

Abstract
Arginyltransferase1 (ATE1) is a conserved enzyme in eukaryotes mediating posttranslational arginylation, the addition of an extra arginine to an existing protein. In mammals, the dysregulations of the ATE1 gene (ate1) is shown to be involved in cardiovascular abnormalities, cancer, and aging-related diseases. Although biochemical evidence suggested that arginylation may be involved in stress response and/or protein degradation, the physiological role of ATE1 in vivo has never been systematically determined. This gap of knowledge leads to difficulties for interpreting the involvements of ATE1 in diseases pathogenesis. Since ate1 is highly conserved between human and the unicellular organism Schizosaccharomyces pombe (S. pombe), we take advantage of the gene-knockout library of S. pombe, to investigate the genetic interactions between ate1 and other genes in a systematic and unbiased manner. By this approach, we found that ate1 has a surprisingly small and focused impact size. Among the 3659 tested genes, which covers nearly 75% of the genome of S. pombe, less than 5% of them displayed significant genetic interactions with ate1. Furthermore, these ate1-interacting partners can be grouped into a few discrete clustered categories based on their functions or their physical interactions. These categories include translation/transcription regulation, biosynthesis/metabolism of biomolecules (including histidine), cell morphology and cellular dynamics, response to oxidative or metabolic stress, ribosomal structure and function, and mitochondrial function. Unexpectedly, inconsistent to popular belief, very few genes in the global ubiquitination or degradation pathways showed interactions with ate1. Our results suggested that ATE1 specifically regulates a handful of cellular processes in vivo, which will provide critical mechanistic leads for studying the involvements of ATE1 in normal physiologies as well as in diseased conditions.
Keywords: posttranslational modification, arginylation, arginyltransferase1, double-knockout screening, genetic interactions
Introduction
Protein posttranslational modifications (PTM) change protein properties without requiring de novo synthesis. Thus, PTMs are frequently relied upon to respond to acute stress or cellular signaling, and are often used to activate lateral response factors such as transcription or epigenetic modulations. For these reasons, dysregulation of PTMs are often indicated in cardiovascular diseases where stress response or cellular signaling play critical roles.
In eukaryotes, many proteins appear to be subjected to N-terminal arginylation (referred to as arginylation in this proposal), a ribosome-independent addition of one extra arginine on the N-terminus of a protein. Arginylation is catalyzed by the family of arginyltransferase (ATE). While plants contain ATE1 and ATE2, fungi and metazoans only contain ATE1, which is highly conserved across different species. Multiple lines of genetic studies have shown an important role of the ATE1 gene (ate1) in the cardiovascular and/or metabolic diseases in animals. For example, a genomic deletion of ATE1 was found to result in complete embryonic lethality in mice during the mid-gestation stage (E9-14) with severe defects in angiogenetic remodeling and cardiac development (Kwon et al., 2002). Moreover, organ/tissue-specific deletions of ATE1 in animals were found to cause a variety of abnormalities in morphology and function. Examples include a progressive dilated cardiomyopathy in mice when ate1 knockout is driven by the cardiac myosin heavy chain promoter (Kurosaka et al., 2010; Kaji and Kaji, 2012; Wang et al., 2017b). Also, inducible systematic deletion of ate1 appears to cause rapid weight loss, damaged spermatogenesis, neurological perturbations, and early lethality in adult mice (Brower and Varshavsky, 2009). In addition to these involvement in cardiovascular and metabolic abnormalities, a dysregulation of ATE1 is indicated in cancer as well. For example, reports from us and other groups showed that ATE1 is often downregulated in high grade cancer cases and is associated with poorer outcomes (Rai et al., 2015; Birnbaum et al., 2019), and that an inhibition of ATE1-mediated arginylation confers cancer cell resistance to apoptosis-induced by radiation (Masdehors et al., 2000). Furthermore, mounting evidence is also starting to indicate the involvement of ATE1 in aging-related conditions (Brower et al., 2013; Wang et al., 2017a). Unfortunately, the physiological role of ATE1 (and its arginylation activity) remains poorly understood, which adds to the difficulty of interpreting its involvements in normal conditions or diseases.
The studies about ATE1 and arginylation are still relatively scarce. Our understandings for these topics are being continuously reshaped with emerging new evidence. Arginylation has been found to take place on nearly a hundred eukaryotic proteins and the list is still expanding on a daily basis (Decca et al., 2006; Wong et al., 2007; Piatkov et al., 2012). Considering that a wide range of proteins are substrates of arginylation, it is reasonable to speculate that ATE1 may act as a root regulator of multiple cellular processes.
A popular theory about arginylation is its involvement in global protein degradation. Arginylation was shown to promote hyper-ubiquitination of the substrate proteins, which is then shown to be degraded by proteasome or autophagy (Saha and Kashina, 2011; Varshavsky, 2011). Based on mostly artificial substrates and in vitro data, arginylation was proposed to take place on proteins bearing certain amino acids on the 2nd residue on the N-terminus. These include the amino acids Asp, Glu, Asn, Gln (in fungi and mammals), and Cys (in mammal only) (Varshavsky, 2011). By this rule, in any given eukaryotic organism, at least 20–25% of its proteome would be estimated to be degraded by arginylation. Based on this assumption, arginylation was proposed as a central component in the generic protein degradation machinery (Varshavsky, 2011). However, the exact impact of arginylation on global homeostasis of proteins in vivo remain undetermined. In a more recent report, based on comparison of the sizes of protein spots on 2D-gels, the knockout of ate1 in mouse cells appears to affect ∼20% of those spots on the steady-state levels. However, much of these effects appear to be derived from transcriptional changes. Also, proteasome inhibitors can only reverse the ate1-dependent reduction of less than 3% of the observed protein spots (Wong et al., 2007). Such a small impact size is inconsistent with the proposed role of arginylation as a global degradation machinery.
In addition to protein degradation, arginylation was also suggested to be involved in many other processes. These may include cellular response to various types of stressors such as those that are closely related to cardiovascular stresses. For example, the activity of arginylation in cells or animal tissues is altered during injury, high temperature, or exposures to high concentrations of oxidants or salt (Zanakis et al., 1984; Chakraborty et al., 1986; Shyne-Athwal et al., 1986, 1988; Luo et al., 1990; Jack et al., 1992; Chakraborty and Ingoglia, 1993; Xu et al., 1993; Wang and Ingoglia, 1997; Bongiovanni et al., 1999; Kumar et al., 2016). Furthermore, other lines of studies also suggested that ATE1 may regulate the dynamics of cytoskeleton (Karakozova et al., 2006; Saha et al., 2010, 2011; Kaji and Kaji, 2012).
While multiple lines of researches are starting to establish ATE1 as a root regulator for multiple processes in the cell, contradicting conclusions are very often seen among reports from different groups. While many of these discrepancies may arise from differences in test conditions, variations may also be generated because many past studies were focused on the role of individual arginylation substrates without considering the effects of arginylation on the other known or potential substrates. To further understand the function of ATE1 in normal or diseased conditions, it is desirable to investigate the physiological role of ate1 in a systematic manner. Among the currently available tools for studying functional genomics, approaches based on a yeast gene-deletion library remain as one of the most robust and straight-forward methods (Boone et al., 2007; Giaever and Nislow, 2014). However, the role of ATE1 gene (ate1) has never been studied with such an unbiased manner, either as a specific query subject or as part of a comprehensive screening.
In this study, we took advantage of the Schizosaccharomyces pombe (S. pombe) single-gene knockout library. S. pombe is a commonly used test model for eukaryotic genes due to its similarity to metazoan organisms, while preserving its ease of genetic operation as a microbe. The meiosis and mating process of this organism also greatly facilitate the combination of different gene deletions to examine their synthetic effects, which can be quantitated by monitoring the growth rates of the resulting cells to deduce the genetic interactions between these two genes (Spirek et al., 2010; Wiley et al., 2014). The advantage of S. pombe as a model system is further demonstrated by the fact that nearly 70% of its gene have orthologs in the human genome, which is higher than S. cerevisiae, the other commonly used yeast as test model (Yanagida, 2002; Hoffman et al., 2015). The conserved genes including ate1, which is nearly 70% homologous in amino acid sequence in the core domain (∼200 residues) compared to its counterpart in human. However, unlike mammalian cells, S. pombe allows > 75% of its genome to be individually knocked out for functional tests (Spirek et al., 2010). These facts made S. pombe a desirable test model to determine the physiological role of ate1 in vivo.
In this study, we examined the effects of combining ate1-deletion with the deletions of other genes in S. pombe. The library we employed covered 3659 genes in S. pombe, which account for nearly 75% of the predicted open reading frames (up to 4940) in this organism (Decottignies et al., 2003). Surprisingly, only 173 of these genes, which is less than 5% of the effective library size, showed significant genetic interactions with ate1. Furthermore, many of these genes can be clustered into a few discrete groups in relation to cellular pathways. These include ribosomal component and translation regulation, gene transcription, oxidative stress response, cytoskeletal/structural components, mitochondrial function, and synthesis/metabolism of organic molecules and amino acids (such as histidine). Also, unexpectedly no significant interactions were observed between ate1 and genes involved in the ubiquitin/proteome system (UPS). Our results indicate that ATE1 may specifically regulate several physiological pathways in vivo. Many of these interactions can be used to provide satisfactory explanations for many observed involvements of ATE1-mediated arginylation in cardiovascular/metabolic abnormalities and other diseased conditions. These novel findings will also provide clues for designing approaches intervening ATE1-related phenotypes in various diseases including cancer and metabolic dysregulations.
Results
The Arginyltransferase Gene ate1 Showed Interactions With Only a Small Subset of Genes
While ate1 is an essential gene for mammals, it can be knocked out in S. pombe without causing lethality. Although S. pombe usually exists in a haploid form and reproduces by symmetric division (fission), they can also be induced to perform mating, during which chromosome recombination proceeds in a relatively high rate. These unique properties allow the knockout of ate1 to be easily combined with the knockout of other individual genes in the library, provided that different selection markers are being used to trace the knockout library and the query knockout (of ate1). Based on the growth of the products, compared to the parental strains, at least two types of interactions can be measured: (1) phenotype-enhancement, in which the crossing of the query results in a lower growth rate (also referred to as synthetic lethal); (2) phenotype-suppression, with a faster growth rate (also known as synthetic suppression).
By using the above-mentioned synthetic knockout approach, we examined the effects of combining ate1-knockout with 3721 individual knockouts in a S. pombe library, in which most of these genes were functionally annotated by either experimental evidence or prediction based on known orthologs. After excluding knockouts that lead to sterilization of the yeast, totally 3659 effective crossings were examined. Out of these crossings, we only found 173 of them resulted in significant phenotype enhancement (see Table 1A) or suppression (see Table 1B). This number is less than 5% of the library size.
TABLE 1.
Genes whose knockout showing genetic interaction, either phenotype enhancement (1A) or suppression (1B) with ate1 in S. Pombe.
|
A. Gene knockouts showing phenotype-enhancing effects (synthetic lethality) with ate1- knockout in S. pombe | ||||||
| Gene | Systematic | Product | S. cerevisiae | Human | ||
| name | Gene ID | p-Value | z-Score | description | ortholog | ortholog |
| igo1 | SPAC10F6.16 | 8.1414E-07 | −4.93195 | Endosulfine (ENSA) serine/threonine protein kinase Igo1 | IGO2, IGO1 | ARPP19, ENSA |
| gor2 | SPBC1773.17c | 0.000027474 | −4.19346 | Glyoxylate reductase (predicted) | GOR1 | GRHPR |
| SPBC19G7.04 | 0.000041554 | −4.09867 | HMG box protein | FYV8 | GCNA | |
| lsd90 | SPBC16E9.16c | 0.000079902 | −3.94469 | Lsd90 protein | ||
| puc1 | SPBC19F5.01c | 0.000083432 | −3.93432 | cyclin Puc1 | ||
| pun1 | SPAC15A10.09c | 0.000109954 | −3.86751 | SUR7 family protein Pun1 (predicted) | PUN1 | |
| mas5 | SPBC1734.11 | 0.00013273 | −3.82134 | DNAJ domain protein Mas5 (predicted) | YDJ1 | DNAJA2,DNAJA4,DNAJA1 |
| tps2 | SPAC3G6.09c | 0.000147704 | −3.7949 | Trehalose-phosphate synthase Tps2 (predicted) | TPS2 | |
| spn1 | SPAC4F10.11 | 0.00028086 | −3.63235 | Mitotic septin Spn1 | CDC3 | SEPTIN7,SEPTIN1,SEPTIN2, SEPTIN5,SEPTIN4 |
| sut1 | SPAC2F3.08 | 0.00034674 | −3.57762 | Plasma membrane sucrose/maltose:proton symporter Sut1 | SLC45A2,SLC45A3 | |
| plc1 | SPAC22F8.11 | 0.00046324 | −3.50116 | Phosphoinositide phospholipase C Plc1 | PLC1 | PLCB1,PLCH2,PLCH1,PLCB2, PLCB3,PLCB4,PLCD1,PLCD3, PLCD4,PLCL1,PLCL2,PLCG1 PLCG2 |
| gid5 | SPAC26H5.04 | 0.00068454 | −3.3957 | GID complex armadillo repeat subunit Gid5 (predicted) | VID28 | ARMC8 |
| pcr1 | SPAC21E11.03c | 0.00083756 | −3.34008 | Transcription factor Pcr1 | ||
| est1 | SPBC2D10.13 | 0.00090084 | −3.3198 | Telomerase regulator Est1 | EST1 | SMG6 |
| puf4 | SPAC6G9.14 | 0.00091582 | −3.31519 | Pumilio family RNA-binding protein Puf4 (predicted) | PUF4,MPT5 | |
| msa1 | SPAC13G7.13c | 0.00092698 | −3.3118 | RNA-binding protein Msa1 | RIM4 | |
| med1 | SPAC2F7.04 | 0.00099746 | −3.29124 | Mediator complex subunit Med1 | MED1 | MED1 |
| SPBCPT2R1.01c | 0.00106888 | −3.27174 | S. pombe specific DUF999 protein family 9 | |||
| coq7 | SPBC337.15c | 0.0010739 | −3.27041 | Ubiquinone biosynthesis protein Coq7 | CAT5 | COQ7 |
| spn4 | SPAC9G1.11c | 0.00109358 | −3.26527 | Mitotic septin Spn4 | CDC12 | |
| Mam3 | SPAP11E10.02c | 0.00146666 | −3.1812 | Cell surface adhesion protein for conjugation Mam3 | ||
| tif51 | SPAC26H5.10c | 0.0016832 | −3.14109 | Translation elongation and termination factor eIF5A (predicted) | HYP2,ANB1 | EIF5A, EIF5A2 |
| prp17 | SPBC6B1.10 | 0.0021378 | −3.07038 | Prp19 complex WD repeat protein Prp17 | CDC40 | CDC40 |
| scs7 | SPAC19G12.08 | 0.002199 | −3.06194 | ER sphingosine hydroxylase Scs7 | SCS7 | FA2H |
| pet1 | SPAC22F8.04 | 0.0022328 | −3.05739 | Golgi phosphoenolpyruvate transmembrane transporter Pet1 | SLC35C1 | |
| vas2 | SPAP27G11.06c | 0.002345 | −3.04265 | AP-1 adaptor complex sigma subunit Aps1 | APS1 | AP1S3,AP1S1,AP1S2 |
| rgs1 | SPAC22F3.12c | 0.002421 | −3.03305 | Regulator of G-protein signaling Rgs1 | SST2 | |
| SPAC186.08c | 0.0038618 | −2.88923 | L-lactate dehydrogenase (predicted) | |||
| ric1 | SPAC1851.04c | 0.0041792 | −2.86431 | Ypt/Rab-specific guanyl-nucleotide exchange factor (GEF) subunit Ric1 | RIC1 | RIC1 |
| qcr8 | SPAC1782.07 | 0.0043588 | −2.85095 | Ubiquinol-cytochrome-c reductase complex subunit 7 | QCR8 | UQCRQ |
| ubi5 | SPAC589.10c | 0.0051636 | −2.79665 | Ribosomal-ubiquitin fusion protein Ubi5 (predicted) | RPS31 | RPS27A |
| mex67 | SPBC1921.03c | 0.006664 | −2.71318 | mRNA export receptor, Tap, nucleoporin Mex67 | MEX67 | NXF1, NXF3 |
| rpl4302 | SPBC83.02c | 0.0073432 | −2.68086 | 60S ribosomal protein L37a (predicted) | RPL43B, RPL43A | RPL37A |
| klp8 | SPAC144.14 | 0.0073454 | −2.68076 | Kinesin-like protein Klp8 | KIF13B,KIF13A,KIF16B, KIF14,KIF1C,KIF1A | |
| rps1102 | SPAC144.11 | 0.0077726 | −2.66179 | 40S ribosomal protein S11 (predicted) | RPS11B,RPS11A | RPS11 |
| SPBC56F2.05c | 0.007884 | −2.657 | Transcription factor (predicted) | |||
| SPAC17C9.11c | 0.0080866 | −2.64843 | zf-C2H2 type zinc finger protein/UBA domain protein | UBXN1 | ||
| ppr5 | SPAC1093.01 | 0.0088172 | −2.61906 | Mitochondrial PPR repeat protein Ppr5 | ||
| rrp16 | SPAC22F8.09 | 0.0092822 | −2.60148 | rRNA processing protein Rrp16 (predicted) | NOP53 | NOP53 |
| fsv1 | SPAC6F12.03c | 0.0098672 | −2.58045 | SNARE Fsv1 | SYN8 | STX8 |
| SPBC354.07c | 0.010096 | −2.57252 | Sterol intermembrane transfer protein (predicted) | OSH7,OSH6,HES1,KES1 | OSBPL9,OSBPL10,OSBPL11 | |
| SPAC2F3.16 | 0.0101944 | −2.56916 | Ubiquitin-protein ligase E3, implicated in DNA repair (predicted) | RCHY1 | ||
| msy1 | SPCC1183.11 | 0.010212 | −2.56857 | MS calcium ion channel protein Msy1 | ||
| SPAC22G7.03 | 0.0102284 | −2.56801 | Schizosaccharomyces specific protein | |||
| alp13 | SPAC23H4.12 | 0.0103838 | −2.56278 | MRG family Clr6 histone deacetylase complex subunit Alp13 | EAF3 | MORF4,MORF4L2, MORF4L1 |
| cwf11 | SPBC646.02 | 0.0109488 | −2.54433 | U2-type spliceosomal complex ATPase Cwf11 | AQR | |
| rpl26 | SPBC29B5.03c | 0.01175 | −2.51956 | 60S ribosomal protein L26 (predicted) | RPL26B,RPL26A | RPL26,RPL26L1 |
| rpl29 | SPBC776.01 | 0.0128654 | −2.48747 | 60S ribosomal protein L29 | RPL29 | RPL29 |
| SPAC144.01 | 0.0142774 | −2.45021 | Schizosaccharomyces specific protein | |||
| rsn1 | SPBC354.08c | 0.0150762 | −2.43054 | Golgi to plasma membrane transport protein Rsn1 (predicted) | RSN1 | TMEM63B,TMEM63C, TMEM63A |
| clr3 | SPBC800.03 | 0.0176608 | −2.37265 | Histone deacetylase (class II) Clr3 | HDA1 | HDAC6,HDAC10 |
| tef103 | SPBC839.15c | 0.0180524 | −2.36454 | Translation elongation factor EF-1 alpha Ef1a-c | TEF2,TEF1 | EEF1A1,EEF1A2 |
| hos2 | SPAC3G9.07c | 0.0188808 | −2.34788 | Histone deacetylase (class I) Hos2 | HOS2 | HDAC1,HDAC2 |
| pab1 | SPAC227.07c | 0.0193724 | −2.33829 | Protein phosphatase PP2A regulatory subunit B-55 Pab1 | CDC55 | PPP2R2D,PPP2R2A,PPP2R2B, PPP2R2C |
| SPCC553.12c | 0.0195392 | −2.33508 | Transmembrane transporter (predicted) | |||
| mug183 | SPAC6G9.03c | 0.0197084 | −2.33185 | Histone H3.3 H4 heterotetramer chaperone Rtt106-like (predicted) | RTT106 | |
| rpl1102 | SPBC17G9.10 | 0.020086 | −2.32475 | 60S ribosomal protein L11 (predicted) | RPL11B,RPL11A | RPL11 |
| puf1 | SPBC56F2.08c | 0.020354 | −2.31974 | Pumilio family RNA-binding protein Puf1 (predicted) | JSN1,PUF2 | |
| SPBC1703.13c | 0.021252 | −2.30346 | Mitochondrial carrier, inorganic phosphate (predicted) | PIC2,MIR1 | SLC25A3 | |
| eaf7 | SPBC16A3.19 | 0.021428 | −2.30036 | Histone acetyltransferase complex subunit Eaf7 | EAF7 | MRGBP |
| gcd1 | SPCC794.01c | 0.023688 | −2.26214 | Glucose dehydrogenase Gcd1 | ZWF1 | H6PD |
| sft1 | SPAC31A2.13c | 0.024076 | −2.25592 | SNARE Sft1 (predicted) | SFT1 | BET1,BET1L |
| SPAC14C4.01c | 0.024618 | −2.24736 | DUF1770 family protein | |||
| nod1 | SPAC12B10.10 | 0.024672 | −2.24651 | Medial cortical node Gef2-related protein Nod1 | ||
| mad1 | SPBC3D6.04c | 0.025142 | −2.23922 | Mitotic spindle checkpoint protein Mad1 | MAD1 | MAD1L1 |
| syp1 | SPBC4C3.06 | 0.025222 | −2.238 | F-BAR domain protein Syp1 (predicted) | SYP1 | FCHO2,SGIP1,FCHO1 |
| rho2 | SPAC16.01 | 0.025592 | −2.23235 | Rho family GTPase Rho2 | RHO2 | RHOA,RHOB,RHOC |
| tpp1 | SPAC19G12.15c | 0.026072 | −2.22515 | Trehalose-6-phosphate phosphatase Tpp1 | TPS2 | |
| SPBC1711.15c | 0.027244 | −2.208 | Schizosaccharomyces pombe specific protein | |||
| rps101 | SPAC13G6.02c | 0.02798 | −2.19757 | 40S ribosomal protein S3a | RPS1A,RPS1B | RPS3A |
| cuf1 | SPAC31A2.11c | 0.028144 | −2.19529 | Nutritional copper sensing transcription factor Cuf1 | CUP2,MAC1, HAA1 | |
| apl6 | SPAC23H3.06 | 0.029952 | −2.17073 | AP-3 adaptor complex subunit Apl6 (predicted) | APL6 | AP3B1,AP3B2 |
| jmj1 | SPAC25H1.02 | 0.02998 | −2.17036 | Histone demethylase Jmj1 (predicted) | JMJD4 | |
| nrl1 | SPBC20F10.05 | 0.032478 | −2.13847 | RNAi-mediated silencing protein, human NRDE2 ortholog Nrl1 | NRDE2 | |
| atp3 | SPBC1734.13 | 0.032508 | −2.13812 | F1-FO ATP synthase gamma subunit (predicted) | ATP3 | ATP5F1C |
| oma1 | SPAP14E8.04 | 0.033356 | −2.12778 | Metallopeptidase Oma1 (predicted) | OMA1 | OMA1 |
| ace2 | SPAC6G10.12c | 0.034516 | −2.11399 | Transcription factor Ace2 | ACE2 | |
| SPACUNK4.13c | 0.036738 | −2.08865 | Mitochondrial NTPase Obg family, human OLA1 ortholog, implicated in mitochondrial translation, ribosome assembly, or tRNA metabolism (predicted) | YLF2 | OLA1 | |
| rtn1 | SPBC31A8.01c | 0.037292 | −2.08255 | Reticulon Rtn1 | RTN2,RTN1 | RTN1,RTN2,RTN3,RTN4 |
| mpn1 | SPAC23C11.10 | 0.037338 | −2.08206 | poly(U)-specific exoribonuclease, producing 3’ uridine cyclic phosphate ends Mpn1 | USB1 | USB1 |
| rud3 | SPBC119.12 | 0.03747 | −2.08061 | Golgi matrix protein Rud3 (predicted) | RUD3 | TRIP11 |
| laf1 | SPAC14C4.12c | 0.037782 | −2.07722 | Clr6 L associated factor 1 Laf1 | FUN19, YOR338W | |
| mbx1 | SPBC19G7.06 | 0.038252 | −2.07215 | MADS-box transcription factor Mbx1 | ARG80,MCM1 | MEF2A,MEF2B,MEF2C, MEF2D |
|
B. Gene knockouts showing phenotype-suppressing effects (synthetic rescue) with ate1- knockout in S. pombe | ||||||
| cgr1 | SPAC1556.05c | 7.7134E-08 | 5.37371 | Ribosome biogenesis CGR1 family (predicted) | CGR1 | CCDC86 |
| kap123 | SPBC14F5.03c | 3.4032E-07 | 5.09963 | Karyopherin/importin beta family nuclear import signal receptor Kap123 | KAP123 | IPO4 |
| rpl2802 | SPCC5E4.07 | 3.7196E-07 | 5.08278 | 60S ribosomal protein L27/L28 | RPL28 | RPL27A |
| gcn1 | SPAC18G6.05c | 7.1728E-07 | 4.95663 | Translation initiation regulator Gcn1 | GCN1 | GCN1 |
| pap1 | SPAC1783.07c | 1.35728E-06 | 4.83118 | Transcription factor Pap1/Caf3 | YAP1 | |
| SPAC3F10.09 | 1.82054E-06 | 4.77239 | 1-(5-phosphoribosyl)-5-[(5-phosphoribosylamino) methylideneamino]imidazole-4-carboxamide isomerase (predicted) | HIS6 | ||
| mcl1 | SPAPB1E7.02c | 0.000002246 | 4.72993 | DNA polymerase alpha accessory factor Mcl1 | CTF4 | WDHD1 |
| mtq1 | SPAC29B12.05c | 4.4576E-06 | 4.58881 | Mitochondrial N(5)-glutamine methyltransferase (predicted) | MTQ1 | HEMK1 |
| atg14 | SPAC25A8.02 | 0.00002277 | 4.23584 | Autophagy associated protein Atg14 | ATG14 | ATG14 |
| byr3 | SPAC13D6.02c | 0.000032138 | 4.15776 | Translational activator, zf-CCHC type zinc finger protein (predicted) | GIS2 | CNBP, ZCCHC13 |
| mms19 | SPAC1071.02 | 0.000037518 | 4.12226 | CIA machinery protein Mms19 | MET18 | MMS19 |
| imt2 | SPCC4F11.04c | 0.000041484 | 4.09906 | Mannosyltransferase Imt2 | CSH1, SUR1 | |
| atd1 | SPAC9E9.09c | 0.000043798 | 4.08647 | Aldehyde dehydrogenase (predicted) | ALD5, ALD4, ALD6 | ALDH1A2, ALDH1A1, ALDH2, ALDH1B1, ALDH1A3 |
| crf1 | SPAC22H10.11c | 0.000048624 | 4.06214 | Transcriptional corepressor for ribosomal proteins via TOR signaling pathway Crf1 (predicted) | CRF1, IFH1 | |
| SPCC61.05 | 0.000099978 | 3.89064 | Schizosaccharomyces specific multicopy membrane protein family 1 | |||
| SPAC29A4.09 | 0.00010776 | 3.87242 | rRNA exonuclease Rrp17 (predicted) | RRP17 | NOL12 | |
| his1 | SPAC25G10.05c | 0.000112986 | 3.86086 | ATP phosphoribosyltransferase | HIS1 | |
| brl1 | SPCC1919.15 | 0.000122818 | 3.84043 | Ubiquitin-protein ligase E3 Brl1 | BRE1 | RNF40 |
| elf1 | SPAC3C7.08c | 0.000131224 | 3.82415 | AAA family ATPase Elf1 | NEW1 | |
| met10 | SPCC584.01c | 0.000142772 | 3.80332 | Sulfite reductase NADPH flavoprotein subunit (predicted) | MET10 | |
| fil1 | SPCC1393.08 | 0.000195024 | 3.72538 | Transcription factor, zf-GATA type | ||
| rps1802 | SPCC1259.01c | 0.000195418 | 3.72487 | 40S ribosomal protein S18 (predicted) | RPS18A,RPS18B | RPS18 |
| atp1 | SPAC14C4.14 | 0.000258 | 3.65419 | F1-FO ATP synthase alpha subunit | ATP1 | ATP5F1A |
| ctp1 | SPCC338.08 | 0.00038412 | 3.55076 | CtIP-related endonuclease | SAE2 | RBBP8 |
| cox19 | SPCC1672.04c | 0.00050878 | 3.47609 | Mitochondrial copper chaperone for cytochrome c oxidase Cox19 (predicted) | COX19 | COX19 |
| rep2 | SPBC2F12.11c | 0.0007297 | 3.37817 | MBF transcription factor activator Rep2 | ||
| rpl3602 | SPBC405.07 | 0.00082778 | 3.34334 | 60S ribosomal protein L36 | RPL36A,RPL36B | RPL36 |
| ght8 | SPCC548.06c | 0.00086908 | 3.3298 | Plasma membrane hexose:proton symporter, unknown specificity Ght8 (predicted) | HXT15,HXT7,HXT6,STL1, HXT13, MAL11,HXT4, HXT1,HXT5,HXT8,HXT9, HXT16, GAL2,HXT2,HXT14, HXT17,HXT11 | |
| rpl35b | SPBC1921.01c | 0.00110496 | 3.26234 | 60S ribosomal protein L35a (predicted) | RPL33B,RPL33A | RPL35A |
| rpl901 | SPAC4G9.16c | 0.00138818 | 3.1971 | 60S ribosomal protein L9 | RPL9A,RPL9B | RPL9 |
| rpl1801 | SPBC11C11.07 | 0.00140148 | 3.19435 | 60S ribosomal protein L18 | RPL18B,RPL18A | RPL18 |
| rpl3702 | SPCC1223.05c | 0.00161788 | 3.15266 | 60S ribosomal protein L37 (predicted) | RPL37B,RPL37A | RPL37 |
| SPAC17G8.06c | 0.00198848 | 3.09195 | Dihydroxy-acid dehydratase (predicted) | ILV3 | ||
| his7 | SPBC29A3.02c | 0.0021838 | 3.06403 | Phosphoribosyl-AMP cyclohydrolase/phosphoribosyl- ATP pyrophosphohydrolase His7 | HIS4 | |
| lys9 | SPBC3B8.03 | 0.0023364 | 3.04376 | Saccharopine dehydrogenase | LYS9 | AASS |
| his5 | SPBC21H7.07c | 0.002481 | 3.02565 | Imidazoleglycerol-phosphate dehydratase His5 | HIS3 | |
| rpl15 | SPCC576.11 | 0.0028468 | 2.98382 | 60S ribosomal protein L15 (predicted) | RPL15A,RPL15B | RPL15 |
| dhm1 | SPCP1E11.10 | 0.0029186 | 2.97618 | Ankyrin repeat protein, unknown biological role | YCR051W | |
| zfs1 | SPBC1718.07c | 0.0033112 | 2.93727 | zf-CCCH tandem zinc finger protein, human Tristetraprolin homolog Zfs1, involved in mRNA catabolism | CTH1,TIS11 | ZFP36L1, ZFP36L2, ZFP36 |
| trm112 | SPAC31A2.02 | 0.0035714 | 2.91373 | eRF1 methyltransferase complex and tRNA (m2G10) methyltransferase complex regulatory subunit Trm112 (predicted) | TRM112 | TRMT112 |
| his2 | SPBC1711.13 | 0.006125 | 2.74102 | Histidinol dehydrogenase His2 (predicted) | HIS4 | |
| hmt2 | SPBC2G5.06c | 0.0065568 | 2.71855 | Sulfide-quinone oxidoreductase | SQOR | |
| rps1201 | SPCC962.04 | 0.0066316 | 2.7148 | 40S ribosomal protein S12 (predicted) | RPS12 | RPS12 |
| mms1 | SPAC3H8.05c | 0.0075438 | 2.67184 | Cul8-RING ubiquitin ligase complex subunit Mms1 (predicted) | MMS1 | 0 |
| rpl902 | SPCC613.06 | 0.0100146 | 2.57533 | 60S ribosomal protein L9 | RPL9A,RPL9B | RPL9 |
| pnk1 | SPAC23C11.04c | 0.0101678 | 2.57007 | DNA kinase/phosphatase Pnk1 | TPP1 | PNKP |
| dml1 | SPAC30C2.06c | 0.0109928 | 2.54293 | Mitochondrial inheritance GTPase, tubulin-like (predicted) | DML1 | MSTO1 |
| SPAC732.02c | 0.0127802 | 2.48984 | Fructose-2,6-bisphosphate 2-phosphatase activity (predicted) | FBP26 | PFKFB1, PFKFB2, PFKFB3, PFKFB4 | |
| ser2 | SPBC3H7.07c | 0.0131908 | 2.47858 | Phosphoserine phosphatase Ser2 (predicted) | SER2 | PSPH |
| ifa38 | SPAC4G9.15 | 0.0135804 | 2.46818 | Ketoreductase involved in fatty acid elongation (predicted) | IFA38 | HSDL1, HSD17B12, HSD17B3 |
| rps2801 | SPAC25G10.06 | 0.0137516 | 2.46369 | 40S ribosomal protein S28 (predicted) | RPS28B,RPS28A | RPS28 |
| lip2 | SPAC4F10.05c | 0.0162584 | 2.40306 | Mitochondrial lipoate-protein ligase Lip2 | LIP2 | LIPT2 |
| clg1 | SPBC1D7.03 | 0.0179078 | 2.36752 | Cyclin-like protein involved in autophagy Clg1 (predicted) | CLG1 | |
| arp5 | SPBC365.10 | 0.018288 | 2.35974 | Ino80 complex actin-like protein Arp5 | ARP5 | ACTR5 |
| mre11 | SPAC13C5.07 | 0.018601 | 2.35343 | Mre11 nuclease | MRE11 | MRE11 |
| met14 | SPAC1782.11 | 0.0203 | 2.32077 | Adenylyl-sulfate kinase (predicted) | MET14 | PAPSS1, PAPSS2 |
| rps1502 | SPAC1071.07c | 0.022698 | 2.27847 | 40S ribosomal protein S15 (predicted) | RPS15 | RPS15 |
| SPAC3C7.04 | 0.024088 | 2.25573 | Transcription factor (predicted) | |||
| rps2802 | SPCC285.15c | 0.026352 | 2.22097 | 40S ribosomal protein S28, Rps2802 | RPS28B,RPS28A | RPS28 |
| git1 | SPBC21C3.20c | 0.027796 | 2.20016 | C2 domain protein Git1 | ||
| rpa12 | SPCC1259.03 | 0.028074 | 2.19625 | DNA-directed RNA polymerase complex I subunit Rpa12 | RPA12 | ZNRD1 |
| cys2 | SPBC106.17c | 0.028446 | 2.19109 | Homoserine O-acetyltransferase (predicted) | ||
| SPBC1A4.04 | 0.030716 | 2.16072 | Schizosaccharomyces specific protein | |||
| lys7 | SPAC17C9.02c | 0.03157 | 2.14981 | Alpha-aminoadipate reductase phosphopantetheinyl transferase Lys7 | LYS5 | AASDHPPT |
| ade10 | SPCPB16A4.03c | 0.033428 | 2.1269 | Bifunctional IMP cyclohydrolase/phosphoribosylaminoimidazole- carboxamide formyltransferase | ADE16,ADE17 | ATIC |
| rpl3001 | SPAC9G1.03c | 0.036378 | 2.09268 | 60S ribosomal protein L30 (predicted) | RPL30 | RPL30 |
| SPBC1271.14 | 0.037576 | 2.07945 | Acetyl-CoA:L-glutamate N-acetyltransferase (predicted) | ARG7 | ||
| ftp105 | SPAC17A5.16 | 0.04206 | 2.03293 | Golgi localized protein, human HID1 ortholog 3, implicated in vesicle-mediated transport | ECM30 | HID1 |
| ppa2 | SPBC16H5.07c | 0.044676 | 2.00769 | Serine/threonine protein phosphatase Ppa2 | PPH21,PPH22 | PPP2CA,PPP2CB |
| sod2 | SPAC1486.01 | 0.044806 | 2.00647 | Mitochondrial superoxide dismutase Sod2 | SOD2 | SOD2 |
| gpd1 | SPBC215.05 | 0.046392 | 1.99181 | Glycerol-3-phosphate dehydrogenase Gpd1 | GPD1 | GPD1L,GPD1 |
The genetic interaction was determined by the effects of combing two gene knockouts on the growth rate of the resulting yeast colony compared to the individual knockouts. Sexual mating of S. pombe was used to generate the crossings between the ate1-knockout and the other gene-knockouts in a library. While the original library contains 3721 different knockout strains, some of these deletion strains appear to be sterile and cannot be mated. Thus, the effective library size was 3659 in our tests. Four replicates were used to determine the effects on growth of the crossed, double-knockout colony compared to the parental strain with a single knockout (without ate1 deletion). Based on the comparison, a result of slower growth is considered as phenotype-enhancement (in Table 1A). Vice versa, those that grow faster than the parental strain is considered phenotype-suppression (Table 1B). A confidence value (p-values) of 0.05 is the minimum for significance.
Genes That Genetically Interact With ate1 Can Be Clustered by Biological Function
Among the genes that showed significant interactions with ate1, we found specific enrichment of several types of functional relevancies. For example, simply by applying functional annotations such as gene ontology (GO), gene expression category, or Fission Yeast Phenotype Ontology (FYPO), we found that several terms are enriched or present in higher frequencies among genes interacting with ate1 compared to the whole library (Figure 1 and Supplementary Table S1). As a further validation of the functional interactions observed between ate1 and these genes, many of them also have known physical interactions or associations of their products among each other (Figure 2).
FIGURE 1.

Gene features that are enriched in the ate1-interacting hit list compared to the library pool. The features of genes were determined by either gene ontology (GO) terms, expression category, or fission yeast phenotype ontology (FYPO).
FIGURE 2.
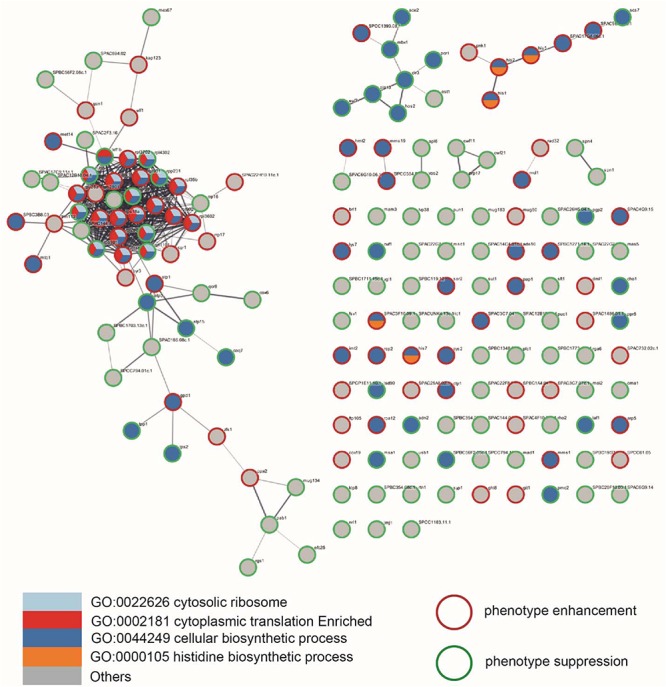
Known protein–protein interactions (PPI) between products of genes that have genetic interactions with ate1 in S. pombe. The PPI database being utilized is Version 11 of STRING (https://string-db.org). The interaction map was generated with Cytoscape using the STRING App for creating the PPI information and enrichment mapping. The thickness of the connecting line represents the confidence of the experimental protein-protein interaction. A thicker line represents a higher confidence (a minimum of 0.4% confidence was used). Each rounded shape represent the product of a gene (with gene name labeled on the side). The assigned color of the rounded shape represents gene category (by GO terms), while the color of the ring (red or green) represent the direction of the genetic interaction (phenotype enhancing or suppressing).
The enriched or increased functional annotations can be grouped into several clustered categories. Based on the frequency of presence in the hit list, the most abundant genes (nearly 50% of the hits) are those related to biosynthesis of biological molecules or proteins (Supplementary Table S1A). These include many regulators of translation or transcription (17–39%). Interestingly, among these categories, those related to histidine/amidazole synthesis/metabolism appear to be particularly impacted by ate1, as most of the genes associated with these pathways (5 out of 7∼9) showed interaction with ate1 (Figure 1 and Supplementary Table S1A).
The second most abundant genre (up to 40%) is those related to cell morphology as well as cellular dynamics (fusion, conjugation, or division). These include many known regulators of cytoskeleton (Figure 1 and Supplementary Table S1B).
The third type (10∼15%) are those related to oxidative stress response, which include many highly expressed genes coding for chaperones or redox regulators (Figure 1 and Supplementary Table S1C).
The fourth are (10∼15%) ribosome components or ribosome-associated factors (Figure 1 and Supplementary Table S1A).
The fifth are those related to nutrient or metabolite stresses, such as genes related to nitrogen starvation, and genes involved in responses to metabolism interferers (caffeine, rapamycin) or metabolite analogs of lipids or nucleotides (Figure 1 and Supplementary Tables S1B,C).
In addition to the above analysis by functional annotations, the genes that have genetic interactions with ate1 can also be grouped into a handful of clustered functional pathways or categories when being analyzed with PANTHER Classification System1 (Mi et al., 2013, 2019), as shown in Supplementary Table S2.
As an example to validate the observations reached in the above non-biased study, we examined the sensitivity of S. pombe to exogenous histidine, which is not an essential amino acid for this organism. While a lower amount of exogenous histidine usually promotes the growth of yeasts, at high concentrations it is known to generate cytotoxicity and requires the actions of the histidine/amidazole synthesis/metabolism pathway for mitigation (Winkler and Ramos-Montanez, 2009; Watanabe et al., 2014; Duncan et al., 2018). When we compared the growth rates of the yeasts hosting the ate1-deletion to the control cells, we found that, although there is no apparent difference in their proliferation in the absence of exogenous histidine (Figure 3A), ate1-deleted yeast grow much slower than the control in high concentrations of histidine (Figures 3B–E). To ensure these observations are not specific to the colony of yeast used in the test, we repeated these experiments with two additional clones of the ate1-deleted S. pombe and reached similar conclusions (Figure 3F). Therefore, these data suggested that ate1-deletion indeed possesses an interaction with the histidine/amidazole synthesis/metabolism pathway.
FIGURE 3.
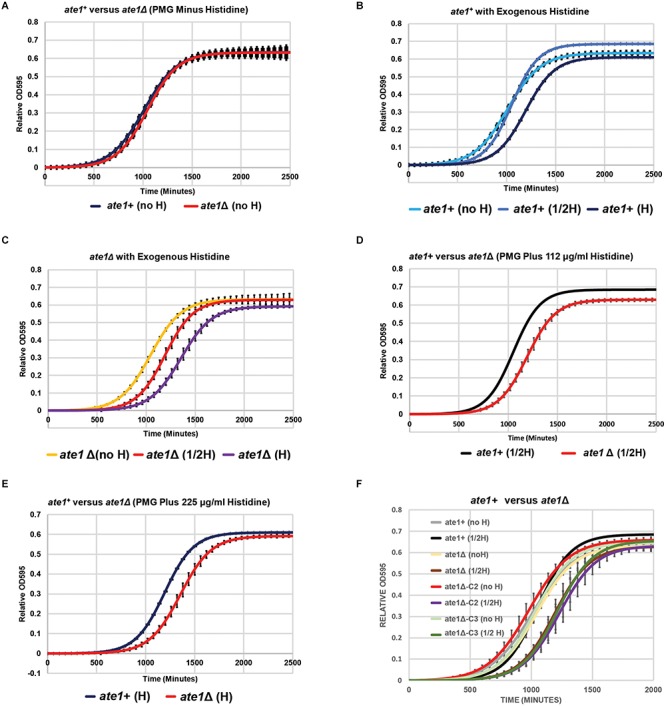
The deletion of ate1 leads to lower tolerance of exogenous histidine in the media. The yeast strains were grown in PMG liquid media supplemented with adenine, leucine, and uracil plus histidine as indicated (no H = no histidine; 1/2H = 112 μg/ml histidine; H = 225 μg/ml histidine), while the culture density was monitor by absorbance (OD 595 nm). In the absence of histidine (referred as to “no H”), both the ate1Δ and the control (ate1 +) strains grow in a similar rate (see A). The addition of moderate amount of histidine (112 μg/ml; “1/2H”) increases the growth of control strain (ate1 +), while a higher concentration (225 μg/ml; “H”) exhibits cytotoxic effect as anticipated (see B). However, in ate1Δ strain, the addition of either 112 or 225 mg/ml of exogenous histidine both lead to slower growth in a dose-dependent manner (see C). Direct comparisons of the growth of ate1Δ and the control strain (ate1 +) at different concentrations of histidine are presented at panel (D) and (E). To exclude the possibility of clone-specific, two different clones of ate1Δ strain (ate1Δ−C2, and −C3) isolated from the same knockout process were subjected to the challenge of 112 μg/ml exogenous histidine, compared to when no histidine was added (see F). The curves were generated with non-linear regression. Error Bars represent standard deviation from 6 replicates.
Many Genes Related to Mitochondria or Energy Production Genetically Interact With ate1
Compared to the relative abundance of literatures showing the effects of arginylation in protein degradation or cytoskeletal regulations, the potential involvements of arginylation or ATE1 in other cellular processes are less known. Interestingly, many of the observed phenotypes in animals resulted from ate1 dysregulation may be at least partly attributed to a disruption of mitochondrial function, which supplies the majority of energy molecules (ATP, NADH, and NADPH, etc.) required for many biosynthesis processes, and also constitute a major source of oxidative stressors in the cell. For example, postnatal systemic knockout of ate1 appears to lead to drastic loss of fat and infertility, which are common consequences expected from a compromise of mitochondrial function. Consistent to this possibility, we observed a significant number (at least 19) of genes related to mitochondria or energy production showing interactions with ate1 (Table 2). These genes represent more than 10% of the total hits, although they were not present as “enriched” because the frequency of them in the hit-list is not higher than that in the library.
TABLE 2.
Mitochondria-related genes, as determined by GO terms, which are genetically interacting with ate1.
| Gene | # of non-sterile | # of genes in | Gene name/systematic ID | |
| description | GO_Term | genes in library | the hits | in the hits list |
| Mitochondrion (mitochondria) | GO:0005739 | 428 | 19 | mpn1, SPACUNK4.13c, lip2, gor2, hmt2, sod2, SPAC14C4.01c, qcr8, SPAC17G8.06c, cox6, oma1, cys2, SPBC1271.14, SPBC1703.13c, atp3, atp15, coq7, rps1802, ppr5 |
| Mitochondria inner membrane | GO:0005743 | 71 | 4 | oma1, SPBC1703.13c, coq7, atp15 |
The numbers of those genes in the effective library (non-sterile) and the hit list are being shown. The names (or systematic ID) of the genes in the hit list are also shown.
Few Genes in Global Protein Degradation Pathways Showed Genetic Interactions With ate1
Another surprising finding in the interacting partners of ate1 is the rarity of genes involved in global protein ubiquitination or degradation machineries.
In principle, the uncertainties about the role of arginylation in proteome homeostasis can at least be partly addressed by the genetic interaction screening. If ate1 is involved in global protein ubiquitination or degradation, it would be expected to have genetic interactions with the other components in these pathways. However, while many genes with known roles in global ubiquitination or degradation are present in the S. pombe knockout library we employed (Supplementary Table S3), only two (ubi5 and atg14) showed significant interactions (either enhancing or suppressing) with ate1 (Table 3). Among these two genes, ubi5 is a fusion gene of ubiquitin and ribosomal component. As such, its genetic interaction with ate1 may derive from the ribosomal component and not necessarily the ubiquitin, since no interaction was observed between ate1 and other genes coding for ubiquitin. The rarity of interactions between ate1 and degradation-related genes is actually consistent with the selectivity of the interacting partners of ate1 as described above (Figure 2 and Tables 1–3). If arginylation is a generic degradation pathway for up to 20% proteins as predicted, then the expected interactions between ate1 and other genes should be much larger than the observed number (< 5%), because many components in the pathways regulated by these arginylation-target proteins would be expected to show genetic interaction with ate1. Interestingly, the small impact size (< 5%) of ate1 in genetic interactions is well consistent with the impact size (∼3%) of arginylation on the degradation of endogenous proteins as estimated in 2D-gels (Wong et al., 2007). These evidences suggest that ATE1-mediated arginylation may not be a major degradation pathway in vivo in the experimental condition we employed.
TABLE 3.
Genes related to global ubiquitination and degradation, as determined by GO terms, that are in the screen library or genetically interacting with ate1.
| # of genes | |||
| Gene | in library | # of genes in | |
| description | GO_term | (non-sterile) | the hits |
| Protein ubiquitination | GO:0016567 | 19 | 1 (ubi5) |
| Ubiquitin binding | GO:0043130 | 25 | 0 |
| Ubiquitin (protein tag) | GO:0031386 | 7 | 1 (ubi5) |
| Ubiquitin ligase complex | GO:0000151 | 20 | 0 |
| Proteasome complex | GO:0000502 | 1 | 0 |
| Lysosome | GO:0005764 | 5 | 0 |
| Autophagy | GO:0006914 | 18 | 0 |
| Autophagosome | GO:0005776 | 4 | 1 (atg14) |
| Protein catabolic process | GO:0030163 | 5 | 0 |
The numbers of those genes in the effective library (non-sterile) and the hit list are being shown. The names of the genes in the hit list are also shown.
Discussion
Despite that ate1 gene has been identified for 30 years, it’s in vivo role has remained poorly defined. For the first time, by using a systematic approach, our results showed that ate1 possesses significant genetic interactions with a small and focused subset of genes concerning multiple critical cellular processes. The results from this study can also provide important leads for mechanistic investigations about the role of ate1 or arginylation in normal or diseased conditions.
The power of the systematic approach employed in our study is demonstrated by the fact that many results from this unbiased investigation are highly consistent or complementing to data from past reports. For example, the observed genetic interactions between ate1 and regulators of cell morphology are well consistent with previous reports showing the impact of arginylation on many cytoskeletal proteins (Wong et al., 2007) and cytoskeletal dynamics (Karakozova et al., 2006; Rai et al., 2008; Saha et al., 2010; Zhang et al., 2010, 2012; Kurosaka et al., 2012). The interactions between ate1 and genes involved in oxidative stress response also support the proposed role of arginylation in stress response process (Zanakis et al., 1984; Chakraborty et al., 1986; Shyne-Athwal et al., 1986, 1988; Luo et al., 1990; Jack et al., 1992; Chakraborty and Ingoglia, 1993; Xu et al., 1993; Wang and Ingoglia, 1997; Bongiovanni et al., 1999; Kumar et al., 2016). Overall, it appears that our non-biased screening study was able to recapitulate many past observations performed in cell or animal.
Many results of this study will provide new clues for investigate the role of arginylation in physiological processes. Arginylation was shown to be involved in stress response, but the exact mechanism still awaits clarification and may benefit from the genetic interactions revealed in our study. For example, while several redox regulators, including manganese superoxide dismutase (SOD) appear to have phenotype-enhancing relationship with ate1, many genes related to mitochondria, the main source of reactive oxygen species (ROS) in the cell, display synthetic suppression relationship. These differences imply that ATE1 may act as a scavenger of oxidatively damaged proteins in stress response. Also, the broad interactions between ate1 and many transcription regulators and histone modulators may help to elucidate the observed but unexplained effects of ATE1 on global transcriptional landscape (Lee et al., 2012; Eisenach et al., 2014; Deka et al., 2016). Particularly, many genes of histone modulators appear to have a phenotype-suppression relationship with ate1. This is highly intriguing considering that previous studies have found that histone proteins are subjected to arginylation modification, which may also affect the other PTMs on histone (Wong et al., 2007; Saha et al., 2011). The interactions between ate1 and genes related to biomolecule synthesis/metabolism are also of high interest because emerging evidence indicated a potential involvement of ATE1 in metabolism. As an example, the enrichment of the genes in histidine synthesis/metabolism pathways in principle is consistent with one of our previous findings about arginylation of phosphoribosyl pyrophosphate synthetase (PRPPS). This is because PRPPS is responsible for production of Phosphoribosyl Diphosphate, which serves as a precursor substrate for biosynthesis of histidine (Zhang et al., 2015). Due to the conserved nature of ATE1-mediated arginylation in eukaryotes (McGary et al., 2010), many of these findings may provide mechanistic insights for the role of ATE1/arginylation in cardiovascular diseases, metabolic dysregulations, and cancer in human. These exciting possibilities will become important directions for future studies.
The lack of interactions between ate1 and genes related to global ubiquitination or protein degradation, as revealed in our study, is quite surprising. Based on the loose consensus on amino acid sequence of known substrates, arginylation was long hypothesized as a signal for the ubiquitination and degradation of at least 20–25% members of the proteome in yeast or metazoan. Such a broad range of substrates would predict a very large impact size of genetic interactions of ate1. However, less than 5% of the tested genes, which cover nearly 75% of the genome in S. pombe yeast, showed interactions with ate1. Consistently, we found that ate1 has very few interactions with components of the ubiquitination and degradation pathways. While these findings appear to be at odds with popular theories, it is worthy pointing out that the exact nature of arginylation in the degradation of endogenous proteins has not been decisively determined and the current theories about arginylation were mainly built on studies with artificial substrates (Bachmair et al., 1986; Varshavsky, 2011). It is possible that the substrate preference of ATE1 is more complexed than originally expected. For example, recent evidence suggested that the efficiency of arginylation may be affected by at least 11 residues on the N-terminus of a peptide, and the substrate preference of ATE1 may also be influenced by additional in vivo factors (Wang et al., 2011, 2018). Also, since the majority of arginylation takes place on the N-terminus of a protein, it must compete with N-terminally acetylation in vivo, which is a dominant PTM in most eukaryotes (Wang et al., 2011). Furthermore, while arginylation in many cases indeed promotes ubiquitination and degradation, exceptions are also abundant on endogenous arginylated proteins (Karakozova et al., 2006; Zhang et al., 2010, 2012, 2015). Maybe not coincidentally, past experimental attempts examining the impact of arginylation on the whole proteome also revealed a relatively small size. In one of such reports, the usage of 2D gels showed that less than 3% of individual proteins appear to be affected by arginylation on proteasome-dependent degradation (Karakozova et al., 2006; Wong et al., 2007). As such, alternative interpretation for the role of arginylation in protein ubiquitination/degradation may be needed and our study may provide clues for that. It is possible that arginylation is not activated under resting state but is specifically utilized for protein degradation during certain conditions such as stress response or nutrient deprivation. This possibility is at least indirectly supported by existing evidence showing the activation of arginylation during stress response and that arginylation preferentially takes place on oxidatively damaged proteins (Zhang et al., 1998; Kumar et al., 2016). It is also supported by the extensive genetic interactions between ate1 and genes related to oxidative and metabolic stress as revealed in our study. However, this current study was conducted in a non-stressed condition and therefore cannot directly test this possibility, which will require further investigations for validation.
The other unexpected finding is the extensive interaction between ate1 and ribosome-related genes. While the biological meaning of this interaction is still unclear, existing literatures may provide a few clues. Particularly, the ATE1 protein was found to at least partially co-localize with ribosome during cellular fractionation (Wang et al., 2011). Also, many ribosome-associated proteins were found to be arginylated (Wong et al., 2007; Wang et al., 2011). Based on this evidence, it is likely ATE1 may regulate the function of ribosome. In addition, current evidence suggest that arginylation may take place during co-translational stage and the outcome in arginylation-mediated degradation may be dependent on the dynamic of co-translational folding (Zhang et al., 2010). As such, it is also likely that arginylation is part of the mechanism for quality check of nascent peptide synthesis. This unexpected connection between arginylation and ribosome, as well as many other newly information about arginylation uncovered in this study, constitute intriguing directions for future research endeavors.
Conclusion
By using a systematic approach, we found that the gene for arginyltransferase1 has specific interactions with a small subset of genes in the eukaryotic genome, which fall into a few clustered functional categories. Our data suggest that ATE1 may specifically regulate a few cellular pathways in vivo. These results will provide novel mechanistic clues to understand the role of protein arginylation in the development of cardiovascular system and the pathogenesis of related diseases.
Materials and Methods
Query Yeast Strain Creation
Growth conditions and genetic manipulations of S. pombe were performed as previously described (Moreno et al., 1991). The query strain containing ate1-deletion (ate1Δ) was prepared by targeted deletion with a linear DNA containing a nourseothricin-resistance gene flanked with sequence derived from the ate1 gene (systemic ID: SPAC3C7.07c) loci. The 5′-region (394 bp) flanking the resistance gene was cloned with these primers, with underline to indicate the regions matching the genome sequence while the rest primes to the resistance cassette:
5′-FWD: TAGAACTTGGTGGATGGTATCGTGG
5′-REV: GGGGATCCGTCGACCTGCAGCGTACGAACTATTGTTTGAAAATTTCCCTGTTTAAT
The 3′-region (385 bp) flanking the resistance gene was cloned with these primers:
3′-FWD: GTTTAAACGAGCTCGAATTCATCTTATATTATCTGTCTACGTGTTTTATTTGC
3′-REV: TCCTTTCTCACCTACTATGCACTGTTTTG
The knockout was performed in an h- leu1-32 ura4-D18 Ade6-M210 S. pombe strain (PN572) as the parental strain with published protocols (Krawchuk and Wahls, 1999). The success of knockout is confirmed with this primer specific for the ate1 gene: TCTTTGGATTGACAAGTTGAGAGTTG.
Yeast Synthetic Genetic Array
The ate1Δ strain was grown to exponential phase in liquid PMG media (Sunrise Scientific Cat. #2060) supplemented with adenine, leucine and uracil. It was then pinned to agar plates in a 384-matrix format using a robotic platform RoToR HDA (Singer Instruments). The query strain was then crossed with individual strains in the S. pombe haploid deletion library containing individual gene deletions marked by a kanamycin resistance gene (Bioneer, version 4.0 equivalent) utilizing a modified SGA procedure (Dixon et al., 2008). This procedure was described in detailed in our previous publication (Wiley et al., 2014). In brief, the crossing was induced on a SPAS mating media (1% glucose, 7.3mM KH2PO4, with 45mg/L supplements of adenine, histidine, leucine, uracil and lysine-HCl, and vitamin supplement for pantothenic acid, nicotinic acid, inositol, and biotin; see this website for detailed recipe: https://dornsife.usc.edu/pombenet/media/). For germination, four replicates of each crossing product were pinned to a 1536 format on selective PAU + G418 media, which is the PMG media containing adenine (225 mg/L, Sigma Cat. #A8751), leucine (225 mg/L, Sigma Cat. #L8912), uracil (225 mg/L, Sigma Cat. #U0750), and the antibiotics G418 and nourseothricin. Colony growth was monitored for 3 days utilizing a flatbed scanner, and then quantified and compared using ScreenMill according to published protocols (Dittmar et al., 2010). Double mutants with a significant growth rate difference compared to the corresponding library gene deletion alone were scored as either slowed (referred to as “phenotype-enhancement”) or accelerated (referred to as “phenotype-suppression”) growth.
Hit Analysis and Bio-Informatics
The gene feature enrichments (over-representation) in the hit list versus the library were examined by AnGeLi2 (Bitton et al., 2015). The hit list was compared directly to the genes screened in our assay using the false discovery rate setting for multiple testing with a p-value setting of 0.01. In addition, for this enrichment we performed pairwise interaction enrichment with 1000 permutations and allowed for the p-value to adjust. As a negative control, we also looked for underrepresented terms and we cannot find significant underrepresented entries based on inputs, which suggesting that our analysis did not create artifacts.
The PANTHER Classification System3 used in this study is Version 14.1, released 2019-03-12 (Mi et al., 2019).
The image of protein-protein interaction was created using Cytoscape with the STRING App (Szklarczyk et al., 2019). The PPI database being utilized is Version 11 of STRING4.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
DW performed most of the experiments and the analysis of data. GD’U was involved in supervising the performance of the experiments. FZ conceptualized and designed the project and also supervised the analysis of the data.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Daniel Isom and Fulvia Verde (University of Miami) for providing critical reading of this manuscript.
Funding. This project was partly supported by a pilot funding from the Florida Breast Cancer Foundation, and an American Cancer Society Institutional Research Grant 98-277-13, to FZ.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00427/full#supplementary-material
References
- Bachmair A., Finley D., Varshavsky A. (1986). In vivo half-life of a protein is a function of its amino-terminal residue. Science 234 179–186. 10.1126/science.3018930 [DOI] [PubMed] [Google Scholar]
- Birnbaum M. D., Zhao N., Moorthy B. T., Patel D. M., Kryvenko O. N., Heidman L., et al. (2019). Reduced Arginyltransferase 1 is a driver and a potential prognostic indicator of prostate cancer metastasis. Oncogene 38 838–851. 10.1038/s41388-018-0462-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitton D. A., Schubert F., Dey S., Okoniewski M., Smith G. C., Khadayate S., et al. (2015). AnGeLi: a tool for the analysis of gene lists from fission yeast. Front. Genet. 6:330 10.3389/fgene.2015.00330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongiovanni G., Fissolo S., Barra H. S., Hallak M. E. (1999). Posttranslational arginylation of soluble rat brain proteins after whole body hyperthermia. J. Neurosci. Res. 56 85–92. [DOI] [PubMed] [Google Scholar]
- Boone C., Bussey H., Andrews B. J. (2007). Exploring genetic interactions and networks with yeast. Nat. Rev. Genet. 8 437–449. 10.1038/nrg2085 [DOI] [PubMed] [Google Scholar]
- Brower C. S., Piatkov K. I., Varshavsky A. (2013). Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol. Cell. 50 161–171. 10.1016/j.molcel.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower C. S., Varshavsky A. (2009). Ablation of arginylation in the mouse N-end rule pathway: loss of fat, higher metabolic rate, damaged spermatogenesis, and neurological perturbations. PLoS One 4:e7757. 10.1371/journal.pone.0007757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty G., Ingoglia N. A. (1993). N-terminal arginylation and ubiquitin-mediated proteolysis in nerve regeneration. Brain Res. Bull. 30 439–445. 10.1016/0361-9230(93)90276-h [DOI] [PubMed] [Google Scholar]
- Chakraborty G., Leach T., Zanakis M. F., Ingoglia N. A. (1986). Posttranslational protein modification by amino acid addition in regenerating optic nerves of goldfish. J. Neurochem. 46 726–732. 10.1111/j.1471-4159.1986.tb13032.x [DOI] [PubMed] [Google Scholar]
- Decca M. B., Bosc C., Luche S., Brugiere S., Job D., Rabilloud T., et al. (2006). Protein arginylation in rat brain cytosol: a proteomic analysis. Neurochem. Res. 31 401–409. 10.1007/s11064-005-9037-z [DOI] [PubMed] [Google Scholar]
- Decottignies A., Sanchez-Perez I., Nurse P. (2003). Schizosaccharomyces pombe essential genes: a pilot study. Genome Res. 13 399–406. 10.1101/gr.636103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka K., Singh A., Chakraborty S., Mukhopadhyay R., Saha S. (2016). Protein arginylation regulates cellular stress response by stabilizing HSP70 and HSP40 transcripts. Cell Death Discov. 2:16074. 10.1038/cddiscovery.2016.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar J. C., Reid R. J. D., Rothstein R. (2010). ScreenMill: a freely available software suite for growth measurement, analysis and visualization of high-throughput screen data. BMC Bioinformatics 11:353. 10.1186/1471-2105-11-353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J., Fedyshyn Y., Koh J. L., Prasad T. S., Chahwan C., Chua G., et al. (2008). Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 105 16653–16658. 10.1073/pnas.0806261105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan C. D. S., Rodriguez-Lopez M., Ruis P., Bahler J., Mata J. (2018). General amino acid control in fission yeast is regulated by a nonconserved transcription factor, with functions analogous to Gcn4/Atf4. Proc. Natl. Acad. Sci. U.S.A. 115 E1829–E1838. 10.1073/pnas.1713991115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenach P. A., Schikora F., Posern G. (2014). Inhibition of arginyltransferase 1 induces transcriptional activity of myocardin-related transcription factor A (MRTF-A) and promotes directional migration. J. Biol. Chem. 289 35376–35387. 10.1074/jbc.M114.578674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G., Nislow C. (2014). The yeast deletion collection: a decade of functional genomics. Genetics 197 451–465. 10.1534/genetics.114.161620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman C. S., Wood V., Fantes P. A. (2015). An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics 201 403–423. 10.1534/genetics.116.187088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack D. L., Chakraborty G., Ingoglia N. A. (1992). Ubiquitin is associated with aggregates of arginine modified proteins in injured nerves. Neuroreport 3 47–50. 10.1097/00001756-199201000-00012 [DOI] [PubMed] [Google Scholar]
- Kaji H., Kaji A. (2012). Global cellular regulation including cardiac function by post-translational protein arginylation. J. Mol. Cell Cardiol. 53 314–316. 10.1016/j.yjmcc.2012.06.009 [DOI] [PubMed] [Google Scholar]
- Karakozova M., Kozak M., Wong C. C., Bailey A. O., Yates J. R., III, Mogilner A., et al. (2006). Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science 313 192–196. 10.1126/science.1129344 [DOI] [PubMed] [Google Scholar]
- Krawchuk M. D., Wahls W. P. (1999). High-efficiency gene targeting in Schizosaccharomyces Pombe using a modular, PCR-based approach with long tracts of flanking homology. Yeast 15, 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Birnbaum M. D., Patel D. M., Morgan W. M., Singh J., Barrientos A., et al. (2016). Posttranslational arginylation enzyme Ate1 affects DNA mutagenesis by regulating stress response. Cell Death Dis. 7:e2378. 10.1038/cddis.2016.284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka S., Leu N. A., Pavlov I., Han X., Ribeiro P. A., Xu T., et al. (2012). Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J. Mol. Cell Cardiol. 53 333–341. 10.1016/j.yjmcc.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka S., Leu N. A., Zhang F., Bunte R., Saha S., Wang J., et al. (2010). Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet. 6:e1000878. 10.1371/journal.pgen.1000878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. T., Kashina A. S., Davydov I. V., Hu R. G., An J. Y., et al. (2002). An essential role of N-terminal arginylation in cardiovascular development. Science 297 96–99. 10.1126/science.1069531 [DOI] [PubMed] [Google Scholar]
- Lee M. J., Kim D. E., Zakrzewska A., Yoo Y. D., Kim S. H., et al. (2012). Characterization of arginylation branch of N-end rule pathway in G-protein-mediated proliferation and signaling of cardiomyocytes. J. Biol. Chem. 287 24043–24052. 10.1074/jbc.M112.364117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D., Chakraborty G., Ingoglia N. A. (1990). Post-translational modification of proteins by arginine and lysine following crush injury and during regeneration of rat sciatic nerves. Restor. Neurol. Neurosci. 2 53–61. 10.3233/RNN-1990-2201 [DOI] [PubMed] [Google Scholar]
- Masdehors P., Glaisner S., Maciorowski Z., Magdelenat H., Delic J. (2000). Ubiquitin-dependent protein processing controls radiation-induced apoptosis through the N-end rule pathway. Exp. Cell Res. 257 48–57. 10.1006/excr.2000.4870 [DOI] [PubMed] [Google Scholar]
- McGary K. L., Park T. J., Woods J. O., Cha H. J., Wallingford J. B., Marcotte E. M. (2010). Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc. Natl. Acad. Sci. U.S.A. 107 6544–6549. 10.1073/pnas.0910200107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Casagrande J. T., Thomas P. D. (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8 1551–1566. 10.1038/s41596-019-0128-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Huang X., Ebert D., Mills C., Guo X., et al. (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14 703–721. 10.1038/s41596-019-0128-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S., Klar A., Nurse P. (1991). Molecular genetic-analysis of fission yeast Schizosaccharomyces-pombe. Methods Enzymol. 194 795–823. [DOI] [PubMed] [Google Scholar]
- Piatkov K. I., Brower C. S., Varshavsky A. (2012). The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. U.S.A. 109 E1839–E1847. 10.1073/pnas.1207786109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R., Wong C. C., Xu T., Leu N. A., Dong D. W., Guo C., et al. (2008). Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development 135 3881–3889. 10.1242/dev.022723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R., Zhang F., Colavita K., Leu N. A., Kurosaka S., Kumar A., et al. (2015). Arginyltransferase suppresses cell tumorigenic potential and inversely correlates with metastases in human cancers. Oncogene 35 4058–4068. 10.1038/onc.2015.473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., Kashina A. (2011). Posttranslational arginylation as a global biological regulator. Dev. Biol. 358 1–8. 10.1016/j.ydbio.2011.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., Mundia M. M., Zhang F., Demers R. W., Korobova F., Svitkina T., et al. (2010). Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Mol. Biol. Cell 21 1350–1361. 10.1091/mbc.e09-09-0829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., Wong C. C., Xu T., Namgoong S., Zebroski H., Yates J. R., III, et al. (2011). Arginylation and methylation double up to regulate nuclear proteins and nuclear architecture in vivo. Chem. Biol. 18 1369–1378. 10.1016/j.chembiol.2011.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyne-Athwal S., Chakraborty G., Gage E., Ingoglia N. A. (1988). Comparison of posttranslational protein modification by amino acid addition after crush injury to sciatic and optic nerves of rats. Exp. Neurol. 99 281–295. 10.1016/0014-4886(88)90148-3 [DOI] [PubMed] [Google Scholar]
- Shyne-Athwal S., Riccio R. V., Chakraborty G., Ingoglia N. A. (1986). Protein modification by amino acid addition is increased in crushed sciatic but not optic nerves. Science 231 603–605. 10.1126/science.3080804 [DOI] [PubMed] [Google Scholar]
- Spirek M., Benko Z., Carnecka M., Rumpf C., Cipak L., Batova M., et al. (2010). S-pombe genome deletion project An update. Cell Cycle 9 2399–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A. L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 D607–D613. 10.1093/nar/gky1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. (2011). The N-end rule pathway and regulation by proteolysis. Protein Sci. 20 1298–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Han X., Leu N. A., Sterling S., Kurosaka S., Fina M., et al. (2017a). Protein arginylation targets alpha synuclein, facilitates normal brain health, and prevents neurodegeneration. Sci. Rep. 7:11323. 10.1038/s41598-017-11713-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Pavlyk I., Vedula P., Sterling S., Leu N. A., Dong D. W., et al. (2017b). Arginyltransferase ATE1 is targeted to the neuronal growth cones and regulates neurite outgrowth during brain development. Dev. Biol. 430 41–51. 10.1016/j.ydbio.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Han X., Saha S., Xu T., Rai R., Zhang F., et al. (2011). Arginyltransferase is an ATP-independent self-regulating enzyme that forms distinct functional complexes in vivo. Chem. Biol. 18 121–130. 10.1016/j.chembiol.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Pejaver V. R., Dann G. P., Wolf M. Y., Kellis M., Huang Y., et al. (2018). Target site specificity and in vivo complexity of the mammalian arginylome. Sci. Rep. 8:16177. 10.1038/s41598-018-34639-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. M., Ingoglia N. A. (1997). N-terminal arginylation of sciatic nerve and brain proteins following injury. Neurochem. Res. 22 1453–1459. 10.1023/a:1021998227237 [DOI] [PubMed] [Google Scholar]
- Watanabe D., Kikushima R., Aitoku M., Nishimura A., Ohtsu I., Nasuno R., et al. (2014). Exogenous addition of histidine reduces copper availability in the yeast Saccharomyces cerevisiae. Microb. Cell 1 241–246. 10.15698/mic2014.07.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley D. J., Juan I., Le H., Cai X., Baumbach L., Beattie C., et al. (2014). Yeast Augmented Network Analysis (YANA): a new systems approach to identify therapeutic targets for human genetic diseases. F1000Research 3:121. 10.12688/f1000research.4188.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler M. E., Ramos-Montanez S. (2009). Biosynthesis of Histidine. EcoSal Plus 3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. C., Xu T., Rai R., Bailey A. O., Yates J. R., III, Wolf Y. I., et al. (2007). Global analysis of posttranslational protein arginylation. PLoS Biol. 5:e258. 10.1371/journal.pbio.0050258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N. S., Chakraborty G., Hassankhani A., Ingoglia N. A. (1993). N-terminal arginylation of proteins in explants of injured sciatic nerves and embryonic brains of rats. Neurochem. Res. 18 1117–1123. 10.1007/bf00978361 [DOI] [PubMed] [Google Scholar]
- Yanagida M. (2002). The model unicellular eukaryote, Schizosaccharomyces pombe. Genome Biol. 3 comment2003.1–comment2003.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanakis M. F., Chakraborty G., Sturman J. A., Ingoglia N. A. (1984). Posttranslational protein modification by amino acid addition in intact and regenerating axons of the rat sciatic nerve. J. Neurochem. 43 1286–1294. 10.1111/j.1471-4159.1984.tb05385.x [DOI] [PubMed] [Google Scholar]
- Zhang F., Patel D. M., Colavita K., Rodionova I., Buckley B., Scott D. A., et al. (2015). Arginylation regulates purine nucleotide biosynthesis by enhancing the activity of phosphoribosyl pyrophosphate synthase. Nat. Commun. 6:7517. 10.1038/ncomms8517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Saha S., Kashina A. (2012). Arginylation-dependent regulation of a proteolytic product of talin is essential for cell-cell adhesion. J. Cell Biol. 197 819–836. 10.1083/jcb.201112129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Saha S., Shabalina S. A., Kashina A. (2010). Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 329 1534–1537. 10.1126/science.1191701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Donnelly R., Ingoglia N. A. (1998). Evidence that oxidized proteins are substrates for N-terminal arginylation. Neurochem. Res. 23 1411–1420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.