

Abstract
The mechanical properties of synthetic hydrogels traditionally have been controlled with the concentration, molecular weight, or stoichiometry of the macromolecular building blocks used for hydrogel formation. Recently, the rate of formation has been recognized as an important and effective handle for controlling the mechanical properties of these water-swollen polymer networks, owing to differences in network heterogeneity (e.g., defects) that arise based on the rate of gelation. Building upon this, in this work, we investigate a rate-based approach for controlling mechanical properties of hydrogels both initially and temporally with light. Specifically, synthetic hydrogels are formed with visible light-initiated thiol-ene ‘click’ chemistry (PEG-8-norbornene, dithiol linker, LAP photoinitiator with LED lamp centered at 455 nm), using irradiation conditions to control the rate of formation and the mechanical properties of the resulting hydrogels. Further, defects within these hydrogels were subsequently exploited for temporal modulation of mechanical properties with a secondary cure using low doses of long wavelength UV light (365 nm). The elasticity of the hydrogel, as measured with Young’s and shear moduli, was observed to increase with increasing light intensity and concentration of photoinitiator used for hydrogel formation. In situ measurements of end group conversion during hydrogel formation with magic angle spinning (MAS 1H NMR) correlated with these mechanical properties measurements, suggesting that both dangling end groups and looping contribute to the observed mechanical properties. Dangling end groups provide reactive handles for temporal stiffening of hydrogels with a secondary UV-initiated thiol-ene polymerization, where an increase in Young’s modulus by a factor of ~ 2.5x was observed. These studies demonstrate how the rate of photopolymerization can be tuned with irradiation wavelength, intensity, and time to control the properties of synthetic hydrogels, which may prove useful in a variety of applications from coatings to biomaterials for controlled cell culture and regenerative medicine.
Introduction
Synthetic hydrogels, water-swollen polymer networks, are of great interest owing to the ease of property control that they afford and their utility for interfacing with biological systems with applications ranging from antifouling coatings to extracellular matrix mimics and therapeutic delivery vehicles. Control of the mechanical properties is highly desirable for directing biological response and, traditionally, has been achieved by using different concentrations, molecular weights, or stoichiometries of macromolecular building blocks for hydrogel formation.1–3 With these approaches, the formation of materials with different mechanical properties in the presence of biological systems (e.g., live cells) has been demonstrated with a variety of ‘click’ chemistries. Commonly, the end groups of multifunctional polymers (e.g., multiarm poly(ethylene glycol) [PEG]) are modified with complementary ‘click’ reactive pairs for a rapid network formation, including oxime ligation, azide-alkyne, Michael addition reactions that occur upon mixing, or radically-mediated ‘thiol-ene’ reactions that allow photoinitiation.4 Notably, the rate of formation of these types of step-growth networks recently has been established as a handle for controlling the mechanical properties of these materials. For example, in non-photoinitiated, step-growth polymerized hydrogels (i.e., acid catalyzed oxime ligation), the kinetics or rate of hydrogel formation has been shown to influence the number of defects within the resulting hydrogel network structure, including dangling end groups and elastically ineffective loops.5 These defects in the network structure have been shown to produce a decrease in the observed crosslink density, and therefore modulus, of the resulting hydrogels, demonstrating a kinetically-controlled strategy for tuning hydrogel mechanical properties that goes beyond altering monomer composition or controlling total reaction time/functional group conversion.5–8 With this approach, pH and buffer strength have been adjusted to control rate and thereby elastic modulus while maintaining constant polymer concentration, stoichiometry, and molecular weight.1,5 However, there are few examples of utilizing this rate-based approach for controlling the mechanical properties of photopolymerized step-growth networks, which would allow on-demand, user-control of both the timing and rate of gelation.
A variety of methods previously have been used for photoinitiation of step-growth polymerizations to form robust hydrogels, particularly by photoinitiated thiol-ene click chemistry. For example, the type I photoinitiator lithium acylphosphinate (LAP) has been widely used for the formation of thiol-ene hydrogels in the presence of live cells owing to the favorable water solubility, cytocompatibility, and rapid and efficient initiation that LAP affords with low doses of long wavelength UV light (365 nm).9 Further, by increasing the concentration of LAP, broad spectrum visible light (400–700 nm) has been demonstrated for the formation of thiol-ene hydrogels, although utilized to a lesser extent.10 The type II photoinitiator eosin-Y with visible light (400–700 nm) also has been utilized, particularly in the presence of sensitive cell types.10–13 Based on these observations, we hypothesized that the rate of formation for these types of hydrogels could be controlled not only with the light intensity and exposure time, but also selection of the light wavelength in comparison to the molar absorptivity of the photoinitiator. For example, LAP has a significant molar absorptivity at 365 nm, allowing rapid polymerization and gelation upon the application of long wavelength UV light, and eosin-Y has a significant molar absorptivity at 515 nm, allowing rapid polymerization with broad spectrum visible light (400–700 nm).14 Moving to a wavelength of light with less significant absorption by the photoinitiator results in a slower rate of formation of the hydrogel, such as using LAP with visible light irradiation.10 While this decrease in rate traditionally has been seen as undesirable, the resulting increase in polymerization time that it imparts has the potential to further increase user control over not only the rate of hydrogel formation, but also the mechanical and network properties of the resulting hydrogels through rate-controlled defect formation. The incorporation of defects and control of their formation is of particular interest in three-dimensional (3D) cell culture applications: for example, recent work has demonstrated that local heterogeneities in hydrogel network structure are beneficial for promoting the connectivity of the extracellular matrix that is deposited by encapsulated cells and aids in neotissue formation.15
Beyond initial mechanical properties, stiffening of hydrogels at desired time points is of particular interest for controlled cell culture applications, where both the initial modulus of the matrix and temporal changes in it have been observed to influence cell functions and fates, such as phenotypic switching of wound healing cells and differentiation of stem cells.2 Light-triggered increases in crosslink density, and thereby stiffening, enable user control for temporal tuning of mechanical properties, which previously has been achieved by two overarching approaches, (i) photoisomerization16,17 or (ii) secondary polymerization. Owing to its chemical simplicity the latter strategy often is used, where a stoichiometric excess of relevant functional groups is included in the hydrogel based on the composition of the original precursor solution and later reacted by a secondary polymerization. With this strategy, approaches have been developed that utilize two different polymerization mechanisms for hydrogel formation and subsequent stiffening: for example, (i) base-catalyzed Michael addition reaction followed by secondary photoinitiated chain growth polymerization or (ii) radically-mediated step-growth thiol-ene followed by secondary enzyme ligation reaction.11,18,19 Alternatively, the same polymerization mechanism can be used for both hydrogel formation and stiffening: for example, multi-arm PEG hydrogels have been formed off stoichiometry by photoinitiated thiol-ene step-growth polymerization providing excess functional groups to facilitate subsequent stiffening upon the addition of more multifunctional macromers, LAP, and a second dose of light.20,21 This secondary polymerization approach for hydrogel stiffening may afford opportunities for modulating hydrogel properties more broadly, such as for subsequent crosslinking of dangling end defects within hydrogels formed on stoichiometry.
Building upon these seminal advances, we hypothesized that (i) the rate of photopolymerization during initial network formation could be used to control the modulus of hydrogels and (ii) the resulting dangling end defects could be used to temporally increase modulus with a second photopolymerization. To test this, we investigated the use of visible light (LED centered at 455 nm) with the photoinitiator LAP for controlling the rate of formation and thereby the mechanical properties of photopolymerized thiol-ene hydrogels (Figure 1). Different light intensities, irradiation times, and concentrations of photoinitiator were used to control the rate of gelation, and the mechanical properties of the resulting hydrogels were measured in situ and after equilibrium swelling. To better understand the source of defects that contributed to differences in hydrogel mechanical properties, end group conversion was monitored with magic angle spinning nuclear magnetic resonance (MAS 1H NMR) spectroscopy and compared to mechanical properties over the time of polymerization, which suggested both dangling end groups and looping were present. Control of reactive end group availability was exploited to stiffen hydrogels with a secondary thiol-ene crosslinking reaction initiated with 365 nm light. Overall, these studies demonstrated the high level of mechanical property tunability afforded by visible light initiation for controlling the rate of hydrogel formation and the potential utility of using dangling end defects generated with this approach for post-polymerization modification.
Figure 1. Overview of hydrogel formation.
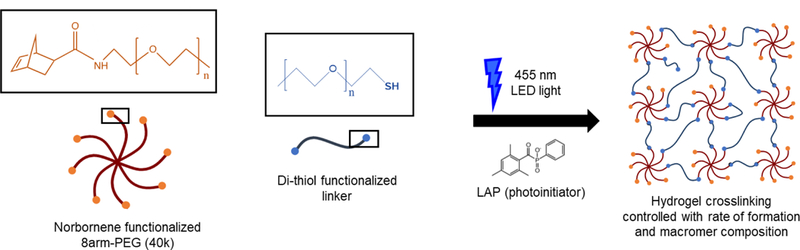
Synthetic hydrogels with tunable properties were formed by photoinitiated, step-growth thiol-ene ‘click chemistry’ with a 40-kDa 8-arm norbornene-functionalized poly(ethylene glycol) macromer (PEG-8-Nb), 3.4-kDa di-thiol functionalized poly(ethylene glycol) linker (PEG-2-SH; for mechanical property studies) or di-thiol enzymatically-degradable linker (GCRDVPMS↓MRGGDRCG; for cell encapsulation studies), LAP photoinitiator, and visible light LED (centered at 455 nm). Both macromer composition and rate of formation were used to tune the mechanical properties of the resulting covalent networks.
Experimental
PEG functionalization
Amine-terminated 8-arm PEG (PEG-8-NH2, 8arm PEG Amine (hexaglycerol), HCl Salt, 40kDa; JenKem) was functionalized with norbornene end groups to form norbornene-terminated 8-arm PEG (PEG-8-Nb) as previously described.22 Briefly, PEG-8-NH2 (10 g, 1 eq) was dissolved in anhydrous dimethylformamide (45 mL, DMF; Fisher Scientific) in a 100 mL round bottom flask. Separately, norbornene carboxylic acid (0.608 g, 2.2 eq; Sigma Aldrich), 4-methylmorpholine (0.99 mL, 36 eq; Sigma Aldrich), and HATU (1.521 g, 2 eq; ChemPep) were dissolved in 7 mL of anhydrous DMF in a 250 mL round bottom flask. The PEG solution was added drop-wise to the norbornene solution and allowed to react overnight. Polymer was precipitated into diethyl ether (500 mL; Fisher Scientific) and collected by vacuum filtration. The conjugated polymer was dissolved in deionized water, dialyzed (MWCO 1000 g/mol; Spectrum Laboratories) for 48 hours, and recovered by lyophilization (FreeZone 4.5 Plus, Labconco). Product purity was confirmed by 1H NMR in DMSO-d6. Modification was determined by integration of norbornene peaks (~ 78%). (Figure S1)
Hydrogel preparation
Monomer stocks were prepared by dissolving each in Dulbecco’s phosphate buffer saline (PBS; ThermoFisher; Paisley, Scotland, UK): (i) PEG-8-Nb (50 mM Nb), (ii) LAP (25.5 mM) synthesized as previously described,14 and (iii) 3.4 kDa PEG di-thiol linker (PEG-2-SH; Laysan Bio, Arab, AL) (100 mM thiol). Stock solutions were aliquoted and stored at −80 °C until use.
Hydrogel precursor solutions were prepared by diluting stock solutions to final concentrations for forming hydrogels at various PEG wt% concentrations. Most hydrogels were formed on stoichiometry (norbornene:thiol stoichiometry of 1:1). For example, 6 wt% PEG hydrogels on stoichiometry were formed with 1.4 mM PEG-8-Nb (6 wt%; 8.8 mM Nb functional groups), 4.4 mM PEG-2-SH linker (8.8 mM SH functional groups), and 4 mM LAP in PBS containing 50 U/mL penicillin, 50 μg/mL streptomycin, and 0.2% fungizone.
For cell encapsulations, a di-thiol cell-degradable linker (GCRDVPMS↓MRGGDRCG) was used and 2 mM CGRGDS pendant peptide incorporated to promote cell adhesion. Both GCRDVPMS↓MRGGDRCG and CGRGDS sequences were synthesized using standard FMOC-chemistry on an automated peptide synthesizer (PS3 Peptide Synthesizer; Protein Technologies, Inc, Tucson, AZ). The peptides were synthesized on Rink Amide MBHA resin (Novabiochem), and all amino acids were double coupled. Peptides were cleaved from resin for 4 hours in 95% trifluoroacetic acid (Acros Organics), 2.5% triisopropylsilane (Acros Organics), and 2.5% water (all percentages v/v) supplemented with 50 mg/mL dithiothreitol (Research Products International). After cleavage, all peptides were precipitated in cold diethyl ether (9x excess volume) overnight at 4 °C and purified by reverse-phase high performance liquid chromatography (HPLC; XBridge BEH C18 OBD 5 μm column; Waters, Milford, MA) with a linear water-acetonitrile (ACN) gradient (Water:ACN 95:5 to 45:5; 1.17% change in water per minute). Purified peptides were lyophilized, and their molecular weights were verified by mass spectrometry (Figures S2). Peptide stocks were dissolved in PBS, aliquoted, and stored at −80 °C. Ellman’s assay was performed to determine the thiol concentration for each peptide stock. Briefly, peptide stock concentrations were diluted 100x in Ellman’s reaction buffer (0.1 M sodium phosphate, 1 mM ethylenediaminetetraacetic acid at pH 7.5–8), and 20 μL of this solution was added to a 96-well plate (n=3). Ellman’s reagent (3.6 μL, 4 mg in 1 mL Ellman’s reaction buffer) was diluted in 180 μL of Ellman’s reaction buffer, then added to wells containing diluted peptide or standard (0–100 mM cysteine in PBS for generation of a calibration curve). Absorbance was measured at 405 nm (Synergy H4 plate reader; BioTek), and the thiol concentration for peptide stocks was calculated using the standard calibration curve.
In situ gelation
In situ rheometry was performed on a Discovery Hybrid Rheometer 3 (DHR; TA Instruments, New Castle, DE) with a curing LED plate with output centered at 455 nm. Hydrogel precursor solutions (10 μL) were pipetted onto the rheometer (8-mm flat plate geometry with a gap of 200 μm). The effects of different LED intensities and concentrations of PEG-8-Nb and LAP were examined: 2 mWcm−2 or 10 mWcm−2 LED intensity, 2 wt% or 10 wt% PEG-8-Nb, and 0.5 or 2 mM LAP. Hydrogel crosslinking was monitored by measuring storage (G’) and loss (G”) moduli at 0.5% applied strain and 2 rads−1 frequency upon irradiation. The time to complete gelation, or gelation time, was assessed, which was defined as when the modulus of the hydrogel was no longer changing (i.e., when the rate of change of the modulus was within 1% for consecutive points) and observed to be within 2 minutes of commencing irradiation for all compositions.23 Frequency sweeps at 1% strain were performed after complete gelation to measure the final moduli of hydrogels formed in situ on the rheometer. All rheometric measurements were performed within the linear viscoelastic regime.
Bulk hydrogel formation
A visible light LED lamp with output centered at 455 nm (M455L3-C1; ThorLabs) was clamped to the top of a ring stand, and the distance between the LED and collimating lens was adjusted to achieve a uniform light intensity across the spot size. The intensity of the light was measured using a programmable photometer (455 nm, IL1400A Radiometer/Photometer; International Light Technologies), and the LED driver was adjusted to achieve the desired light intensity (either 70 mWcm−2 (the lowest intensity observed to give consistent gel formation) or 90 mWcm−2 (the highest intensity achieved with this apparatus)). Syringe molds (e.g., 1 mL syringes with ends cut off) were filled with 20 μL of polymer solution and then placed uncovered under the LED for irradiation. The resulting hydrogels were placed in well plates and twice rinsed with PBS. After the final rinse, the hydrogels were swollen in PBS at room temperature overnight.
Bulk hydrogel dynamic mechanical analysis
Dynamic mechanical analysis (DMA) was performed to measure the moduli of equilibrium swollen hydrogels. Hydrogels swollen at room temperature were placed between flat plates on a dynamic mechanical analyzer (RSA-G2; TA Instruments) such that the axial (normal) force on each gel was 0.01 N. Frequency sweeps were conducted from 0.1 to 10 Hz, at 1.0% strain and room temperature, to obtain Young’s modulus (E).
Human mesenchymal stem cell culture
Human mesenchymal stem cells (hMSCs; Lonza, Walkersville, MD) were selected as a model human primary cell type and cultured in complete growth medium (Dulbecco’s Modified Eagle Medium (DMEM) low glucose, sodium pyruvate, L-Glutamine (11885092 Gibco, Grand Island, US) supplemented with 10% fetal bovine serum, certified, US origin (Gibco, Grand Island, US), 50 U/mL penicillin, 50 μg/mL streptomycin, and 20 μM fibroblast growth factor (bFGF)). Cells were propagated in tissue culture treated T-175 flasks incubated at 37°C with 5% CO2. Cells were passaged at confluency of approximately 85% using Trypsin-EDTA (0.5%, no phenol red (Gibco, Grand Island, US)) for cell detachment.
Assessing cell viability and metabolic activity in hydrogels formed with visible light LED lamp
To assess any impact of photopolymerization with visible light LED lamp on the viability of human primary cells, hMSCs (passage 10) were encapsulated as a single cell suspension (5 × 106 hMSCs per mL) in hydrogel precursor solution (100,000 hMSCs per 20 μL hydrogel). Briefly, a bulk hydrogel precursor solution as described earlier was prepared for the formation of 9 20-μL hydrogels. hMSCs were dissociated with 5 mL of Tryspin-EDTA and resuspended in complete media. Cells were counted using a hemocytometer, and hMSCs aliquots for 9 gels (at 5 × 106 hMSCs per mL) were spun down at 1200 RPM for 3 minutes. Cells were re-suspended in precursor solution for forming 6 wt% PEG hydrogels, and encapsulated in hydrogels formed in syringe molds (irradiation with 70 or 90 mWcm−2 at 455 nm for 5 minutes using the ThorLabs 455 nm LED). One gel was polymerized at a time and each placed into a well of a 48-well non-tissue culture treated plate with 500 μL of growth medium. Initial culture medium was replaced after 1 hour of incubation. Cell-gel constructs in culture medium, which was replaced every 2–3 days, were incubated at 37°C with 5% CO2 to support cell growth.
The viability of encapsulated hMSCs was assessed using a LIVE/DEAD Viability/Cytotoxicity Kit (ThermoFisher Scientific) (days 1, 3, and 7 after encapsulation). Calcein AM detects esterase activity of cells, producing a fluorescent green dye (ex/em ~ 495 nm/515 nm) in the cytosol of living cells, whereas ethidium homodimer-1 is a fluorescent red dye (ex/em ~ 495 nm/635 nm) that binds to nucleic acids and labels the nuclei of cells with damaged membranes, indicating dead cells. Briefly, at time points of interest, cell-gel constructs (n = 3) were washed 2x with 500 μL of PBS for 5 minutes followed by a 30-minute incubation (37 °C at 5% CO2) with 400 μL of PBS containing calcein AM (2 μM) and ethidium homodimer-1 (4 μM). After staining, hydrogels were again washed (2× 500 μL of PBS for 5 minutes) before imaging. Hydrogels were transferred to a chamber slide (Nunc Lab-Tek II Chamber Slide, Glass, 1 well) and imaged with a confocal microscope (Zeiss LSM 800, 10x objective, 200-μm z-stack with frame size of 1024 × 1024 for each image, 3 images per hydrogel sample). Orthogonal projections were made of each z-stack, and live (green) and dead (red) cells were counted using ImageJ. The percentage of viable cells was calculated by the number of green cells/total number of cells x 100%.
AlamarBlue® cell viability reagent (Thermo Fisher) was used to examine hMSC metabolic activity in hydrogels following a modified version of a previously published protocol.22 hMSCs were encapsulated in hydrogels (n = 3) and cultured for up to 7 days. At time points of interest (days 1, 3, and 7), alamarBlue® reagent (10x) was diluted 1:10 in phenol red-free growth medium. The culture medium for each hydrogel was replaced with this solution (500 μL per hydrogel cultured in 48-well plate) and incubated for 4 hours (37°C at 5% CO2). Conditioned culture media were collected from each well, and hydrogels were replenished with fresh standard culture medium. Conditioned media (100 μL from each well) were transferred to a black 96-well plate, and fluorescence was measured (BioTek Synergy H4 Hybrid Reader, ex/em ~ 560 nm/590 nm).
Magic angle spinning NMR
Hydrogel precursor solutions (6 wt% PEG-8-Nb, PEG-2-SH (1:1 Nb:SH), 4 mM LAP) were prepared in D2O, placed into rotor inserts, and exposed to light using the same method as described for bulk hydrogel formation (70 mWcm−2 at 455 nm ThorLED) or using light conditions that previously had been shown to reach full conversion as a control (2 mM LAP, 365 nm at 10 mWcm−2; Omnicure S2000; Excelitas, 365 nm bandpass filter)24, for various times of light exposure. 1H spectra were acquired using a 4.0 mm HRMAS probe on a 600 MHz spectrometer, tuned to a 1H frequency of 600.323 MHz. For all samples, the spin frequency was set to 6000 Hz, and spectra were obtained using a zg pulse program with 90° pulse and 128 scans. The absolute value of the norbornene peak integration, measured by MestreNova software (Mestrelab Research), was used to monitor the consumption of norbornene end groups, where peak integration was normalized to t = 0 and then subtracted from 1 to calculate the percentage of reacted norbornene end groups.
Bulk hydrogel stiffening
Fresh monomer solutions were prepared for use in hydrogel stiffening experiments. Based on a modified version of published protocols,20,21 ‘stiffening solution’ was prepared with PEG-8-Nb (40kDa, 4 mM Nb), PEG-4-SH (10 kDa, 4 mM SH), and LAP (2 mM) in PBS. After swelling overnight in PBS, hydrogels were incubated with stiffening solution (150 μL) at room temperature for ≥1 hour on a rocker. To examine stiffening upon irradiation, hydrogels swollen in stiffening solution were placed on parallel plate rheometer (axial force 0.1 N; AR-G2; TA Instruments) with a UV-visible light attachment connected to a light source (365 nm bandpass filter; Omnicure S2000; Excelitas). The shear modulus was monitored, measuring at a strain of 1% and frequency of 6.0 rad s−1, as the hydrogels were exposed to light (10 mWcm−2 at 365 nm). To evaluate the bulk properties of the hydrogels post-stiffening, hydrogels were irradiated with light (10 mWcm−2 at 365 nm for 2 minutes) and then measured by DMA.
Statistics
All experiments included at least three replicates of each condition. Data are reported as the mean ± standard error. Statistical significance was determined by p-value (p < 0.05) using two-sided Student’s t-test or one-way ANOVA with Tukey pairwise comparisons.
Results and discussion
In situ polymerization with visible light LED for probing effects of photopolymerization conditions on hydrogel properties
We first investigated how the rate of gelation and resulting mechanical properties of hydrogels could be controlled with the conditions used for photopolymerization, including light intensity and concentrations of photoinitiator and PEG-8-Nb (Figure 2). Ideal step-growth thiol-ene photopolymerizations are known to undergo a mechanism where free-radicals generated upon light exposure abstract a hydrogen from a thiol to generate a thiyl radical that reacts with an electron-rich alkene; through chain-transfer and radical propagation, thioether bonds are formed until termination.24,25 In this context, we expected that increasing the light intensity, photoinitiator concentration, and PEG-8-Nb concentration would increase the rate of photopolymerization and decrease the gelation time. While this difference in rate of formation was expected to impact the final modulus of the hydrogel, as noted in the Introduction, the potential magnitude of this impact was unknown. To examine this, low and high conditions for key variables were investigated, including PEG-8-Nb macromer concentration (2 wt% and 10 wt%), LAP photoinitiator concentration (0.5 and 2 mM), and LED intensity (2 and 10 mWcm−2), using a rheometer with a LED bottom plate attachment (output centered at 455 nm) for in situ monitoring of gelation and mechanical properties.
Figure 2. In situ rheometry of hydrogels formed with visible light LED lamp.
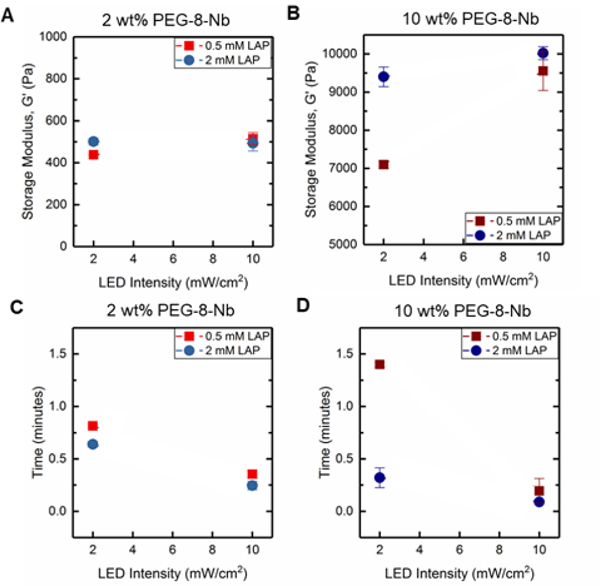
Storage moduli were measured in situ on a rheometer with LED attachment (with output centered at 455 nm), and the impact of macromer concentration, initiator concentration, and light intensity assessed with low and high values of each variable: G’ for a) 2 wt% and b) 10 wt% PEG-8-Nb and gelation time for c) 2 wt% and d) 10 wt% PEG-8-Nb.
At low concentration of PEG-8-Nb and low LED intensity, increased LAP concentration resulted in an increased storage modulus, G’ from 438 ± 3 to 501 ± 16 Pa (*p-value < 0.05) (Figure 2A). However, with high LED intensity, increased LAP concentration did not have a significant impact on hydrogel modulus, G’ = 438 ± 3 to G’ = 515 ± 29 Pa (low LAP) and G’ = 501 ± 16 to G’ = 493 ± 37 Pa (high LAP), respectively. The time to complete gelation, or gelation time, also was measured, defined as when the modulus of the hydrogel was no longer changing (i.e., when the rate of change of the modulus was within 1% for consecutive points). When comparing gelation times, there was a significant decrease in the gelation time with increased light intensity (Figure 2C). For example, at the high LAP concentration, increased LED intensity resulted in a decreased gelation time from t = 0.64 ± 0.03 to t = 0.25 ± 0.04 min (**p-value < 0.01). These data suggest that, at a low concentration of PEG-8-Nb, increased light intensity or photoinitiator concentration, which should produce more free-radicals, statistically increased the rate of step-growth polymerization with modest increases in modulus.
Notably, similar and more significant trends in mechanical properties were observed when forming hydrogels at the higher concentration of PEG-8-Nb (10 wt%) (Figure 2B). At lower LAP concentration (0.5 mM) and LED intensity (2 mWcm−2), mechanical properties were significantly lower, G’ = 7098 ± 101 Pa, than for hydrogels formed with either increased LAP concentration, G’ = 9401 ± 257 Pa (**p-value < 0.01), or increased light intensity, G’ = 9556 ± 513 Pa (*p-value < 0.05). Additionally, these changes corresponded with significant decreases in gelation time (Figure 2D). Smaller, non-statistical differences in moduli were measured upon increasing light intensity for hydrogels formed with a high LAP concentration, with storage moduli of G’ = 9401 ± 257 and G’ = 10021 ± 170 Pa, respectively.
Overall, these studies provide insights into handles for controlling the rate of hydrogel formation and hydrogel mechanical properties. As expected, changes in PEG-8-Nb concentration resulted in large changes in mechanical properties, where 2 wt% PEG-8-Nb hydrogels have storage moduli on the order of ~ 100 Pa and 10 wt% PEG-8-Nb on the order of ~ 10,000 Pa. Indeed, increased polymer density during hydrogel formation has been widely used as a handle for controlling the mechanical properties of the resulting hydrogel, where it has been demonstrated that increasing the polymer density during formation decreases the number of looping defects present in the final network structure, thus increasing the final modulus.7,26,27 More interestingly, significant changes in modulus, which correlated with inverse changes in gelation time, were achieved by either increasing the LAP concentration or increasing the light intensity. This result is supported by previous findings, which have correlated the increase in photoinitiator concentration and light intensity with increased final modulus and reaction rates in photoinitiated systems.28,29 Further, these trends were more substantial and pronounced at the high PEG-8-Nb concentration (10 wt%), suggesting that tuning of modulus with these rate-based handles may be better achieved with higher macromer concentrations.
Modulus of bulk hydrogels controlled with visible light LED irradiation intensity and macromer concentration
In situ rheometry provides important information for identifying trends in handles for controlling hydrogel mechanical properties, especially with regards to measuring rate of gelation. However, synthesis of bulk hydrogels within molds off the rheometer is desirable for scale up and use these materials in a wide variety of applications, including cell culture. To translate the findings of in situ rheometry to the formation of bulk hydrogels, we utilized an accessible, stand-alone visible light LED. The selected LED centered at 455 nm has a spectral output range between 420–500 nm, where LAP has low levels of absorbance between 420–455 nm (Figure S3). The molar absorptivity of LAP spans 1 – 4 M−1cm−1 within this range of wavelengths (455 – 420 nm, respectively) (Figure S4) and is comparable to the molar absorptivity of the water soluble photoinitiator Irgacure 2959 at 365 nm (reported as 4 M−1cm−1),14 which has been commonly used for hydrogel formation. An additional advantage of utilizing a photoinitiator off peak absorbance is reduced light attenuation: at 365 nm, a 2 mM LAP solution is estimated to have ~ 90% transmittance at a thickness ~ 1 mm, whereas light from the visible light LED is estimated to have greater that 99% transmittance in a solution of 4 mM LAP at the same thickness (Table S1). Moving to visible light affords opportunities to polymerize thicker geometries without attenuation in addition to enabling precise control of the rate of hydrogel formation. LED systems are increasingly available and can be easily mounted to a ring stand for sample irradiation. With this approach, hydrogels were formed in molds (5-mm diameter cylinders) and swollen in buffer (PBS) overnight, and the mechanical properties of hydrogels after swelling were measured by DMA.
Hydrogels were formed at a low and high macromer concentration (6 and 14 wt% PEG-8-Nb), shifting to a higher range of concentrations than probed with in situ rheometry toward having more significant control of modulus with the rate-based handles provided by different photoinitiation conditions. To observe any rate dependence of the resulting modulus for these bulk hydrogels, two different LED light intensities (70 and 90 mWcm−2 at 455 nm) were used with different total times of light exposure (from 2 to 10 minutes), and the Young’s modulus (E) of resulting hydrogels were measured after equilibrium swelling (Figure 3A and 3B). Hydrogels rapidly formed for all conditions, with less than 2 minutes of irradiation, and increasing the total irradiation time was observed to increase modulus until a plateau as reached. Specifically, for the low or high PEG-8-Nb concentration (6 and 14 wt%), the swollen moduli of the resulting hydrogels for each composition were not statistically different after 4 or 3 minutes of irradiation, respectively, indicating completion of hydrogel formation. Consequently, for all subsequent investigations, all compositions were irradiated for 5 minutes.
Figure 3. Gelation and mechanical properties of bulk hydrogels formed by irradiation with visible light LED.
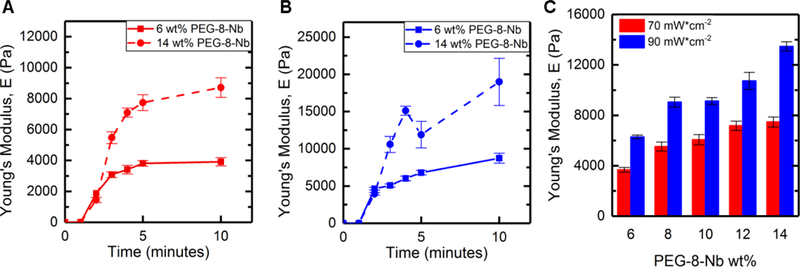
The moduli of equilibrium-swollen hydrogels (6 wt% and 14 wt%) formed with different irradiation times and intensities was measured with DMA: a) 70 mWcm−2 and b) 90 mWcm−2 for up to 10 minutes. Statistical increases in modulus (*p-value < 0.05) were observed until approximately 4 minutes and 3 minutes of irradiation at 70 mWcm−2 and 90 mWcm−2, respectively. c) Further, increasing PEG-8-Nb concentration and light intensity resulted in increased Young’s moduli after equilibrium swelling, where all compositions were irradiated for 5 minutes during hydrogel formation.
The trends in mechanical properties of equilibrium swollen bulk hydrogels correlate with those of the in situ formed hydrogels, where increased light intensity or PEG-8-Nb concentration resulted in increased moduli. Hydrogels were formed at a range of polymer concentrations (from 6 wt% to 14 wt% PEG-8-Nb) with five minutes of irradiation with visible light LED lamp at two different intensities (70 and 90 mWcm−2 at 455 nm) (Figure 3C). Hydrogel modulus increased with increased polymer concentration and increased LED intensity. Hydrogels formed with a light intensity of 70 mWcm−2 ranged in moduli from E = 3700 ± 200 Pa to E = 7500 ± 400 Pa for the given polymer concentration range, compared to the hydrogels formed with the higher intensity of 90 mWcm−2, which ranged in moduli from E = 6300 ± 100 Pa to E =13500 ± 400 Pa. Moduli of gels formed at different light intensities were significantly different for all wt% PEG-8-Nb (**p < 0.01) (Table S2). These results supported that different light intensities could be used to control the moduli of hydrogels independent from hydrogel composition, even after equilibrium swelling, with bulk hydrogel formation using a visible light LED lamp.
Visible light polymerization allows hydrogel formation in the presence of live human primary cells
To evaluate the suitability of this approach to hydrogel formation for use with biological systems, the viability and metabolic activity of human primary cells, specifically human mesenchymal stem cells (hMSCs) as a model cell type, were monitored over one week after encapsulation within hydrogels formed with the visible light LED lamp. hMSCs broadly are of interest in biological and biomedical research because of their multipotency, permitting differentiation into many cell types for tissue regeneration, and their roles in matrix remodeling and cell-cell signaling during both wound healing and disease.30,31 These cells were encapsulated in cell-degradable hydrogels (6 wt% PEG-8-Nb 1:1 stoichiometry Nb:SH) using a linker that is known to degrade in response to a variety of matrix metalloproteinases (GCRDVPMS↓MRGGDRCG) and incorporating a pendant peptide that is known to bind relevant integrins for promoting cell adhesion (2 mM CGRGDS). Hydrogels were polymerized with visible light (70 mWcm−2 at 455 nm, 5 minutes), and a live/dead membrane integrity assay was used to determine viability, staining live cells green and dead cell red and imaging by confocal microscopy. High hMSC viability was observed with > 90% viability (Figure 4A–C) at days 1, 3, and 7. Additionally, after one week in culture, hMSCs spread, exhibiting an elongated, spindle-shaped morphology (Figure 4B). Increased metabolic activity over time was measured for hMSCs formed in hydrogels at both light intensities (Figure 4D). Metabolic activity nearly doubled in 7 days in all conditions, suggesting successful three-dimensional (3D) culture of hMSCs within these hydrogels formed with visible light. Overall, these results supported that visible light formation of these bulk hydrogels was permissive to use with biological systems, including the encapsulation and growth of hMSCs in 3D culture.
Figure 4. Cytocompatibility of visible light conditions.
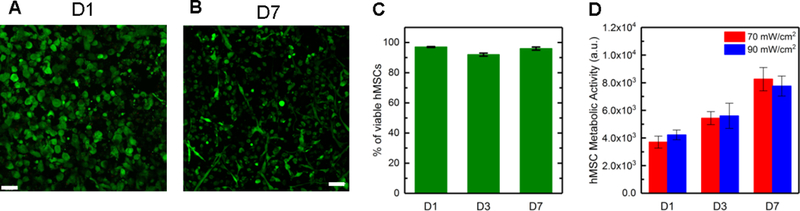
High viability was observed for hMSCs encapsulated in hydrogels formed with 455 nm visible light (70 mWcm−2 for 5 minutes): a) day 1 and b) day 7 (live (green) and dead (red) cells; example confocal projections; scale bar, 100 μm) and c) quantified (n = 3). d) Further, increased metabolic activity was observed over one week, supporting cell viability and suggesting cell growth during 3D culture within these hydrogels formed with visible light.
Correlation of functional group conversion with resulting hydrogel mechanical properties
To more deeply understand the relationship between polymerization rate and the resulting mechanical properties for this system, we selectively performed MAS 1H NMR to monitor functional group conversion during hydrogel formation. We aimed to compare functional group conversion with rheometric measurements of modulus for the resulting hydrogel for observations of any correlations. Specifically, we examined functional group conversion and modulus within the low macromer concentration composition (6 wt% PEG-8-Nb) for hydrogels formed with the low intensity of the visible light LED (70 mWcm−2 at 455 nm, irradiation times of 1, 2, 5, 10, and 20 minutes), which was observed to have the lowest modulus after complete gelation amongst the conditions probed and thus hypothesized to have the potential for the most defects. Functional group conversion as compared to a control photopolymerization condition (10 mWcm−2 at 365 nm). As noted in the Introduction, 365 nm light is commonly used for photopolymerization of hydrogels with the initiator LAP, and complete functional group conversion previously has been reported for similar conditions to the photopolymerization conditions used here.24 In addition to providing a positive control for functional group conversion, we postulated that the irradiation wavelength also could serve as a handle for influencing the rate of photopolymerization and ultimately modulus of the hydrogels. Since the molar absorptivity of LAP is greater in the UV light range compared to the visible light range, the generation of free radicals should be more efficient for the UV condition compared to the visible light condition. Based on the trends observed with in situ and bulk rheometry, we expected that the increased efficiency in free radical generation with 365 nm light would lead to an increased rate of photopolymerization relative to visible light that would correlate with differences in the moduli of the resulting hydrogels.
Norbornene end group conversion was monitored by MAS 1H NMR spectra at different irradiation times (0 to 5 minutes for 365 nm light and 0 to 20 minutes for visible light LED lamp) (Figure 5). Protons characteristic of the norbornene were observed from 5.9 to 6.2 ppm (t = 0 minutes, Figure 5A). After 5 minutes of irradiation, the length of time used for photopolymerization of bulk hydrogels with the LED, complete disappearance of peaks associated with the norbornene end groups was observed with 365 nm light, whereas these norbornene peaks remained present with visible light. The remaining norbornene peaks observed by MAS 1H NMR after 5 minutes of irradiation, which is when changes in mechanical properties previously had been observed to stop, suggested the presence of dangling norbornene end groups within these hydrogels formed with visible light. Given the geometry of the MAS-NMR rotor insert, some light attenuation through the sample depth with 2 mM LAP and 365 nm light is expected (51% transmittance at the bottom of the sample), and consequently, the rate of conversion observed with MAS-NMR may be slightly slower than the rate of gelation observed with in situ rheometry; regardless, full functional group conversion was observed with 365 nm light. No significant attenuation is expected in this geometry with 4 mM LAP and the visible light LED (98–99% transmittance) (Table S1).
Figure 5. Functional group conversion during hydrogel formation.
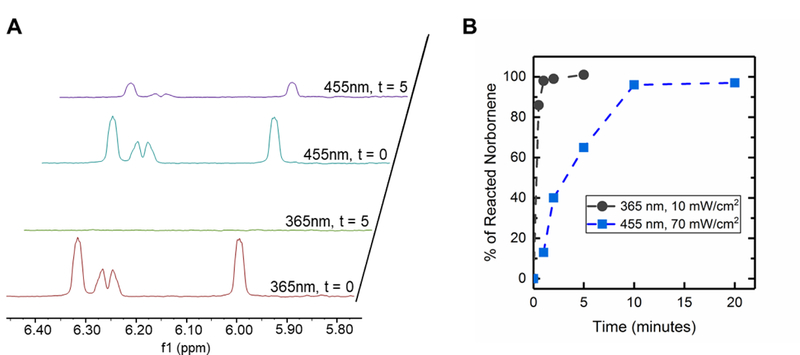
a) MAS 1H NMR spectra obtained before and after 5 min of light exposure for hydrogels polymerized with visible light (70 mWcm−2 at 455 nm, 4 mM LAP) or 365nm (10 mWcm−2, 2 mM LAP) (control), focusing on chemical shifts associated with norbornene end groups. b) Norbornene conversion (% reacted norbornene) over time was quantified by subtracting the integration of these peaks (normalized to t=0) from 1 for spectra obtained after different irradiation times.
Conversion of the norbornene end groups was quantified over time by integrating these characteristic peaks within NMR spectra at different irradiation times (Figure 5B). For 365 nm light, nearly 100% conversion of norbornene groups was observed after 2 minutes of irradiation, which is consistent with the polymerization time observed by in situ rheometry for these PEG-8-Nb hydrogels with 365 nm irradiation (Figure S5a), as well as literature reports.24 In contrast, after 2 minutes of irradiation with visible light, significantly lower conversion of norbornene groups (~ 40%) was observed. However, at longer time points, full conversion of norbornene end groups was observed: specifically, ~ 70% conversion was observed at 5 minutes and 100% conversion at 10 minutes. Note, the moduli of these 6 wt% PEG-8-Nb hydrogels did not increase with increased irradiation time after 5 minutes (Figure 3A); yet, conversion of the remaining ~ 30% of the norbornene end groups was observed during this same time frame (continued irradiation between 5 and 10 minutes).
These observations suggested that a large fraction of norbornene end groups were present as dangling ends after 5 minutes of irradiation and likely reacted with local, unreacted thiols to form looping defects (rather than crosslinks that contribute to modulus) between 5 and 10 minutes of irradiation. The 30% of norbornene end groups that were reacted between 5 and 10 minutes did not contribute to the final modulus, meaning hydrogels from the same precursor solution can be formed with the same final mechanical properties while having different concentrations of dangling end groups available for subsequent exploitation. During hydrogel formation, as end groups are reacted, chain mobility becomes increasingly restricted, and thus, especially at high conversion, free end groups of different macromers are less likely to meet than free end groups of the same macromer.32,33 Indeed, the final moduli of equilibrium swollen hydrogels formed with visible light were significantly lower than those formed with 365 nm light (Figure S5b), suggesting that differences in photopolymerization rate and efficiency between these irradiation wavelengths contributed to differences in defect formation and ultimately moduli of the resulting hydrogels.
Previously, light-based methods for controlling the moduli of ‘click’ hydrogel systems during hydrogel formation largely have focused on controlling end group conversion with the duration of irradiation (e.g., achieving a lower modulus by turning off the light to stop the polymerization before full functional group conversion is reached).33,34 Complementary to this, recent work using an initiator-free, non-light based chemistry (e.g., oxime ligation) has demonstrated a rate-based approach for controlling hydrogel modulus, in which pH was used to control the rate of hydrogel formation and thereby network defects (dangling end groups and looping) and modulus.5 The work presented here demonstrates a photopolymerized system in which, although full conversion is reached, moduli can still be controlled by an increased presence of defects. By controlling the rate of gelation, with light wavelength and intensity, the formation of defects can be controlled to produce moduli of interest.
The slower reaction kinetics for thiol-ene photopolymerization with visible light demonstrated here provides a unique opportunity to control the hydrogel modulus and retain functional groups as reactive handles for later modification. While the rate of photopolymerization was slower than with 365 nm, the onset of gelation (e.g., bulk hydrogels observed after 2 minutes of irradiation) is adequate for a variety of applications, including in situ formation in the presence of biological systems. With this approach, the availability of reactive end groups can be modulated by the length of time of LED light exposure, ultimately influencing mechanical properties independent of the hydrogel composition. This provides an alternative handle to control hydrogel modulus in addition to initial polymer concentration, stoichiometry, and light intensity. This unique feature can be harnessed for post-polymerization modifications, including the temporal addition of crosslinks to modulate mechanical properties and dynamically ‘stiffen’ hydrogels as demonstrated below.
Hydrogel stiffening through secondary photopolymerization
Dynamic hydrogel stiffening previously has been utilized to ask specific questions about how changes in the modulus of the native matrix that occur during specific biological events influence cell responses, such as during remodelling of the extracellular matrix upon injury or disease progression.11,20,35 The visible light LED approach, demonstrated above, yielded a robust polymerization that also provided facile control over the extent of end group availability within hydrogels after formation simply based on tuning of irradiation time, with largely dangling end groups present after 5 minutes of irradiation with 70 mWcm−2 at 455 nm. The resulting dangling end groups are available for additional reaction, with the potential for further crosslinking of the hydrogel network to modulate mechanical properties when (or where) desired. To test this, we investigated reaction of these end groups with a secondary photopolymerization: additional macromer and initiator were diffused into the hydrogels followed by irradiation for crosslinking with the existing network, as well as more broadly increasing polymer density within the hydrogel (Figure 6A). Following a modified version of published protocols,20,21 we incubated equilibrium swollen hydrogels (6 wt% PEG-8-Nb formed on stoichiometry with PEG-2-SH and 70 mWcm−2 at 455 nm, 5 minutes) in a buffer solution containing additional PEG-8-Nb (4 mM, 40 kDa), LAP (2 mM), and PEG-4-SH (4 mM, 10 kDa). Two incubation times were investigated: 1 hour and 6.5 h, times roughly estimated for diffusion of macromer into the hydrogels based on free or hindered diffusion, respectively (Figure S6), and consistent with incubation times previously published for stiffening via a secondary polymerization with PEG-8-Nb.21 As the mesh size of hydrogels formed with either visible or 365 nm light was estimated to be ~ 2 fold greater than the hydrodynamic diameter of the PEG-8-Nb, macromer in the stiffening solution was expected to infiltrate these hydrogels. After incubation with this ‘stiffening solution’, the hydrogels were irradiated (10 mWcm−2 at 365 nm, 2 minutes) to initiate a rapid secondary photopolymerization (Figure 6B), which was monitored by in situ rheometry (Figure 6C). Increases in modulus occurred rapidly, plateauing within 30 seconds of commencing irradiation.
Figure 6. Temporal increase in hydrogel modulus with secondary polymerization.
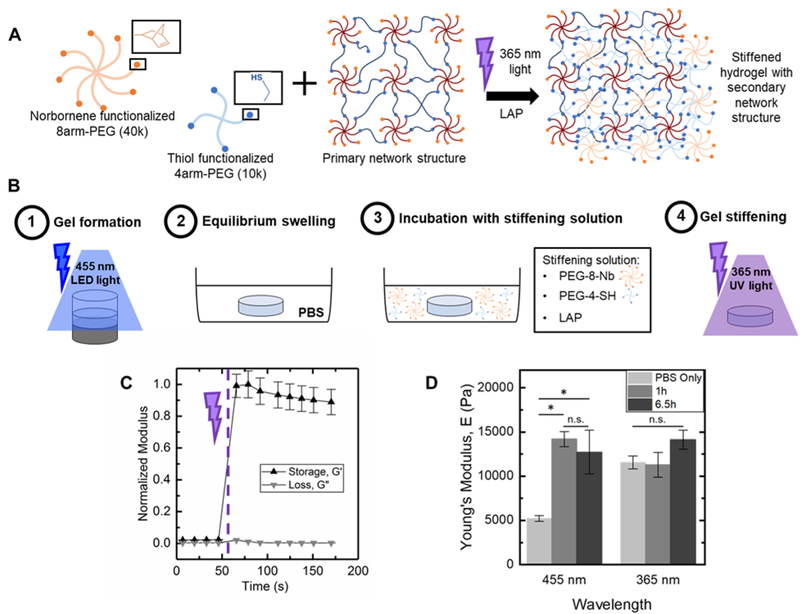
a) To harness dangling end groups present within hydrogels, secondary photopolymerization was performed to increase modulus. b) After 1) visible light formation, hydrogels (6 wt% PEG-8-Nb 1:1 Nb:SH with PEG-2-SH) were 2) equilibrium swollen in PBS for 24 hours and subsequently 3) incubated with additional macromer and initiator (PEG-8-Nb [4 mM Nb], SH-PEG-SH [4 mM SH], LAP [2 mM] for 1 hour) followed by 4) irradiation with 365 nm light (10 mWcm−2). c) A rapid increase modulus was observed upon irradiation (irradiation commenced at t = 60 s on rheometer with light attachment). d) Moduli of hydrogels incubated with PBS only or ‘stiffening’ solution for 1 h or 6.5 h and irradiated for the secondary polymerization were measured with DMA. Statistically significant increases in modulus (*p-value < 0.05) were observed for hydrogels formed with visible light, whereas no significant change was observed for control hydrogels formed with 365 nm.
Bulk mechanical properties of ‘stiffened’ hydrogels were measured by DMA immediately after stiffening. Moduli of these stiffened hydrogels that were originally formed with visible light were compared to controls: (i) same hydrogels before stiffening (‘PBS only’) and (ii) hydrogels originally formed with 365 nm light (10 mWcm−2 for 2 minutes) and similarly stiffened with a second photopolymerization (Figure 6D). A large and statistically significant increase in modulus was observed for hydrogels formed with visible light and subsequently stiffened, from E ~ 5,200 ± 300 Pa after original formation to E ~ 14,200 ± 900 Pa after 1 h incubation and stiffening and E ~ 12,700 ± 2,500 Pa after 6.5 h incubation and stiffening (*p-value < 0.05) (Figure 6D). Further, these hydrogels were observed to maintain their ‘stiffened’ modulus after equilibrium swelling (Figure S7). In contrast, no significant change in Young’s modulus was observed for hydrogels originally formed with 365 nm light and ‘stiffened’ under the same conditions, E ~ 11,600 ± 700 Pa after original formation and E ~ 11,300 ± 1,400 Pa after 1 h incubation and secondary stiffening and E ~ 14,100 ± 1,100 Pa after 6.5 h incubation and stiffening. These data supported that 1 h of incubation with stiffening solution was sufficient for achieving consistent stiffened moduli in hydrogels initially polymerized with both visible and UV light and, more importantly, the significant change in modulus that could be achieved upon stiffening of the hydrogels formed with visible light.
Taken together, these studies demonstrate how the rate-based approach of controlling defect formation with visible light polymerization to create dangling end groups can be combined with post-polymerization modification methods to allow hydrogel stiffening, establishing a complementary approach to other stiffening methods that incorporate free functional groups for later modification by altering the composition of the original hydrogel precursor solution (e.g., formation off stoichiometry).20 We suspect that dangling end groups present after hydrogel formation with visible light, which were observed by MAS 1H NMR (Figure 5), were reacted during the secondary photopolymerization and contribute to the observed increase the crosslink density and thereby modulus of the hydrogel. The lack of change in modulus upon the secondary polymerization for the hydrogel formed by 365 nm light, which lacked measurable dangling end groups from MAS 1H NMR, further suggests that the presence of free end groups within the primary network, as we observe in visible light formed hydrogels, may be important for subsequent stiffening of hydrogels with a secondary polymerization.
This method of initial gel formation with a visible light LED lamp offers precise control over initial mechanical properties and functional group availability while holding macromer composition constant. Precise control of reactive end group availability provides a key handle to impart dynamic stiffening; here, similarly large changes in modulus were observed upon stiffening to those reported for PEG-8-Nb hydrogels formed off stoichiometry and were achieved using lower concentrations of macromer in the ‘stiffening solution’ than demonstrated previously.20 Further, this method is able to achieve an increase in modulus comparable to other recently reported methods utilizing a different polymerization mechanism for secondary stiffening such as secondary photocrosslinking of cyclooctyne hydrogels.36 Although not explicitly examined in this work, similar incubation and irradiation conditions to those used here for hydrogel stiffening have been shown to be cytocompatible for a variety of cell types, including hMSCs.9,20 Of note, the range of Young’s modulus demonstrated before and after stiffening with this approach (Figure 6D) is relevant for mimicking the modulus of a variety of human tissues.37 Thus, this approach that utilizes visible light formation and secondary temporal modification of hydrogels has a variety of potential applications, including probing and directing cell function in response to dynamic changes in their microenvironment within controlled cell culture.
Conclusions
In summary, we have demonstrated an approach for using LED visible-light (centered at 455 nm) for photopolymerization of synthetic thiol-ene hydrogels. With this approach, we established that hydrogel modulus could be tuned with a variety of parameters, including the macromer concentration, photoinitiator concentration, and intensity, duration, and wavelength of light used for hydrogel formation. Further, the conditions established were shown to be cytocompatible, enabling the encapsulation of live human primary cells. Comparison of observations of functional group conversion and resulting hydrogel modulus provided insights into the mechanism of modulus control afforded with this visible light system, where dangling end and looping defects were observed owing to the less-efficient photopolymerization achieved with visible light relative to traditional approaches with 365 nm. Despite these defects, hydrogels with robust mechanical properties were successfully formed with this visible light approach, and the resulting dangling end groups present after gelation were harnessed to temporally stiffen these hydrogels with a secondary thiol-ene photopolymerization. Overall, these studies provide insight into the range of handles available for controlling hydrogel modulus at formation or temporally with light, where defects within the network can be viewed in some circumstances to be beneficial rather than detrimental for control of bulk properties. Heterogeneity in hydrogel network structure also recently has been shown to influence cell behaviour in 3D cell culture,15 and the approaches established here could be used in future work to control network heterogeneity and probe specific hypotheses about cell response. This approach and findings of this work may prove useful for the formation and modulation of the properties of synthetic hydrogels in the presence of biological systems, from fundamental biological studies to applied work in coatings and biomaterials.
Supplementary Material
Acknowledgements
The authors would like to acknowledge TA instruments for lending the 455 nm LED plate and DHR rheometer for measurements. We also thank the NMR Core facility for their assistance with magic angle spinning and the Mass Spectrometry Core facility at the University of Delaware, which are partly funded by the Delaware COBRE programs supported by grants from the National Institute of General Medical Sciences - NIGMS (5 P30 GM110758-02 and P20GM104316) from the National Institutes of Health. The authors would like to acknowledge support, for related work, from the Delaware COBRE program (P20GM104316) from the National Institutes of Health. The authors would like to acknowledge support, for related work, from the Delaware COBRE program (P20GM104316) from the National Institutes of Health and from the National Science Foundation (NSF) DMR BMAT (1253906).
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Lutolf MP and Hubbell JA, Biomacromolecules, 2003, 4, 713–722. [DOI] [PubMed] [Google Scholar]
- 2.Vining KH and Mooney DJ, Nat. Rev. Mol. Cell Biol, 2017, 18, 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan D, Ding Y, Dauskardt RH and Appel EA, ACS Appl. Mater. Interfaces, 2017, 9, 42217–42224. [DOI] [PubMed] [Google Scholar]
- 4.Kharkar PM, Kiick KL and Kloxin AM, Chem. Soc. Rev, 2013, 42, 7335–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zander ZK, Hua G, Wiener CG, Vogt BD and Becker ML, Adv. Mater, 2015, 27, 6283–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsunaga T, Sakai T, Akagi Y, Chung U and Shibayama M, Macromolecules, 2009, 42, 1344–1351. [Google Scholar]
- 7.Kawamoto K, Zhong M, Wang R, Olsen BD and Johnson JA, Macromolecules, 2015, 48, 8980–8988. [Google Scholar]
- 8.Zhong M, Wang R, Kawamoto K, Olsen BD and Johnson JA, Science, 2016, 353, 1264–1268. [DOI] [PubMed] [Google Scholar]
- 9.Sawicki LA and Kloxin AM, Biomater. Sci, 2014, 2, 1612–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greene T, Lin TY, Andrisani OM and Lin CC, J. Appl. Polym. Sci, 2017, 134, 1–10. [Google Scholar]
- 11.Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL, Wells RG and Burdick JA, Sci. Rep., 2016, 6, 21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shih H and Lin C-C, Macromol. Rapid Commun, 2013, 34, 269–273. [DOI] [PubMed] [Google Scholar]
- 13.Shih H, Greene T, Korc M and Lin CC, Biomacromolecules, 2016, 17, 3872–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairbanks BD, Schwartz MP, Bowman CN and Anseth KS, Biomaterials, 2009, 30, 6702–6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider MC, Chu S, Sridhar SL, de Roucy G, Vernerey FJ and Bryant SJ, ACS Biomater. Sci. Eng, 2017, 3, 2480–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosales AM, Mabry KM, Nehls EM and Anseth KS, Biomacromolecules, 2015, 16, 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kehe GM, Mori DI, Schurr MJ and Nair DP, ACS Appl. Mater. Interfaces, 2019, 11, 1760–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guvendiren M and Burdick JA, Nat. Commun, 2012, 3, 792–799. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen HD, Liu H-Y, Hudson BN and Lin C-C, ACS Biomater. Sci. Eng, 2019, 5, 1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mabry KM, Lawrence RL and Anseth KS, Biomaterials, 2015, 49, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiedler CI, Aisenbrey EA, Wahlquist JA, Heveran CM, Ferguson VL, Bryant SJ and McLeod RR, Soft Matter, 2016, 12, 9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ovadia EM, Colby DW and Kloxin AM, Biomater. Sci, 2018, 6, 1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKinnon DD, Kloxin AM and Anseth KS, Biomater. Sci, 2013, 1, 460–469. [DOI] [PubMed] [Google Scholar]
- 24.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN and Anseth KS, Adv. Mater, 2009, 21, 5005–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoyle CE and Bowman CN, Angew. Chemie Int. Ed, 2010, 49, 1540–1573. [DOI] [PubMed] [Google Scholar]
- 26.Wang R, Johnson JA and Olsen BD, Macromolecules, 2017, 50, 2556–2564. [Google Scholar]
- 27.Metters A and Hubbell J, Biomacromolecules, 2004, 6, 290–301. [DOI] [PubMed] [Google Scholar]
- 28.Sarvestani AS, Xu W, He X and Jabbari E, Polymer, 2007, 48, 7113–7120. [Google Scholar]
- 29.Kilambi H, Reddy SK and Bowman CN, Macromolecules, 2007, 40, 6131–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engler AJ, Sen S, Sweeney HL and Discher DE, Cell, 2006, 126, 677–89. [DOI] [PubMed] [Google Scholar]
- 31.Rehmann MS, Luna JI, Maverakis E and Kloxin AM, J. Biomed. Mater. Res. Part A, 2016, 104, 1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou H, Schön E-M, Wang M, Glassman MJ, Liu J, Zhong M, Díaz Díaz D, Olsen BD and Johnson JA, J. Am. Chem. Soc, 2014, 136, 9464–9470. [DOI] [PubMed] [Google Scholar]
- 33.Johnson PM, Stansbury JW and Bowman CN, Polymer, 2007, 48, 6319–6324. [Google Scholar]
- 34.Sheth S, Jain E, Karadaghy A, Syed S, Stevenson H and Zustiak SP, Int. J. Polym. Sci, 2017, 2017, 1–9. [Google Scholar]
- 35.Vats K and Benoit DSW, Tissue Eng. Part B Rev, 2013, 19, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown TE, Silver JS, Worrell BT, Marozas IA, Yavitt FM, Günay KA, Bowman CN and Anseth KS, J. Am. Chem. Soc, 2018, 140, 11585–11588. [DOI] [PubMed] [Google Scholar]
- 37.Handorf AM, Zhou Y, Halanski MA and Li WJ, Organogenesis, 2015, 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.