

Key Points
Diagnostic and relapse diffuse large B-cell lymphoma (DLBCL) biopsies reveal increased mutational burden/loss of heterozygosity in HLA-A.
Serially sampled tumor biopsies provide insight into therapeutic targets and evolutionary divergence in relapsed/refractory DLBCL.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma, representing ∼30% of all cases in western countries. It is an aggressive and molecularly heterogeneous form of lymphoma, with >18 different subtypes classified by the World Health Organization (WHO).1,2 Primary refractory disease or relapse DLBCL (rrDLBCL), in which the disease progresses after an apparent effective treatment or clinical response to standard of care (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone [R-CHOP]), has long been recognized as a major contributor to the cancer’s mortality. The 3-year progression-free survival (PFS) in rrDLBCL cases is only 14%.3 Moreover, with the introduction of personalized and targeted therapies as viable therapeutic options, the appearance of multiple resistance mechanisms represents a major identified challenge. Thus, understanding the common features of drug resistance in DLBCL will not only contribute to our understanding of the leading cause of death in lymphoma, but may also assist in future drug design.
Next-generation-sequencing studies have refined the complexity of DLBCL subtypes and provided new categorizations of de novo DLBCL.4-7 The genetics of rrDLBCL have been less studied, and only a limited number of studies have focused on tumor genetic evolution upon relapse by analyzing paired diagnostic and relapsed biopsies.8-11 In recent years, the majority of sequencing studies in DLBCL have focused on providing a genetic “lymphopanel” to define the underpinnings of diversity and improve overall survival (OS) in this disease. We aimed to enhance DLBCL genomic profiling with a relapse-rich cohort of Nordic DLBCL cases, including 18 serially sampled cases, to contribute substantial information on progression and relapse.
Methods
Patient samples
A total of 83 cases and 102 biopsies (n [cases] = 83; b [biopsies] = 102) of DLBCL were included in this study. The whole-exome sequencing (WES) cohort consisted of 45 fresh-frozen biopsies from a Nordic cohort of 37 de novo DLBCL cases with matched normal DNA from peripheral blood, and 11 biopsies without matched normal from 5 patients. Patients were included from Oslo University Hospital (OUH) (n = 40; b = 54) and the Helsinki University Central Hospital Comprehensive Cancer Center (HUCHCCC) in Finland (n = 2; b = 4) (supplemental Tables 1-3; supplemental Figure 1).
A validation cohort using custom panel sequencing of 46 formalin-fixed paraffin-embedded biopsies was collected at OUH (n = 35) or at HUCHCCC (n = 11) (supplemental Tables 2 and 4; supplemental Figure 1), excluding cases with low DNA quality.
This study was approved by the Norwegian South Eastern Regional Ethics Committee (REK 2014/127). The study was performed according to the Declaration of Helsinki. All patients consented to written patient information.
WES and variant detection
Genomic DNA from fresh-frozen tissue and EDTA-treated blood was extracted using the Maxwell 16 DNA isolation automated magnetic bead instrument (Promega). Library preparation was carried out using SureSelectXT Human All Exon V5 (Agilent) per the manufacturer’s instructions at the OUH Genomics Core Facility, and whole exome capture and paired-end sequencing was performed using the Illumina HighSeq 2500 platform (average depth 289X, range 178-377X for tumor, 126X [range 88-173X] for normal; supplemental Figure 5). A benchmarked bioinformatics pipeline12 was used to process the sequencing reads and perform somatic variant calling (supplemental Materials and methods; supplemental Figure 4).
Copy-number analysis
Copy-number aberration analysis was performed using FACETS13 version 0.5.14. Recurrent copy-number alterations by arm level and focal, <50% of the chromosome arm, peaks were analyzed by GISTIC2.0 (version 2.0.22). Regions of alteration with a q value of <0.25 were reported as significant. The WES cohort was also subjected to SNP6.0 analysis using the Affymetrix Genome Wide SNP6.0 Array platform (Gene Chip lot 4296976).14,15
HLA class I allele and variant detection
POLYSOLVER software (v1; Broad Institute, Cambridge, MA) was used to infer the HLA class I alleles for each individual, and POLYSOLVER mutation calling and annotation was performed16 on WES cases with matched normal. Loss of heterozygosity (LOH) was determined by FACETS13 and POLYSOLVER.
Data analysis
Driver genes were identified with MutSig2CV (Broad Institute)4,17 and OncodriveFM and OncodriveCLUST programs18 (version 2.4.1). Genes with a q value of <0.1 were selected as harboring potential driver mutations.17
Clonal structure was inferred with sciClone (version 1.1)19 with loss-of-heterozygosity information from SNP6.0 array inferred by ASCAT. We used ClonEvol (https://github.com/hdng/clonevol) and Fishplot20 (R version 3.4.1) to build tumor phylogenies. Targeted cancer drugs were identified through the Open Targets Platform (downloaded 09.19),21 with additional filtering collected from OncoTree (http://oncotree.mskcc.org/#/home; phenotypes available upon request). Variant deleterious effect was inferred through a combined annotation-dependent depletion (CADD)22 phred score of over 10.
Immunohistochemical staining
Immunohistochemical staining (IHC) for class I antigens was performed applying antibodies against HLA-A (EP1395Y; Abcam) and β2-microglobulin (B2M) (2M2; Lifespan BioSciences). Details on IHC staining are provided in supplemental Materials and methods.
Statistical analysis
GraphPad Prism (Version 8.01) was used to perform an unpaired 2-tailed Student t test with Welch correction, contingency χ2, and the paired Student t test. R (version 3.4.1) was used for linear regression on time to relapse.
Results and discussion
To address the underlying genomics of acquired and/or intrinsic resistance in DLBCL, WES was performed on 42 de novo DLBCL cases consisting of 56 biopsies; 13 cases had available serial biopsies and 34% of all biopsies were taken at relapse (supplemental Figures 1-4; supplemental Tables 1-3). Custom targeted sequencing, of 139 genes (supplemental Table 5), was performed on a second cohort of formalin-fixed paraffin-embedded samples representing 41 cases of which 23% were relapse biopsies (supplemental Figures 1 and 3; supplemental Tables 2, 4, and 5).
Given the unique enrichment of relapsed DLBCL in our cohort, we categorized biopsies as diagnostic DLBCL (from patients who did not experience relapse), diagnostic rrDLBCL, and relapse biopsies from rrDLBCL (Figure 1A), and analyzed multiple case sets stratified by disease subtype, survival, and relapse status. This approach allowed a comprehensive investigation into the heterogeneous subtypes included in our analysis (ABC, GCB, PMBCL, T-cell/histiocyte-rich; and more, supplemental Tables 1-4) and exploration based on resistance and progression. Mutation signatures and overall mutation burden in this Nordic cohort were similar to previously published western and Chinese cohorts (median of 125 coding mutations per case; supplemental Figures 5-7).4,8,10,23-27 Using 2 different approaches for cancer driver gene prediction, we identified 118 potential driver genes including previously reported genes as well as multiple genes involved in antigen presentation (supplemental Tables 6 and 7). Four previously unidentified DLBCL driver genes were discovered: TMEM199, TBC1D24, IQSEC3, and SYNGR1 (supplemental Tables 6 and 7). Comparison of driver genes found in diagnostic biopsies from cases with a durable treatment response and cases that ultimately went on to relapse revealed an increased number of mutations in genes involved in antigen presentation, including HLA-A, HLA-B, CD74, and LILRA4 in the latter set of cases (Figure 1B; supplemental Figure 8; supplemental Table 6), implicating immune escape as a notable feature of DLBCL-intrinsic resistance.
Figure 1.
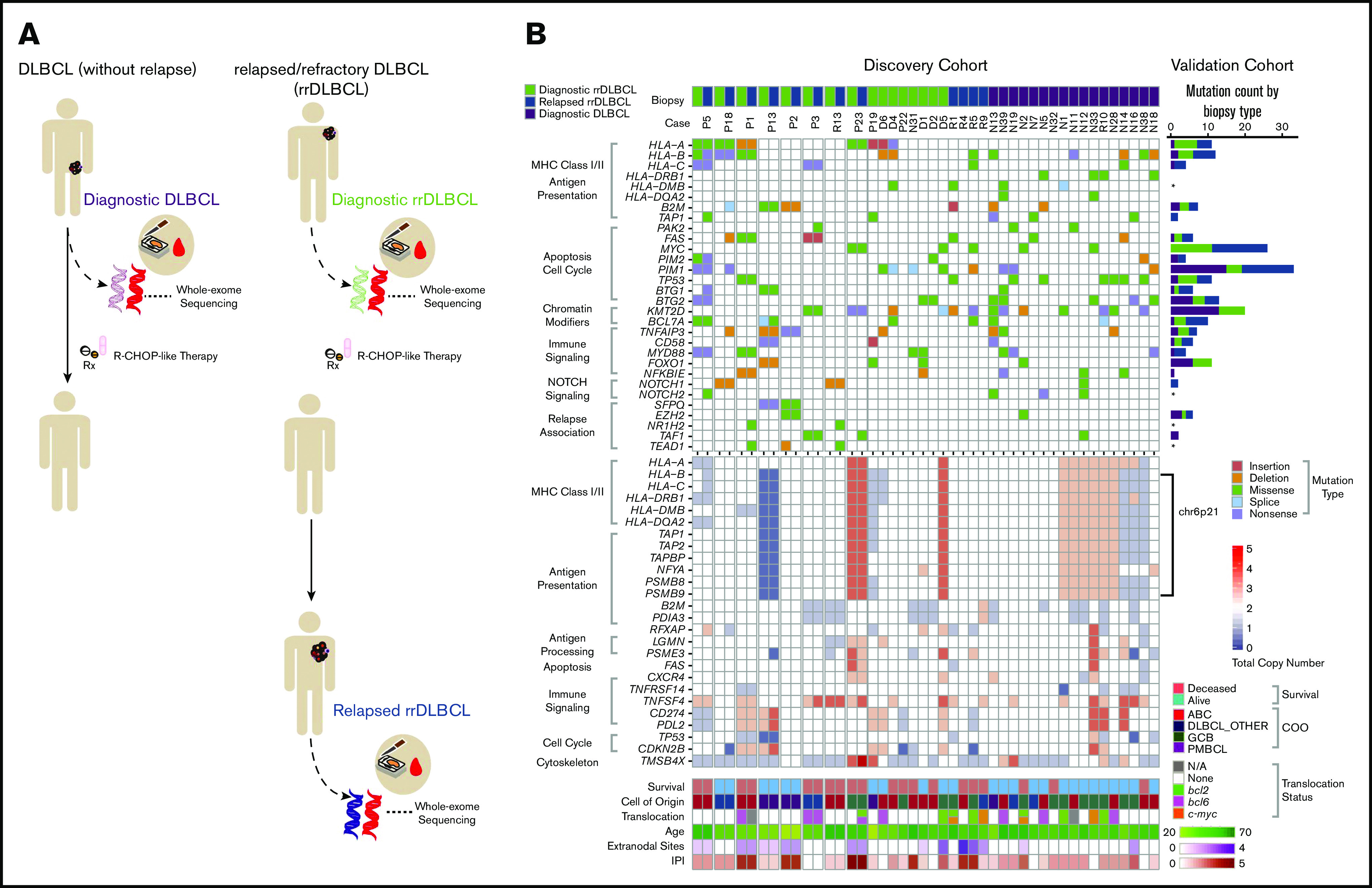
Recurrently altered genes identified in diagnostic and rrDLBCL. Collection of 2 DLBCL cohorts representing various chemo-immunotherapy treatment profiles allowed for the division into 3 biopsy types prior to genomic alteration analyses by WES (discovery cohort) or by targeted sequencing (validation cohort): Diagnostic DLBCL, representing initial diagnostic biopsies of patients that go on to have a durable response to RCHOP-like therapies; Diagnostic rrDLBCL, representing initial diagnostic biopsies of patients that progress or relapse after RCHOP-like therapies; and Relapsed rrDLBCL, representing relapse biopsies of patients who experienced refractory or relapsed disease. (A) Schematic overview of the sampling procedures for the 3 defined biopsy types of DLBCL. (B) Genes of interest identified by multiple mutation analyses including the MutSig2CV and IntOgen programs (n [cases] = 37; b [biopsies] = 45). Genes of interest have been organized into signaling/pathway categories (gray brackets). Mutations are shown as colored boxes. In cases where a gene had multiple mutations in the same patient, the mutation resulting in the most severe change to the protein structure is shown. Biopsies are listed horizontally and divided by biopsy type with multiple biopsies from the same patient placed adjacently. The number of mutations for each gene in the validation cohort is displayed as a histogram on the far right (n = 41; b = 46). Genes of interest identified through recurrent copy number alteration analysis (GISTIC2.0) (n = 42 in initial analysis, n = 37 displayed). Multiple genes involved in antigen processing and presentation were identified in the 6p21 cytoband. Each box represents a gene's total copy number state. Survival, subtype (including activated B-cell [ABC] like, germinal center B-cell [GCB] like, and primary mediastinal B-cell lymphoma [PMBCL]), translocation status, age, International Prognostic Index (IPI), and number of extranodal sites for each biopsy are displayed in the bottom rows.
In contrast to the initial high sensitivity of DLBCL to standard-of-care treatment, patients who experience relapse often have astonishingly aggressive and treatment-refractory disease. We observed that of the coding variants appearing at relapse, 8 recurrently mutated genes (ie, genes mutated in at least 17% of the tested relapse biopsies) were not mutated in any diagnostic pretreatment tumors: CACNA1B, FLRT1, KRAS, LRFN5, MPRIP, WNT8A, and NR1H2 (Figure 1B; supplemental Figure 12). However, all are represented in diagnostic biopsies in previously studied cohorts. Nevertheless, NR1H2 was reported as exclusively mutated in relapse biopsies by Morin et al, providing potential evidence for an association with resistance.8 NR1H2, a nuclear receptor for LXR-β, is ubiquitously expressed in tissues and is a key regulator of macrophage function, with a small molecule agonist currently in clinical trial.28,29
Furthermore, copy-number variation analysis revealed recurrent loss (either focal or arm level) of a region of chromosome 6p21 containing multiple antigen-presentation and antigen-processing genes (Figure 1B; supplemental Figure 9). Association of copy-number variations in 6p21 to clinical outcomes and immune-privileged site dissemination has been shown previously.30,31 We detected focal losses of immune-processing genes LGMN and PSME3, in addition to genes involved in antigen presentation: B2M, PDIA3, and RFXAP (Figure 1B; supplemental Figure 9). These findings provide further support for the idea that immune cloaking may be important in DLBCL resistance.
We quantified variants in HLA-A and revealed an increased incidence in cases that ultimately relapsed (Figure 2; supplemental Table 8). Given the highly polymorphic nature of the MHC regions, we performed further in-depth alignments and investigations into the mutational prevalence, timing, impact on prognosis, and LOH. We revealed HLA-A mutagenesis and LOH as a plausible method of immune evasion in cases that went on to relapse (Figure 2A-C; supplemental Figure 10; χ2 test; P < .02). Given that 2 of our cases were not treated with R-CHOP–like therapies, we retested this association without these 2 cases, and it remained significant (χ2 test; P < .04). To determine whether these genomic changes in immune-related genes result in altered protein expression patterns, we performed IHC staining of HLA-A and B2M on 58 biopsies representing 55 cases of DLBCL pooled from all of our cohorts (Figure 2D-E). The majority of HLA-A losses were observed in the rrDLBCL diagnostic and relapse biopsies, but the difference did not reach statistical significance (Figure 2D; χ2 test; P < .1). B2M expression was variable between the groups (supplemental Figure 11). Host immunogenetics, particularly HLA haplotypes and MHC class II variations, has emerged as a potential contributor to survival in DLBCL and follicular lymphoma patients.32-34 HLA class I protein deficiencies have been reported in up to 43% of DLBCL cases.35 We look forward to future investigations to unravel whether HLA-A mutational and protein status correlate with neoantigen profiles or the immune microenvironment of patients.
Figure 2.
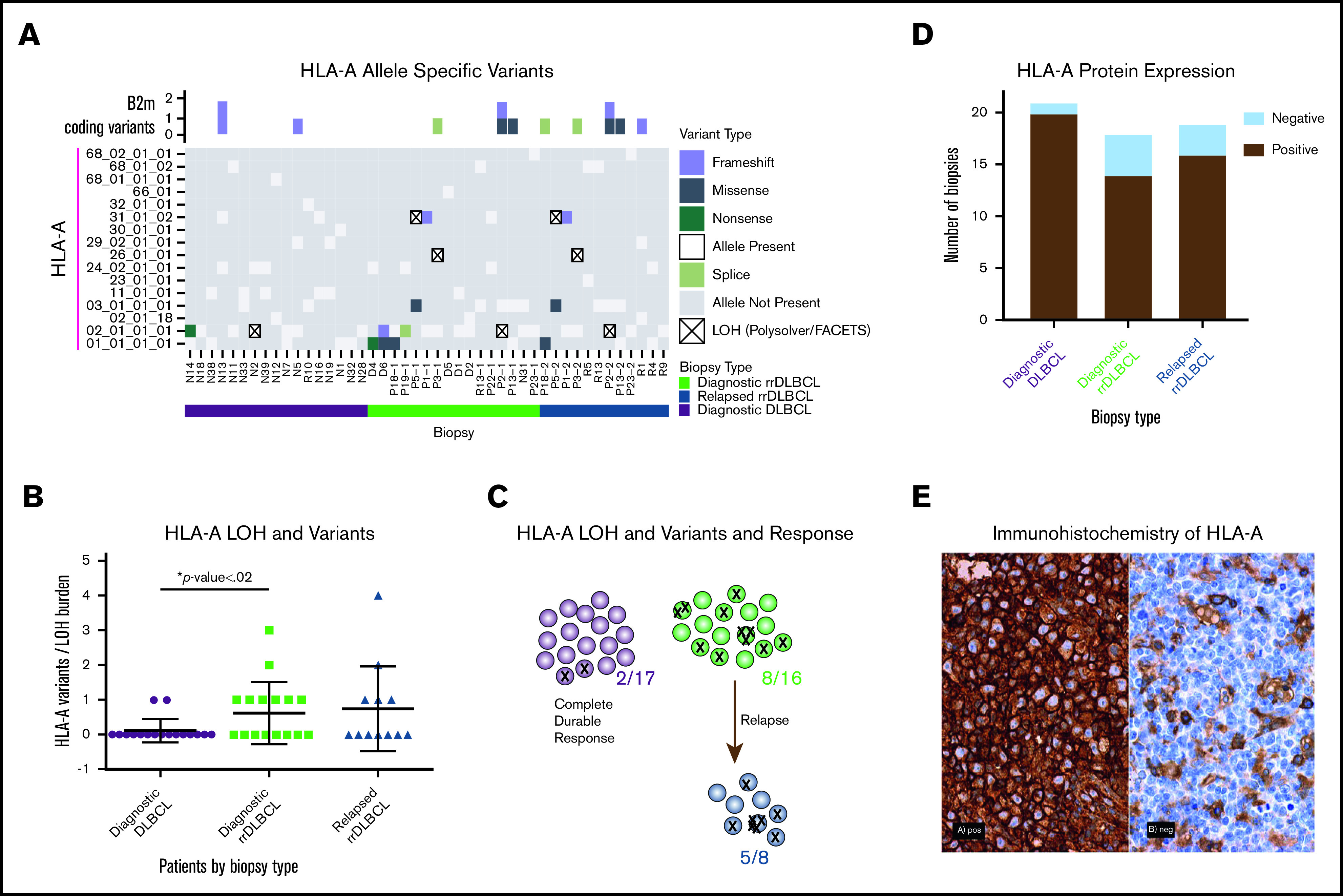
Frequent alterations of HLA-A in rrDLBCL. (A) For each biopsy type (diagnostic DLBCL, diagnostic rrDLBCL and relapse rrDLBCL), MHC class I allele inference for HLA-A was performed using POLYSOLVER. Depicted along the vertical axis are all inferred alleles from the WES with matched normal cohort (n = 37; b = 45). Each box is colored to represent absence, presence, loss, or mutation of each allele in each biopsy. Above the grid, B2M protein-coding variants in each biopsy are shown as boxes colored by mutation type. (B) Mutation burden in HLA-A per sample by biopsy type. Comparing initial diagnostic biopsies that do (green) or do not (purple) go on to relapse, the relapsing cases show a significantly higher burden of HLA-A alterations (P < .02, unpaired Student t test with Welch correction). (C) Increased mutational burden in HLA-A in cases that go on to progress or relapse from R-CHOP–like therapies. Each circle represents a case where analysis was available, each X represents a mutation or LOH event. (D) Immunohistochemical analysis of HLA-A protein expression in the combined cohort of DLBCL samples from the WES and targeted sequencing cohorts. Classification into HLA-A negative and positive staining was performed for each biopsy type, n = 55, b = 58. (E) Representative images of IHC staining of HLA-A; HLA-A+ and HLA-A− biopsies obtained on a Nikon Eclipse Ci at 60× objective.
Thus far, our analyses of the 3 biopsy types have suggested that immune cloaking may be an intrinsic-resistance mechanism, as antigen-presentation alterations already exist at diagnosis and may predict response. Our relapse-rich cohort should provide significant information in the search for acquired relapse mechanisms and thereby potentially contribute to development of improved salvage therapies. Due to clinical practices, serial biopsies are rare, but we were able to assemble 18 serially sampled cases with corresponding detailed clinical annotations. Patient-wise, we compared mutations identified in the initial diagnostic biopsy to mutations identified in the relapse biopsy and integrated the molecular results with the clinical parameters including biopsy location (supplemental Figure 13). As time to relapse increased, we observed a strong trend of decreasing similarity between biopsies representing evolutionary divergence over time (Figure 3A-B). However, this observation was independent of lesion location. Leveraging the availability of matched blood and of copy-number information, we applied 2 clonality analysis tools (sciClone19 and ClonEvol20) on 7 cases with serial biopsies available (Figure 3C; supplemental Figures 13 and 14). At each time point, all cases had clones that contained variants unique to the biopsy at ∼50% variant allelic fraction, thus suggesting 2 different dominant clones at diagnosis and relapse (Figure 3C; supplemental Figures 13 and 14), with a substantial overlap of variants between biopsies. This indicates branched evolution with late divergence of the tumors, and suggests that the dominant clone at diagnosis is eliminated by the given therapy and a unique clone, albeit derived from a common progenitor clone, dominates at relapse (Figure 3C). Founding clones frequently had alterations in antigen-presentation and -processing genes such as HLA genes and B2M. In 1 case, P5, multiple variants in HLA genes were gained throughout the oncogenic progression of the tumor. However, as in previous investigations, we were unable to find a recurrent mutation clearly responsible for resistance to R-CHOP–like therapies. This may suggest that resistance is intrinsic, or that multiple resistance mechanisms appear in each patient and are diverse among DLBCL cases.
Figure 3.
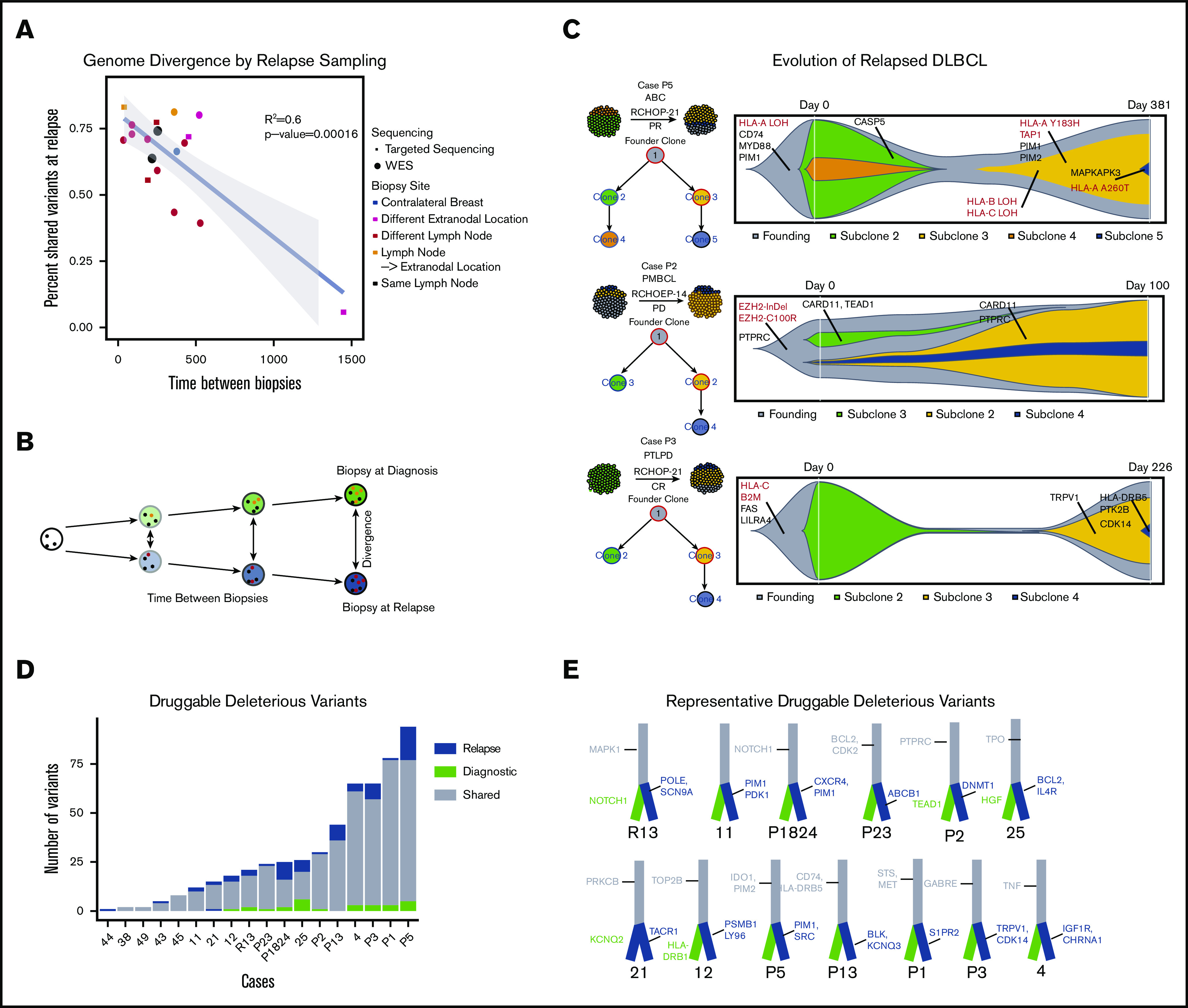
Genomic evolution and druggability. Mutational dynamics and evolutionary divergence in serially sampled rrDLBCL cases. (A) rrDLBCL cases that were serially sampled between treatment regimens were analyzed to compare the genomic alterations before and after treatment. Variants were classified as shared (present in both biopsies) or private (present in only 1 biopsy). For each case, the percent of shared variants between the diagnostic and relapse biopsies was calculated and plotted against time between biopsies. Biopsy site location and type of sequencing (WES or targeted) were integrated as the color and shape of each point, respectively. A negative linear correlation between genetic similarity and time between biopsies was observed (P < .00016). (B) Below the plot, a schematic example is shown: over time, a founding clone (white circle with black founding mutations) evolves into the clones present at diagnosis (green with orange private mutations) and at relapse (blue, with red private mutations). Clonal evolutionary analysis and identification of druggable alterations was performed, n = 18. (C) Schematic images of representative biopsies are shown with inferred clonal population percentages and branching evolutionary trees. Clinical information including subtype and treatment are displayed. To the right of each case, “fish plots” illustrate the clonal architecture of the 2 tumor biopsies, inferred from clonal fractions of mutation clusters and response to treatment. Mutations in genes of interest are shown in clones when they occurred, with genes involved in antigen presentation highlighted in red. (D) Druggable somatic mutations were identified using the Open Targets Platform, and oncogenic potential was estimated using CADD. Histogram of mutation counts of those variants identified as druggable and oncogenic in serially biopsied cases colored by shared vs private status, relapse vs diagnostic (n = 18, b = 37) paired Student t test (n = 16 (excluded relapse-relapse serial pairs), P = .0221). (E) Illustrative branched evolutionary trees representing founding clone mutations (gray trunk) and mutations private to diagnostic biopsies and relapsed biopsies (green and blue, respectively). Druggable mutations in genes of interest are displayed.
Given the strong similarities between diagnostic and relapse biopsies and the observed evolutionary divergence with time, we sought to understand whether relapse sampling advances our understanding of treatment options enough to warrant rebiopsies. We assessed the evolutionary timing of mutations in druggable targets. There were a median of 23 mutations per biopsy in genes targetable by known antineoplastic drugs as listed in the Open Targets Platform.21 To incorporate the functional impact of each variant on driving cancer, we analyzed all druggable variants using CADD22 (Figure 3D-E). Private variants in druggable genes were found more frequently in relapse biopsies than in initial diagnostic biopsies (Figure 3D; P < .0221). Although our results indicate that relapse sampling is important for guiding treatment, obtaining relapse biopsies can present ethical and practical issues for clinicians. Therefore, sampling of the peripheral blood for circulating tumor DNA or circulating tumor cells may provide an alternative investigational route for relapse sampling.36-38
Two of the previous studies on serially sampled rrDLBCL cases revealed a novel increase in mutations associated with the NF-κB pathway and that immune surveillance was disrupted by indels and copy number variations in CD58 and B2M.23 In contrast, in our paired samples, these alterations were seen in both the diagnostic and relapse biopsies. Our results suggest that immune-surveillance targeting also occurs at diagnosis, and should be taken into consideration when making clinical decisions on the use of the growing arsenal of immune therapies. Theoretically, these patients may be able to develop resistance to checkpoint blockade and neoantigen therapy, but may be sensitive to engineered immune therapies such as chimeric antigen receptor–T-cell therapies and NK-cell therapies. CD19-engineered chimeric antigen receptor T cells have shown long-term durable response rates in up to 58% in rrDLBCL.39-43 More broadly, our findings yield insight into the development of resistance in DLBCL, and highlight antigen processing and presentation as key targets for genomic aberrations in rrDLBCL cases. Moreover, our findings provide a clear investigational route into the effect of HLA-A mutations on relapse status in future clinical correlative studies.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Merete Hektoen for training and support in DNA sample preparation, and Jinchang Sun and Anna Lid from the Genomics Core Facility at the Oslo University Institute for Cancer Research for their contributions.
This work was supported by the Norwegian Research Council (grant nos. 221580 and 218241).
J.F.W., S.N., D.V., and L.B.A. are associated members, L.A.M.-Z., E.H., R.A.L., and H.H. are members of the leadership group, and O.M. is the head of the Norwegian Cancer Genomics Consortium.
Footnotes
In accordance with Norwegian legislation and the ethical approval for the study, all sensitive data will be stored in protected ICT systems under the control of Oslo University Hospital. On request, the data will be made available for other institutions and researchers. This will require necessary approvals and formalization of a data transfer agreement.
Authorship
Contribution: J.F.W., J.H.M., E.B.S., and H.H. conceptualized and designed the study; J.F.W., G.T., B.B., V.H., S. Leppä, A.P., and H.H. collected and organized samples; J.F.W. analyzed data and drafted the article; J.F.W., S.N., C.B.S., M.S.L., L.B.A., D.V., E.H., and O.C.L. performed bioinformatics analyses; B.J., A.S., and R.A.L. analyzed and provided supportive design for HLA allele inference; O.C.L. and M.S.L. provided statistical analysis support; Y.N.B., S. Leppä, and H.H. collected clinical data; L.A.M.-Z. and S. Lorenz provided sequencing technologies and expertise, as well as generation of the sequencing data; K.B. provided pathology support and IHC evaluation; J.F.W., S.N., C.B.S., M.S.L., E.B.S., and H.H. provided data interpretation; O.M., J.H.M., S.N., D.V., E.H., O.C.L., M.S.L., Y.N.B., C.B.S., E.B.S., and H.H. provided critical revision of the article; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jillian F. Wise, University of Oslo, Ullernchausseen 70, Montebello, 0310 Oslo, Norway; e-mail: jillian.wise@medisin.uio.no; and Harald Holte, Oslo University Hospital, Institute for Cancer Research, PB 4953 Nydalen, 0424 Oslo, Norway; e-mail: hhe@ous-hf.no.
References
- 1.Beham-Schmid C. Aggressive lymphoma 2016: revision of the WHO classification. Memo. 2017;10(4):248-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martelli M, Ferreri AJ, Agostinelli C, Di Rocco A, Pfreundschuh M, Pileri SA. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2013;87(2):146-171. [DOI] [PubMed] [Google Scholar]
- 3.Ayers EC, Li S, Medeiros LJ, et al. Outcomes in patients with aggressive B-cell non-Hodgkin lymphoma after intensive frontline treatment failure. Cancer. 2020;126(2):293-303. [DOI] [PubMed] [Google Scholar]
- 4.Chapuy B, Stewart C, Dunford AJ, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes [published corrections appear in Nat Med. 2018;24(8):1292 and Nat Med. 2018;24(8):1290-1291]. Nat Med. 2018;24(5):679-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy A, Zhang J, Davis NS, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171(2):481-494.e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morin RD, Assouline S, Alcaide M, et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22(9):2290-2300. [DOI] [PubMed] [Google Scholar]
- 9.Greenawalt DM, Liang WS, Saif S, et al. Comparative analysis of primary versus relapse/refractory DLBCL identifies shifts in mutation spectrum. Oncotarget. 2017;8(59):99237-99244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Miranda NF, Georgiou K, Chen L, et al. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. 2014;124(16):2544-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nijland M, Seitz A, Terpstra M, et al. Mutational evolution in relapsed diffuse large B-cell lymphoma. Cancers (Basel). 2018;10(11): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alioto TS, Buchhalter I, Derdak S, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015;6(1):10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44(16):e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Loo P, Nordgard SH, Lingjærde OC, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. 2010;107(39):16910-16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shukla SA, Rooney MS, Rajasagi M, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33(11):1152-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10(11):1081-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller CA, White BS, Dees ND, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLOS Comput Biol. 2014;10(8):e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller CA, McMichael J, Dang HX, et al. Visualizing tumor evolution with the fishplot package for R. BMC Genomics. 2016;17(1):880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho-Silva D, Pierleoni A, Pignatelli M, et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 2019;47(D1):D1056-D1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886-D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Y, Redmond D, Nie K, et al. Deep sequencing reveals clonal evolution patterns and mutation events associated with relapse in B-cell lymphomas. Genome Biol. 2014;15(8):432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melchardt T, Hufnagl C, Weinstock DM, et al. Clonal evolution in relapsed and refractory diffuse large B-cell lymphoma is characterized by high dynamics of subclones. Oncotarget. 2016;7(32):51494-51502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morin RD, Mungall K, Pleasance E, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122(7):1256-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohr JG, Stojanov P, Lawrence MS, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA. 2012;109(10):3879-3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013;110(4):1398-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Derangère V, Chevriaux A, Courtaut F, et al. Liver X receptor β activation induces pyroptosis of human and murine colon cancer cells. Cell Death Differ. 2014;21(12):1914-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Y, Zhuang L, Fan KY, et al. Discovery of a novel, orally efficacious liver X receptor (LXR) β agonist. J Med Chem. 2016;59(7):3264-3271. [DOI] [PubMed] [Google Scholar]
- 30.Booman M, Szuhai K, Rosenwald A, et al. Genomic alterations and gene expression in primary diffuse large B-cell lymphomas of immune-privileged sites: the importance of apoptosis and immunomodulatory pathways. J Pathol. 2008;216(2):209-217. [DOI] [PubMed] [Google Scholar]
- 31.Monti S, Chapuy B, Takeyama K, et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell. 2012;22(3):359-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang SS, Abdou AM, Morton LM, et al. Human leukocyte antigen class I and II alleles in non-Hodgkin lymphoma etiology. Blood. 2010;115(23):4820-4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rimsza LM, Roberts RA, Miller TP, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103(11):4251-4258. [DOI] [PubMed] [Google Scholar]
- 34.Möller P, Herrmann B, Moldenhauer G, Momburg F. Defective expression of MHC class I antigens is frequent in B-cell lymphomas of high-grade malignancy. Int J Cancer. 1987;40(1):32-39. [DOI] [PubMed] [Google Scholar]
- 35.Ennishi D, Takata K, Béguelin W, et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 2019;9(4):546-563. [DOI] [PubMed] [Google Scholar]
- 36.Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8(364):364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurtz DM, Esfahani MS, Scherer F, et al. Dynamic risk profiling using serial tumor biomarkers for personalized outcome prediction. Cell. 2019;178(3):699-713.e619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurtz DM, Scherer F, Jin MC, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol. 2018;36(28):2845-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20(1):31-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuster SJ, Bishop MR, Tam CS, et al. ; JULIET Investigators . Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45-56. [DOI] [PubMed] [Google Scholar]
- 42.Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545-2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kochenderfer JN, Somerville RPT, Lu T, et al. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol Ther. 2017;25(10):2245-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.