin levels, are regulated by treating fibroblasts with the indicated MMP inhibitors. Two different cell lines show similar results. (C and D) Levels of acetyl-tubulin increase after treatment with integrin inhibitors. Acetyl-tubulin levels increase in the presence of integrin inhibitors in both 293T and fibroblasts. Glu-tubulin levels are constant and are not affected by treatment with any of these integrin inhibitors. (E) 293T cells overexpressing MMP9 were transfected with increasing levels of short RECK to release MMP9. Effects on tubulin acetylation and detyrosination were monitored with immunoblotting. As short RECK levels increased, acetyl-tubulin levels increased and Glu-tubulin levels decreased. Two independent experiments showed similar results. (F) Working model. In reduced cell migration conditions, the long RECK isoform interacts with MMP9 and suppresses MMP9 activity to inhibit cell migration. In contrast, when short RECK is generated under conditions of active cell migration, MMP9 liberated from long RECK by short RECK can affect tubulin acetylation levels, possibly through modulation of integrin activity. In addition, short RECK expression can also reduce tubulin detyrosination. Fibroblast migration is enhanced by tubulin PTMs modified by short RECK. Dashed lines show a pathway yet to be elucidated. Numbers on the western blots indicate relative protein expression level compared to control.
To further understand how MMP9 inhibition might affect tubulin PTMs, we considered integrin signaling. MMPs are expected to affect the amount and quality of ECM proteins, and these proteins signal through integrins [20]. We treated 293T cells or fibroblasts with integrin-specific inhibitors and monitored the levels of acetyl-tubulin and Glu-tubulin. Each of the different integrin inhibitors individually increased levels of acetylated tubulin but did not change levels of Glu-tubulin (Fig. 4C and D). These results are consistent with a model in which inhibiting MMP9 results in higher levels of ECM proteins, more integrin signaling, and reduced acetylated tubulin. In Fig. 1, we observed that RECK isoforms can regulate both acetylated tubulin and Glu-tubulin levels, but neither MMP9 nor integrin inhibitors affect Glu-tubulin levels. Thus, we hypothesize that in addition to MMP9 and integrins, there are also other pathways through which RECK isoforms regulate Glu-tubulin independent of MMP9-integrin pathways.
3.6. Short and long RECK have opposing effects on tubulin PTMs.
In our previous study, we observed that increased expression levels of short RECK disrupted MMP9 binding to long RECK on the cell surface as a result of short RECK binding to long RECK and releasing MMP9 from long RECK [15]. To further understand the effects of short RECK on tubulin PTMs, we tested for changes in tubulin acetylation and Glu-tubulin levels when short RECK protein is induced. We transfected 293T cells with increasing concentrations of short RECK while also overexpressing MMP9 to induce MMP9 liberation from long RECK by short RECK. Increased short RECK resulted in an increase in tubulin acetylation and a decrease in tubulin detyrosination (Fig. 4E). While the changes in tubulin acetylation could result from its effects on MMP9, the changes in Glu-tubulin are expected to reflect additional roles of the short RECK. Further studies will be needed to clarify the pathway that results in changes in Glu-tubulin levels in response to RECK isoforms and the connection between integrins and tubulin acetylation state. Taken together, we propose a novel model of isoform-specific RECK-mediated migration through tubulin PTM modulation. We hypothesize that MMP9 released from long RECK by competition with short RECK digests ECM proteins and reduces integrin-ECM interaction. Changes in integrin signaling and other pathways can then modulate tubulin PTMs, which will, in turn, affect fibroblast migration rates (Fig. 4F).
Highlights.
RECK isoforms differentially regulate tubulin post-translational modifications
Fibroblast migration can be regulated by tubulin post-translational modifications
RECK affects cell migration through tubulin post-translational modifications
MMP9 liberated by short RECK expression can affect tubulin acetylation
Acknowledgments
We thank the Eli & Edythe Broad Center for Regenerative Medicine & Stem Cell Research for Core Facilities that house and Incucyte instrument. We acknowledge Jeff Long (UCLA) for sharing equipment for real-time RT-PCR. HAC was the Milton E. Cassel scholar of the Rita Allen Foundation (http://www.ritaallenfoundation.org). HNL acknowledges a Whitcome Fellowship from the UCLA Molecular Biology Institute and a dissertation year fellowship from the Graduate Program, UCLA. HAC acknowledges funding from the National Institute of General Medical Sciences R01 GM081686, National Institute of General Medical Sciences R01 GM0866465, the Eli & Edythe Broad Center for Regenerative Medicine & Stem Cell Research, the Iris Cantor Women’s Health Center, and the UCLA Life Science Innovation Fund, The authors thank Luisa Iruela-Arispe, Tracy Johnson, William Lowry, Jorge Torres, and Jose Rodriguez for helpful input.
Footnotes
Competing interests
The authors declare no competing or financial interests.
References
- [1].Tang DD, Gerlach BD, The roles and regulation of the actin cytoskeleton, intermediate filaments and microtubules in smooth muscle cell migration, Respiratory research 18 (2017) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG, EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration, Nature cell biology 6 (2004) 820–830. [DOI] [PubMed] [Google Scholar]
- [3].Gundersen GG, Bulinski JC, Selective stabilization of microtubules oriented toward the direction of cell migration, Proc Natl Acad Sci U S A 85 (1988) 5946–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gundersen GG, Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Gomes ER, Regulation of microtubules by Rho GTPases in migrating cells, Novartis Found Symp 269 (2005) 106–116; discussion 116–126, 223–130. [PubMed] [Google Scholar]
- [5].Gundersen GG, Kim I, Chapin CJ, Induction of stable microtubules in 3T3 fibroblasts by TGF-beta and serum, J Cell Sci 107 (Pt 3) (1994) 645–659. [DOI] [PubMed] [Google Scholar]
- [6].Cook TA, Nagasaki T, Gundersen GG, Rho guanosine triphosphatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid, J Cell Biol 141 (1998) 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Janke C, Bulinski JC, Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions, Nat Rev Mol Cell Biol 12 (2011) 773–786. [DOI] [PubMed] [Google Scholar]
- [8].Ersfeld K, Wehland J, Plessmann U, Dodemont H, Gerke V, Weber K, Characterization of the tubulin-tyrosine ligase, The Journal of cell biology 120 (1993) 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nieuwenhuis J, Adamopoulos A, Bleijerveld OB, Mazouzi A, Stickel E, Celie P, Altelaar M, Knipscheer P, Perrakis A, Blomen VA, Brummelkamp TR, Vasohibins encode tubulin detyrosinating activity, Science 358 (2017) 1453–1456. [DOI] [PubMed] [Google Scholar]
- [10].Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J, MEC-17 is an alpha-tubulin acetyltransferase, Nature 467 (2010) 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP, HDAC6 is a microtubule-associated deacetylase, Nature 417 (2002) 455–458. [DOI] [PubMed] [Google Scholar]
- [12].North BJ, Marshall BL, Borra MT, Denu JM, Verdin E, The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase, Molecular Cell 11 (2003) 437–444. [DOI] [PubMed] [Google Scholar]
- [13].Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, Ide C, Horan TP, Arakawa T, Yoshida H, Nishikawa S, Itoh Y, Seiki M, Itohara S, Takahashi C, Noda M, The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis, Cell 107 (2001) 789–800. [DOI] [PubMed] [Google Scholar]
- [14].Morioka Y, Monypenny J, Matsuzaki T, Shi S, Alexander DB, Kitayama H, Noda M, The membrane-anchored metalloproteinase regulator RECK stabilizes focal adhesions and anterior-posterior polarity in fibroblasts, Oncogene 28 (2009) 1454–1464. [DOI] [PubMed] [Google Scholar]
- [15].Lee HN, Mitra M, Bosompra O, Corney DC, Johnson EL, Rashed N, Ho LD, Coller HA, RECK isoforms have opposing effects on cell migration, Molecular biology of the cell 29 (2018) 1825–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Legesse-Miller A, Elemento O, Pfau SJ, Forman JJ, Tavazoie S, Coller HA, let-7 Overexpression leads to an increased fraction of cells in G2/M, direct down-regulation of Cdc34, and stabilization of Wee1 kinase in primary fibroblasts, The Journal of biological chemistry 284 (2009) 6605–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boggs AE, Vitolo MI, Whipple RA, Charpentier MS, Goloubeva OG, Ioffe OB, Tuttle KC, Slovic J, Lu Y, Mills GB, Martin SS, alpha-Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration, Cancer Res 75 (2015) 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Janke C, Montagnac G, Causes and Consequences of Microtubule Acetylation, Current biology : CB 27 (2017) R1287–R1292. [DOI] [PubMed] [Google Scholar]
- [19].Takahashi C, Sheng ZQ, Horan TP, Kitayama H, Maki M, Hitomi K, Kitaura Y, Takai S, Sasahara RM, Horimoto A, Ikawa Y, Ratzkin BJ, Arakawa T, Noda M, Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK, Proceedings of the National Academy of Sciences of the United States of America 95 (1998) 13221–13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stefanidakis M, Koivunen E, Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression, Blood 108 (2006) 1441–1450. [DOI] [PubMed] [Google Scholar]