

Key Points
iMC activation in NK cells enhances antitumor cytotoxicity through increased cytokine production and degranulation.
Coupling IL-15/iMC-mediated activity with CAR expression increases NK cell antitumor efficacy.
Abstract
Natural killer (NK) cells expressing chimeric antigen receptors (CARs) are a promising anticancer immunotherapy, leveraging both innate NK cell antitumor activity and target-specific cytotoxicity. Inducible MyD88/CD40 (iMC) is a potent, rimiducid-regulated protein switch that has been deployed previously as a T-cell activator to enhance proliferation and persistence of CAR-modified T cells. In this study, iMC was extended to CAR-NK cells to enhance their growth and augment cytotoxicity against tumor cells. iMC-activated NK cells substantially increased cytokine and chemokine secretion and displayed higher levels of perforin and granzyme B degranulation. In addition, iMC activation could be coupled with ectopic interleukin-15 (IL-15) to further enhance NK cell proliferation. When coexpressed with a target-specific CAR (CD123 or BCMA), this IL-15/iMC system showed further augmented antitumor activity through enhanced CAR-NK cell expansion and cytolytic activity. To protect against potential toxicity from engineered NK cells, an orthogonal rapamycin-regulated Caspase-9 (iRC9) was included in a 4-gene, dual-switch platform. After infusion of dual-switch NK cells, pharmacologic iRC9 dimerization led to rapid elimination of a majority of expanded transduced NK cells. Thus, CAR-NK cells utilizing dual molecular switches provide an innovative and effective approach to cancer immunotherapy with controlled specificity, efficacy, and safety.
Visual Abstract
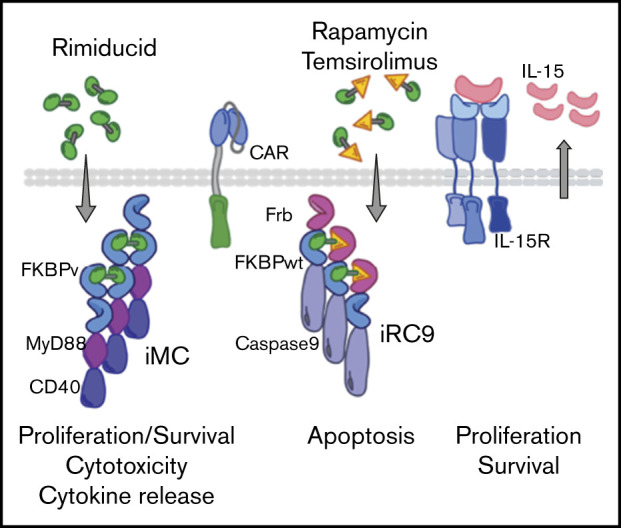
Introduction
Natural killer (NK) cells possess innate mechanisms to target and kill tumor cells when released from inhibition by major histocompatibility (MHC) class 1 molecules through receptor-mediated targeting of stress-induced ligands, production of cytotoxic and inflammatory cytokines, and antibody-directed cellular cytotoxicity.1,2 These properties prompted clinical trials exploring the use of NK cells as an antitumor immunotherapy.3-5 To further improve antitumor activity, expression of chimeric antigen receptors (CARs) in NK cells (CAR-NK–based cell therapy) augments the targeting of hematologic and solid malignancies with antigen specificity,6 as reported in recent clinical trials that relied on CD19-directed CAR-NK cells. Because CAR-NK cells retain their innate tumor-targeting mechanisms in the absence of CAR engagement, it is hypothesized that, relative to autologous CAR T-cell (CAR-T) therapy, the unique graft-versus-tumor effects of CAR-NK cell therapies may also reduce the risk of tumor relapse resulting from antigen escape.7-9
Additionally, the absence of a polyclonal T-cell receptor (TCR) in NK cells minimizes the risk of a graft-versus-host (GVH) response, translating to an increased margin of safety relative to allogeneic adoptive T-cell therapy.3,10,11 In clinical studies using NK cells derived from haploidentical donors or HLA-disparate third-party cord blood products for the treatment of hematologic or solid malignancies, increased risk of GVH disease (GVHD) has not generally been observed.4,12-14
Despite broad antitumor targeting and a low GVHD risk in off-the-shelf applications, CAR-NK cells have historically exhibited poor expansion and persistence after infusion in vivo, which limits their clinical efficacy.15,16 Mature human NK cells have a limited lifespan, with an estimated half-life of 14 days.17 Recent studies have shown increased cytotoxicity and persistence in NK cells implanted in vivo, following expansion ex vivo after activation with a cocktail of interleukin-12 (IL-12), IL-15, and IL-18.18-20 In mice, IL-18 and Toll-like receptor (TLR) signaling are essential for the maintenance of NK cells as a barrier against solid tumor formation.21,22 TLRs, IL-1, IL-18, and IL-37 each signal intracellularly through the scaffolding node MyD88.
We have developed inducible MyD88/CD40 (iMC) as a regulated mimetic of TLR activation in dendritic cells and more recently as a potent costimulatory moiety that enhances CAR-T proliferation, survival, and cytokine production.23-25 The potency of IL-18 signaling through MyD88 in NK cells prompted us to investigate whether iMC may activate and improve the antitumor function of NK cells engineered to also express a CAR. Here, we demonstrate that activation of iMC in NK cells with the small-molecule dimerizing ligand rimiducid augments CAR-NK tumor killing by increasing cytotoxic function, cytokine secretion, and proliferation. Furthermore, autocrine IL-15 secretion in engineered NK cells complements iMC to drive CAR-NK cell proliferation and survival in vivo. Lastly, to offset any increased toxicity risk associated with enhanced efficacy, we incorporated an orthogonally regulated, proapoptotic switch, rapamycin-inducible Caspase-9 (iRC9).24,26
Materials and methods
Standard immunological methods are described in the supplemental Data.
Transduction of NK cells
Retroviral supernatants were produced by transient transfection of 293T cells as previously described.23 Human NK cells derived from peripheral blood buffy coats were stimulated with recombinant human IL-15 (15 ng/mL) for 1 day. The following day, they were further activated with irradiated (100 Gy) K562 cells at the ratio of 2:1 feeder/NK cells and 200 U/mL of recombinant human IL-2 (all cytokines from Miltenyi Biotec, Inc., San Diego, CA). Four days later, NK cells were transduced via spinfection on RetroNectin-coated (Takara Bio, Mountain View, CA) plates and subsequently restimulated with K562 cells. For modification with 2 vectors, NK cells were sequentially transduced on days 4 and 5. Nontransduced and gene-modified NK cells were expanded for 14 days and then used for in vitro and in vivo experiments. Transduction efficiency was determined by flow cytometry (supplemental Methods).
Assessing cytotoxicity of NK cells
Coculture assays were performed with unmodified and transduced NK cells against an enhanced green fluorescent protein-firefly luciferase (eGFPFFluc)–modified HPAC and THP-1 tumor cells in the presence or absence of 1 nM of rimiducid in short (24 hours) and extended (6 days) assays. Short-term cytotoxicity was measured by luciferase activity following the manufacturer’s protocol (Thermo Scientific) and reported as specific lysis relative to target cells alone. Additional long-term coculture assays were analyzed by flow cytometry for the frequency of tumor cell GFP+ populations or by real-time fluorescent microscopy to measure tumor cell (GFP) and NK cell (red fluorescent protein) proliferation (IncuCyte; Essen Biosciences).
In vivo studies
NOD/SCID/IL-2γR−/− (NSG) mice27 (Jackson Laboratory, Bar Harbor, ME) were maintained in the Bellicum Pharmaceuticals AAALAC-approved vivarium. These studies were approved by the Bellicum Pharmaceuticals Institutional Animal Care and Use Committee. For NK persistence studies, 107 doubly transduced NK cells (indicated transgenes and eGFPFfluc) in 100 μL of phosphate-buffered saline were injected into the tail vein of NSG mice (8-week-old female mice). Rimiducid or vehicle alone was administered via intraperitoneal (IP) injection at a dose of 1 mg/kg weekly. In the NK persistence study with THP-1 tumor targets, 5 × 106 doubly transduced NK cells (with the indicated transgenes and Orange Nanolantern) were tail vein injected 5 days before and 12 days after engraftment with 1 × 106 THP-1–eGFPFfluc cells. For CAR-NK experiments, 1 × 107 gene-modified NKs were tail vein injected 3 days after engraftment with 1 × 106 THP-1–eGFPFfluc cells. In vivo NK cell presence and tumor growth were measured by weekly bioluminescence imaging (BLI) by IP injection of 150 mg/kg of D-luciferin (firefly luciferase) or 150 ng of coelenterazine-h (renilla luciferase; Perkin Elmer, Waltham, MA) and imaged using the IVIS imaging system (Perkin Elmer). Photon emission was accessed by whole-body region of interest. Signal quantitation was measured as average radiance (photons per second per cm2 per steradian). Weight measurements were performed at least once per week. End point analysis involved flow cytometry of splenocytes, bone marrow, or peripheral blood.
Statistics
All statistical tests (noted in figure legends) were carried out and analyzed using GraphPad Prism software (version 8.0; GraphPad). Data are presented as means ± standard errors of the mean. All Student t tests were 2 tailed. All analyses of variance were 2 way. P < .05 was considered significant.
Results
Inducible MC signaling enhances NK cell proliferation and cytotoxicity
To evaluate the effect of iMC expression and activation on NK cell proliferation, K562-activated NK cells from 9 donors were transduced with γ-retroviral vectors28 encoding iMC, iRC9, and the signaling-defective surface marker ΔCD19 or a control version lacking iMC (Figure 1A). Transduction levels between gene-modified NK cells were not statistically different (iRC9-ΔCD19, 67.8% ± 56%; iRC9-ΔCD19-iMC, 73.7% ± 43.3%; n = 9; Figure 1B-C). Subsequently, transduced NK cells were stimulated with irradiated K562 cells and cultured with or without the addition of 1 nM of rimiducid for 8 days. iMC-modified NK cells demonstrated increased proliferation compared with control iRC9-expressing NK cells, and this basal signaling effect was further augmented by activating iMC with rimiducid (Figure 1D). This enhanced proliferation with iMC activity was confirmed by examining the dilution of cell trace dyes in the whole population of NK cells after K562 stimulation in the presence or absence of rimiducid (supplemental Figure 1A-B).
Figure 1.

iMC augments NK cell proliferation in vitro. (A) Schematic of the retroviral vector design for iRC9-ΔCD19-iMC encoding iRC9, signaling-defective truncated human CD19, and iMC with the transgenes separated by T2A and P2A ribosomal skipping sequences and the control vector iRC9-ΔCD19 encoding iRC9 and truncated human CD19. (B-C) Flow cytometric analysis to determine transduction efficiency using anti-CD19 and anti-CD56 antibodies 8 days after transduction. (D) At day 5 posttransduction, iMC.iRC9- or iRC9-modified NK cells were cultured in the presence or absence of 1 nM of rimiducid (Rim). At day 10 posttransduction, the live cells were counted using acridine orange and propidium iodide staining. Paired Student t test was used to compare indicated groups. *P < .05. NS, not significant.
We assessed whether iMC activation of NK cell proliferation also enhanced antitumor responses. Targeting of innate NK cytotoxicity is repressed by MHC class 1 through inhibitory KIR receptors and activated, in part, by MHC-like stress-activated ligands (ie, MICA/B and ULBP1-3).2 Expression of MHC class 1 and MICA/B in a variety of human tumor cell lines, including K562, OE19, THP-1, HPAC, and SKOV3, was measured by flow cytometry (Figure 2A-B), and HPAC (pancreatic adenocarcinoma) and THP-1 (monocytic leukemia) were selected for short-term cytotoxicity assays with engineered NK cells by their high levels of both MHC class 1 and MICA/B. Control NK cells (expressing iRC9-ΔCD19) were unreactive against HPAC cells with or without rimiducid (Figure 2C). In contrast, NK cells expressing iMC demonstrated enhanced killing that was further augmented by iMC activation with rimiducid. Although control NK cells were cytotoxic against the NK-sensitive THP-1 cell line at moderate effector/target ratios, iMC transduction and activation further enhanced tumor killing (P < .001; Figure 2D). Interestingly, mere transduction with the iRC9-ΔCD19-iMC vector increased cytotoxicity compared with iRC9-ΔCD19–modified NK cells, suggesting that basal signaling from high-level iMC expression can contribute to NK cell potency (Figure 2D).
Figure 2.
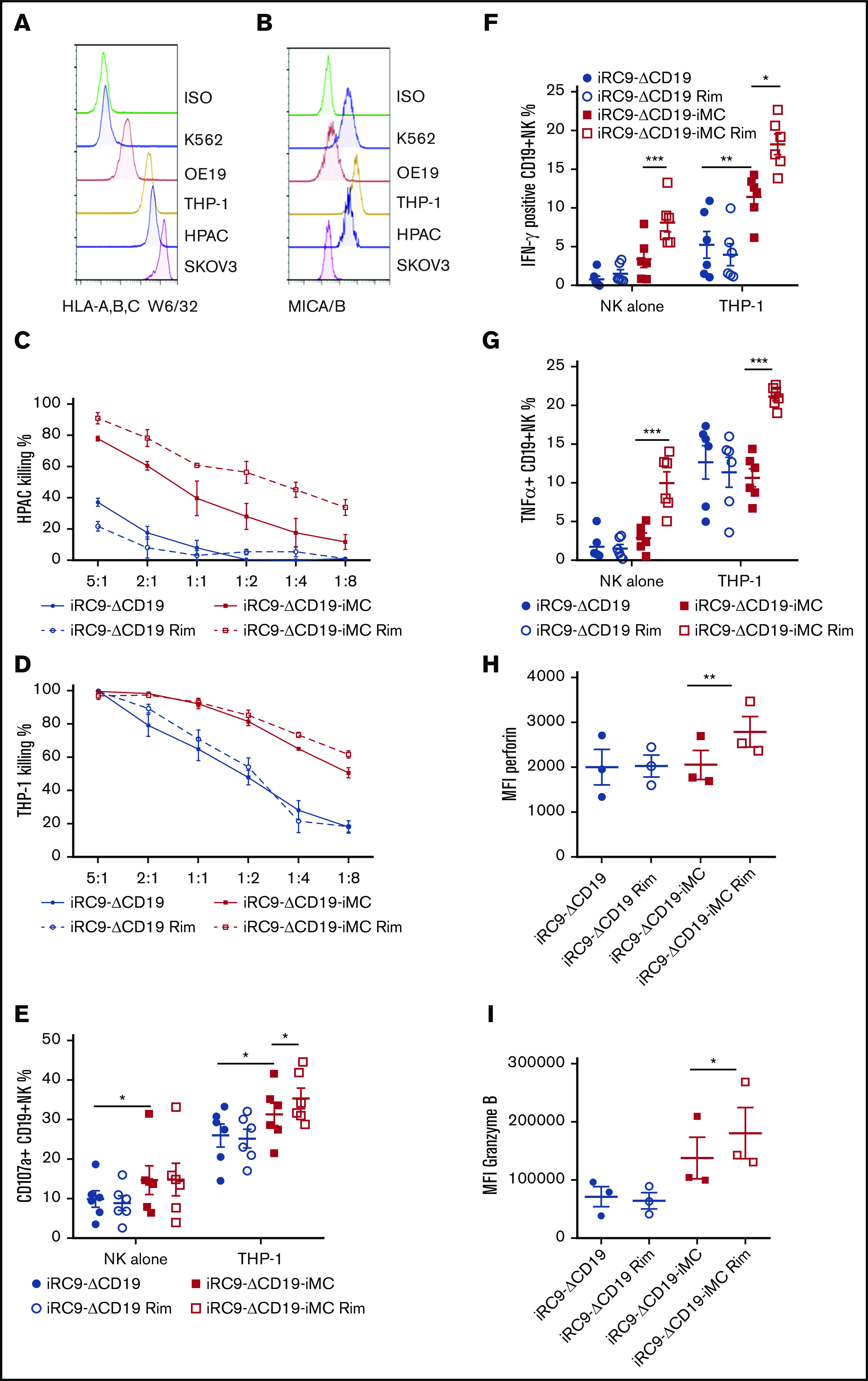
iMC enhances NK cell targeting of tumor cells. (A-B) Flow cytometric analysis to assess MHC class 1 surface expression of tumor cell lines using anti–HLA-A,B,C antibody (clone w6/32) and surface expression of the NKG2D ligands using MICA/B-specific antibody. (C-D) Potency of NK cells modified with iRC9-iMC or iRC9 was tested in 24-hour coculture assays with HPAC-eGFPFfluc or THP-1–eGFPFfluc at decreasing effector/target ratios. Tumor cell killing percentages were calculated by luciferase activity relative to tumor cells alone; 2-way analysis of variance statistical analysis (n = 4; P < .0001). (E-I) iRC9- or iRC9-iMC–modified NKs were incubated with or without THP-1 targets for 4 hours (E) or overnight (F-I) in the presence or absence of 1 nM of rimiducid (Rim). Percentages of cells expressing surface CD107a (E), intracellular interferon-γ (IFN-γ) (F), and tumor necrosis factor α (TNF-α) (G) were measured by flow cytometry. Mean fluorescence intensities (MFIs) of perforin (H) and granzyme B (I) were measured in NK cells cocultured with THP-1 overnight. Transduced NK cells were first gated as CD56+CD19+ population. Paired Student t test was used to compare indicated groups. *P < .05, **P < .01, ***P < .001.
To understand how iMC enhances NK cell antitumor potency, we determined NK cell receptor expression, cytokine production, and degranulation in coculture assays against THP-1 targets in the presence and absence of rimiducid. Interestingly, although retroviral modification (or the transduction process itself) led to increased levels of some NK receptors (ie, DNAM1, NKp30, NKp44, and FasL, but not NKp46, NKG2D, CD16, or CD95/Fas), iMC expression or activation with rimiducid did not seem to significantly modulate receptor levels (supplemental Figure 2A-B). In contrast, the proportion of degranulated NK cells in response to THP-1 cell exposure (measured by CD107a/LAMP1 surface expression) was increased by iMC expression and further enhanced by prior overnight stimulation with 1 nM of rimiducid (Figure 2E). Similarly, overnight iMC activation resulted in markedly increased expression of perforin and granzyme B expression compared with control NK cells and those not exposed to rimiducid (Figure 2H-I). These data suggest that the enhanced short-term cytotoxicity by iMC-enhanced NK cells is likely due to increased degranulation and release of proapoptotic granzymes into target cells. Interestingly, the proportion of NK cells producing IFN-γ and tumor necrosis factor α was also markedly increased by iMC activation with rimiducid, and not surprisingly, this effect was further enhanced by coculture with THP-1 target cells (Figure 2F-G). Similar to iMC-enhanced T cells,23 rimiducid activation of iMC increased production of many proinflammatory cytokines, in some cases by several orders of magnitude relative to control NK cells, with or without target cell exposure (supplemental Figure 3A-B).
The observation of target cell independence for NK cell cytokine secretion and proliferation in response to iMC activity prompted us to examine signal transduction in response to iMC dimerization. Natural MyD88 signaling from activated IL-1 receptor family members and Toll-like receptors and from CD40 signaling each lead to the recruitment and activation of TRAF ubiquitin ligases and ultimately activation of AP1 and NF-κB transcription factors that drive cytokine gene transcription. NK cells transduced with or without iMC-encoding γ-retrovirus were treated with rimiducid, and total MyD88 (including iMC) was immunoprecipitated from lysates harvested over a 30-minute time course. iMC dimerization led to rapid association with TRAF2 and TRAF6, but not TRAF1 or TRAF3 (supplemental Figure 4A). Rimiducid treatment of iMC-enabled NK cells also led to rapid (5-30 minute) phosphorylation of the mitogen-activated kinases JNK, ERK, and p38 that drive AP1/ATF activation, activating phosphorylation of the NF-κB component p65, and activating phosphorylation of the progrowth and prosurvival kinase Akt (supplemental Figure 4B-C). Thus, iMC signaling recapitulated signaling by native MyD88 and CD40 signaling molecules.
NK cell cytotoxicity against target cells is directed by actin cytoskeletal alterations directed by Rho family GTPases activated by the guanine nucleotide exchange factor Vav.29,30 Activating phosphorylation of Vav-1 was induced rapidly by iMC activation (supplemental Figure 4D). Interestingly, reduction of Vav-1 levels with anti-Vav short hairpin RNA resulted in the elimination of the enhanced cytotoxicity directed by iMC activation but did not influence iMC expression (supplemental Figure 4F-G). Furthermore, enhancement of cytokine production driven by iMC activation was also reduced by reduction of Vav-1 expression (supplemental Figure 4H). Together, these results indicate that Vav-1 is essential for the enhanced NK activation driven by iMC signaling (supplemental Figure 4I).
Transgenic IL-15 synergizes with iMC activation to drive NK cell expansion
Although iMC-mediated NK cell activation enhances NK cell proliferation, iMC activation itself is not sufficient for high-level secretion of growth-promoting cytokines, such as IL-2 or IL-15 (supplemental Figure 3A).31 To further increase NK proliferation after iMC activation, the iRC9-ΔCD19-iMC vector was modified to express a fourth gene, IL-15 (iRC9-IL15-ΔCD19-iMC; Figure 3A). IL-15 production was minimal at basal level (13.04 ± 10.02 pg/mL per 106 NK cells; Figure 3B); however, secretion was significantly increased by transgenic IL-15 expression, presence of tumor cells, and activation of iMC with 1 nM of rimiducid (41.26 ± 16.3 pg/mL per 106 cells with targets; 63.86 ± 20.62 pg/mL per 106 cells with rimiducid and targets; Figure 3B), in keeping with the enhanced proliferation of NK cells in culture. Overall expression of the ΔCD19 marker (and iMC) was decreased in iRC9-IL15-ΔCD19-iMC relative to iRC9-ΔCD19-iMC, presumably because of the larger open reading frame; however, iMC activation with 1 nM of rimiducid still significantly increased the expansion of NK cells expressing IL-15 (52% ± 8.2% to 75.7% ± 4.6%; P = .002; Figure 3C-D). In 3-day coculture assays against THP-1 target cells, the enhanced killing potency of iMC-expressing NK cells by rimiducid activation was not altered by IL-15 incorporation into the vector (supplemental Figure 5A-C). These studies suggest that IL-15 expression provides a readily accessible NK cell growth factor after iMC-based cellular activation to promote cellular expansion while retaining cytotoxic potential.
Figure 3.
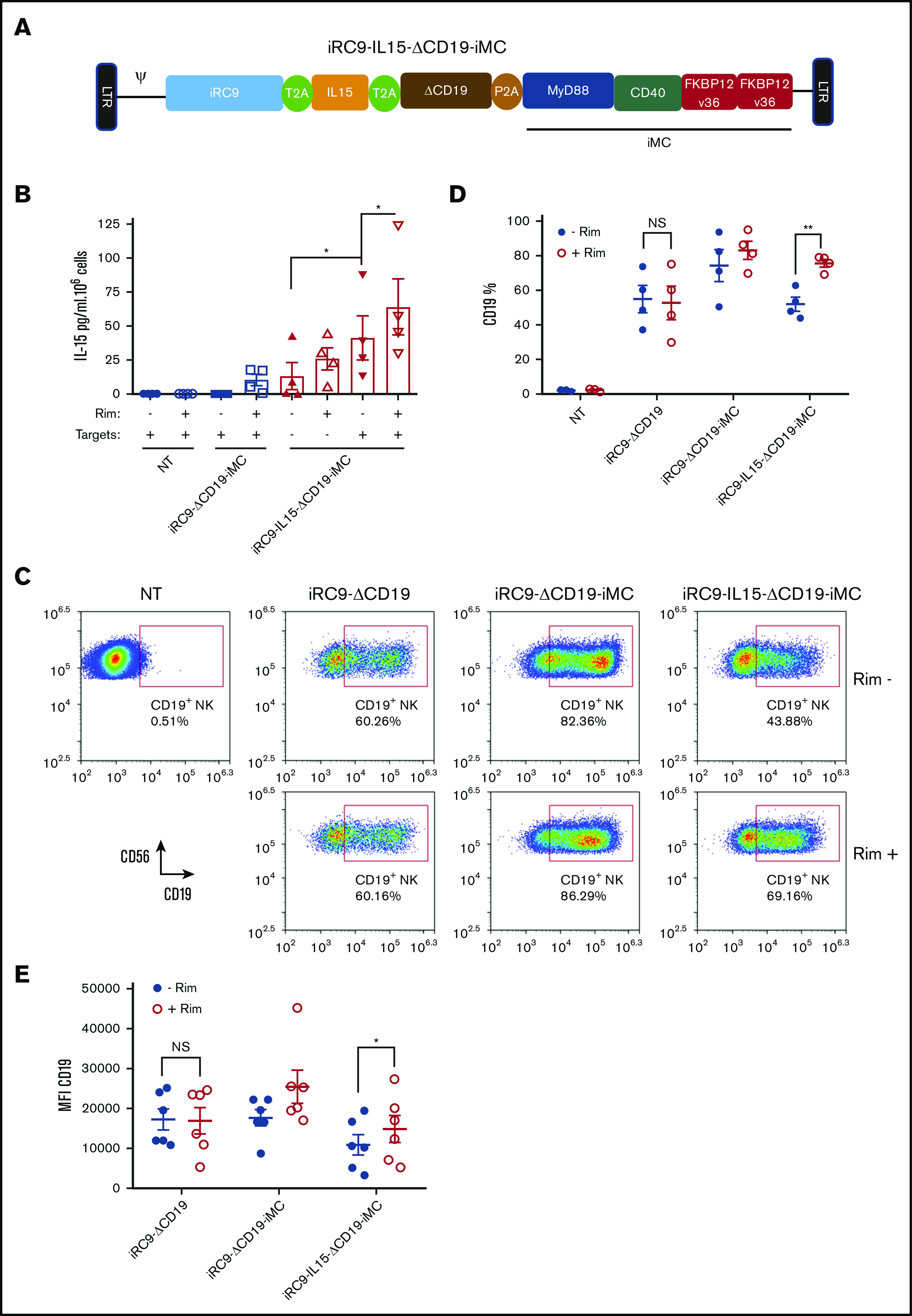
IL-15 synergizes with iMC to enhance NK cell proliferation. (A) Schematic retroviral vector design iRC9-IL15-ΔCD19-iMC encoding iRC9, human IL-15, truncated human CD19, and iMC with the transgenes separated by T2A and P2A ribosomal skipping sequences. (B) IL-15 production from supernatant of nontransduced (NT) or iRC9-ΔCD19-iMC– or iRC9-IL15-ΔCD19-iMC–modified NK cells cocultured with or without K562 targets at an effector/target ratio of 3:1 for 72 hours in the absence or presence of 1 nM of rimiducid (Rim) was determined by enzyme-linked immunosorbent assay. Paired Student t test was used to compare the indicated groups. (C-D) NT or iRC9-ΔCD19–, iRC9-ΔCD19-iMC–, or iRC9-IL15-ΔCD19-iMC–modified NKs treated with or without 1 nM of Rim for 6 days. Transgene expression as indicated by CD19+ percentage. Paired Student t test was used to compare indicated groups. (E) CD19 transgene expression in iRC9-ΔCD19–, iRC9-ΔCD19-iMC–, or iRC9-IL15-ΔCD19-iMC–modified NKs was determined with or without pretreatment with 1 nM of Rim. Paired Student t test was used to compare indicated groups. *P < .05, **P < .01.
The growth-promoting effect of the combination of iMC, activation with rimiducid, and autocrine IL-15 expression was most dramatic when NK cells were grown in vivo as xenografts in tumor-free, immunodeficient NSG mice (Figure 4). Dual-switch NK cells with and without transgenic IL-15 were cotransduced to express eGFPFfluc and engrafted IV without tumor targets, and growth was observed over 7 weeks by BLI. NK cells modified with iRC9-ΔCD19 or iRC9-ΔCD19-iMC proliferated poorly after engraftment, even with weekly administration of rimiducid. However, the combination of iMC activation and IL-15 secretion resulted in sustained NK cell engraftment, expansion, and persistence (BLI area under the curve, 2.28 × 107 vs 3.86 × 105 or 3.65 × 105 photons per second per cm2 per steradian for iRC9-IL15-ΔCD19-iMC with rimiducid, iRC9-IL15-ΔCD19-iMC without rimiducid, and iRC9-ΔCD19 with rimiducid, respectively; P < .01; Figure 4B). These data indicate a synergy between IL-15 and iMC signaling to promote NK cell expansion in vivo.
Figure 4.

IL-15 synergizes with iMC to enhance NK persistence in vivo. (A) NSG mice were engrafted with 1 × 107 NK cells transduced with iRC9-ΔCD19 iRC9-ΔCD19-iMC or iRC9-IL15-ΔCD19-iMC and eGFPFfluc retroviral vectors. Mice subsequently received weekly IP vehicle only (Veh) or rimiducid (Rim) injections of 1 mg/kg in the vehicle. (B) Gene-modified NK cells levels were assessed by BLI weekly; 2-way analysis of variance (ANOVA) was used to compare differences among groups (P < .01). (C) Mice weights were assessed weekly; 2-way ANOVA was used to compare differences among groups (P = .30). p, photon; sr, steradian.
Excessive IL-15 has been associated with NK cell anergy or exhaustion,32 and we examined the effect of continuous exposure to rimiducid during expansion in iMC only and iMC/IL-15–transduced NK cells. At the <100 pg/mL levels of IL-15 secreted into culture supernatant, no significant compromise in the cytotoxicity of NK cells by IL-15 was observed (supplemental Figure 6A). Expression of NK receptors DNAM-1, CD16, and NKG2D and NK cell exhaustion markers T-bet and eomesodermin32,33 was also not significantly different between iRC9-IL15-ΔCD19-iMC– and iRC9-ΔCD19-iMC–modified NK in the presence or absence of rimiducid (supplemental Figure 6B-C). Moreover, NK functionality indicated by CD107a degranulation, TRAIL, and FasL surface expression was not affected by IL-15 gene expression (supplemental Figure 6D-E).
IL-15/iMC enhances antitumor activity of CD123-specific CAR-NK cells
We next evaluated whether the IL-15/iMC system could improve antitumor efficacy of NK cells expressing a target-specific CAR. NK cells were either nontransduced or modified with a combination of retroviral vectors to express the first-generation CAR CD123.ζ (Figure 5A), iRC9-IL15-ΔCD19-iMC, or both vectors sequentially. Transduction of NK cells from 4 donors was highly efficient, with >65% efficiency for the dual-switch vector (Figure 5B). In short-term cytotoxicity assays targeting CD123-expressing THP-1 cells, addition of the anti-CD123 CAR significantly increased specific lysis relative to control NK cells (P < .01; Figure 5C). Gene-modified NK cells were then evaluated for efficacy in vivo in NSG mice harboring established THP-1.eGFPFfluc tumors, subsequently engrafted with 1 × 107 NK cells, and treated weekly with rimiducid (1 mg/kg IP) or vehicle alone (Figure 5D). NK cells expressing the CAR or iRC9-IL15-ΔCD19-iMC alone failed to control THP-1 tumor growth, even with iMC/rimiducid activation, but coexpression of the CAR and iMC/IL-15 resulted in significant tumor control, but only upon rimiducid activation of iMC (P < .001 from day 11 to 35 after NK cells; Figure 5E-F). Moreover, iMC-activated NK cells could be detected at higher levels in the spleen, bone marrow, and blood compared with NK cells modified with vehicle alone (Figure 5G; supplemental Figure 7A). These data further support that IL-15/iMC/rimiducid enhances proliferation in vivo, leading to synergistic antitumor efficacy.
Figure 5.
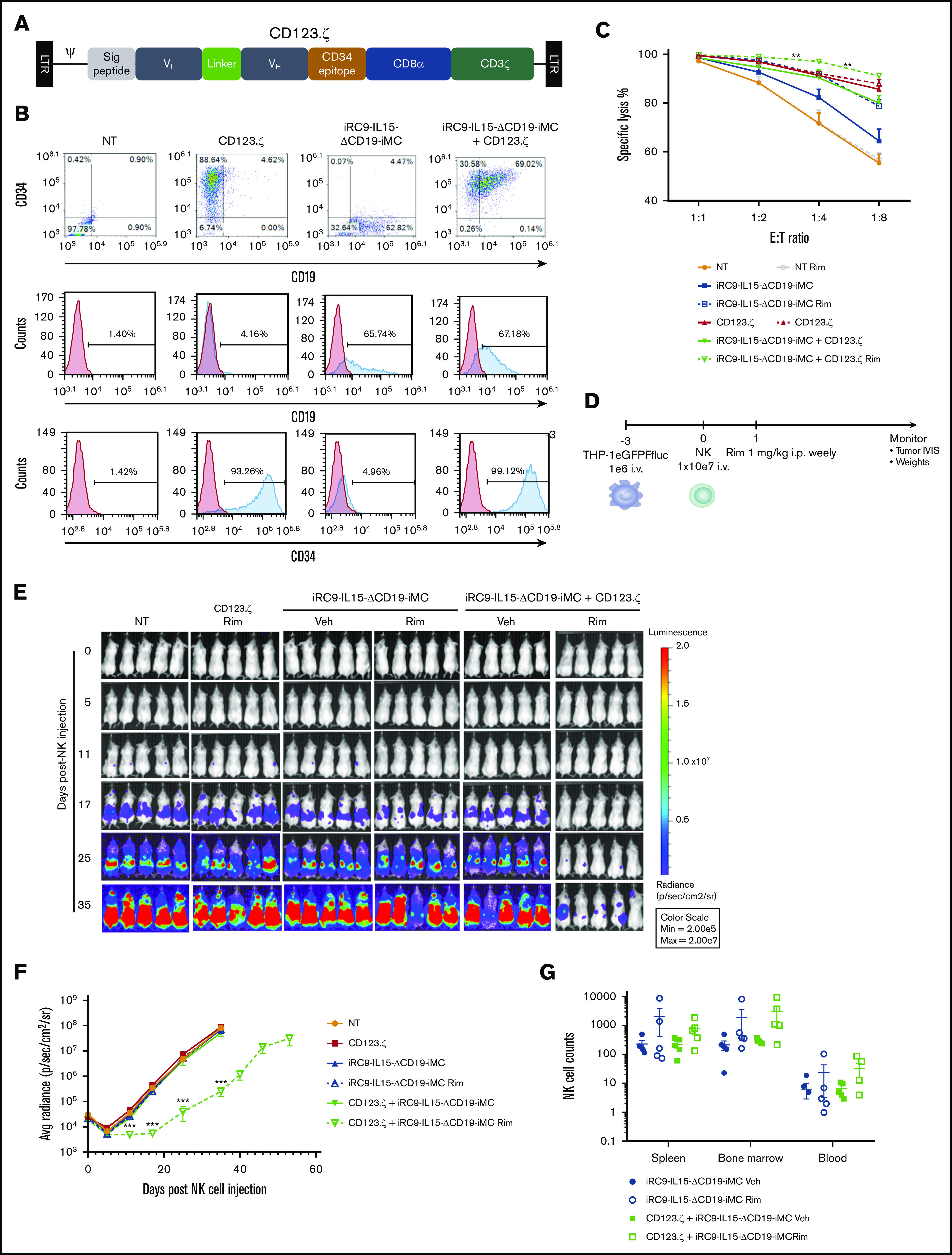
Generation of CAR-NK cells with iMC and IL-15 targeting CD123. (A) Schematic retroviral vector encoding signal peptide, CD123-targeting single-chain variable fragment variable light (VL) and heavy (VH) domains, the minimal CD34 epitope, CD8α stalk and transmembrane region, and the CD3ζ signaling domain. (B) Activated NK cells were doubly transduced with γ-retroviral vector encoding CD123.ζ (CAR) and/or dual-switch iRC9-IL15-ΔCD19-iMC. Flow cytometric analysis to determine transduction efficiency using anti-CD34 (CD123.ζ) and anti-CD19 (iRC9-IL15-ΔCD19-iMC) antibodies compared with nontransduced (NT) or single transduced NK cells. NK cells were first gated out as CD56+ populations. (C) Gene-modified NK cells (n = 4 donors) were cocultured with THP-1–eGFPFfluc cells at dilutive effector/target (E:T) ratios in the presence or absence of 1 nM of rimiducid (Rim) for 24 hours. Tumor-cell killing percentages were calculated by luciferase activity relative to tumor cells alone. Multiple Student t tests were used to compare iRC9-IL15-ΔCD19-iMC Rim and iRC9-IL15-ΔCD19-iMC plus CD123.ζ Rim. (D-F) NSG mice (n = 5 per group) were engrafted with 106 THP-1–eGFPFFluc tumor cells and, 3 days later, treated with 107 NK cells, NT or transduced with CD123.ζ, DS.IL15, or CD123.ζ plus DS.IL15. Mice were subsequently administered weekly IP vehicle (Veh) or 1 mg/kg of Rim. Tumor BLI was assessed by IVIS. Multiple Student t tests were used to compare CD123.ζ plus iRC9-IL15-ΔCD19-iMC Rim group with NT group. (G) At day 53 after NK therapy, CD123.ζ plus DS.IL15 Rim group was euthanized. Human NK cells were identified in spleen, bone marrow, and peripheral blood by flow cytometric analysis as hCD56+mCD45− populations. All groups were euthanized at time point day 35, except for the DS.IL15 plus CD123.ζ Rim group that was obtained at day 53; 2-way analysis of variance was performed for comparisons (P = .059). **P < .01, ***P < .001.
IL-15/iMC improves antitumor activity of BCMA-expressing CAR-NK cells
Adoptive transfer of NK cells to treat multiple myeloma has shown clinical efficacy without significant toxicities; however, antitumor effects have been modest.34,35 To further improve efficacy against multiple myeloma, we constructed a tricistronic vector encoding a B-cell maturation antigen (BCMA)–targeted CAR along with IL-15 and iMC (iMC-BCMA.ζ-IL15). Cells were further transduced with the iRC9-ΔCD19 safety switch–expressing vector to generate dual-switch NK-CAR cells constitutively expressing IL-15 (Figure 6A-B). Despite characterization as a monocytic leukemia, we targeted THP-1 cells because of their high levels of surface BCMA (Figure 6C). Similar to IL-15/iMC–enhanced CD123-specific CAR-NK cells in the alternative vector platform, 1 × 107 BCMA-targeted CAR-NK cells showed significant rimiducid-dependent antitumor efficacy (P < .001; Figure 6D-E). Rimiducid-enhanced clearance of tumor from the spleen and bone marrow (bone marrow, P < .001; spleen, P < .05; Figure 6F-G) correlated with increased NK cell expansion (Figure 6H-I). Thus, in 2 distinct vector platforms and with 2 CAR models, IL-15/iMC–modified CAR-NK cells controlled tumor growth in a rimiducid-dependent manner that was associated with an increase in persistence and expansion of the adoptively transferred cells.
Figure 6.
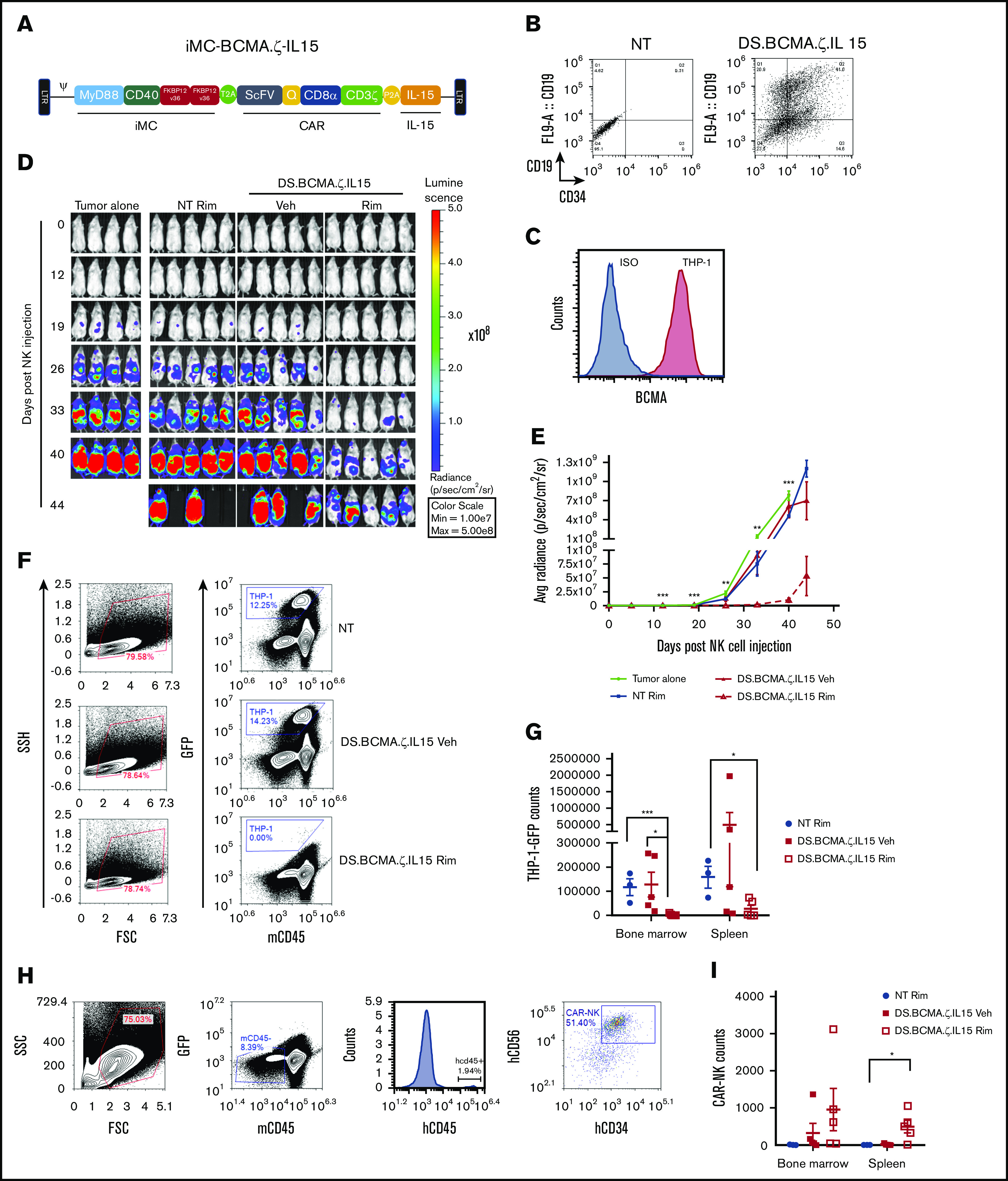
iMC enhanced antitumor efficacy of BCMA-CAR NK cells against THP-1 tumors in NSG mice. (A) Schematic retroviral vector encoding iMC; CAR including BCMA-targeting single-chain variable fragment, the minimal CD34 epitope (Q), CD8α stalk and transmembrane region, and the CD3ζ signaling domain; and human IL-15 with the transgenes separated by T2A and P2A ribosomal skipping sequences. (B) Activated NK cells were doubly transduced with γ-retroviral vectors encoding iMC-BCMA.ζ-IL15 and iRC9-ΔCD19 to generate DS.BCMA.ζ/IL15-modified NK cells. Flow cytometric analysis to determine transduction efficiency is displayed using anti-CD34 (iMC-BCMA.ζ-IL15) and anti-CD19 (iRC9-ΔCD19) antibodies compared with nontransduced (NT) NK cells. (C) BCMA expression in THP-1 tumor cells was determined by flow cytometric analysis with anti-BCMA antibody or isotype control. (D-E) NSG mice (n = 5 per group) were engrafted with 107 NK cells NT or transduced with DS.BCMA.ζ.IL15 NKs 3 days after IV implantation of 106 THP-1–eGFPFFluc tumor cells; 1 mg/kg of rimiducid (Rim) or vehicle (Veh) was administrated IP 5 times per week for the first week and 3 times per week thereafter. BLI was monitored by IVIS. Multiple Student t tests were used to compare DS.BCMA.ζ/IL15 NK Rim group with tumor alone group. (F-G) From day 40 to 48, mice from NT, DS.BCMA.ζ.IL15 NK vehicle, and Rim groups were euthanized. THP-1–eGFPluc cells were identified in bone marrow and spleen as GFP+ populations. (H-I) Human NK cells were identified in spleen and bone marrow as mCD45−GFP−hCD45+hCD34+ populations. Student t test was used for comparisons. *P < .05, **P < .01, ***P < .001. FSC, forward scatter; SSC, side scatter.
iRC9 eliminates dual-switch NK cells
Augmentation of NK cells with IL-15/iMC carries the theoretical risk of also increasing toxicities from NK cell cytokine production and/or constitutive secretion of IL-15.32,36 Furthermore, expression of CAR-targeted antigen on nonmalignant tissue can lead to toxicity. To minimize this risk, we verified that the iRC9 transgene present in the dual-switch platform could eliminate transduced NK cells by iRC9 dimerization with rapamycin or its analog, temsirolimus. In short-term killing assays, iRC9 dimerization produced a dose-dependent induction of annexin V surface expression, indicative of apoptosis. Complete cell death revealed by permeability to actinomycin D was subsequently observed within 24 hours, even at low dimerizer drug concentrations. Cell ablation of Orange Nanolantern–labeled37 dual-switch NK cells in tumor-bearing NSG mice with 1 mg/kg of temsirolimus (bolus, IP) was also rapid and efficient (P = .17 vs .82, untreated;Figure 7C-D). Most of the residual human NK cells observed in the spleen and bone marrow of treated mice were untransduced with the dual-switch vector (Figure 7E-G). Overall, these data indicate that, similar to our experience with gene-modified T cells,24 the iRC9 safety switch can also rapidly induce apoptosis in NK cells.
Figure 7.
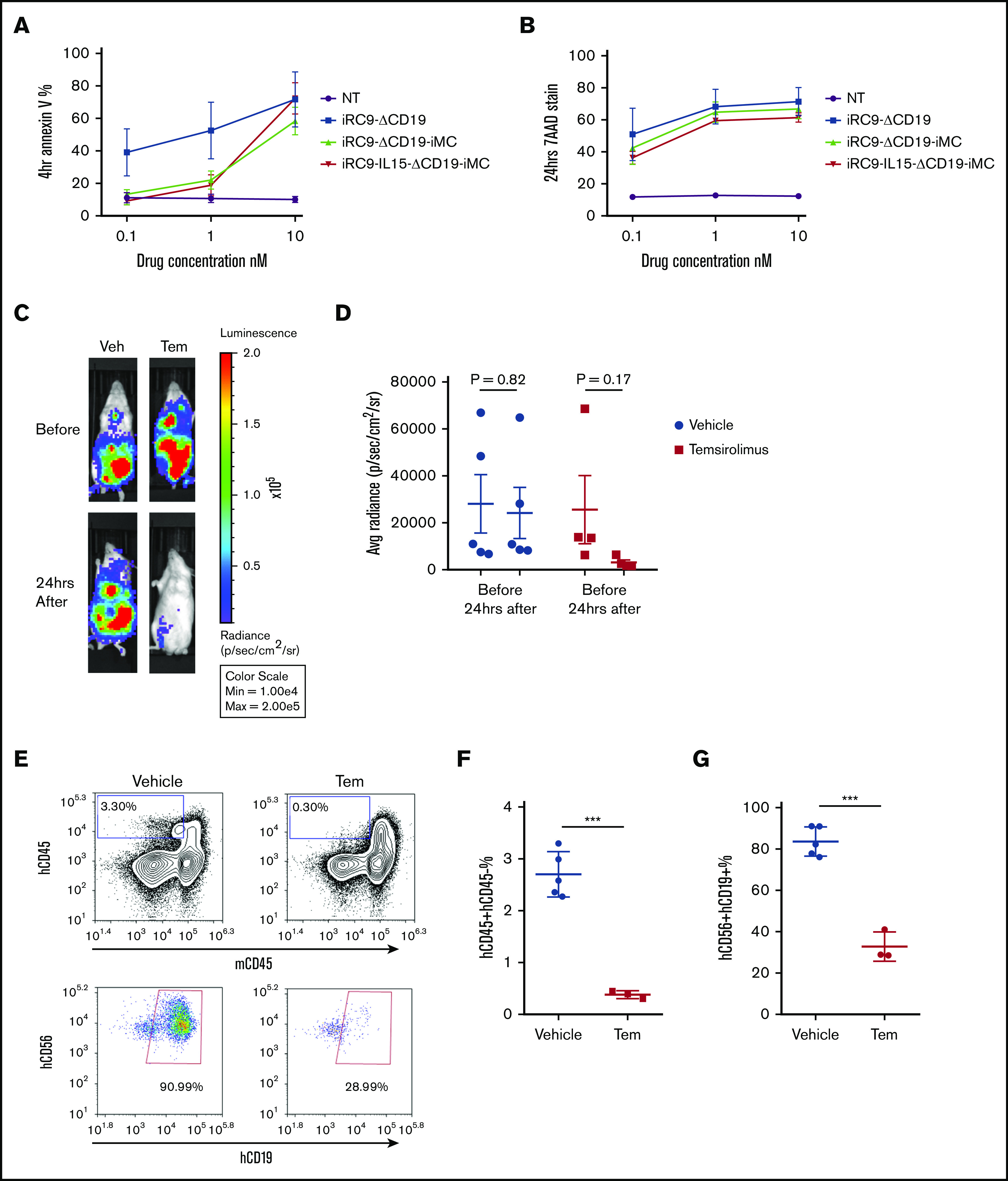
Elimination of NK cells with the iRC9 safety switch. (A) Nontransduced (NT) NK cells or iRC9-ΔCD19–, iRC9-ΔCD19-iMC–, or iRC9-IL15-ΔCD19-iMC–modified NK cells were administrated with temsirolimus (Tem) at concentrations 0, 0.1, 1, and 10 nM for 4 hours. Annexin V staining was assessed by flow cytometry. (B) Tem at increasing concentrations was administrated for 24 hours. 7AAD permeability was assessed by flow cytometry. Cells were first gated as CD56+CD19+ for transduced NK cells and CD56+ for NT NK cells. (C-G) Nine NSG mice engrafted with iRC9-IL15-ΔCD19-iMC–modified NK cells were randomly divided into 2 groups. Four mice received 1 mg/kg of Tem IP, whereas 5 mice had the same volume of vehicle (Veh) administered. (C-D) Before injection and 24 hours later, BLI was determined for NK presence in vivo. (E-G) Mice were then euthanized, and spleens were analyzed for the presence of NK cells by flow cytometry. Human cells were identified as hCD45+mCD45− populations. Transduced NKs were identified as hCD45+mCD45−hCD56+hCD19+ populations. Student t test was used for comparisons. ***P < .001.
Discussion
CAR-T therapies produce dramatic antitumor responses against B-cell malignancies,8,38 and 2 autologous CD19-directed CAR-T products are currently US Food and Drug Administration approved. Although numerous clinical studies have tested CAR-T efficacy in other hematological malignancies and some solid tumors,23,39,40 development of CAR-NK cells as a clinical cell therapy is less mature, despite key advantages of NK cells. NK cells eradicate neoplastic cells via several mechanisms: recognition of stress receptors on transformed cells, antigen-specific targeting through antibody-dependent cellular cytotoxicity, and expression of proapoptotic ligands.15,41 This innate cytotoxicity provides a mechanism for CAR-NK cells to target tumor cells that evade CAR targeting by heterogeneous expression of the CAR-specific antigen, a noted means of tumor escape from CAR-T therapy.7,8,42 Additionally, iMC-enhanced CAR-NK cells abundantly produce cytokines and chemokines (MIP-1α, MIP-1β, RANTES, IFN-γ, IL-13, IL-8, MCP-1) that shape the adaptive immune responses against tumors by recruiting and activating dendritic cell, macrophage, and T-cell responses.43,44 Moreover, because NK cells lack a TCR, they have greatly reduced capacity to elicit GVHD, raising the potential to develop standardized, donor-derived, off-the-shelf therapies.3,10,11 In fact, very promising clinical results have now been reported for the targeting of B-cell neoplasms with allogeneic second-generation CD19.CD28.ζ CAR-NK cells augmented with IL-15 and iCaspase-9.6,15,45 Another recent preclinical study using CD19.4-1BB.ζ CAR-NK cells expanded from peripheral blood via a feeder-free method showed antileukemia activity superimposable to that of CAR-T cells with a lower toxicity profile in xenografted animals.46
Several attributes of NK cells as a cell therapy are enhanced by engineered iMC expression. Tonic signaling from iMC expression and activation of the switch with rimiducid leads to substantial increases in innate NK cell cytotoxicity. The proportion of NK cells capable of degranulation increased against cells expressing high or low levels of MHC class 1, and their cytotoxicity was further associated with enhanced levels of perforin and granzyme B in cytotoxic granules. These findings predict that iMC-enhanced NK cells are more prone to act as so-called tumor cell serial killers before the depletion of granules reduces cytotoxic capacity. In this sense, iMC-enhanced NK cells function similarly to NK cells activated ex vivo with cytokines, including IL-18, that signal through MyD88 and display a mature phenotypic signature with high cytotoxic potential.19
Concomitant with the potent activation of NK cell cytotoxicity, iMC signaling also promotes NK cell proliferation and survival both when tumor targets are present to activate NK receptors and when they are absent. The cell signaling pathways activated by iMC costimulation in T cells,23 including NF-κB, Akt, JNK, and p38 pathways, are also activated in iMC-enhanced NK cells. The mitogen-activated protein kinase pathways are essential to NK cell cytotoxicity and IFN-γ and tumor necrosis factor α production and expansion.47-51 Interestingly, we also observed rapid ERK1 phosphorylation with rimiducid activation of iMC in NK cells, although this pathway seemed to require additional CAR engagement in T cells to be activated.23 Vav activation is essential to NK cell cytotoxicity.52 Although this activator of the Rac and Rho pathways has been described to be downstream of NK receptor activation through Fyn,30 we observed that iMC stimulation with rimiducid led to rapid Vav-1 and that Vav-1 reduction inhibited iMC-stimulated NK cell cytotoxicity and reduced IFN-γ production. The intersection of Vav-1 activation with IKKα has been shown to underlie activation of NF-κB by CD28-mediated costimulation in T cells,53 and a similar mechanism may partially direct iMC-induced cytokine release. It will be interesting to examine the similarities and differences in NK cell signaling introduced by the addition of CD28 and 4-1BB signaling domains in a second-generation CAR relative to iMC. Although there are some similarities in signaling outcomes (eg, NK-κB and mitogen-activated protein kinase activation) shared between iMC and these domains in CAR-T cells,23 how CD28, for example, signals in CAR-NK cells is less well defined. Furthermore, antigen-independent signaling from iMC may drive the expansion and persistence of CAR-NK cells without CAR engagement, and although speculative, this periodic separation of ITAM engagement (which drives exhaustion in T cells) from auxiliary activation may be important for long-term functionality of iMC CAR-NK cells.
iMC activation by rimiducid stimulates NK cell growth ex vivo, and T-cell expansion is potently stimulated by iMC/rimiducid in vivo.23 iMC was insufficient to promote engraftment and persistence of NK cells in NSG mice alone, but a combination vector expressing IL-15 with iMC promoted the engraftment and survival of NK cells beyond 6 weeks. This effect was synergistic, because iMC/IL-15–enhanced NK cells failed to engraft without the administration of rimiducid to activate iMC signaling. IL-15 is a critical cytokine to NK development survival and function and has been an accessory to several NK cell adoptive therapy protocols, although with considerable toxicity when administered systemically.32,54 Incorporation of IL-15 into CAR-NK–based cell therapies permits relatively low-level IL-15 secretion and autocrine signaling with the potential for local paracrine effects along with other iMC-CAR–directed cytokines in the tumor microenvironment. We have not observed excessive cytokine release resulting in toxicity from the administration of iMC-enhanced NK cells in our animal models.
To further guard against the possibility of toxicity generated by cytokine release syndrome or mistargeting of CAR-modified effector cells to healthy tissue, a proapoptotic safety switch, iRC9, has been incorporated into the dual-switch CAR-NK platform directed by 2 vector configurations.24 One vector system expresses the safety switch separately from iMC and the CAR, whereas the other expresses both the activation and safety switch in a vector separated from a cotransduced first-generation CAR. In either strategy, a marker protein, ΔCD19, permits positive selection of transduced NK cells containing iRC9. iRC9 is not activated by rimiducid and is orthogonally activated by rapamycin or its prodrug temsirolimus to drive apoptosis within hours both in vitro and in vivo.24
In conclusion, we have developed a platform that addresses several of the current challenges of allogeneic approaches to adoptive CAR-based cell therapy. Dual-switch CAR-NK cells provide antigen-specific cytotoxicity with the potentially important benefit of potent cytotoxicity against tumor cells with reduced or completely absent CAR target antigen expression, each of which is enhanced by rimiducid-directed iMC activation. NK cells have greatly reduced risk of eliciting GVHD, enabling the sourcing of allogeneic, donor-derived cells rather than autologous cells potentially compromised by the malignancy and previous therapeutic regimens. Finally, controlled iMC signaling combined with autocrine IL-15 has the potential to overcome the relatively poor engraftment and survival observed with NK-based therapies while stimulating CAR-NK cytokine production that may further ignite a host-derived antitumor response.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Eric Yvon for his insights and expertise during the development phase of this project and all employees of Bellicum Pharmaceuticals, particularly MyLinh Duong, Christine Gagliardi, Peter Chang, Alan Guerrero, and Kelly Sharp for important insights into and contributions to the NK program.
Footnotes
The authors are willing to share data and resources via e-mail to corresponding authors, J. Henri Bayle (jhbayle@bellicum.com) and Aaron E. Foster (afoster@bellicum.com).
Authorship
Contribution: X.W. designed and performed experiments described in each figure and drafted the manuscript; D.L.J. designed aspects of the NK cell isolation, growth, and phenotyping processes; J.L.M. assisted in molecular cloning and animal experiments, particularly in Figure 4; D.M.S. designed and supervised aspects of the project; and A.E.F. and J.H.B. designed and supervised the experiments and wrote the manuscript.
Conflict-of-interest disclosure: Aspects of this work have been filed in a patent, WO/2019/217327 A1, applied for by Bellicum Pharmaceuticals. The authors declare no competing financial interests.
Correspondence: J. Henri Bayle, Bellicum Pharmaceuticals, 2130 W Holcombe Blvd, Suite 850, Houston, TX 77030; e-mail: jhbayle@bellicum.com; and Aaron E. Foster, Bellicum Pharmaceuticals, 2130 W Holcombe Blvd, Suite 850, Houston, TX 77030; e-mail: afoster@bellicum.com.
References
- 1.Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat Rev Immunol. 2011;11(10):645-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raulet DH, Marcus A, Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol Rev. 2017;280(1):93-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097-2100. [DOI] [PubMed] [Google Scholar]
- 4.Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051-3057. [DOI] [PubMed] [Google Scholar]
- 5.Shah N, Li L, McCarty J, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017;177(3):457-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. 2018;51:146-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115(21):4293-4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285(5426):412-415. [DOI] [PubMed] [Google Scholar]
- 12.Locatelli F, Moretta F, Brescia L, Merli P. Natural killer cells in the treatment of high-risk acute leukaemia. Semin Immunol. 2014;26(2):173-179. [DOI] [PubMed] [Google Scholar]
- 13.Rubnitz JE, Inaba H, Ribeiro RC, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. 2010;28(6):955-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah NN, Baird K, Delbrook CP, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125(5):784-792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. 2018;9:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J. 2015;21(6):486-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lutz CT, Karapetyan A, Al-Attar A, et al. Human NK cells proliferate and die in vivo more rapidly than T cells in healthy young and elderly adults. J Immunol. 2011;186(8):4590-4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351-2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romee R, Rosario M, Berrien-Elliott MM, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo). 2014;2014:205796. 10.1155/2014/205796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molgora M, Bonavita E, Ponzetta A, et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature. 2017;551(7678):110-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vigneron C, Mirouse A, Merdji H, et al. Sepsis inhibits tumor growth in mice with cancer through Toll-like receptor 4-associated enhanced natural killer cell activity. OncoImmunology. 2019;8(11):e1641391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster AE, Mahendravada A, Shinners NP, et al. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol Ther. 2017;25(9):2176-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duong MT, Collinson-Pautz MR, Morschl E, et al. Two-dimensional regulation of CAR-T cell therapy with orthogonal switches. Mol Ther Oncolytics. 2018;12:124-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narayanan P, Lapteva N, Seethammagari M, Levitt JM, Slawin KM, Spencer DM. A composite MyD88/CD40 switch synergistically activates mouse and human dendritic cells for enhanced antitumor efficacy. J Clin Invest. 2011;121(4):1524-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stavrou M, Philip B, Traynor-White C, et al. A rapamycin activated Caspase 9-based suicide gene. Mol Ther. 2018;26(5):1266-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477-6489. [DOI] [PubMed] [Google Scholar]
- 28.Rivière I, Brose K, Mulligan RC. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci USA. 1995;92(15):6733-6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riteau B, Barber DF, Long EO. Vav1 phosphorylation is induced by beta2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J Exp Med. 2003;198(3):469-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong Z, Davidson D, Pérez-Quintero LA, Kurosaki T, Swat W, Veillette A. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity. 2012;36(6):974-985. [DOI] [PubMed] [Google Scholar]
- 31.Wagner J, Pfannenstiel V, Waldmann A, et al. A two-phase expansion protocol combining interleukin (IL)-15 and IL-21 improves natural killer cell proliferation and cytotoxicity against rhabdomyosarcoma. Front Immunol. 2017;8:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Felices M, Lenvik AJ, McElmurry R, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. 2018;3(3):96219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gill S, Vasey AE, De Souza A, et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood. 2012;119(24):5758-5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garg TK, Szmania SM, Khan JA, et al. Highly activated and expanded natural killer cells for multiple myeloma immunotherapy. Haematologica. 2012;97(9):1348-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szmania S, Lapteva N, Garg T, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother. 2015;38(1):24-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fehniger TA, Suzuki K, Ponnappan A, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193(2):219-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takai A, Nakano M, Saito K, et al. Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA. 2015;112(14):4352-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Chen LJ, Yang SS, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci USA. 2019;116(19):9543-9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gargett T, Yu W, Dotti G, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24(6):1135-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413(6852):165-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219-1226. [DOI] [PubMed] [Google Scholar]
- 43.Bigley AB, Simpson RJ. NK cells and exercise: implications for cancer immunotherapy and survivorship. Discov Med. 2015;19(107):433-445. [PubMed] [Google Scholar]
- 44.Böttcher JP, Bonavita E, Chakravarty P, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022-1037.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quintarelli C, Sivori S, Caruso S, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia. 2019;34(4):1102-1115. [DOI] [PubMed] [Google Scholar]
- 47.Trotta R, Fettucciari K, Azzoni L, et al. Differential role of p38 and c-Jun N-terminal kinase 1 mitogen-activated protein kinases in NK cell cytotoxicity. J Immunol. 2000;165(4):1782-1789. [DOI] [PubMed] [Google Scholar]
- 48.Yang G, Kong Q, Wang G, et al. Low-dose ionizing radiation induces direct activation of natural killer cells and provides a novel approach for adoptive cellular immunotherapy. Cancer Biother Radiopharm. 2014;29(10):428-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y, Wang Y, Zhuang Y, Zhou F, Huang L. Mifepristone increases the cytotoxicity of uterine natural killer cells by acting as a glucocorticoid antagonist via ERK activation. PLoS One. 2012;7(5):e36413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferrari de Andrade L, Ngiow SF, Stannard K, et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014;74(24):7298-7308. [DOI] [PubMed] [Google Scholar]
- 51.Gross O, Grupp C, Steinberg C, et al. Multiple ITAM-coupled NK-cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-kappaB and MAPK activation to selectively control cytokine production. Blood. 2008;112(6):2421-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cella M, Fujikawa K, Tassi I, et al. Differential requirements for Vav proteins in DAP10- and ITAM-mediated NK cell cytotoxicity. J Exp Med. 2004;200(6):817-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piccolella E, Spadaro F, Ramoni C, et al. Vav-1 and the IKK alpha subunit of I kappa B kinase functionally associate to induce NF-kappa B activation in response to CD28 engagement. J Immunol. 2003;170(6):2895-2903. [DOI] [PubMed] [Google Scholar]
- 54.Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.