

Abstract
Neuroendocrine prostate cancer (NEPC) is the most virulent form of prostate cancer. Importantly, our recent work examining metastatic biopsy samples demonstrates NEPC is increasing in frequency. In contrast to prostate adenocarcinomas that express a luminal gene expression program, NEPC tumors express a neuronal gene expression program. Despite this distinction, the diagnosis of NEPC is often challenging, demonstrating an urgent need to identify new biomarkers and therapeutic targets. Our prior work demonstrated that the histone demethylase LSD1 (KDM1A) is important for survival of prostate adenocarcinomas, but little was known about LSD1’s role in NEPC. Recently, a neural-specific transcript variant of LSD1—LSD1+8a—was discovered and demonstrated to activate neuronal gene expression in neural cells. The splicing factor SRRM4 was previously shown to promote LSD1+8a splicing in neuronal cells, and SRRM4 promotes NEPC differentiation and cell survival. Therefore, we sought to determine if LSD1+8a might play a role in NEPC and whether LSD1+8a splicing was linked to SRRM4. To investigate a potential role for LSD1+8a in NEPC, we examined a panel of prostate adenocarcinoma and NEPC patient-derived xenografts and metastatic biopsies. LSD1+8a was expressed exclusively in NEPC samples and correlated significantly with elevated expression of SRRM4. Using SRRM4-overexpressing cell lines, we determined that SRRM4 mediates alternative splicing of LSD1+8a. Finally, using gain of function studies, we confirmed that LSD1+8a and SRRM4 co-regulate target genes distinct from canonical LSD1. Our findings suggest further study of the interplay between SRRM4 and LSD1+8a and mechanisms by which LSD1+8a regulates gene expression in NEPC is warranted.
Abbreviations: NEPC, Neuroendocrine Prostate Cancer; AR, Androgen Receptor; SCLC, Small Cell Lung Cancer; CRPC, Castration-Resistant Prostate Cancer; PDX, Patient-Derived Xenograft
Keywords: Neuroendocrine prostate cancer, LSD1, LSD1+8a, SRRM4, Epigenetics
Introduction
Treatment options for men with prostate cancer are increasing; however, prostate cancer remains the second leading cause of cancer-related deaths in the United States with over 33,000 men predicted to die from this disease in 2020 [1]. Nearly all prostate cancers are adenocarcinomas at diagnosis, while <1% are neuroendocrine prostate cancers (NEPC)—an aggressive form of the disease with an alternate differentiation program [2]. Importantly, the frequency of NEPC appears to be increasing in frequency due to more widespread use of drugs that block the function of the androgen receptor (AR)—a transcription factor that promotes a luminal lineage program [3]. Thus, there is an urgent need to identify biomarkers that indicate the presence of NEPC and to identify new therapeutic targets in NEPC.
Our prior work demonstrates that lysine specific demethylase 1 (LSD1, alternatively known as KDM1A) promotes growth of adenocarcinoma prostate cancer cells—including those that are castration-resistant or AR null—and that these effects are independent of LSD1’s canonical demethylase function [4]. Recently, it was determined that inclusion of a 12 nucleotide microexon (exon 8a) between exons 8 and 9 of the canonical LSD1 transcript results in an mRNA splice variant called LSD1+8a [5]. This LSD1+8a transcript variant has distinct histone demethylation substrate specificity and unique protein–protein interactions vs. the canonical LSD1 [6]. Importantly, activity of the LSD1+8a isoform has been linked to modulation of neuronal gene transcription in neuronal cells [6], [7]. Additionally, LSD1+8a has recently been shown to regulate expression of genes linked with neuroendocrine differentiation in small cell lung cancer (SCLC) tumors that share many features with NEPC tumors [8], [9], [10]. Given the role for LSD1 in castration-resistant prostate cancer (CRPC) as well as the finding of LSD1+8a in neuronal cells and in SCLC, we hypothesized that LSD1+8a might be linked to NEPC.
Multiple groups have recently identified an RNA splicing factor, Serine/Arginine Repetitive Matrix 4 (SRRM4, alternatively known as nSR100), as a driver of NEPC progression [11], [12], [13]. SRRM4 was demonstrated to induce an adenocarcinoma to NEPC transition, especially when cells were deficient in TP53 and treated with the AR antagonist enzalutamide [10]. Development of NEPC xenografts from adenocarcinoma cell lines over sequential passaging also revealed SRRM4 was required for neural gene expression [12]. SRRM4 is known to splice a cryptic 62 bp exon into mRNA of the neuronal gene silencing protein REST that leads to production of an inactive REST isoform and subsequent activation of neuronal genes [13]. This REST splice variant has also recently been identified in NEPC, demonstrating that SRRM4 promotes neuroendocrine gene expression through alternative splicing of specific transcripts [11]. Finally, SRRM4 has been demonstrated to mediate alternative splicing of LSD1+8a in mouse models of epilepsy [14]. These observations led us to investigate the connection between SRRM4 and LSD1+8a in NEPC.
In this report, we determined that LSD1+8a expression was limited to human NEPC tissue samples and patient-derived xenograft (PDX) samples but was not detectable in prostate adenocarcinoma samples. Importantly, LSD1+8a expression correlated with elevated SRRM4 expression in these samples. Using SRRM4 gain of function experiments, we confirmed that SRRM4 overexpression induced alternative splicing of LSD1+8a. Furthermore, we determined that LSD1+8a and SRRM4 co-regulate many genes whose expression was not induced by canonical LSD1 overexpression, including genes previously implicated in cancer progression. Altogether, our results demonstrate that LSD1+8a may be a useful biomarker of NEPC and targeting LSD1+8a may block expression of genes linked to cancer progression.
Results
LSD1+8a expression is only detected in neuroendocrine prostate cancer patient-derived xenografts and metastases and correlates with elevated SRRM4 expression
LSD1+8a was previously demonstrated to promote neuronal differentiation in neural cells [6]. Therefore, we sought to determine the role of LSD1+8a in the prostate cancer subset linked with neuronal or neuroendocrine differentiation—NEPC. Using qRT-PCR and exon junction-specific primers [6], we measured mRNA expression of LSD1+8a in 11 PDX samples (9 from the LuCaP series [15] and two from the LTL series [16]) categorized as either adenocarcinoma or NEPC. We also measured expression of SRRM4, which was previously implicated in NEPC [11], [12], [17] Lee, et al and was shown to influence splicing of LSD1+8a in neuronal cells [14]. LSD1+8a was detectable in all of the NEPC PDX samples but not in the adenocarcinoma PDXs, and SRRM4 was highly expressed in all NEPC PDX samples compared to adenocarcinoma PDX samples (Fig. 1a). Importantly, we determined that there was a significant positive correlation between SRRM4 and LSD1+8a expression in these samples (Supplementary Fig. S1a).
Fig. 1.

LSD1+8a mRNA expression is only observed in neuroendocrine prostate cancer PDXs and metastases. qRT-PCR was used to determine LSD1+8a and SRRM4 expression in (a) 11 CRPC PDX samples, including 5 adenocarcinoma and 6 NEPC tumors, and in (b) 19 CRPC patient tumors, including 10 adenocarcinoma and 9 NEPC tumors. Absolute quantification of mRNA was determined using standard curve of known copy numbers. Error bars represent SD.
To corroborate these results, we next examined LSD1+8a and SRRM4 expression in CRPC tumors categorized as adenocarcinoma or NEPC from 19 patients. LSD1+8a expression was only detected in NEPC tumors and was not observed in any of the adenocarcinoma tumors (Fig. 1b). Additionally, SRRM4 expression was observed in 100% of NEPC tumors. Conversely, SRRM4 expression was only detectable in 30% of the adenocarcinoma tumors we tested, and SRRM4 was more highly expressed in the NEPC tumors vs. the adenocarcinoma tumors (Fig. 1b). Again, we found a significant positive correlation between SRRM4 and LSD1+8a expression in these patient tumor samples (Supplementary Fig. S1b). It should be noted that we examined a previously published human CRPC RNA-seq dataset with both adenocarcinoma and NEPC tumors [3] but were unable to detect the LSD1+8a transcript. This may be due to a small number of reads containing the 12 base-pair LSD1+8a transcript, low expression in the sample, or low NEPC tumor content in the biopsy samples.
Importantly, expression levels of canonical LSD1 (described hereafter simply as LSD1) were similar between adenocarcinoma and NEPC PDXs or patient tumor samples (Supplementary Fig. S1c-d). Altogether, these data suggest that LSD1+8a expression was only detected in the NEPC tumors we examined and that LSD1+8a expression correlates with increased SRRM4 expression.
SRRM4 mediates splicing of LSD1+8a in prostate cancer cell line models
Because we observed a correlation with SRRM4 upregulation and LSD1+8a expression in NEPC PDX and patient samples, we sought to determine if SRRM4 was responsible for LSD1+8a alternative splicing. Thus, we examined expression of LSD1+8a in several cell line models with stable overexpression of SRRM4 described previously [17]. These models include prostate adenocarcinoma cancer cell lines (LNCaP, PC3, DU145, 22RV1), benign prostate epithelial cell lines (BPH1 and RWPE1), a prostate stromal cell line (WPMY1), and a uterine smooth muscle cell line (hTERT-Myo). LSD1+8a was detectable in 6 of 8 of SRRM4-overexpressing cell lines but was undetectable in all control cell lines (Fig. 2a). These data suggest that ectopic expression of SRRM4 causes inclusion of the 8a exon in the LSD1 transcript, resulting in expression of LSD1+8a.
Fig. 2.

SRRM4 directly mediates alternative splicing of LSD1+8a. (a) Absolute quantification of LSD1+8a and SRRM4 mRNA was determined using a standard curve of know copy numbers. Error bars represent SD. (b) Schematic of the LSD1 transcript focused on the region around the 8a exon. (c) An RNA immunoprecipitation assay was performed from LNCaP cells stably overexpressing FLAG-SRRM4 (LNCaP-SRRM4) using an anti-FLAG antibody. IgG was used as a negative control. Immunoprecipitated and input RNA were reverse transcribed to cDNA. qRT-PCR was performed using indicated primers. The input RNA without reverse transcription was used to control for DNA contamination. Data is expressed as the percentage of reverse transcribed input sample. Error bars represent SD. (d) An LSD1 minigene splicing reporter was designed to constitutively express mRNA from human LSD1 exons 8, 8a, and 9, along with 300 base pairs of intronic nucleotides directly flanking each exon. Horizontal arrows indicate annealing sites for exon junction-specific primers used to assay mRNA splicing from the reporter. (e) An empty control vector or the LSD1 minigene depicted in (D) was transiently transfected into control LNCaP cells (Control) or LNCaP cells stably overexpressing SRRM4 (+SRRM4). Splicing of LSD1+8a or LSD1 was assayed at 72 hours post-transfection by qRT-PCR using exon junction-specific primers at the regions indicated in (d). mRNA copy number for each condition was determined using standard curves of known copy numbers. (f) Splicing assay experiment was performed as in (E) except control 22RV1 (Control) or 22RV1 cells stably overexpressing SRRM4 (+SRRM4) were used. Splicing of LSD1+8a or LSD1 was assayed at 96 hours post-transfection by qRT-PCR.
Next, we sought to determine if SRRM4 directly mediates alternative splicing of LSD1+8a in prostate cancer. To determine if SRRM4 directly binds the LSD1 pre-mRNA transcript in the region of the 8a exon, we used RNA immunoprecipitation (RNA-IP) using an anti-FLAG antibody (Fig. 2b). Following immunoprecipitation, we performed qRT-PCR using primers matching the putative UGC motif upstream of the splice acceptor site (8a 5′, previously shown to be important for SRRM4-dependent splicing [18]), and the exon–intron junction of exon 8a and intron 9 (8a 3′) (Fig. 2c). SRRM4 has been shown previously to interact with and alternatively splice REST [10]. Therefore, we included REST as a positive control in addition to the GAPDH negative control to demonstrate specificity of SRRM4 interactions. To control for genomic DNA contamination, qPCR was performed on input samples treated with or without reverse transcriptase. Compared to a control IgG antibody immunoprecipitation, eluates from the SRRM4 immunoprecipitation were enriched for RNA corresponding to the 8a 5′ and 8a 3′ regions. No immunoprecipitation of RNA from the 1st intron of LSD1 was observed, demonstrating that SRRM4 binds specifically to LSD1 pre-mRNA proximal to exon 8a.
To further confirm that SRRM4 mediates splicing of LSD1+8a in prostate cancer cells, we next designed an LSD1 minigene splicing reporter to quantify the amount of exon 8a inclusion in two prostate cancer cell lines stably overexpressing SRRM4 vs their corresponding control cell lines. Importantly, this minigene was designed to constitutively express mRNA from human LSD1 exons 8, 8a, and 9, along with 300 base pairs of intronic nucleotides directly flanking each exon (Fig. 2d, see also Supplementary Fig. S2). The flanking intronic nucleotides upstream of exon 8a included the aforementioned putative UGC motif upstream of the splice acceptor site. The LSD1 minigene was transiently transfected into 22RV1-SRRM4 and LNCaP-SRRM4 cells, as well as corresponding control cells (22RV1-CTL and parental LNCaP, respectively) [17]. qRT-PCR using exon junction-specific primers were used to quantify LSD1 vs LSD1+8a splicing of the minigene [6]. In cells transfected with the minigene, LSD1+8a transcripts were increased in both SRRM4-overexpressing cell lines compared to their corresponding control cell lines lacking SRRM4 overexpression (right panels, Fig. 2e & f). Additionally, SRRM4 overexpression did not increase levels of the canonical LSD1 transcript; in fact, levels of the canonical isoform were decreased in LNCaP-SRRM4 cells compared to control LNCaP cells (left panels, Fig. 2e & f). This, in combination with the RNA-IP data shown in Fig. 2c showing a specific SRRM4-exon 8a interaction, indicates that SRRM4 specifically promotes splicing of the LSD1+8a transcript rather than splicing of the canonical LSD1, generally. Altogether, these data demonstrate that the alternative splicing of the LSD1+8a isoform is indeed directly mediated by SRRM4 in these prostate cancer cell line models.
LSD1+8a regulates a unique transcriptional program from LSD1 in prostate cancer cell lines
Having established that LSD1+8a is expressed specifically in a subset of NEPC PDXs and patient tumors and that SRRM4 is capable of inducing LSD1+8a splicing, we next sought to determine the functional relevance of LSD1+8a expression in prostate cancer. To test whether LSD1+8a can recapitulate effects of SRRM4 or LSD1, we performed a series of gain-of-function experiments. First, we transiently overexpressed FLAG-tagged LSD1+8a, LSD1, or SRRM4 in LNCaP cells (Fig. 3a). We then performed qRT-PCR analysis to measure expression of several markers of NEPC differentiation (CHGA, SCG3, CHGB, SYP) [19], [20] previously shown to be up-regulated by SRRM4 [10]. As expected, SRRM4 overexpression strongly activated expression of these neuroendocrine marker genes. However, only minor changes in expression of these genes were seen with overexpression of either LSD1 isoform, demonstrating that LSD1+8a or LSD1 overexpression alone was not sufficient to activate these genes’ expression (Fig. 3b).
Fig. 3.
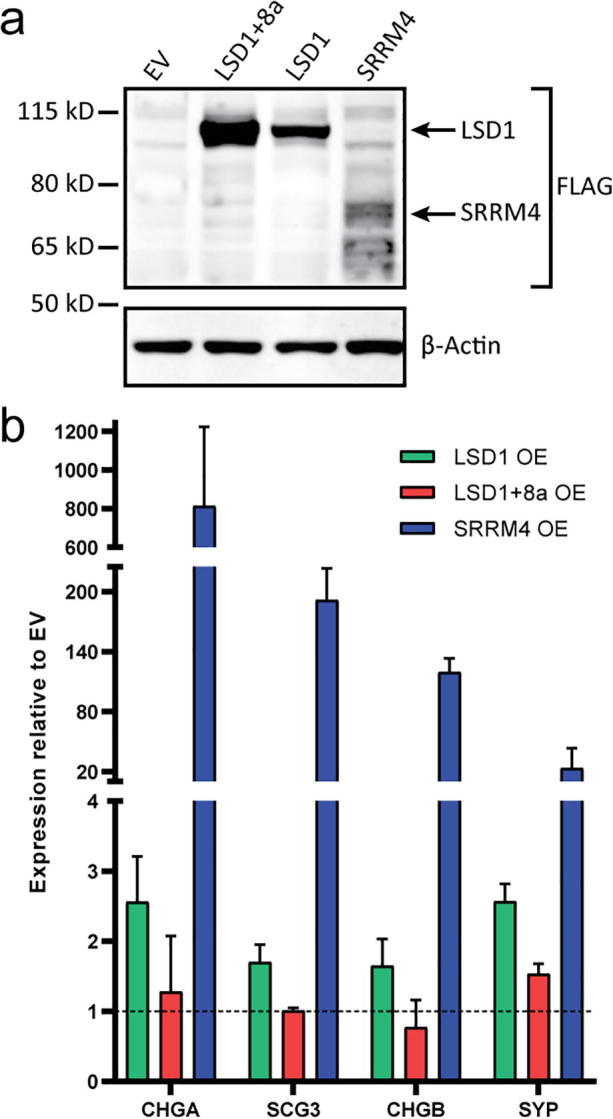
LSD1+8a alone is not sufficient to activate expression of markers of NEPC differentiation (a) Western blot to confirm overexpression of FLAG-tagged proteins LSD1+8a, LSD1, or SRRM4 in LNCaP cells. (b) qRT-PCR was used to measure the expression of several markers of neuroendocrine differentiation in cells over-expressing LSD1, LSD1+8a, or SRRM4. Error bars represent SD.
To understand gene expression changes induced by SRRM4, LSD1+8a, or LSD1, we overexpressed SRRM4, LSD1+8a, LSD1, or empty vector in LNCaP cells and performed RNA-seq (Fig. 4a, Supplementary Table 1). To identify differentially-expressed genes, we used a significance cutoff of p = 0.05 and a fold change cutoff of ±1.5. Interestingly, LSD1+8a overexpression modulated the expression of a larger set of genes (142 genes) when compared to the canonical LSD1 (88 genes). SRRM4 overexpression modulated expression of the largest number of genes—424—of which 84.9% (360/424) were upregulated. The majority of differentially-expressed genes with either LSD1 (67.0%, 59/88) or LSD1+8a, (69.7%, 99/142) overexpression were also upregulated. LSD1 overexpression caused fewer gene expression changes, and the magnitude of the gene expression changes was lower vs. either SRRM4 or LSD1+8a overexpression (Fig. 4a).
Fig. 4.

LSD1+8a regulates a unique transcriptional program from LSD1 in prostate cancer cell lines. LSD1+8a, LSD1, or SRRM4 were transiently overexpressed in LNCaP cells as in Fig. 3. RNA was harvested 96 hours post-transfection, and RNA-seq was performed. (a) Volcano plots of fold change vs significance of differentially expressed genes in each condition. All comparisons were made between overexpression and empty vector conditions. (b) Venn diagram of up-(left) and down-(right) regulated genes show the overlap of common significant, differentially expressed genes identified in LSD1+8a, LSD1, or SRRM4 overexpressing LNCaP cells. (c) qRT-PCR was performed to measure the mRNA expression of select differentially expressed genes identified by RNA-seq in the LSD1+8a/SRRM4 overlap shown in (b). n = 3, error bars represent SD. *p ≤ 0.05, **p ≤ 0.01 (two-tailed t-test). (d) Gene set enrichment analysis of LSD1+8a/SRRM4-specific up-regulated genes in NEPC vs adenocarcinoma patient tumor samples. Normalized Enrichment Score = 1.466, nominal p = 0.043, FDR q = 0.027.
Because we determined that SRRM4 induces LSD1+8a splicing, we were particularly interested in gene expression changes induced by both SRRM4 and LSD1+8a overexpression that might represent target genes induced by SRRM4-splicing of LSD1+8a (Fig. 4b, Supplementary Table 1). Importantly, most gene expression changes (83.1%, 118/142) induced by overexpression of LSD1+8a were not shared by LSD1 overexpression. There was much greater overlap of genes altered by both SRRM4 and LSD1+8a overexpression in both the up (n = 44, p = 2.0264 × 10−58) and down (n = 7, p = 4.0476 × 10−12) directions than those altered by both SRRM4 and LSD1 overexpression (up: n = 26, p = 9.7702 × 10−35; down: n = 1, p = 0.059482).
Next, we focused on the shared genes upregulated by both SRRM4 and LSD1+8a overexpression. Interestingly, a Gene Ontology (GO) analysis of genes in the LSD1+8a/SRRM4 overlap was not enriched for neuronal or neuroendocrine-related biological processes. Rather, we observed a strong enrichment for biological processes related to interferon signaling and immune response among the overlapping genes (Supplementary Table 3). Importantly, several of the most upregulated genes in both SRRM4 and LSD1+8a overexpression conditions have been previously implicated in promoting cancer progression and/or therapeutic resistance. These genes include: Interferon Induced Protein with Tetratricopeptide Repeats 3 (IFIT3), Bone Marrow Stromal Cell Antigen 2 (BST2), Tripartite Motif Containing 22 (TRIM22), HECT and RLD Domain Containing E3 Ubiquitin Protein Ligase 5 (HERC5), and Sterile Alpha Motif Domain Containing 9 (SAMD9) [22], [23], [24], [25], [26], [27], [28], [29], [21]. We used qRT-PCR to verify that these genes were uniquely and significantly upregulated in SRRM4 or LSD1+8a overexpressing cells but not LSD1 overexpressing cells (Fig. 4c). To further validate the relevance of the 31 genes co-upregulated by SRRM4 and LSD1+8a (Fig. 4b) in human NEPC tumors, we determined if this 31 gene signature was activated in NEPC vs. adenocarcinoma patient tumors in the Aggarwal, et al. dataset [3]. The LSD1+8a/SRRM4 co-upregulated gene signature was significantly activated in NEPC patient tumors (Fig. 4d). Furthermore, we confirmed that this signature was also activated in NEPC PDXs (Supplementary Fig. S3) that we confirmed to co-express LSD1+8a and SRRM4 (Fig. 1a). Altogether, these results demonstrate a functional role for SRRM4 and LSD1+8a in co-regulating expression of important prostate cancer genes linked to cancer aggressiveness in NEPC—genes that are distinct from those regulated by canonical LSD1.
Discussion
NEPC appears to be increasing in frequency clinically, and NEPC tumors are associated with much worse outcomes than adenocarcinoma tumors [3]. Thus, there is an urgent need to identify targetable mechanisms involved in progression to NEPC as well as biomarkers of this aggressive form of prostate cancer. Previously, we identified LSD1 as a key driver of adenocarcinoma CRPC growth [4]. Here, we determined that a transcript variant of LSD1—LSD1+8a—is expressed specifically in NEPC PDXs and in NEPC patient tumors but not in adenocarcinomas.
Not all of the NEPC patient tumor biopsies we examined expressed LSD1+8a by qRT-PCR (Fig. 1b). We hypothesize that NEPC tumor biopsies that did not have appreciable LSD1+8a expression may have had low tumor-cell content relative to other cell types. We were also not able to detect 8a-containing LSD1 transcripts in NEPC patient biopsies by RNA-seq analysis of a published dataset [3]. Importantly, we did determine that target genes activated by LSD1+8a overexpression in our cell line experiments (Fig. 4b) were highly expressed in NEPC tumors from that same dataset (Fig. 4d). This strongly suggests that LSD1+8a may have been important for activation of those genes in those NEPC tumors and that our inability to detect LSD1+8a expression by RNA-seq may have been due to the limited number of RNA-seq reads containing the 12 base-pair cryptic 8a exon. Additional studies with greater numbers of NEPC patient tumors—ideally with single cell sequencing analysis—and the generation of LSD1+8a-specific commercial antibodies—that are not currently available—will also help to determine the prevalence of LSD1+8a expression in NEPC tumors. Nonetheless, our results demonstrate that expression of LSD1+8a is associated with the NEPC phenotype. Importantly, LSD1+8a expression can be quickly and specifically detected using exon junction-specific primers. Thus, if our results are validated on a larger number of samples, it is possible that measuring LSD1+8a expression could help confirm the diagnosis of NEPC in cases where the histology is not definitive using tumor biopsies or circulating nucleic acids in the blood [30].
SRRM4 is an RNA-splicing factor known to mediate inclusion of short “microexons” [31]. SRRM4 has been previously shown to promote alternative splicing of transcripts commonly found in human NEPC patient tumors, including the REST gene; SRRM4 has also been shown to regulate the expression of genes linked with neuroendocrine differentiation [10]. In this report, we demonstrated that SRRM4 expression is higher in both NEPC vs. adenocarcinoma PDXs and patient tumors (Fig. 1). Using SRRM4-overexpressing cell lines, RNA-immunoprecipitation, and minigene splicing experiments (Fig. 2), we demonstrate that SRRM4 is responsible for the inclusion of the 8a exon into a mature LSD1+8a mRNA. It should be noted that while we demonstrate that SRRM4 is sufficient to induce LSD1+8a splicing, there may be other splicing factors that contribute to LSD1+8a splicing or stabilization. Future studies will be necessary to answer that question.
LSD1+8a was not sufficient to activate expression of genes linked to NEPC differentiation induced by SRRM4 (Fig. 3). However, our RNA-seq results demonstrate that LSD1+8a does indeed activate expression of multiple genes previously implicated in driving cancer progression and/or treatment resistance that are regulated by SRRM4 but not LSD1. Our results demonstrating that many of the LSD1+8a-induced gene expression changes were shared by SRRM4 overexpression suggests that these two factors may co-regulate similar genes. Importantly, we confirmed that the shared genes co-upregulated by LSD1+8a and SRRM4 (Fig. 4b) were activated in human NEPC tumors (Fig. 4d) and PDXs (Supplementary Fig. S3), suggesting that SRRM4-induced splicing of LSD1+8a may contribute to activation of these genes. Among these genes are IFIT3, whose overexpression has been linked to pancreatic cancer progression via induction of inflammatory cytokines [21]; SAMD9, whose upregulation is associated with increased lymphatic invasion and metastasis in esophageal squamous cell carcinoma [28]; TRIM22, whose overexpression has been associated with poor prognosis in non-small cell lung cancer (NSCLC) patients [27]; HERC5, linked to poor outcomes in NSCLC patients [26]; and BST2, which is upregulated in bone metastases [24] and whose overexpression is correlated with poor survival in esophageal, gastric, and colorectal cancers [25], resistance to cisplatin in nasopharyngeal cancer [22], and tamoxifen resistance in breast cancers [23]. Further studies are necessary to determine mechanisms by which LSD1+8a and SRRM4 induce these genes’ expression and to determine whether LSD1+8a regulates important cancer hallmarks. Completion of those studies may provide the rationale for targeting LSD1+8a in NEPC and other cancers in which this transcript variant is expressed.
Methods
Prostate cancer clinical specimens and patient-derived xenografts
Adenocarcinoma patient tumors or NEPC patient tumors—the latter defined as those expressing chromogranin A and synaptophysin—were obtained from the UW Rapid Autopsy Program [32]. Biospecimens were obtained within eight hours of death from patients who died of metastatic CRPC. Visceral metastases were identified at the gross level; bone biopsies were obtained according to a previously described template from 16 to 20 different sites and metastases identified at a histological level [32]. Prostate adenocarcinoma or NEPC LuCaP PDX lines were established from specimens acquired at either radical prostatectomy or at rapid autopsy, implanted, and maintained by serial passage in immune compromised male mice [15]. All rapid autopsy tissues were collected from patients who signed written informed consent under the aegis of the Prostate Cancer Donor Program at the University of Washington. The Institutional Review Board of the University of Washington approved this study. LTL NEPC patient-derived xenograft lines have been described previously [16]. All patients signed an informed consent form approved by the Institutional Review Board of the University of British Columbia. All methods were performed in accordance with relevant guidelines and regulations.
Cell culture
Parental LNCaP cells (clone FGC) were obtained from the American Type Culture Collection (ATCC). All SRRM4-overexpressing cell lines and their corresponding controls have been described previously [10], [17]. PC3, DU145, and hTERT-Myo cell models were cultured in Dulbecco Modified Eagle Medium/High Glucose (DMEM; Gibco; Waltham, MA, USA; Cat# 11995-065) with 10% fetal bovine serum (FBS) whereas LNCaP, BPH1, and 22RV1 cells were cultured in RPMI-1640 medium with 10% FBS. WPMY1 cells were maintained in DMEM medium containing 10% FBS and 1% penicillin–streptomycin (Hyclone; Cat# SV30010). RWPE1 cells were grown in Keratinocyte-SFM (1X) media with human recombinant epidermal growth factor 1–53 and bovine pituitary extract supplements (Gibco; Cat# 17005042). Transduced cells were selected by and cultured under blasticidin (Gibco; Cat# A11139-03) with the respective concentrations for the following cells: RWPE1, 2 μg/ml; BPH1, 2.5 μg/ml; LNCaP, 22RV1, and DU145, 5 μg/ml; PC3, WPMY1, and hTERT-Myo, 10 μg/ml. Mycoplasma testing was performed on all cell lines and found to be negative.
For cell line experiments involving the minigene and those involving FLAG overexpression constructs, cells were cultured as above but supplemented with 6 µg/mL blasticidin (Sigma-Aldrich #15205) to maintain overexpression constructs for the course of the experiments.
RNA extraction
Total RNA was extracted from 19 CRPC metastases frozen in Optimal Cutting Temperature compound (OCT; Tissue-Tek) with RNA STAT-60 (Tel-Test). Using an H&E stained slide for each metastatic site for orientation, one millimeter core punches of tumor were obtained, or where appropriate multiple sections enriched for tumor were cut using a Leica CM3050S cryostat. The purity and yield of the RNA were determined on a NanoDrop 2000 (Thermo Scientific). RNA integrity was assessed on a 2100 Bioanalyzer (Agilent). Total RNA was extracted from all PDX tissue and cell lines using Trizol (Thermo Fisher Scientific) according to manufacturer’s protocol. Purity and yield were determined using a NanoDrop instrument.
qRT-PCR
For data in Fig. 2a, 2 µg of total RNA extracted from stable SRRM4-overexpressing cell lines and corresponding controls was reverse-transcribed using random hexamers and SuperScript IITM Reverse Transcriptase (Invitrogen) according to manufacturer’s instructions. For all other samples, 1 µg of total RNA was reverse-transcribed using random hexamers and a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. PCR for LSD1+8a and canonical LSD1 transcripts was performed using SYBR Green ER qPCR SuperMix (Thermo Fisher Scientific) with 100 nmol of previously-described exon junction-specific LSD1 primers [6] (sequences in Supplementary Table 4). All other PCRs were performed with TaqMan reagents (Thermo Fisher Scientific) as previously described [33], using probes for Human SRRM4 (Product No. Hs00916552_m1), CHGA (Hs00900370_m1), CHGB (Hs01084631_m1), SCG3 (Hs00203076_m1), SYP (Hs00300531_m1), BST2 (Hs00171632_m1), CLU (Hs00156548_m1), HERC5 (Hs00180943_m1), IFIT3 (Hs01922752_s1), SAMD9 (Hs00539471_s1), and TRIM22 (Hs01001179_m1). For absolute quantitation of expression, serial dilutions of LSD1+8a, LSD1, or SRRM4 plasmids (described in detail below) were performed to generate standard curves of log copy number vs. Ct value for each gene. Ct values of unknown samples were then fitted to the curves’ linear regression equations to determine copy number. Human ACTB TaqMan probe (Hs99999903_m1) was used as an endogenous control for all relative expression experiments. cDNA equivalent of 10 ng of RNA was used for each PCR. All qRT-PCR experiments were performed using a QuantStudio3 thermocycler (Thermo Fisher Scientific).
RNA immunoprecipitation
The RNA immunoprecipitation assay was performed as described previously [10]. Briefly, LNCaP-SRRM4 cells were treated with 1% formaldehyde for 10 minutes to cross-link RNA-protein complex. Lysates prepared from cross-linked cells were sonicated using Diagenode Bioruptor sonicator to fragment RNA. FLAG-SRRM4 was immunoprecipitated from the sonicated lysates using monoclonal anti-FLAG antibody in mouse (Sigma-Aldrich F4042). IgG was used as a negative control for immunoprecipitation. RNA fragments immunoprecipitated with FLAG-SRRM4 were used as templates for one step real-time qPCR using One-Step SYBR GreenER kit (Thermo Fisher Scientific). We designed primers to amplify a region covering a UGC motif predicted to be an SRRM4 recognition site in intron 8 (8a 5′), a region at the junction of the LSD1 exon 8-intron 9 (8a 3′), and a negative control region lacking an SRRM4 binding site in intron 1 (Intron 1). Data were calculated as a percentage of input. Primer sequences are listed in Supplementary Table 4.
LSD1 minigene design and assay
A detailed description of the minigene construction, including complete sequence, is included in Supplementary Fig. S2. Briefly, the minigene was designed to constitutively express mRNA (as driven by CMV promoter) from human LSD1 exons 8, 8a, and 9, along with 300 base pairs of intronic nucleotides directly flanking each exon. Human LSD1 exon and intron sequences were obtained from Ensembl [34]. After design, the minigene sequence was synthesized and cloned into the pcDNA3.1(+) vector (GenScript). For the assay, the minigene construct or pcDNA3.1 empty vector control was transiently transfected into cells using Lipofectamine 2000 reagent as previously described [35]. LNCaP and LNCaP-SRRM4 cells were harvested 96 hours post-transfection, 22RV1-CTL and 22RV1-SRRM4 were harvested 72 h post-transfection. RNA was extracted and then converted into cDNA as described above. qRT-PCR was then performed using exon junction-specific LSD1 primers [6] as described above to quantify alternative splicing of 8a+ or canonical LSD1 conformations.
Transient overexpression
Plasmid overexpressing FLAG-tagged canonical LSD1 cDNA (pTP6-LSD1) was described previously [36]. To generate a FLAG-tagged LSD1+8a overexpressing plasmid, we inserted 12 nucleotides corresponding to exon 8a (5′-gacactgtcaag-3′) into pTP6-LSD1 between exons 8 and 9 (Mutagenex). A constitutively overexpressing FLAG-SRRM4 plasmid (pCMV6-SRRM4) was obtained from Origene (Cat No. RC219268). LNCaP cells were transiently transfected with the aforementioned plasmids, or an empty pTP6 control vector, using Lipofectamine 2000 reagent as previously described [35]. After 96 h, RNA was extracted as described above, and protein lysates were extracted from samples transfected in parallel using RIPA buffer. qRT-PCR and Western blotting were used to confirm transfection.
Western blotting
Protein lysates were run on NuPAGE 4–12% Bis-Tris protein gels (Thermo Fisher Scientific) and transferred to PVDF membranes per manufacturer’s instructions. Primary FLAG-M5 antibody was used to probe tagged overexpressed proteins. Appropriate fluorescent secondary antibodies (Licor) were used to detect proteins, and images were captured using a Licor Odyssey CLx instrument.
RNA-seq
RNA was extracted using Trizol as described above, and integrity was assessed on a TapeStation 2200 (Agilent Technologies) with Analysis Software A.02.02. Library preparation was performed using a TruSeq library preparation kit (Illumina) with poly(A)+ stranded selection. Libraries were sequenced on a HiSeq 2500 (Illumina) as paired-end 100 base pair reads by the OHSU Massively Parallel Sequencing Shared Resource.
RNA-seq reads were mapped to hg19 reference transcripts in RSEM [37] using the STAR aligner. All genes with read counts in at least one of case were kept for subsequent differential expression gene (DEG) analyses. DEG analyses were performed using DESeq2 according to the corresponding phenotypes [38]. Gene expression changes as a result of overexpression of LSD1+8a, LSD1, and SRRM4 were determined in comparison to cells transfected with an empty control vector. DEGs were called with Wald test p-values less than 0.05 and absolute fold-change cutoff larger than 1.5. Due to the limited number of DEGs, we did not use false discovery rate corrections for further filtration. Gene Ontology (GO) enrichment analyses [39], [40], [41] of DEGs for each overexpression condition and LSD1+8a/SRRM4 shared DEGs (Supplementary Table 3) were performed using an online tool (http://software.broadinstitute.org/gsea/msigdb/annotate.jsp).
Gene set enrichment analysis (GSEA)
In this study, GSEA version 3.0 [41] was used to validate the relevance of human NEPC with the shared 31 genes co-upregulated by SRRM4 and LSD1+8a. The Aggarwal, et al. [3] expression data normalized by variance stabilizing transformation in DESeq2 [38] was used as the input of GSEA, and the default metric Signal2Noise in GSEA was applied to calculate the differential expression with respect to the adenocarcinoma tumors and NEPC tumors. LuCaP PDX RNA-seq analysis has been described previously [42]. Differential expression analysis was performed using transcript abundances as inputs to the limma [43] bioinformatic package and using the Benjamin-Hochberg false discovery rate (FDR) adjustment. Gene expression results were ranked by their limma statistics and used to conduct GSEA.
Acknowledgments
Acknowledgements
We thank the OHSU Massively Parallel Sequencing Shared Resource for performing short read sequencing assays. We thank the patients and their families who donated their samples, Drs. Celestia Higano, Evan Yu, Elahe Mostaghel, Heather Cheng, Bruce Montgomery, Mike Schweizer, Daniel Lin, Funda Vakar-Lopez, Lawrence True, and the rapid autopsy teams for their contributions to the University of Washington Medical Center Prostate Cancer Donor Rapid Autopsy Program and the Development of the LuCaP PDX models.
Author contributions
D.J.C., D.A.S., A.S., and J.J.A. designed the research; Y.W., A.R.L, X.D, C.M., and E.C. provided samples, D.J.C., D.A.S., A.S., J.S., and J.U. performed the experiments, D.J.C., D.A.S., A.S., A.K., D.S., J.S., J.U., I.M.C., C.M., Z.X., J.A.Y, and J.J.A. analyzed the data, D.J.C., D.A.S., I.M.C, P.S.N, X.D., C.M. E.C., J.A.Y., and J.J.A. provided critical analysis of the manuscript, D.J.C., D.A.S., A.K., J.A.Y., and J.J.A. wrote the paper.
Competing interests
Dr. Alumkal has performed consulting or held an advisory role with Astellas Pharma, Bayer, and Janssen Biotech, Inc. Dr. Nelson has performed consulting or held an advisory role with Astellas Pharma, Janssen Biotech, Inc and Bristol Myers Squibb. OHSU has received institutional research funding from Aragon Pharmaceuticals Inc., Astellas Pharma, Novartis, Zenith Epigenetics Ltd, and Gilead Sciences Inc. These interests had no role in the design or conduct of this study.
Data availability
RNA-seq data depicted in Fig. 4 and Supplementary Tables 1–4 have been deposited to the Gene Expression Omnibus (GEO) [44], [45] with accession number GSE132062.
Funding
We thank the following funding sources: (1) National Institutes of Health (NIH)/National Cancer Institute (NCI) R01 award CA178610, (2) Pacific Northwest Prostate Cancer SPORE/NCI SPORE/NCI (P50 CA097186), (3) Michigan Prostate SPORE/NCI (P50 CA186786), (4) Cancer Center Support Grants CCSG (P30 CA069533 and CA046592), (5) Wayne D. Kuni and Joan E. Kuni Foundation, (6) The Department of Defense Prostate Cancer Biorepository Network (PCBN) (W81XWH-14-2-0183), (7) Department of Defense Award (W81XWH-17-1-0415), (8) NIH PO1 grant (PO1 CA163227), (9) the Richard M. LUCAS Foundation, (10) the Institute for Prostate Cancer Research (IPCR), (11) Canadian Institute of Health Research (CIHR, PJT-156150), and (12) the Prostate Cancer Foundation, (13) University of Michigan Rogel Scholar Award.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neo.2020.04.002.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590 [DOI] [PubMed]
- 2.Grigore A.D., Ben-Jacob E., Farach-Carson M.C. Prostate cancer and neuroendocrine differentiation: more neuronal, less endocrine? Front Oncol. 2015;5:37. doi: 10.3389/fonc.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal R., Huang J., Alumkal J.J., Zhang L., Feng F.Y., Thomas G.V., Weinstein A.S., Friedl V., Zhang C., Witte O.N. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J Clin Oncol. 2018;36:2492–2503. doi: 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sehrawat A., Gao L., Wang Y., Bankhead A., 3rd, McWeeney S.K., King C.J., Schwartzman J., Urrutia J., Bisson W.H., Coleman D.J. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci U S A. 2018;115:E4179–E4188. doi: 10.1073/pnas.1719168115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zibetti C., Adamo A., Binda C., Forneris F., Toffolo E., Verpelli C., Ginelli E., Mattevi A., Sala C., Battaglioli E. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J Neurosci. 2010;30:2521–2532. doi: 10.1523/JNEUROSCI.5500-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurent B., Ruitu L., Murn J., Hempel K., Ferrao R., Xiang Y., Liu S.C., Garcia B.A., Wu H., Wu F.Z. A Specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol Cell. 2015;57:957–970. doi: 10.1016/j.molcel.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toffolo E., Rusconi F., Paganini L., Tortorici M., Pilotto S., Heise C., Verpelli C., Tedeschi G., Maffioli E., Sala C. Phosphorylation of neuronal Lysine-Specific Demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. J Neurochem. 2014;128:603–616. doi: 10.1111/jnc.12457. [DOI] [PubMed] [Google Scholar]
- 8.Jotatsu T., Yagishita S., Tajima K., Takahashi F., Mogushi K., Hidayat M., Wirawan A., Ko R., Kanemaru R., Shimada N. LSD1/KDM1 isoform LSD1+8a contributes to neural differentiation in small cell lung cancer. Biochem Biophys Rep. 2017;9:86–94. doi: 10.1016/j.bbrep.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balanis N.G., Sheu K.M., Esedebe F.N., Patel S.J., Smith B.A., Park J.W., Alhani S., Gomperts B.N., Huang J., Witte O.N. Pan-cancer convergence to a small-cell neuroendocrine phenotype that shares susceptibilities with hematological malignancies. Cancer Cell. 2019;36(17–34) doi: 10.1016/j.ccell.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Donmez N., Sahinalp C., Xie N., Wang Y., Xue H., Mo F., Beltran H., Gleave M., Wang Y. SRRM4 drives neuroendocrine transdifferentiation of prostate adenocarcinoma under androgen receptor pathway inhibition. Eur Urol. 2017;71:68–78. doi: 10.1016/j.eururo.2016.04.028. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X.T., Coleman I.M., Brown L.G., True L.D., Kollath L., Lucas J.M., Lam H.M., Dumpit R., Corey E., Chery L. SRRM4 expression and the loss of REST activity may promote the emergence of the neuroendocrine phenotype in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:4698–4708. doi: 10.1158/1078-0432.CCR-15-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y., Chen R., Bowden M., Mo F., Lin Y.Y., Gleave M., Collins C., Dong X. Establishment of a neuroendocrine prostate cancer model driven by the RNA splicing factor SRRM4. Oncotarget. 2017;8:66878–66888. doi: 10.18632/oncotarget.19916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raj B., O'Hanlon D., Vessey John P., Pan Q., Ray D., Buckley Noel J., Miller Freda D., Blencowe Benjamin J. Cross-regulation between an alternative splicing activator and a transcription repressor controls neurogenesis. Mol Cell. 2011;43:843–850. doi: 10.1016/j.molcel.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Rusconi F., Paganini L., Braida D., Ponzoni L., Toffolo E., Maroli A., Landsberger N., Bedogni F., Turco E., Pattini L. LSD1 neurospecific alternative splicing controls neuronal excitability in mouse models of epilepsy. Cereb Cortex. 2015;25:2729–2740. doi: 10.1093/cercor/bhu070. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen H.M., Vessella R.L., Morrissey C., Brown L.G., Coleman I.M., Higano C.S., Mostaghel E.A., Zhang X., True L.D., Lam H.M. LuCaP prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease and serve as models for evaluating cancer therapeutics. Prostate. 2017;77:654–671. doi: 10.1002/pros.23313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin D., Wyatt A.W., Xue H., Wang Y., Dong X., Haegert A., Wu R., Brahmbhatt S., Mo F., Jong L. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74:1272–1283. doi: 10.1158/0008-5472.CAN-13-2921-T. [DOI] [PubMed] [Google Scholar]
- 17.Lee A.R., Gan Y., Tang Y., Dong X. A novel mechanism of SRRM4 in promoting neuroendocrine prostate cancer development via a pluripotency gene network. EBioMedicine. 2018;35:167–177. doi: 10.1016/j.ebiom.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raj B., Irimia M., Braunschweig U., Sterne-Weiler T., O'Hanlon D., Lin Z.Y., Chen G.I., Easton L.E., Ule J., Gingras A.C. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol Cell. 2014;56:90–103. doi: 10.1016/j.molcel.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies A.H., Beltran H., Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol. 2018;15:271–286. doi: 10.1038/nrurol.2018.22. [DOI] [PubMed] [Google Scholar]
- 20.Li Y., Zhang Q., Lovnicki J., Chen R., Fazli L., Wang Y., Gleave M., Huang J., Dong X. SRRM4 gene expression correlates with neuroendocrine prostate cancer. Prostate. 2019;79:96–104. doi: 10.1002/pros.23715. [DOI] [PubMed] [Google Scholar]
- 21.Niess H., Camaj P., Mair R., Renner A., Zhao Y., Jackel C., Nelson P.J., Jauch K.W., Bruns C.J. Overexpression of IFN-induced protein with tetratricopeptide repeats 3 (IFIT3) in pancreatic cancer: cellular “pseudoinflammation” contributing to an aggressive phenotype. Oncotarget. 2015;6:3306–3318. doi: 10.18632/oncotarget.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuang C.M., Fu X., Hua Y.J., Shuai W.D., Ye Z.H., Li Y., Peng Q.H., Li Y.Z., Chen S., Qian C.N. BST2 confers cisplatin resistance via NF-kappaB signaling in nasopharyngeal cancer. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi E.H., Yoo H., Noh K.H., Han S., Lee H., Lee J.K., Won C., Kim B.H., Kim M.H., Cho C.H. BST-2 is a potential activator of invasion and migration in tamoxifen-resistant breast cancer cells. Biochem Biophys Res Commun. 2013;435:685–690. doi: 10.1016/j.bbrc.2013.05.043. [DOI] [PubMed] [Google Scholar]
- 24.Cai D., Cao J., Li Z., Zheng X., Yao Y., Li W., Yuan Z. Up-regulation of bone marrow stromal protein 2 (BST2) in breast cancer with bone metastasis. BMC Cancer. 2009;9:102. doi: 10.1186/1471-2407-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukai S., Oue N., Oshima T., Mukai R., Tatsumoto Y., Sakamoto N., Sentani K., Tanabe K., Egi H., Hinoi T. Overexpression of transmembrane protein BST2 is associated with poor survival of patients with esophageal, gastric, or colorectal cancer. Ann Surg Oncol. 2017;24:594–602. doi: 10.1245/s10434-016-5100-z. [DOI] [PubMed] [Google Scholar]
- 26.Wrage M., Hagmann W., Kemming D., Uzunoglu F.G., Riethdorf S., Effenberger K., Westphal M., Lamszus K., Kim S.Z., Becker N. Identification of HERC 5 and its potential role in NSCLC progression. Int J Cancer. 2015;136:2264–2272. doi: 10.1002/ijc.29298. [DOI] [PubMed] [Google Scholar]
- 27.Liu L., Zhou X.-M., Yang F.-F., Miao Y., Yin Y., Hu X.-J., Hou G., Wang Q.-Y., Kang J. TRIM22 confers poor prognosis and promotes epithelial-mesenchymal transition through regulation of AKT/GSK3β/β-catenin signaling in non-small cell lung cancer. Oncotarget. 2017;8:62069. doi: 10.18632/oncotarget.18911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang S., Zheng X., He L., Qiao L., Jing D., Ni Z., Zeng W., Jiang M. Significance of SAMD9 expression in esophageal squamous cell carcinoma. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2014;30:411–413. [PubMed] [Google Scholar]
- 29.Tanaka M., Shimbo T., Kikuchi Y., Matsuda M., Kaneda Y. Sterile alpha motif containing domain 9 is involved in death signaling of malignant glioma treated with inactivated Sendai virus particle (HVJ-E) or type I interferon. Int J Cancer. 2010;126:1982–1991. doi: 10.1002/ijc.24965. [DOI] [PubMed] [Google Scholar]
- 30.Schwarzenbach H., Hoon D.S.B., Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 31.Scheckel C., Darnell R.B. Microexons-Tiny but mighty. EMBO J. 2015;34:273–274. doi: 10.15252/embj.201490651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roudier M.P., True L.D., Higano C.S., Vesselle H., Ellis W., Lange P., Vessella R.L. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum Pathol. 2003;34:646–653. doi: 10.1016/s0046-8177(03)00190-4. [DOI] [PubMed] [Google Scholar]
- 33.Coleman D.J., Gao L., Schwartzman J., Korkola J.E., Sampson D., Derrick D.S., Urrutia J., Balter A., Burchard J., King C.J. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci Rep. 2019;9:3823. doi: 10.1038/s41598-019-40518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G. Ensembl 2018. Nucleic Acids Res. 2017;46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coleman D.J., Gao L., King C.J., Schwartzman J., Urrutia J., Sehrawat A., Tayou J., Balter A., Burchard J., Chiotti K.E. BET bromodomain inhibition blocks the function of a critical AR-independent master regulator network in lethal prostate cancer. Oncogene. 2019;38:5658–5669. doi: 10.1038/s41388-019-0815-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adamo A., Sese B., Boue S., Castano J., Paramonov I., Barrero M.J., Belmonte J.C.I. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol. 2011;13:652–U265. doi: 10.1038/ncb2246. [DOI] [PubMed] [Google Scholar]
- 37.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.The Gene Ontology Consortium The Gene Ontology Resource: 20 years and still going strong. Nucleic Acids Res. 2018;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Labrecque M.P., Coleman I.M., Brown L.G., True L.D., Kollath L., Lakely B., Nguyen H.M., Yang Y.C., da Costa R.M.G., Kaipainen A. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest. 2019;130:4492–4505. doi: 10.1172/JCI128212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43 doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2012;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further reading
- 46.Nakano Y., Jahan I., Bonde G., Sun X., Hildebrand M.S., Engelhardt J.F., Smith R.J., Cornell R.A., Fritzsch B., Banfi B. A mutation in the Srrm4 gene causes alternative splicing defects and deafness in the Bronx waltzer mouse. PLoS Genet. 2012;8:e1002966. doi: 10.1371/journal.pgen.1002966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data depicted in Fig. 4 and Supplementary Tables 1–4 have been deposited to the Gene Expression Omnibus (GEO) [44], [45] with accession number GSE132062.