

Key Points
Ability to accurately attribute adverse events post–gene therapy is required to describe the benefit-risk of these novel treatments.
A SCD patient developed myelodysplastic syndrome post-LentiGlobin treatment; we show how insertional oncogenesis was excluded as the cause.
Introduction
HGB-206 (registered at www.clinicaltrials.gov as #NCT02140554) is a multicenter, phase 1/2 study evaluating the safety and efficacy of LentiGlobin for sickle cell disease (SCD). The LentiGlobin drug product (DP) contains autologous CD34+ hematopoietic stem cells (HSCs) transduced ex vivo with the BB305 lentiviral vector (LVV) encoding antisickling β-globin, βA-T87Q.1,2 Following myeloablative busulfan conditioning, patients who receive LentiGlobin DP are monitored for 2 years in HGB-206 and for an additional 13 years in a long-term follow-up study.
Although gene therapy has the potential for curative outcome, it may have associated risks. Most existing gene therapy and allogeneic HSC transplantation (HSCT) protocols require myeloablation with an alkylating agent, which carries a risk of secondary malignancy, including acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS).3,4 Hematologic malignancies due to insertional oncogenesis were previously reported following gene therapy with γ-retroviral vectors in several hematologic diseases.5 We have thus used a modified LVV developed to mitigate the potential risk of insertional oncogenesis, with no such cases reported for >110 patients treated with LVV-based HSC gene therapy in bluebird bio–sponsored clinical trials (≤5 years follow-up) or in >200 patients described in the literature who received any LVV-based HSC gene therapy (≤12 years follow-up)6; however, a theoretical risk remains.
This case describes the approaches used to assess whether MDS, diagnosed 36 months post-LentiGlobin infusion in a patient enrolled in HGB-206, was due to vector-mediated insertional oncogenesis.
Methods
The patient, aged 42 years at consent, presented with SCD (βS/βS genotype) and a history of vaso-occlusive pain, asthma, hypertension, iron overload, leg ulcers, depression, transaminitis, and gallbladder disease. For 8 years before study entry, the patient received hydroxyurea (HU), which was discontinued 6 months pre-LentiGlobin treatment. This patient was a part of the initial cohort of 7 HGB-206 patients (group A) for whom the HSCs were collected by bone marrow (BM) harvest and DPs were made using the original manufacturing process, which was later modified for subsequent patients enrolled.7 The LentiGlobin DP for this patient had a vector copy number (VCN) of 1.3 copies per diploid genome (c/dg), a cell dose of 2.8 × 106 CD34+ cells per kilogram, and 29% LVV-containing HSCs. Before LentiGlobin infusion, the patient received myeloablative conditioning with IV busulfan 3.3 mg/kg (200 mg) daily over 4 days. Per protocol, the goal for busulfan exposure is an area under curve (AUC) of 4000 (minimum, 3600; maximum, 5000) μm × minutes. The busulfan pharmacokinetic monitoring was performed locally and a manually calculated AUC was 3460 μm × minutes, based on the partial actual busulfan level (5 of 6 planned busulfan pharmacokinetic measurements are available; the first value is missing). Neutrophil engraftment (absolute neutrophil counts ≥0.5 × 109/L for 3 consecutive days) and platelet engraftment (the first day of 3 consecutive platelet measurements ≥50 × 109/L without platelet transfusions for 7 days) occurred on days 17 and 29, respectively. One grade 3 serious adverse event of iron overload, assessed unrelated to LentiGlobin, occurred ∼9 months post-LentiGlobin infusion.
Due to persistent anemia, HU was restarted and darbepoetin introduced at 1 and 2 years post-LentiGlobin infusion, respectively. Peripheral blood (PB) VCN over 3 years post-LentiGlobin infusion ranged from 0.08 to 0.15 c/dg and the resulting gene therapy–derived hemoglobin was 0.1 to 1.23 g/dL.
During a routine clinical visit 36 months post-LentiGlobin infusion, the patient had a 3.43 × 109/L white blood cell count, 0.79 × 109/L absolute neutrophil counts, a 131 × 109/L platelet count, and 7.3 g/dL hemoglobin. Routine blood tests revealed 3% blast-like cells in the PB, which increased to 6% in 9 days. Per BM biopsy, ∼10% of BM cells were malignant myeloblasts. The patient was diagnosed with MDS with excess blasts (MDS-EB-2), designated a grade 4 serious adverse event. Post-MDS diagnosis, the patient received standard treatment including 5-azacytadine and decitabine, and, while on treatment, was diagnosed with AML. AML was treated by 7+3 induction chemotherapy of idarubicin/cytarabine, followed by reinduction with cladribine, high-dose cytarabine, and granulocyte–colony-stimulating factor. The patient subsequently received myeloablative doses of melphalan, fludarabine, and 200 rad of total-body irradiation, followed by an HLA-haploidentical HSCT and 2 doses of cyclophosphamide posttransplant. At 3 months posttransplant, the patient was in remission with BM findings consistent with posttransplant recovery, normal cytogenetics, no blasts in the PB, and no graft-versus-host disease. At 6 months posttransplant, blasts (confirmed by flow cytometry) were detected in the PB and BM, and treatment with a hypomethylating agent and venetoclax was initiated.
The study protocol was reviewed and approved by the Institutional Review Boards/Ethics Committees at all HGB-206 clinical sites, including the General Medicine 1 Institutional Review Board (Bethesda, MD).
Results and discussion
Multiple cytogenetic and molecular assays were performed to investigate the MDS etiology. Malignant blasts had monosomy 7 and structurally abnormal chromosome 19p in 8 of 20 metaphases. Archived BM samples, collected preconditioning, were negative for monosomy 7 per fluorescence in situ hybridization (NeoGenomics, Fort Myers, FL) and negative for the 54 mutations associated with myeloid disorders per next-generation sequencing–based analysis (NeoTYPE; NeoGenomics, Fort Myers, FL).
Routine integration site (IS) analyses performed at 6-month intervals post-LentiGlobin infusion showed no protocol-defined clonal dominance through 36 months (Figure 1A).2 No single IS represented >30% of the total sites, and the top 10 prevalent clones were transitory (Figure 1B).
Figure 1.
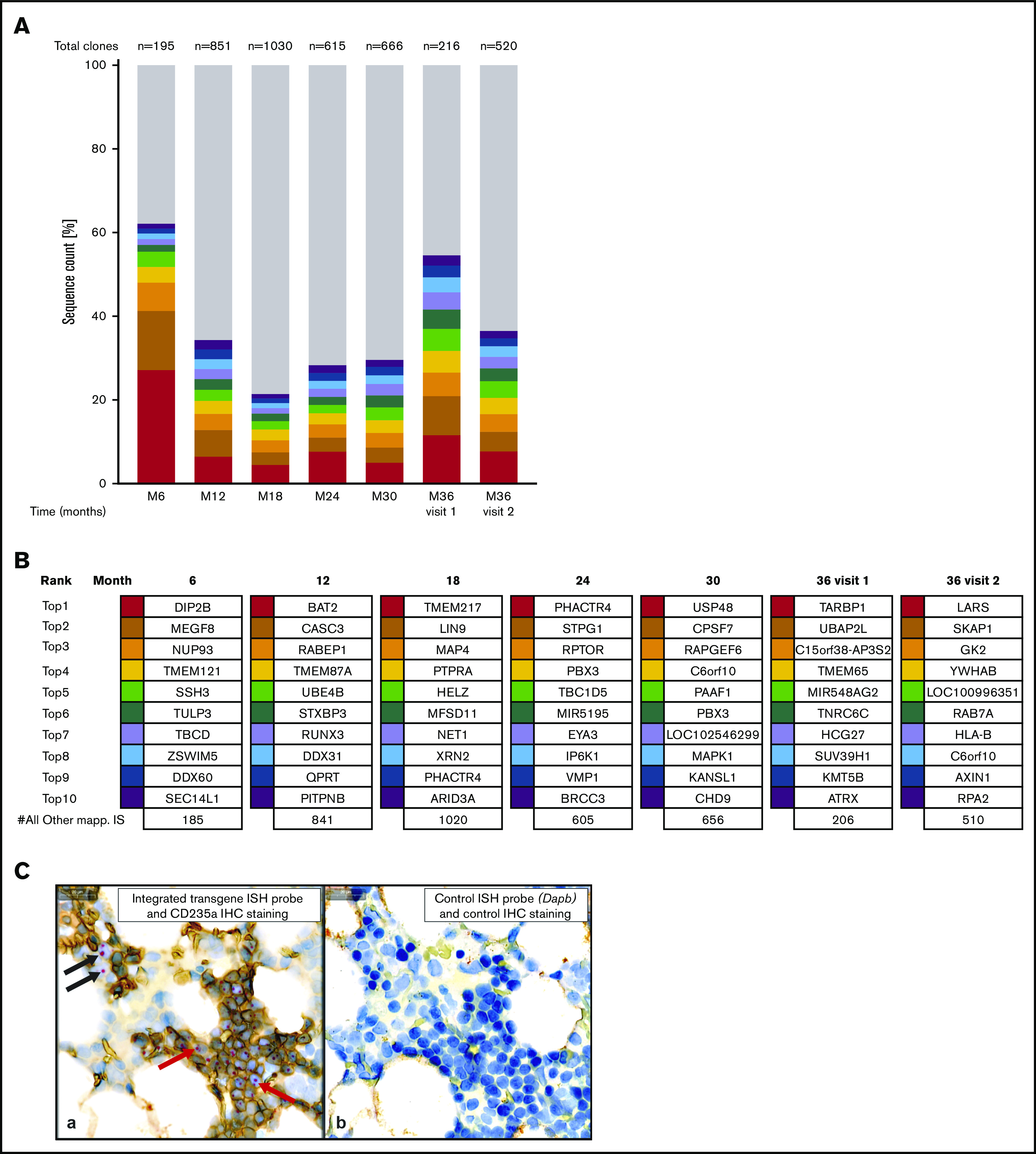
Evaluation of ISs in whole blood and integrated transgene abundance in bone marrow precursors. (A-B) IS analysis was done, using whole blood samples, by nonrestrictive linear amplification-mediated polymerase chain reaction [(nr)LAM PCR] and sequencing as described previously.2 (A) IS analysis for months 6 through 36 showing distribution of gene-marked cell clones. Each of the top 10 most represented unique ISs is indicated by a different color, with gray showing the cumulative proportion of all other unique ISs. The total number of ISs is indicated at the top of each column. None of the clones showed a clonal contribution of >30% of the total retrieved ISs. Month 36 (M36) visit 1 was the regular study follow-up. M36 visit 2 was at the time of MDS diagnosis. (B) IS clonal abundance for months 6 to 36. For individual samples, sequence data form all (nr)LAM-PCR amplicons were combined. RefSeq gene names for the top 10 genes located next to or at the IS are shown. None of the top 10 most prominent clones appeared in >2 samples collected at different time points. (C) A core BM biopsy was collected from the patient 20 days post-MDS diagnosis, fixed in formalin, decalcified, and routinely processed. (a) In situ hybridization (ISH) was performed on a tissue section using a specific probe to detect integrated transgene DNA, and immunohistochemistry (IHC) for CD235a (glycophorin A) protein was used to identify erythroid precursors. Red and black arrows indicate examples of the integrated transgene signal within erythroid (CD235a+) and nonerythroid (CD235a−) precursors, respectively. (b) A serial section stained with a probe specific to an absent gene (the bacterial gene Dapb) to control for nonspecific ISH signals and with the isotype control antibody to assess the background IHC signal. Counterstaining with hematoxylin was also performed. Scale bars, 20 μm.
The frequency of CD34+ cells was stable: 1.83% at baseline in BM and 1.5% in PB at month 21 post-LentiGlobin infusion. RUNX1, KRAS, and PTPN11 mutations were detected by the NeoType analysis in the BM sample at MDS diagnosis (NeoGenomics). Malignant blasts were determined to be CD34+. Therefore, the enrichment of CD34+ cells from BM and PB would be expected to predominantly yield malignant blasts, which would have a VCN ≥1 c/dg if LVV-mediated insertional mutagenesis was causing cell transformation. BM cells were sorted into CD34− (purity 98%) and CD34+ (purity 93%) fractions and compared with unsorted cells in BM aspirate collected post-MDS diagnosis (Table 1). In unsorted and CD34− cells, the VCNs were 0.14 c/dg and 0.21 c/dg, respectively. In CD34+ cells, mostly containing malignant blasts, the VCN was 0.02 c/dg, which was less than the lower limit of quantitation of 0.07 c/dg. This is consistent with the absence of vector integration in the CD34+ blast cells. Similarly, enriched CD34+ cells from PB did not have increased VCN compared with the unsorted sample (Table 1). In the CD34+ and CD34− samples from BM post-AML recurrence after HLA-haploidentical HSCT, no LVV was detected.
Table 1.
VCN analysis of CD34 + and CD34− cells post-MDS diagnosis and post-AML recurrence
| Sample | Post-MDS diagnosis | Post-AML recurrence* | |
|---|---|---|---|
| Purity, % | VCN, c/dg | VCN, c/dg | |
| BM | |||
| Unsorted BM sample | n/a | 0.14 ± 0.0 | nd |
| CD34− selected | 98 | 0.21 ± 0.03 | <LOQ |
| CD34+ selected for myeloblasts | 93 | 0.02 ± 0.01 (<LOQ) | <LOQ |
| PB | |||
| Unsorted PB sample | n/a | 0.10 ± 0.0 | nd |
| CD34− selected | 99 | 0.07 ± 0.01 | <LOQ |
| CD34+ selected for myeloblasts | 53 | 0.08 ± 0.01 | <LOQ |
Cells were collected from the patient at diagnosis (PB), 20 days postdiagnosis of MDS (BM), and 8 days post-AML recurrence (PB and BM). Ficoll-Paque density gradient or hypotonic ammonium chloride solution (ACK Lysing Buffer; Gibco) were used to deplete BM and PB of red blood cells (RBCs). Post-RBC depletion, ∼2 × 105 mononuclear cells (MNCs) were set aside for VCN analysis (unsorted BM sample). For PB, the remaining cells were stained with anti-CD34 BV421 antibody (BD Biosciences) and separated into CD34+ and CD34− fractions using fluorescence-activated cell sorting (SH800 cell sorter; Sony Biotechnology). For BM, remaining MNCs were stained with an anti–CD34-allophycocyanin, washed, and sorted via a Beckman Coulter MoFlo Astrios EQ cell sorter to isolate CD34− (CD34− selected) and CD34+ (with malignant blasts as main contributors) cells. Purity of sorted cell populations was subsequently determined. Additionally, sorted cells were counted by trypan blue exclusion on a hemacytometer and cells from each population were used for VCN analysis, conducted as previously described.32 For post-MDS diagnosis VCN, mean ± standard deviation is shown.
LOQ, limit of quantitation; n/a, not applicable; nd, not determined.
After haploidentical transplant.
DNA in situ hybridization of a BM sample collected post-MDS diagnosis with an integrated transgene-specific probe showed nuclear signals in isolated clusters of CD235+ erythroid precursors and rarely within CD235− nonerythroid cells (Figure 1C).
LVV gene therapy has been safe in animal models,8-11 and there have been no published cases of insertional oncogenesis in patients thus far.6,12-15 The patient received HU for several years, but evidence supporting a relationship between HU use and myeloproliferative disease in SCD is lacking. Although MDS has been reported in 2 individuals with SCD after 14 and 15 years of HU treatment, respectively,16,17 a systematic literature review (with up to 9 years of patient follow-up) concluded that HU use is not leukemogenic in patients with SCD.18 Whether underlying SCD increases the risk of myeloid neoplasms is still under investigation, with recent epidemiology reports in California and the United Kingdom suggesting a higher risk of hematologic malignancies for patients with SCD. In the Seminog et al study, patients in the United Kingdom SCD cohort had significantly higher rate ratios for all cancers combined (2.1; P < .001) and for hematologic malignancies (2.6-10.0; P range from <.001 to .006, depending on type) compared with the non-SCD patient cohort.19 Similarly, the Brunson et al study reported a 72% higher risk of hematologic malignancies for patients with SCD in California compared with the general population of California; the risk of solid tumors was 38% lower.20 High cellular turnover in the BM and chronic inflammation associated with SCD have been discussed as factors that may contribute to leukemic transformation, but confirmatory mechanistic evidence is lacking.20 In the patient described in this case study, the persistence of unmodified progenitor cells postablation and postinfusion may have occurred due to a low proportion of LVV-containing HSCs in the drug product (<30%), preserving a highly proliferative BM characteristic of hemoglobinopathies, which may have contributed to the development of MDS.21,22 Additionally, given that the patient’s actual busulfan AUC was lower than the goal per protocol, it is possible that residual unablated HSCs may have persisted postconditioning. The genomic stability of the patient’s HSCs might have been compromised by hematopoietic stress associated with SCD and further disrupted by chemotherapy, potentially resulting in MDS, given that MDS and secondary AML are identified risks following HSCT using alkylating agents.4 The finding of monosomy 7 in this patient is expected in secondary myeloid neoplasia, including following exposure to alkylating agents such as the busulfan.23 Reports describe MDS in patients with SCD who experienced graft failure and autologous reconstitution post–HLA-haploidentical HSCT using nonmyeloablative conditioning regimens, typically with alkylating agents. One report showed that 2 of 23 patients with hemoglobinopathies (21 with SCD and 2 with thalassemia) who received 400 cGy total-body irradiation and/or posttransplant addition of alkylating agent cyclophosphamide developed high-grade MDS.24 Another report highlighted a patient with SCD who underwent prolonged HU treatment (>5 years) and developed a rapidly progressing MDS that transformed into AML after graft failure following an HLA-haploidentical transplant that included both radiotherapy and alkylating agents.25 Currently, ex vivo HSC gene therapy requires myeloablative chemotherapy to ensure engraftment of gene-modified cells, but alternative nonchemotherapy and nonirradiation approaches aimed at reducing short- and long-term toxicities associated with conditioning warrant further investigation. Early protocols and clinical studies that explored naked antibody-based conditioning for HSCT still required the use of chemotherapy and/or radiation,26 or application to particular disease states.27 More recently, preclinical models were used to investigate approaches coupling cytotoxic conjugates, such as saporin, to antibodies targeting either CD45, expressed exclusively on most hematopoietic cells including HSCs, or CD117 (c-Kit), a marker of HSCs and hematopoietic progenitors.28,29 Additionally, anti–c-Kit antibodies have been tested in mice in combination with antagonists targeting a “don’t eat me” signal CD47, with the goal of activating macrophage-assisted depletion of anti–c-Kit-antibody-marked HSCs.30 Although these methods have enabled varying levels of HSC clearance in animal models, their efficacy and safety in human studies, in particular with respect to off-target effects, the impact on the body’s ability to fight opportunistic infections, and potential to result in long-term multilineage engraftment of transplanted cells, remain to be determined.31
In summary, a patient in HGB-206, who did not have MDS prior to myeloablation, developed MDS post-LentiGlobin treatment. Multiple independent assays demonstrated the absence of vector integration in the CD34+ blasts and excluded LVV-mediated oncogenesis as the MDS cause. The data presented here show that the MDS in this patient was unrelated to LentiGlobin.
Acknowledgments
The authors thank the patients, their families, and caregivers for participating in the study. The authors also acknowledge the following individuals from bluebird bio: Jean-Antoine Ribeil for medical study oversight; Ricky Lane, Gretchen Lewis, Katie Groglio, Erin Meister, Fay Eng, Ken Ganley, Shelby LaBarre, and Lauren Beaudin for assistance with experiments; Briana Deary, Alexandra Miller, Heidi Elliot, Alexandria Petrusich, and Erin Whitney for coordinating sample flow and processing between the study site, sponsor, and laboratories of external vendors; and Iva Kronja for editorial support.
This work was supported by bluebird bio, Inc. Medical writing support was provided by Patrice Ferriola (KZE PharmAssociates, LLC) and was funded by bluebird bio.
Footnotes
Data-sharing requests may be e-mailed to the corresponding author, John F. Tisdale, at johntis@mail.nih.gov.
Authorship
Contribution: A.A.T., M.A., M.B., M.M.H., J.K., F.J.P., J.F.T., and M.C.W. contributed to the conception, design, and planning of the analysis; M.A., M.B., F.J.P., J.R., J.F.T., L.D., M.S., and N.U. contributed to data analysis; M.B., M.M.H., F.J.P., J.R., M.S., and N.U. contributed to data acquisition; A.A.T., M.A., M.B., M.M.H., J.K., F.J.P., J.F.T., and M.C.W. contributed to interpretation of the results; and all authors contributed to critically reviewing or revising the manuscript.
Conflict-of-interest disclosure: A.A.T. provided consultancy services for bluebird bio, Inc, Celgene, and Novartis, and receives research funding from Baxalta, bluebird bio, Inc, Celgene, and Novartis. J.K. provided consultancy services for bluebird bio, Inc, Cowen, Imara, Jeffries, Modus, Novartis, and Sangamo; provides consultancy for Guidepoint Global, GLG, and Novartis; received honoraria from Medscape, Peerview, and Rockpointe; and has membership on an entity’s board of directors or on advisory committees for the National Heart, Lung, and Blood Institute and the Sickle Cell Disease Association of America. M.B., F.J.P., J.R., and L.D. are employees of, and own stock in, bluebird bio, Inc. M.A. was employed by, and owns stock in, bluebird bio, Inc. M.C.W. is consulting for Editas, All Cells, Inc, and Veevo, and provided consulting services for TruCode. The remaining authors declare no competing financial interests.
Correspondence: John F. Tisdale, National Heart, Lung, and Blood Institute, National Institutes of Health, 10 Center Dr, Building 10, Room 9N116, Bethesda, MD 20814; e-mail: johntis@mail.nih.gov.
References
- 1.Negre O, Eggimann AV, Beuzard Y, et al. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the β(A(T87Q))-globin gene. Hum Gene Ther. 2016;27(2):148-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848-855. [DOI] [PubMed] [Google Scholar]
- 3.Leone G, Mele L, Pulsoni A, Equitani F, Pagano L. The incidence of secondary leukemias. Haematologica. 1999;84(10):937-945. [PubMed] [Google Scholar]
- 4.Pagano L, Pulsoni A, Tosti ME, et al. Acute lymphoblastic leukaemia occurring as second malignancy: report of the GIMEMA archive of adult acute leukaemia. Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto. Br J Haematol. 1999;106(4):1037-1040. [DOI] [PubMed] [Google Scholar]
- 5.Booth C, Gaspar HB, Thrasher AJ. Treating immunodeficiency through HSC gene therapy. Trends Mol Med. 2016;22(4):317-327. [DOI] [PubMed] [Google Scholar]
- 6.Cavazzana M, Bushman FD, Miccio A, André-Schmutz I, Six E. Gene therapy targeting haematopoietic stem cells for inherited diseases: progress and challenges. Nat Rev Drug Discov. 2019;18(6):447-462. [DOI] [PubMed] [Google Scholar]
- 7.Walters MC, Tisdale JF, Kwiatkowski JL, et al. Exploring the drivers of potential clinical benefit in initial patients treated in the Hgb-206 study of lentiglobin for sickle cell disease (SCD) gene therapy [abstract]. Blood. 2019;134(suppl 1). Abstract 2061. [Google Scholar]
- 8.Biffi A, Bartolomae CC, Cesana D, et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117(20):5332-5339. [DOI] [PubMed] [Google Scholar]
- 9.Lidonnici MR, Paleari Y, Tiboni F, et al. Multiple integrated non-clinical studies predict the safety of lentivirus-mediated gene therapy for β-thalassemia. Mol Ther Methods Clin Dev. 2018;11:9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montini E, Cesana D, Schmidt M, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24(6):687-696. [DOI] [PubMed] [Google Scholar]
- 11.Negre O, Fusil F, Colomb C, et al. Correction of murine β-thalassemia after minimal lentiviral gene transfer and homeostatic in vivo erythroid expansion. Blood. 2011;117(20):5321-5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. [DOI] [PubMed] [Google Scholar]
- 13.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012;507:187-198. [DOI] [PubMed] [Google Scholar]
- 14.Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6(5):e239-e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479-1493. [DOI] [PubMed] [Google Scholar]
- 16.Baz W, Najfeld V, Yotsuya M, Talwar J, Terjanian T, Forte F. Development of myelodysplastic syndrome and acute myeloid leukemia 15 years after hydroxyurea use in a patient with sickle cell anemia. Clin Med Insights Oncol. 2012;6:149-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aumont C, Driss F, Lazure T, et al. Myelodysplastic syndrome with clonal cytogenetic abnormalities followed by fatal erythroid leukemia after 14 years of exposure to hydroxyurea for sickle cell anemia. Am J Hematol. 2015;90(7):E131-E132. [DOI] [PubMed] [Google Scholar]
- 18.Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med. 2008;148(12):939-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seminog OO, Ogunlaja OI, Yeates D, Goldacre MJ. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J R Soc Med. 2016;109(8):303-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunson A, Keegan THM, Bang H, Mahajan A, Paulukonis S, Wun T. Increased risk of leukemia among sickle cell disease patients in California. Blood. 2017;130(13):1597-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abraham A, Hsieh M, Eapen M, et al. ; Center for International Blood and Marrow Transplant Research . Relationship between mixed donor-recipient chimerism and disease recurrence after hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2017;23(12):2178-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walters MC, Patience M, Leisenring W, et al. ; Multicenter Investigation of Bone Marrow Transplantation for Sickle Cell Disease . Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7(12):665-673. [DOI] [PubMed] [Google Scholar]
- 23.Tanizawa RS, Kumeda CA, de Azevedo Neto RS, Leal AM, Ferreira PB, Velloso ED. Karyotypic and fluorescent in situ hybridization study of the centromere of chromosome 7 in secondary myeloid neoplasms. Rev Bras Hematol Hemoter. 2011;33(6):425-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. 2017;1(11):652-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janakiram M, Verma A, Wang Y, et al. Accelerated leukemic transformation after haplo-identical transplantation for hydroxyurea-treated sickle cell disease. Leuk Lymphoma. 2018;59(1):241-244. [DOI] [PubMed] [Google Scholar]
- 26.George BM, Kao KS, Kwon HS, et al. Antibody conditioning enables MHC-mismatched hematopoietic stem cell transplants and organ graft tolerance. Cell Stem Cell. 2019;25(2):185-192.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal R, Dvorak CC, Prohaska S, et al. Toxicity-free hematopoietic stem cell engraftment achieved with anti-CD117 monoclonal antibody conditioning [abstract]. Biol Blood Marrow Transplant. 2019;25(suppl 3):S92. Abstract 123. [Google Scholar]
- 28.Palchaudhuri R, Saez B, Hoggatt J, et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol. 2016;34(7):738-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czechowicz A, Palchaudhuri R, Scheck A, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun. 2019;10(1):617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chhabra A, Ring AM, Weiskopf K, et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci Transl Med. 2016;8(351):351ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cowan MJ, Kiem HP. Devouring the hematopoietic stem cell: setting the table for marrow cell transplantation. Mol Ther. 2016;24(11):1892-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heffner GC, Bonner M, Christiansen L, et al. Prostaglandin E2 increases lentiviral vector transduction efficiency of adult human hematopoietic stem and progenitor cells. Mol Ther. 2018;26(1):320-328. [DOI] [PMC free article] [PubMed] [Google Scholar]