

Key Points
Targeted busulfan, fludarabine, serotherapy, and HLA-matched hematopoietic stem cell transplantation achieved 100% event-free survival in HLH.
After a median follow-up of 3 years, satisfactory DC on myeloid and T cells, no graft failures, and a low rate of chronic GHVD were observed.
Abstract
Reduced-intensity/reduced-toxicity conditioning and allogeneic T-cell replete hematopoietic stem cell transplantation are curative in patients with hemophagocytic lymphohistiocytosis (HLH). Unstable donor chimerism (DC) and relapses are clinical challenges . We examined the effect of a reduced-intensity conditioning regimen based on targeted busulfan to enhance myeloid DC in HLH. The European Society for Bone and Marrow Transplantation–approved reduced-intensity conditioning protocol comprised targeted submyeloablative IV busulfan, IV fludarabine, and serotherapy comprising IV alemtuzumab (0.5-0.8 mg/kg) for unrelated-donor and IV rabbit anti–T-cell globulin for related-donor transplants. We assessed toxicity, engraftment, graft-versus-host disease (GHVD), DC in blood cell subtypes, and overall survival/event-free survival. Twenty-five patients from 7 centers were treated (median age, 0.68 year). The median total dose and cumulative area under the curve of busulfan was 13.1 mg/kg (6.4-26.4) and 63.1 mg/L × h (48-77), respectively. Bone marrow, peripheral blood stem cell, or cord blood transplants from HLA-matched related (n = 7) or unrelated (n = 18) donors were administered. Donor cells engrafted in all patients (median: neutrophils d+20/platelets d+28). At last follow-up (median, 36 months; range, 8-111 months), the median DC of CD15+ neutrophils, CD3+ T cells, and CD16+56+ natural killer cells was 99.5% (10-100), 97% (30-100), and 97.5% (30-100), respectively. Eight patients (32%) developed sinusoidal obstruction syndrome, resolving after defibrotide treatment. The 3-year overall survival and event-free survival rates were both 100%. None of the patients developed acute grade III to IV GHVD. Limited chronic GVHD was encountered in 4%. This regimen achieves excellent results with stable DC in patients with HLH.
Visual Abstract

Introduction
Primary hemophagocytic lymphohistiocytosis (HLH) comprises a group of genetically determined, life-threatening immune disorders with an estimated incidence of 1.2 per million children per year.1 Deficiencies in the content, assembly, trafficking, or release of cytolytic granules in T cells and natural killer (NK) cells account for the majority of patients, whereas molecular diagnosis may remain unsolved in some individuals with presumed primary HLH.2,3 To date, mutations in 4 different genes (PRF1, UNC13D, STXBP2, and STX11) have been found causative for familial HLH (FHL). Mutations in other genes (RAB27A, LYST, SH2D1A, BIRC4, HAVCR2 (TIM-3), NLRC4, CD48, CDC42, and AP3B3A) were shown to be responsible for primary immunodeficiency and auto-inflammatory syndromes in which HLH is a prominent clinical feature.3-8 Viral infections are important triggers of HLH episodes in patients with a genetic predisposition. Persistent, uncontrolled activation of innate and adaptive immunity in secondary lymphoid organs and liver with cytokine hypersecretion ultimately results in hemorrhage, multiorgan failure, and death.2,4,9 The clinical and laboratory features of HLH include fever, hepatosplenomegaly, pancytopenia, hyponatremia, hyperlipidemia, hyperferritinemia, and severe coagulopathy. Timely diagnosis, systemic/intrathecal treatment with immunosuppressive/anti-inflammatory drugs, and specific treatment of triggering infections, pancytopenia, and coagulopathy are able to halt disease progression in some patients until definitive cure by allogeneic hematopoietic stem cell transplantation (HSCT).10-12
Advances in the pathophysiological understanding have led to improved treatment protocols to achieve remission of HLH episodes.13,14 The analysis of both prospective HLH-1994/HLH-2004 trials showed that only 60% of patients achieved complete remission (CR), and disease progression remained the primary cause of death in patients without HSCT within 12 months of disease onset.15-20 Therapeutic strategies to avoid etoposide as the most toxic component of HLH-2004 comprise alemtuzumab13 or emapalumab,21 which are currently being investigated to prove their efficacy in inducing remission in HLH until HSCT. Currently, allogeneic HSCT remains the only curative treatment modality in HLH. This is mainly achieved by correction of cytotoxicity via adoptive transfer and de novo production of healthy CD3+ T cells and NK cells of donor origin.17,22-29
For a long time, myeloablative conditioning regimens (MAC) based on busulfan/cyclophosphamide (± etoposide) have been considered as the standard of care for patients with HLH, even though transplant-related morbidity and mortality were substantial (up to 64%).22,24,30-33 The introduction of reduced-intensity conditioning (RIC) and reduced-toxicity conditioning protocols helped decrease MAC-associated transplant-related morbidity and mortality; however, graft failure (GF), waning donor chimerism (DC),29,31,34 relapses of HLH,34 and acute/chronic graft-versus-host disease (GHVD)35 remained issues of clinical concern. Because low DC may predispose to disease recurrence,34 posttransplant interventions, including early tapering of immunosuppression, administration of donor lymphocyte infusion (DLI), and stem cell boosts, have been increasingly used to augment DC after RIC transplantations. These approaches clearly increased the risk of inducing acute/chronic GHVD, however, with inacceptable implications on quality of life and long-term outcome.12,29,31,35
A RIC regimen based on targeted busulfan showed excellent engraftment and low toxicity features in high-risk pediatric and adult patients with chronic granulomatous disease (CGD).36 The current study examined the effects of this RIC regimen in patients with primary HLH undergoing HSCT with HLA-matched donors within a collaborative multicenter study of the Inborn Errors Working Party of the European Society for Bone and Marrow Transplantation (IEWP EBMT).
Methods
Study design
From June 2009 to June 2019, seven European pediatric SCT centers agreed to use the protocol in patients with HLH. Functional and molecular evaluation for primary HLH was based on a standardized algorithm (supplemental Data A). For induction of remission, modified HLH-200417 and/or alemtuzumab-based protocols were administered. Central nervous system (CNS) involvement and HLH remission status (complete remission [CR]; partial remission) were defined according to HLH-2004. In line with the tenets of the Declaration of Helsinki, parents and patients provided written informed consent for participation in this transplantation protocol, which is part of the IEWP EBMT guidelines to treat high-risk primary immunodeficiency patients.36
Conditioning regimen, transplantation, and GHVD prophylaxis
RIC conditioning was administered as previously described36 comprising IV fludarabine (30 mg/m2 or in infants <9 kg body weight [bw] 1.2 mg/kg per dose on d-8 to d-3) and pharmacokinetic-adjusted targeted IV busulfan. Busulfan was administered from d-5 to d-2. The recommended infusion modality was twice daily over 3 or 4 hours. Four-times-daily administration over 1 hour was also accepted. The modified starting doses of busulfan depended on a weight-based protocol introduced by Bartelink et al.37,38 The kinetic parameters, including the cumulative area under the curve (cAUC) of busulfan, was calculated by using the two-compartment WinNonlin pharmacokinetic program.37 To prevent GF (previously 5% in patients with CGD), the cAUC for busulfan was recommended to be targeted toward the upper part of the previously reported busulfan exposure (45-65 mg/L × h)36 (supplemental Data B).
Patients with a 9-10/10-HLA–matched unrelated donor (MUD) or 9/10-HLA–matched cord blood (9/10/UCB) underwent serotherapy with IV alemtuzumab (0.17-0.27 mg/kg bw per dose) from d-8 to d-6. To reduce the risk of GVHD by grafts of HLA single-mismatched unrelated donors unwilling to donate bone marrow, study centers were permitted to perform a CD34+ HSC-positive selection in peripheral blood stem cell (PBSC) grafts (CliniMACS technique, Miltenyi Biotec) together with a CD3+ T-cell addback. The CD3+ content of the graft was adjusted to bone marrow conditions by a CD3+ T-cell reduction of approximately one log.39
Serotherapy included rabbit anti-thymocyte globulin (Thymoglobulin [Sanofi-Aventis]; 2.5 mg/kg bw IV per dose on d-5 to d-3) or anti–T-lymphocyte globulin against Jurkat-cells (Grafalon [Neovii Pharmaceuticals AG]; 10 mg/kg bw IV per dose on d-4 to d-1) for 6/6-HLA–matched related sibling/related (MSD/MRD) donor transplants and 10/10-HLA–matched unrelated cord blood (10/10/UCB) donor transplants (Table 1). IV cyclosporin A (CSA) was started at d-3 to maintain trough levels of 200 µg/L. In case of absent GVHD, oral CSA was administered until d+120 and tapered until d+180. IV mycophenolate-mofetil was started at day 0 (3 × 400 mg/m2), later continued orally for 60 to 100 days.36
Table 1.
Patient and transplant characteristics
| UPN | Genetic type | Sex | Age at tx | Pre-tx disease manifestations | CNS+, Y/N | Time from HLH diagnosis to tx, wk | PR/CR at tx | HLH criteria (Y/N)/HLH therapy (Y/N) | Donor/HLA/source | Serotherapy, total mg/kg bw | CD34+/kg bw, ×106 | Total Bu, dose, mg/kg bw | Cum. Bu AUC, mg/L × h | D until ANC >0.5 g/L | D until PLT >50 g/L |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | XLP1/SH2D1A | M | 16 y | Family history | N | NA | CR | N/IVIG | MUD/10/10/BM | Alemtuzumab 0.5 | 2.1 | 6.4 | 59 | 23 | 21 |
| 2 | XLP1/SH2D1A | M | 22 y | HLH episode, IgG low EBV-hepatitis/-CMP | N | 1040 | CR | Y/steroids | MSD/6/6/PBSC | Thymoglobulin 7.5 | 2.4 | 9.7 | 53 | 17 | 14 |
| 3 | XLP1/SH2D1A | M | 7 y | IgG low, DLBCL 2017 | N | N/A | CR | N/DLBCL chemotherapy | MUD/10/10/BM | Alemtuzumab 0.6 | 7.23 | 17 | 67 | 20 | 16 |
| 4 | XLP1/SH2D1A | M | 3.8 y | Developmental regression ADEM | Y | 14 | PR | N/IVIG, steroids, CY | MUD/9/10/BM | Alemtuzumab 0.6 | 15.3 | 26.4 | 54 | 24 | 74 |
| 5 | CHS/LYST | F | 1 y | Nystagmus, oculocutaneous albinism | N* | NA | CR | N/no treatment | MUD/10/10/BM | Alemtuzumab 0.5 | 2.7 | 12 | 64 | 23 | 38 |
| 6 | FHL5/STXBP2 | F | 3 mo | HLH episode, sepsis, mild renal impairment | N | 8 | PR | Y/HLH-2004, IVIG, alemtuzumab | MSD/6/6/BM | Alemtuzumab 0.5 | 13.4 | 14 | 53 | 17 | 38 |
| 7 | FHL5/STXBP2 | M | 8 mo | HLH episode, failure to thrive | N | 25 | CR | Y/HLH-2004, no CSA | MSD/6/6/BM | Thymoglobulin 7.5 | 7 | 10 | 66 | 14 | 47 |
| 8 | FHL5/STXBP2 | M | 4 mo | HLH episode, hepatic/renal failure | N | 15 | CR | Y/HLH-2004 | UCB/9/10/cord | Alemtuzumab 0.6 | 1.15 | 7.2 | 63 | 24 | 30 |
| 9 | FHL5/STXBP2 | F | 3 y | Recurrent HLH episodes | N | 20 | CR | Y/HLH-2004 | MUD/10/10/BM | Alemtuzumab 0.5 | 2.25 | 17.3 | 55 | 22 | 34 |
| 10 | FHL3/UNC13D | M | 10 mo | HLH episode, cardiac effusion | N | 11 | CR | Y/HLH-2004 | MUD/10/10/BM | Alemtuzumab 0.6 | 8.6 | 15.6 | 64 | 20 | 17 |
| 11 | FHL3/UNC13D | M | 5 mo | HLH episode, focal seizures, NEC | Y | 11 | CR | Y/HLH-2004 | MUD/10/10/BM | Alemtuzumab 0.5 | 5.58 | 16 | 77 | 19 | 42 |
| 12 | FHL3/UNC13D | M | 10 mo | HLH episode | N | 31 | CR | Y/steroids, CSA, alemtuzumab | MUD/10/10/PBSC | Alemtuzumab 0.6 | 10 | 16 | 65 | 28 | 14 |
| 13 | FHL3/UNC13D | F | 9.25 y | HLH episode, GBS | Y | 29 | PR | Y/HLH-2004 | MRD/10/10/BM | ATG-N 40 | 4.59 | 11.4 | 56 | 28 | 29 |
| 14 | FHL3/UNC13D | F | 4 mo | HLH episode, CMV+, CMP, neurologically handicapped | Y | 12 | PR | Y/steroids | UCB/10/10/cord | Thymoglobulin 7.5 | 0.9 | 12.08 | 48 | 21 | 28 |
| 15 | FHL3/UNC13D | M | 5 mo | HLH episode, FTT | Y | 20 | CR | Y/HLH-2004 | MSD/6/6/BM | Thymoglobulin 7.5 | 32.2 | 19.04 | 63 | 11 | 37 |
| 16 | FHL3/UNC13D | M | 7 mo | HLH episode, polyserositis | N | 23 | CR | Y/HLH-2004 | MUD/10/10/BM | Alemtuzumab 0.6 | 5.55 | 13.1 | 68 | 18 | 29 |
| 17 | FHL3/UNC13D | F | 3.5 y | HLH episode, thrombosis of the sinus sagittalis superior | Y | 19 | CR | Y/HLH-2004 | MUD/9/10/PBSC† | Alemtuzumab 0.6 | 13.95 | 12.4 | 62 | 15 | 11 |
| 18 | FHL3/UNC13D | F | 5 mo | HLH episode, FTT | N | 20 | CR | Y/HLH-2004, alemtuzumab | MUD/10/10/BM | Alemtuzumab 0.6 | 10.7 | 24.6 | 65 | 15 | 40 |
| 19 | FHL3/UNC13D | F | 5 mo | Family history | N | NA | CR | N/CSA | MUD/10/10/BM | Alemtuzumab 0.6 | 10 | 15.4 | 65 | 15 | 26 |
| 20 | FHL3/UNC13D | F | 2 mo | Family history | N | NA | CR | N/N | MSD/6/6/BM | Thymoglobulin 7.5 | 24.8 | 11.4 | 65 | 28 | 23 |
| 21 | FHL3/UNC13D | F | 3 mo | Recurrent HLH episodes | N | 15 | CR | Y/steroids, CSA, alemtuzumab | MUD/10/10 PBSC† | Alemtuzumab 0.6 | 25 | 11.8 | 62 | 20 | 13 |
| 22 | FHL3/UNC13D | M | 7 mo | HLH episode | N | 20 | CR | N/N | MSD/6/6/BM | Thymoglobulin 7.5 | 6.95 | 14.4 | 55 | 31 | 43 |
| 23 | FHL3/UNC13D | F | 4 mo | HLH episode | N | 10 | CR | Y/HLH-2004 | MUD/10/10/BM | Alemtuzumab 0.6 | 7.3 | 8 | 73 | 15 | 14 |
| 24 | Heterozygous UNC13D | M | 16 mo | Recurrent HLH episodes | N | 32 | CR | Y/steroids, CSA | MUD/10/10/PBSC | Alemtuzumab 0.6 | 11.3 | 17.2 | 59 | 20 | 12 |
| 25 | Unknown | F | 23 mo | Recurrent HLH episodes | Y | 62 | PR | Y/HLH-2004, infliximab | MUD/10/10/PBSC† | Alemtuzumab 0.8 | 15 | 11.1 | 65 | 18 | 24 |
ADEM, acute disseminated encephalomyelitis; ANC, absolute neutrophil count; ATG-N, ATG-Neovii (Grafalon); AUC, area under the curve; BM, bone marrow; Bu, busulfan; bw, body weight; CMP, cardiomyopathy; Cum., cumulative; CY, cyclophosphamide; D, days until engraftment; DLBCL, diffuse large B-cell lymphoma; F, female; FTT, failure to thrive; GBS, Guillain-Barré-Syndrome; IgG, immunoglobulin G; IVIG, intravenous immunoglobulin; M, male; MRD, HLA-matched related donor; MSD, HLA-matched sibling donor; MUD, HLA-matched unrelated donor; N, no; NA, not applicable; NEC, necrotizing enterocolitis; PR, partial remission; tx, hematopoietic stem cell transplantation; UCB, unrelated cord blood; UPN, unique patient number; wk, weeks; XLP1, X-linked lymphoproliferative syndrome 1; Y, yes.
Neurological disease due to Chédiak-Higashi syndrome but not due to CNS involvement or HLH.
Patients treated with PBSC and CD34+ selection (CliniMACS technique) and CD3+ addback.
To prevent sinusoidal obstruction syndrome (SOS), prophylactic measures included IV defibrotide (DF; total 25 mg/kg per day, 4 times a day) or IV heparin (total 100 U/kg per day continuous infusion) and/or intravenous N-acetyl cysteine (total 200 mg/kg, twice daily), according to the center’s preference.40,41 Recommended SOS treatment is IV DF42 starting immediately after fulfillment of the modified Seattle criteria.42,43 The current pediatric EBMT criteria were applied retrospectively to grade the SOS severity.44 SOS diagnostics included abdominal sonography and Doppler scan to detect inversed portal-venous flow for SOS confirmation.40,42 In oxygen-dependent states, echocardiography screens helped to exclude pulmonary hypertension.
GHVD was assessed according to the Glucksberg and National Institutes of Health classifications. The severity of mucositis was assessed according to the World Health Organization score.
Statistical analysis
The overall survival (OS), event-free survival (EFS; event = graft failure with <5% DC in whole blood or <20% CD3+ T cells),34 chronic GHVD–free survival, and neutrophil (≥0.5 × 109/L) and platelet (≥ 50×109/L) engraftment were described by using Kaplan-Meier estimates; countable characteristics (eg, age, cell dose, AUC of busulfan) by using medians and interquartile ratios (IQRs); and comparisons by using two-tailed Student t tests. For analyses, statistics, and graphics, GraphPad Prism (version 6.0a), Microsoft Excel, and Word 2011 software were used.
Results
Patients
Twenty-two consecutive patients from 5 centers and 3 nonconsecutive patients from 2 centers have been treated according the protocol. Transplantations were performed at a median age of 0.68 year (IQR, 0.42-3.07; range, 2 months-22 years). Twenty-three patients had molecularly proven autosomal recessive HLH: 14 with FHL3 (UNC13D), 4 with FHL5 (STXBP2), 4 with X-linked lymphoproliferative syndrome 1 (SH2D1A), and 1 with Chédiak-Higashi syndrome (LYST) (Table 1). Extensive functional and genetic analyses failed to clearly reveal causative mutations in 2 patients (supplemental Data C).
Pretreatment and complications before HSCT
Fifty-two percent (n = 13) of patients received treatment to induce remission of HLH episodes by administration of the HLH-2004 protocol. Of these, 38% (n = 5) needed additional treatment of remission induction that included IV infliximab or IV alemtuzumab. Based on the aim of avoiding administration of VP-16, 36% (n = 9) received steroids, IV immunoglobulins, and/or IV alemtuzumab instead of the HLH-2004 protocol for remission induction. Twenty-four percent (n = 6) with CNS involvement received intrathecal methotrexate. Two patients with SAP deficiency presented initially with non-HLH symptoms: unique patient number 4 (UPN4) developed acute demyelinating encephalomyelitis and was treated with cyclophosphamide. UPN3 presented with malignant diffuse large B-cell lymphoma and received anti-lymphoma chemotherapy. Sixteen percent of patients (UPN1, UPN5, UPN19, and UPN20) were diagnosed by using family history and had not experienced HLH episodes before transplantation. At the time point of HSCT, 20% of patients (n = 5) were in partial remission, and 80% (n = 20) were in CR (Table 1).
Therapeutic drug monitoring of busulfan
Ninety-six percent (n = 24) of patients received twice-daily busulfan infusions except UPN23, who received a four-times-daily regimen. Ninety-two percent (n = 23) were targeted within the target range for the cAUC (45-65 mg/L × h, ±5%). Forty-three percent (n = 12) were targeted around 65 mg/L × h. UPN11 and UPN23 were slightly above the target range (73 and 77 mg/L × h); none was below (Figure 1; Table 1). The median busulfan exposure was 63.1 mg/L × h (IQR, 56.02-65; range, 48-77 mg/L × h) corresponding to 11 693 to 18 757 μM × min. The median total cumulative busulfan dose was 13.1 mg/kg (IQR, 11.4-16; range, 6.4-26.4).
Figure 1.

Interindividual variability of busulfan exposure. Each symbol denotes the cAUC (mg/L × h) in relation to the administered total cumulative dose (mg/kg bw) of busulfan in patients aged <1 year (squares), >1 year and <4 years (circles), and >4 years (triangles) of age. Filled symbols denote patients developing SOS. The target submyeloablative cAUC (45-65 mg/L × h) is depicted in gray.
Stem cell sources and graft composition
Bone marrow (68%; n = 17), PBSC (24%; n = 6), and CB (8%; n = 2) grafts were transfused. UPN17, UPN21, and UPN25 received PBSC grafts after CD34+ HSC-positive selection and CD3+ T-cell addback (0.5-3.0 × 10 e7 CD3+ T cells/kg). Transplants comprised HSCs from MSD/MRD (n = 7) and MUD (n = 18) donors, of which three were 9/10-HLA–matched and 15 were 10/10-HLA–matched and two 9/10-HLA–matched and 10/10-HLA–matched cords (Table 1). The median number of CD34+ HSCs infused was 8.6 (IQR, 5.6-13.7; range, 2.25-32.2) in bone marrow/PBSC and 1.025 × 106/kg (0.9 and 1.15) in CB grafts.
Toxicity, engraftment, chimerism, and immune recovery
Toxicity.
Mucositis was observed in 48% (n = 12) of patients and did not exceed grade II except in UPN3, UPN21, and UPN24 (grade III). DF prophylaxis was administered in 20% of patients (n = 5), and SOS developed in 32% (n = 8) of patients. The SOS incidence did not differ between patients with DF prophylaxis and those without (20% vs 35%; P = .74). Six patients were classified as having moderate SOS and 2 as having severe SOS.44 All patients who developed SOS responded well to DF therapy, with complete resolution of SOS. Three patients needed additional procedures, including paracentesis to drain ascites, and one required ventilator support (Table 2). None developed pulmonary hypertension.33
Table 2.
Outcome Data
| UPN | SOS prophylaxis (DF mg/kg) | SOS, Y/N | SOS severity (moderate/severe) | SOS treatment (DF dose in mg/kg/d) | Cure of SOS, Y/N | Time at last FU, mo post-HSCT | Survival | Karnofsky/Lansky score at last FU, % | CD15+ DC at last FU, % | CD3+ DC at last FU, % | aGVHD (Glucksberg) | cGVHD (NIH) | Infection post-HSCT | Other transplant-related complications post HSCT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Hep | N | 52 | Alive | 100 | 100 | 88 | 0 | 0 | BK | Hemorrhagic cystitis | |||
| 2 | Hep | N | 64 | Alive | 100 | 100 | 100 | 2 | 0 | EBV | Femur head necrosis after 2 mo of steroids due to transient acute GHVD II of the liver | |||
| 3 | NAC | N | 16 | Alive | 100 | 100 | 100 | 2 | 0 | EBV, rhinovirus, BK | Mucositis (grade 3) | |||
| 4 | None | N | 8 | Alive | 40 | 100 | 100 | 2 | 0 | Fungal chest infection, ADV viremia, EBV viremia | Pancreatitis | |||
| 5 | Hep/NAC | N | 36 | Alive | 100 | 100 | 100 | 1 | 0 | Bacterial CVL infection | None | |||
| 6 | DF 25 | N | 31 | Alive | 100 | 90 | 85 | 0 | 0 | Rhinovirus, BCG | Mucositis (grade 2), PH (d+30), gut/renal salt protein losses, renal calcification (successful surgical removal) | |||
| 7 | DF 25 | Y | M | DF/steroids | Y | 24 | Alive | 100 | 98 | 88 | 0 | 0 | EBV, BCG | Hematemesis, rectal bleeding, diarrhea, early reactivation of HLH, renal impairment, arterial hypertension |
| 8 | Hep | Y | M | DF 40 | Y | 61 | Alive | 100 | 98 | 98 | 0 | 0 | N | Mucositis, GI problems, Hashimoto’s thyroiditis |
| 9 | NAC | Y | M | DF 40 paracentesis | Y | 15 | Alive | 100 | 9 | 42 | 0 | 0 | N | None |
| 10 | NAC | N | 51 | Alive | 100 | 100 | 100 | 0 | 0 | N | Mucositis (grade 1), CVL-associated atrial thrombus cured by surgical removal | |||
| 11 | Hep | Y | M | DF/steroids | Y | 88 | Alive | 100 | 90 | 89 | 0 | 0 | N | Mucositis (grade 2) |
| 12 | Hep | N | 111 | Alive | 100 | 94 | 92 | 1 | 0 | Urinary infection | Fever, mucositis (grade 2) | |||
| 13 | None | N | 45 | Alive | 100 | 100 | 100 | 1 | 0 | N | None | |||
| 14 | NAC | N | 63 | Alive | 40 | 100 | 98 | 0 | 0 | N | None | |||
| 15 | NAC | Y | S | DF/steroids, paracentesis, ventilation | Y | 43 | Alive | 100 | 95 | 96 | 0 | 0 | N | Mucositis (grade 2), ventilated, atelectasis left lung, urinary infection |
| 16 | NAC | Y | M | DF/steroids | Y | 31 | Alive | 100 | 100 | 99 | 1 | 0 | N | Recurrent CVL complications |
| 17 | NAC | N | 37 | Alive | 90 | 99,5 | 99,5 | 1 | 1 (oral/scalp) | Vulvitis with Pseudomonas | Mucositis (grade 2), engraftment syndrome, eosinophilic folliculitis of the scalp with partial effluvium, stomatitis, DD cGHVD | |||
| 18 | NAC | Y | M | DF/steroids/paracentesis | Y | 36 | Alive | 100 | 73 | 30 | 0 | 0 | N | Mucositis (grade 2) |
| 19 | NAC, DF 25 | N | 31 | Alive | 100 | 96 | 87 | 0 | 0 | N | None | |||
| 20 | NAC, DF 25 | N | 18 | Alive | 100 | 58 | 79 | 0 | 0 | Metapneumovirus, HHV-6 | None | |||
| 21 | Hep | N | 14 | Alive | 100 | 100 | 98,5 | 0 | 0 | Bacterial CVL infection | Mucositis grade 3 | |||
| 22 | None | Y | M | DF | Y | 22 | Alive | 100 | 100 | 81 | 0 | 0 | N | EBV viremia, rituximab |
| 23 | Hep/DF 25 | N | 8 | Alive | 100 | 67 | 74 | 0 | 0 | N | Mucositis (grade 3), seizure of unknown cause | |||
| 24 | NAC | N | 78 | Alive | 100 | 100 | 100 | 2 | 0 | N | Mucositis (grade 1), arterial hypertension | |||
| 25 | NAC | N | 49 | Alive | 100 | 97 | 97 | 2 | 0 | N | Engraftment syndrome, arthritis of 2 joints (intra-articular steroid injections) |
ADV, adenovirus; aGVHD, acute GVHD; BCG, bacillus Calmette-Guérin; cGVHD, chronic GVHD; CVL, central venous line; DC, donor chimerism; DD, differential diagnosis; DF, defibrotide; EBV, Epstein-Barr virus; FU, follow-up; GI, gastrointestinal; HEP, heparin; HHV-6, human herpesvirus 6; NAC, N-acetyl cysteine; NIH, National Institutes of Health; PH, pulmonary hypertension; SOS, sinusoidal obstruction syndrome.
Engraftment.
The median time until donor neutrophil engraftment was 20 days (IQR, 17-23; range, 11-31); the median time until platelet engraftment was 28 days (IQR, 20-38; range, 11-74) (Figure 2).
Figure 2.

Engraftment. Primary engraftment of neutrophils (time to reach >0.5 g/L; red line) and platelets (time to reach >50 g/L; blue line).
Chimerism.
After a median follow-up time of 36 months (IQR, 22-52; range, 8-111), the median donor CD15+ neutrophil chimerism was 99.5% (IQR, 94-100; range, 10-100) with 18 patients ≥95%, 3 ≥90%, 3 between 58% and 73%, and 1 with 10%. The median donor CD3+ T-cell chimerism was 97% (IQR, 87-100; range, 30-100) with 13 patients ≥95%, 10 at 74% to 92%, 1 with 40%, and 1 with 30% DC. The median donor CD56+ NK cell chimerism was 97.5% (IQR, 87.75-100; range, 30-100; n = 16) with 10 ≥95% and 6 between 30% and 88%. There were no primary or secondary GFs. None received secondary cellular treatments such as DLI, stem cell boosts, or second transplants. None needed precocious tapering of immunosuppressive agents due to decline in myeloid and lymphocyte DC until d+180 (Figure 3; supplemental Figure 1).
Figure 3.
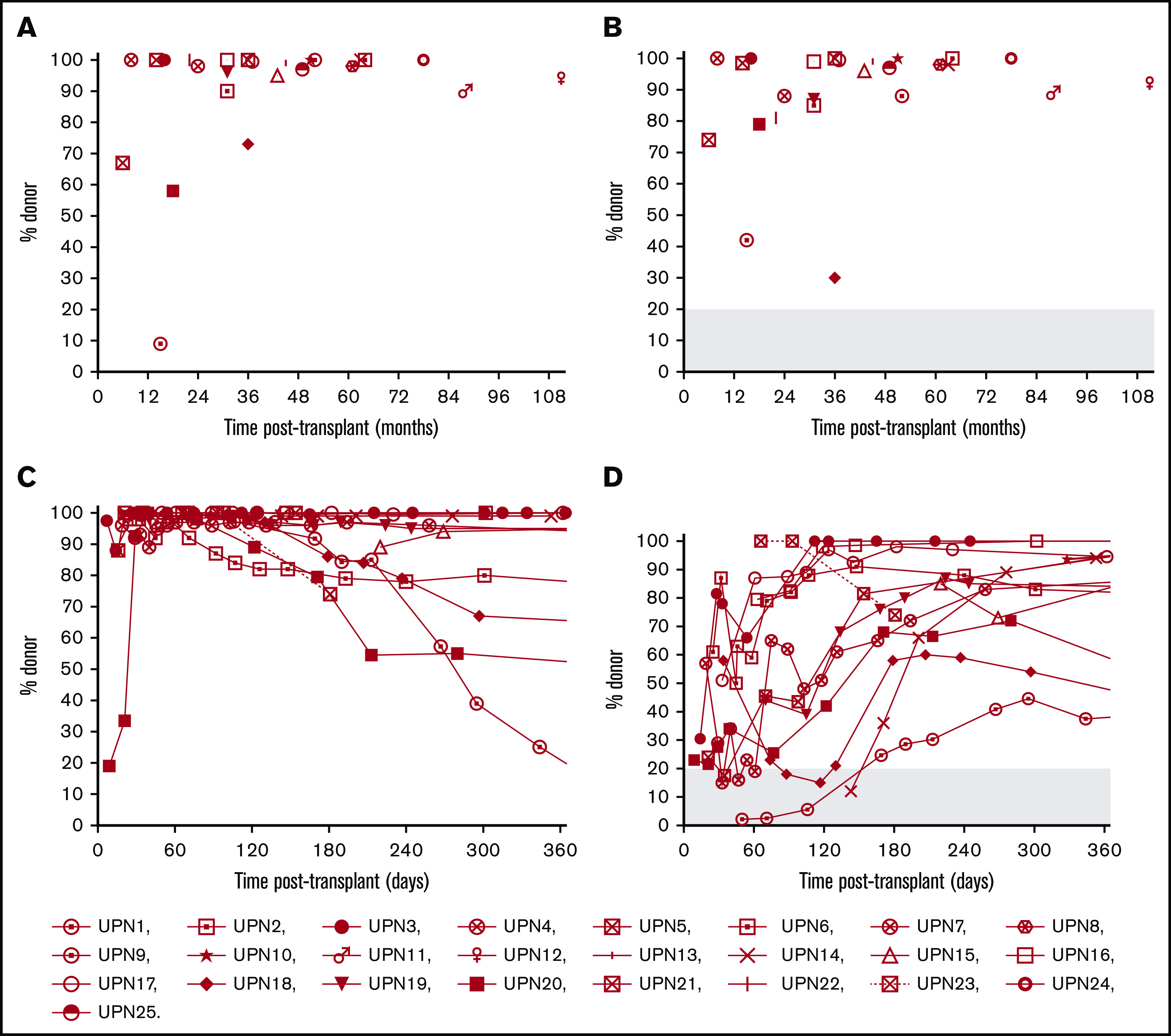
DC analysis in peripheral blood. (A) Percentages of donor CD15+ neutrophils at last follow-up (n = 25). (B) Percentages of donor CD3+ T cells at last follow-up (n = 25). (C) Longitudinal analysis of DC in CD15+ neutrophils in the first year after HSCT (n = 14). UPN9 with continuous decrease of neutrophil DC. (D) Longitudinal analysis of DC in CD3+ T cells in the first year after HSCT (n = 14). UPN9 with continuous increase of CD3+ T-cell DC. CD3+ T-cell DC <20% after 6 months (gray area) increases the risk for relapse of HLH.34 Each symbol depicts 1 patient.
GHVD disease, immune reconstitution, complications after HSCT, and clinical outcome
GHVD.
Forty percent (n = 10) of patients developed grade I to II acute GHVD, affecting mainly the skin and responding well to topical and/or systemic corticosteroids. None developed grade III to IV GHVD (Table 2). UPN3 experienced acute relapsing grade II acute GHVD of the skin. He responded well to extracorporeal photopheresis, topical steroid, sirolimus, and ruxolitinib combination treatment that could be reduced to ruxolitinib monotherapy. UPN17 developed localized lichenoid GHVD, which responded favorably to mammalian target of rapamycin inhibitor treatment and topical steroids, which could be stopped successfully. The other 24 patients became independent of immunosuppression within 4 to 10 months’ post-HSCT. All patients, except UPN3 who is currently been treated with waning ruxolitinib doses, recovered with naive CD4+ T cells reaching normal age-related percentiles and normalized B-cell function (data not shown).
Clinical outcome.
At last follow-up (median, 36 months; range, 8-111 months), all 25 patients were alive and disease free (Figure 4). None developed HLH relapses or progressive CNS disease. Preexisting neurological impairment resolved (UPN13) or remained unchanged (UPN4 and UPN14).
Figure 4.

OS/EFS and chronic GHVD-free survival. The OS/EFS (red line) and 1-year OS/EFS probabilities (100%) and the chronic GHVD-free survival (blue line) and 1-year GFS probability (95% confidence interval, 68.12-99.24) are shown.
Two patients with STXBP2 mutations have persistent mild gastrointestinal dysfunction with no need for parenteral nutrition despite the absence of GHVD. UPN8 developed asymptomatic Hashimoto’s thyroiditis. Patient UPN25 with HLH of unsolved molecular origin (supplemental data C) developed knee arthritis that has responded well to intra-articular steroid injections. She has remained free of HLH and joint inflammation as of publication time. Steroid-induced avascular bone necrosis was observed in 1 patient (UPN2). His quality of life has normalized after bilateral endoprosthetic hip surgery, with normal pain-free walking.
Eleven patients developed a total of 18 episodes of relevant infectious reactivations/complications: Epstein-Barr virus (n = 4), bacillus Calmette-Guérin infection (n = 2), BK virus (n = 2), rhinovirus (n = 2), adenovirus (n = 1), HHV-6 (n = 1), metapneumovirus (n = 1), and fungal (n = 1) as well as bacterial infection, including central line (n = 2), vulva (n = 1), and urinary tract (n = 1). All infectious episodes resolved after specific treatment and/or reduction of immunosuppression (Table 2).
Discussion
In the past 12 years, several studies have investigated the effect of RIC/reduced-toxicity regimens and T-cell replete HSCT in patients with HLH (supplemental Table 1). Despite improved overall outcomes compared with MAC regimens, several complications were encountered, including primary/secondary GF, HLH relapses due to waning DC, and increased incidence of severe acute and chronic GVHD, mainly after DLI administration to prevent imminent GF.12,29,31,45-47
In early retrospective single-center studies, melphalan-fludarabine–based RIC regimens applied mainly in the context of HLA-identical HSCT achieved OS rates between 80% and 89%; however, high rates of mixed DC12,31,46 and GF in up to 27% of patients were observed.45
A reduced-toxicity regimen replacing melphalan with treosulfan in HLA-compatible transplants including cords achieved an OS rate of 44%.47 Treosulfan (mainly combined with thiotepa) was administered in a German multicenter study infusing primarily HLA-compatible grafts from largely MRD/MUD donors. The OS and EFS rates improved to 100% and 89%, respectively, but the rate of secondary cellular treatments remained considerable (42%).29
Finally, in 2018, a prospective US multicenter study investigated 34 treated patients with HLH by using melphalan-fludarabine/RIC conditioning combined with intermediate-dose alemtuzumab.48 The 18-month OS was 68%, with mixed DC in 65%, grade II to IV acute GHVD in 26.5%, and chronic GHVD in 32.9% of patients. The proportion of patients with HLH who were alive with sustained engraftment without needing secondary cellular therapies was 41.2% (supplemental Table 1).
In the current multicenter study led by the IEWP EBMT, we administered a busulfan-based RIC protocol and T-cell replete HLA-identical transplants based on excellent outcome results with this regimen in high-risk patients with CGD.36,49,50 HLH diagnostics, pretreatment findings, and remission status of the current HLH cohort were similar to previous studies.26,29,31,48 Unintentionally, no perforin-, STX11-, or XIAP-deficient patients were among the study population (supplemental Table 1). The diagnosis of HLH was based on proven HLH episodes in 19 patients and according to family history in 3 patients. Two patients with X-linked lymphoproliferative syndrome 1 presented atypically with acute demyelinating encephalomyelitis or B-cell lymphoma before transplant51 and another with Chédiak-Higashi syndrome with neurological symptoms. Collection and treatment of patients in this study led to reproducible results on the multicenter level after adequate follow-up, which is in agreement with the recently postulated criteria for transplant studies in HLH.48 After a median follow-up of 36 months post-HSCT, we have not encountered GF or systemic/CNS HLH relapses. Satisfactory myeloid and CD3+ T-cell DC were achieved with no requirement for additional cellular therapies, including DLI. The 3-year OS/EFS/chronic GHVD–free survival rates were excellent with 100%/100%/95% (CI, 68.12-99.24), respectively (Figure 4).
Our aim was to reduce the previously observed GF rate of 5% with this RIC regimen36 without adding other alkylating myeloablative agents. For this purpose, we modified our previous target range of busulfan toward the upper part of the submyeloablative busulfan exposure range (45-65 mg/L × h).37,38,52 Previous reports showed that myeloablative and toxic properties of busulfan depended on the cAUC, rendering the total administered busulfan dose irrelevant.36,37,52,53 Here, we observed a wide range of total busulfan doses to achieve the target cAUC (Figure 1; Table 1), indicating that real-time therapeutic drug monitoring was important in this protocol. Without real-time therapeutic drug monitoring, full-dose busulfan might have resulted in toxic exposures in some patients with slow drug clearance and insufficient myeloablation in those with rapid drug clearance.52
Fully myeloablative busulfan was previously shown to correspond with a cAUC of 80 to 100 mg/L × h.41 Bartelink et al37 suggested that a cAUC of 78 to 101 mg/L × h was the optimal target for any patient undergoing HSCT. However, the cohort of Bartelink et al was heterogeneous and included children with malignant diseases. We deliberately targeted busulfan to lower exposures to reduce short-term and gonadal toxicity and to ensure donor cell engraftment in fragile patients with HLH. The approach to target busulfan within the described submyeloablative exposure was based on in vitro experiments by Hassan41 and on our previous clinical experience in patients with CGD.36 Some testable male adolescents in the former CGD cohort had exhibited preserved fertility while achieving robust myeloid engraftment (Peter Shaw, Westmead Hospital Sydney and Rachel Hough, University College Hospital London, personal communication by telephone and e-mail, 14 November 2014 and 2 June 2015), indicating a reduced gonadal toxicity in male adolescents receiving this RIC regimen. Whether this finding is also transposable to female recipients of this RIC regimen is currently unclear. We believe that long-term toxicity studies on oocyte/sperm viability after similar submyeloablative busulfan exposures compared with treosulfan and melphalan are mandatory.54
The excellent engraftment presented here has recently been confirmed in a retrospective series of 12 patients with HLH, including 2 patients with perforin deficiency (supplemental Table 1). Richards et al administered once-daily busulfan, fludarabine, and serotherapy and infused T-cell replete MSD/MUD PBSC donor transplants as well as UCBs.55 They performed pharmacokinetic analyses without analyzing the cAUC of busulfan. Retrospective estimation of the busulfan exposure (Stephanie Richards, Department of Allergy and Immunology, The Royal Children’s Hospital, personal communication by e-mail 12 May 2019) of their cohort indicated that the majority of their patients had received busulfan exposures (cAUC, 45-65 mg/L × h) similar to that of the current study patients. In addition to fludarabine, they administered higher dose alemtuzumab (up to 3 mg/kg) not later than d-10 before HSCT. They reported a robust DC >95% at 1 year (one exception was 82%) and did not administer DLI or stem cell boost infusions. The OS and EFS of their cohort was 83% and 83%, respectively. Two patients died of infections early after UCB transplantation.55
Unexpectedly, the major drawback of our RIC protocol was a rather high rate of SOS22,25,30 although the overall toxicity of the protocol was low. Initiated at early clinical signs, therapeutic DF administration was successful and led to a full resolution of SOS in all affected patients (Table 2).42,43 Most at risk for developing SOS were infants <1 year of age (6 of 8 [75%]) who had not received DF prophylaxis. Two of these were formerly premature infants (UPN11 and UPN15) (Figure 1; Tables 1 and 2). A prospective study found that DF prophylaxis in children achieved a 40% reduction of the SOS incidence, whereas the SOS-associated mortality did not differ compared with DF-treated control subjects.40 It is noteworthy that no SOS cases were reported in the cohort of Richards et al,55 who had used a once-daily busulfan protocol. Previous reports indicate that the frequency of busulfan administration per day did not affect the SOS incidence and severity in mainly malignant diseases,56,57 but it is conceivable that once-daily administration34,58 could further reduce the SOS incidence in infants with HLH. Additional preventive measures to reduce the risk of SOS of our regimen could include decreasing the cAUC to 55 to 60 mg/L × h and/or the application of a DF prophylaxis in all infants with HLH.40,59,60
The persistence of host CD3+ T cells after RIC regimens and viral reactivations soon after HSCT are both considered risk factors for the recurrence of HLH.34,61 Hartz et al showed that the percentage of DC, particularly a CD3+ T-cell DC >20% after day +180 post-HSCT, was essential for disease control in primary HLH.34 This is in accordance with animal studies of perforin knockout mice.62 The investigation of Hartz et al included CD3+ T-cell or full blood but not CD15+ myeloid DC analysis.34 Although a minimal threshold level of donor-derived CD3+ T cells was proposed, the minimal level of CD15+ myeloid DC to achieve long-term cure of HLH remains to be determined. Because stable myeloid DC is a prerequisite for long-term CD3+ T-cell production of donor origin, we were encouraged by the overall stable myeloid DC obtained by using this regimen (Figure 3A). Regular analysis of both CD3+ T-cell and CD15+ neutrophil DC in our patients helped us to avoid premature reduction of immunosuppression and infusion of DLI or administration of other secondary cellular therapies.29,48,63 Because of excellent CD15+ myeloid DC results, we could accept an initially mixed donor CD3+ T-cell chimerism in some of our patients (UPN9, UPN18, and UPN23); neither GF nor HLH relapses were encountered. All viral reactivations were treatable and not associated with HLH relapses (Table 2). Regular lineage-specific DC analyses showed that patients with initially mixed CD3+ T-cell DC, CD3+ T cells of donor origin gradually increased over the first year posttransplant, mainly between day +30 and day +180 but also later (Figure 3D). The fastest gains of CD3+ T-cell DC occurred after cessation of MMF and CSA mainly by rising de novo production of naive donor-derived CD3+ T cells.
Except for UPN9, continuous increases in CD3+ T-cell DC and/or stable CD15+ neutrophil DC (Table 2; Figure 3) over time were standard. A very slow decline of CD15+ DC but steadily increasing CD3+ T-cell DC over time were observed in UPN9, with 9% CD15+ neutrophils and 42% CD3+ T cells at last follow-up (15 months post-HSCT). It is noteworthy that even though UPN9 is considered cured of HLH, we cannot completely exclude a further decline of myeloid DC in the future.
Lymphocyte depletion via serotherapy is essential for the prevention of higher grade acute/chronic GVHD. The serotherapy concept of this protocol presented here had previously been shown to successfully prevent acute/chronic GHVD; there was no severe grade II or higher acute GHVD and a 4% incidence of limited chronic GHVD in patients with CGD transplanted with HLA-compatible donors.36,49,50 Considering the relative half-life of alemtuzumab, our dosing regimen (median, 0.6 mg/kg total; d-8 to d-6) was safe and shared properties of the intermediate dosing scheme of Marsh et al.63 Marsh et al showed that alemtuzumab dosing and timing substantially influenced DC as well as acute/chronic GHVD incidence and severity.31,35,63 Intermediate dosing of alemtuzumab (1 mg/kg; d-14 to d-10) led to lower mixed DC rates (31%) than proximal dosing (1 mg/kg; d-8/9 to d-4/5) (72%) or distal dosing (≥2 mg/kg; d-22 to d-19) (75%).35 Given the variability of inflammation and lymphocyte count before transplant, a more precise pharmacokinetic approach toward pretransplant alemtuzumab levels could be helpful to further improve or individualize dosing64 in the future.
Another potential cause for the low prevalence of acute/chronic GHVD observed in the current study was the preference of HSC from bone marrow/CB over PBSC sources. The introduction of T cell–depleted PBSCs with CD3+ T-cell addback in unrelated single allele mismatch PBSC donors may have been additionally GHVD preventive.39 The latter technique has been applied in 3 patients (UPN17, UPN21, and UPN25), adjusting the CD3+ T-cell content of their grafts to average bone marrow CD3+ T-cell contents. Interestingly, the sole event of reversible limited chronic GHVD still occurred in a patient (UPN17) treated with this technique after a 9/10-HLA–matched PBSC transplant. This outcome indicates that patients with single allele mismatch PBSC transplants could benefit from more rigorous in vivo and in vitro CD3+ T-cell depletion techniques; however, this would be at the expense of risking insufficient engraftment due to insufficient graft-versus-marrow effect.
This multicenter investigation found high cure rates and successful prevention of GF and GVHD in patients with various types of HLH. Sufficient myeloid and CD3+ T-cell donor cell engraftment was achieved without applying secondary cellular treatments. Targeted busulfan administration within the analyzed range achieved curative myeloid DC, rendering the administration of other alkylating agents unnecessary. The reduction of the cAUC of busulfan by ∼20% to 35% was sufficiently myeloablative, and the overall toxicity was low. Nevertheless, some infants developed SOS that was successfully treated with DF. Lower cumulative busulfan exposures, modification of busulfan application (once-daily infusion), and DF prophylaxis in infants could further reduce the risk for SOS. After sufficient follow-up, satisfactory donor CD15+ myeloid, CD3+ T-cell, and NK cell DC were achieved, and relapses of HLH in blood and CNS were successfully prevented.65 Because no perforin-, STX11-, and XIAP-deficient patients were treated with the current RIC regimen, more experience with this protocol in these forms of HLH is mandatory. The analysis of long-term cure and fertility should be objectives of future studies.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their families for their cooperation, the nurses and the clinicians for the care of the patients, and Lennart Opitz for his assistance. They also thank Peter Shaw, Westmead Hospital Sydney, and Rachel Hough, University College Hospital London, for communicating the fertility and paternity analyses of previous CGD patients treated with the same RIC regimen. They thank Stephanie Richards, Department of Allergy and Immunology, The Royal Children’s Hospital, for sharing busulfan pharmacokinetic data from her cohort.
Footnotes
Requests for data may be submitted to the corresponding author (Tayfun Güngör; e-mail: tayfun.guengoer@kispi.uzh.ch).
Authorship
Contribution: M.F., J.P.S., M.M.H.-H., and T.G. prepared the manuscript; M.M.H.-H., M.H.A., F.H., and V.H. performed chimerism analyses; M.F., M.M.H.-H., J.P.S., T.G., M.H.A., K.L., B.G., C.G.S., R.F.W., S.P., U.Z., K.K., K.R., P.V., and A.F. contributed to data collection and patients’ care; B.V., J.P.S., S.E., C.G.S., and R.F.W. performed degranulation assays and genetic analyses and contributed to data analyses; M.H. and D.M. performed pharmacokinetic analysis for busulfan; A.C.L., M.H.A., and A.R.G. have approved the protocol for the IEWP EBMT; T.G. designed the protocol and initiated the collaboration; and all authors critically read and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tayfun Güngör, Department of Immunology, Hematology, Oncology and SCT, University Children’s Hospital, Steinwiesstr 75, CH-8032 Zurich, Switzerland, e-mail: tayfun.guengoer@kispi.uzh.ch.
References
- 1.Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015;62(2):346-352. [DOI] [PubMed] [Google Scholar]
- 2.Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr Opin Allergy Clin Immunol. 2011;11(6):512-516. [DOI] [PubMed] [Google Scholar]
- 3.Bode SF, Ammann S, Al-Herz W, et al. ; Inborn Errors Working Party of the EBMT . The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pachlopnik Schmid J, Côte M, Ménager MM, et al. Inherited defects in lymphocyte cytotoxic activity [published correction appears in Immunol Rev. 2010;236(1):276]. Immunol Rev. 2010;235(1):10-23. [DOI] [PubMed] [Google Scholar]
- 5.Gayden T, Sepulveda FE, Khuong-Quang DA, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome [published correction appears in Nat Genet. 2019;51(1):196]. Nat Genet. 2018;50(12):1650-1657. [DOI] [PubMed] [Google Scholar]
- 6.Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volkmer B, Planas R, Gossweiler E, et al. Recurrent inflammatory disease caused by a heterozygous mutation in CD48. J Allergy Clin Immunol. 2019;144(5):1441-1445.e17. [DOI] [PubMed] [Google Scholar]
- 8.Lam MT, Coppola S, Krumbach OHF, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. 2019;216(12):2778-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu L, Wang J, Wei N, et al. Allogeneic hematopoietic stem-cell transplantation for adult and adolescent hemophagocytic lymphohistiocytosis: a single center analysis. Int J Hematol. 2016;104(5):628-635. [DOI] [PubMed] [Google Scholar]
- 10.Lehmberg K, Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013;160(3):275-287. [DOI] [PubMed] [Google Scholar]
- 11.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. 2008;42(suppl 2):S47-S50. [DOI] [PubMed] [Google Scholar]
- 13.Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lounder DT, Bin Q, de Min C, Jordan MB. Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv. 2019;3(1):47-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henter JI, Aricò M, Egeler RM, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH Study Group of the Histiocyte Society. Med Pediatr Oncol. 1997;28(5):342-347. [DOI] [PubMed] [Google Scholar]
- 16.Henter JI, Samuelsson-Horne A, Aricò M, et al. ; Histocyte Society . Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. [DOI] [PubMed] [Google Scholar]
- 17.Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. [DOI] [PubMed] [Google Scholar]
- 18.Mahlaoui N, Ouachée-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622-e628. [DOI] [PubMed] [Google Scholar]
- 19.Trottestam H, Horne A, Aricò M, et al. ; Histiocyte Society . Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577-4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vallurupalli M, Berliner N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood. 2019;134(21):1783-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horne A, Janka G, Maarten Egeler R, et al. ; Histiocyte Society . Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129(5):622-630. [DOI] [PubMed] [Google Scholar]
- 23.Dürken M, Horstmann M, Bieling P, et al. Improved outcome in haemophagocytic lymphohistiocytosis after bone marrow transplantation from related and unrelated donors: a single-centre experience of 12 patients. Br J Haematol. 1999;106(4):1052-1058. [DOI] [PubMed] [Google Scholar]
- 24.Baker KS, DeLaat CA, Steinbuch M, et al. Successful correction of hemophagocytic lymphohistiocytosis with related or unrelated bone marrow transplantation. Blood. 1997;89(10):3857-3863. [PubMed] [Google Scholar]
- 25.Baker KS, Filipovich AH, Gross TG, et al. Unrelated donor hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2008;42(3):175-180. [DOI] [PubMed] [Google Scholar]
- 26.Cooper N, Rao K, Gilmour K, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006;107(3):1233-1236. [DOI] [PubMed] [Google Scholar]
- 27.Cesaro S, Locatelli F, Lanino E, et al. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: a retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP). Haematologica. 2008;93(11):1694-1701. [DOI] [PubMed] [Google Scholar]
- 28.Jabado N, de Graeff-Meeder ER, Cavazzana-Calvo M, et al. Treatment of familial hemophagocytic lymphohistiocytosis with bone marrow transplantation from HLA genetically nonidentical donors. Blood. 1997;90(12):4743-4748. [PubMed] [Google Scholar]
- 29.Lehmberg K, Albert MH, Beier R, et al. Treosulfan-based conditioning regimen for children and adolescents with hemophagocytic lymphohistiocytosis. Haematologica. 2014;99(1):180-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouachée-Chardin M, Elie C, de Saint Basile G, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117(4):e743-e750. [DOI] [PubMed] [Google Scholar]
- 31.Marsh RA, Vaughn G, Kim MO, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010;116(26):5824-5831. [DOI] [PubMed] [Google Scholar]
- 32.Marsh RA, Rao K, Satwani P, et al. Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood. 2013;121(6):877-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel SA, Allewelt HA, Troy JD, et al. Durable chimerism and long-term survival after unrelated umbilical cord blood transplantation for pediatric hemophagocytic lymphohistiocytosis: a single-center experience. Biol Blood Marrow Transplant. 2017;23(10):1722-1728. [DOI] [PubMed] [Google Scholar]
- 34.Hartz B, Marsh R, Rao K, et al. The minimum required level of donor chimerism in hereditary hemophagocytic lymphohistiocytosis. Blood. 2016;127(25):3281-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marsh RA, Lane A, Mehta PA, et al. Alemtuzumab levels impact acute GVHD, mixed chimerism, and lymphocyte recovery following alemtuzumab, fludarabine, and melphalan RIC HCT. Blood. 2016;127(4):503-512. [DOI] [PubMed] [Google Scholar]
- 36.Güngör T, Teira P, Slatter M, et al. ; Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation . Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436-448. [DOI] [PubMed] [Google Scholar]
- 37.Bartelink IH, Lalmohamed A, van Reij EM, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol. 2016;3(11):e526-e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartelink IH, Boelens JJ, Bredius RG, et al. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet. 2012;51(5):331-345. [DOI] [PubMed] [Google Scholar]
- 39.Seitz CM, Eyrich M, Greil J, et al. Favorable immune recovery and low rate of GvHD in children transplanted with partially T cell-depleted PBSC grafts. Bone Marrow Transplant. 2019;54(1):53-62. [DOI] [PubMed] [Google Scholar]
- 40.Corbacioglu S, Cesaro S, Faraci M, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379(9823):1301-1309. [DOI] [PubMed] [Google Scholar]
- 41.Hassan Z. Optimal approach to prevent veno-occlusive disease following hematopoietic stem cell transplantation in children. Pediatr Transplant. 2010;14(6):683-687. [DOI] [PubMed] [Google Scholar]
- 42.Haussmann U, Fischer J, Eber S, Scherer F, Seger R, Gungor T. Hepatic veno-occlusive disease in pediatric stem cell transplantation: impact of pre-emptive antithrombin III replacement and combined antithrombin III/defibrotide therapy. Haematologica. 2006;91(6):795-800. [PubMed] [Google Scholar]
- 43.Corbacioglu S, Greil J, Peters C, et al. Defibrotide in the treatment of children with veno-occlusive disease (VOD): a retrospective multicentre study demonstrates therapeutic efficacy upon early intervention [published correction appears in Bone Marrow Transplant. 2004;33(6):673]. Bone Marrow Transplant. 2004;33(2):189-195. [DOI] [PubMed] [Google Scholar]
- 44.Corbacioglu S, Carreras E, Ansari M, et al. Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in pediatric patients: a new classification from the European society for blood and marrow transplantation. Bone Marrow Transplant. 2018;53(2):138-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishi M, Nishimura R, Suzuki N, et al. Reduced-intensity conditioning in unrelated donor cord blood transplantation for familial hemophagocytic lymphohistiocytosis. Am J Hematol. 2012;87(6):637-639. [DOI] [PubMed] [Google Scholar]
- 46.Hamidieh AA, Pourpak Z, Hashemi S, et al. Fludarabine-based reduced-intensity conditioning regimen for hematopoietic stem cell transplantation in primary hemophagocytic lymphohistiocytosis. Eur J Haematol. 2014;92(4):331-336. [DOI] [PubMed] [Google Scholar]
- 47.Slatter MA, Rao K, Abd Hamid IJ, et al. Treosulfan and fludarabine conditioning for hematopoietic stem cell transplantation in children with primary immunodeficiency: UK experience. Biol Blood Marrow Transplant. 2018;24(3):529-536. [DOI] [PubMed] [Google Scholar]
- 48.Allen CE, Marsh R, Dawson P, et al. Reduced-intensity conditioning for hematopoietic cell transplant for HLH and primary immune deficiencies. Blood. 2018;132(13):1438-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freudenberg F, Wintergerst U, Roesen-Wolff A, et al. Therapeutic strategy in p47-phox deficient chronic granulomatous disease presenting as inflammatory bowel disease. J Allergy Clin Immunol. 2010;125(4):943-946.e1. [DOI] [PubMed] [Google Scholar]
- 50.Güngör T, Halter J, Klink A, et al. Successful low toxicity hematopoietic stem cell transplantation for high-risk adult chronic granulomatous disease patients. Transplantation. 2005;79(11):1596-1606. [DOI] [PubMed] [Google Scholar]
- 51.Lucchini G, Marsh R, Gilmour K, et al. Treatment dilemmas in asymptomatic children with primary hemophagocytic lymphohistiocytosis. Blood. 2018;132(19):2088-2096. [DOI] [PubMed] [Google Scholar]
- 52.Malär R, Sjöö F, Rentsch K, Hassan M, Güngör T. Therapeutic drug monitoring is essential for intravenous busulfan therapy in pediatric hematopoietic stem cell recipients. Pediatr Transplant. 2011;15(6):580-588. [DOI] [PubMed] [Google Scholar]
- 53.Hassan Z, Hellström-Lindberg E, Alsadi S, Edgren M, Hägglund H, Hassan M. The effect of modulation of glutathione cellular content on busulphan-induced cytotoxicity on hematopoietic cells in vitro and in vivo. Bone Marrow Transplant. 2002;30(3):141-147. [DOI] [PubMed] [Google Scholar]
- 54.Faraci M, Diesch T, Labopin M, et al. ; Pediatric and Transplant Complications Working Parties of the European Society for Blood and Marrow Transplantation . Gonadal function after busulfan compared with treosulfan in children and adolescents undergoing allogeneic hematopoietic stem cell transplant. Biol Blood Marrow Transplant. 2019;25(9):1786-1791. [DOI] [PubMed] [Google Scholar]
- 55.Richards S, Choo S, Mechinaud F, Cole T. Single centre results of targeted busulphan, fludarabine and serotherapy conditioning in haematopoietic stem cell transplantation for haemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2018;53(6):784-786. [DOI] [PubMed] [Google Scholar]
- 56.Fernandez HF, Tran HT, Albrecht F, Lennon S, Caldera H, Goodman MS. Evaluation of safety and pharmacokinetics of administering intravenous busulfan in a twice-daily or daily schedule to patients with advanced hematologic malignant disease undergoing stem cell transplantation. Biol Blood Marrow Transplant. 2002;8(9):486-492. [DOI] [PubMed] [Google Scholar]
- 57.Ryu SG, Lee JH, Choi SJ, et al. Randomized comparison of four-times-daily versus once-daily intravenous busulfan in conditioning therapy for hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2007;13(9):1095-1105. [DOI] [PubMed] [Google Scholar]
- 58.de Lima M, Couriel D, Thall PF, et al. Once-daily intravenous busulfan and fludarabine: clinical and pharmacokinetic results of a myeloablative, reduced-toxicity conditioning regimen for allogeneic stem cell transplantation in AML and MDS. Blood. 2004;104(3):857-864. [DOI] [PubMed] [Google Scholar]
- 59.Myers AL, Kawedia JD, Champlin RE, et al. Clarifying busulfan metabolism and drug interactions to support new therapeutic drug monitoring strategies: a comprehensive review. Expert Opin Drug Metab Toxicol. 2017;13(9):901-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mohty M, Malard F, Abecasis M, et al. Prophylactic, preemptive, and curative treatment for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a position statement from an international expert group. Bone Marrow Transplant. 2020;55(3):485-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nikiforow S. The role of hematopoietic stem cell transplantation in treatment of hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 2015;29(5):943-959. [DOI] [PubMed] [Google Scholar]
- 62.Terrell CE, Jordan MB. Mixed hematopoietic or T-cell chimerism above a minimal threshold restores perforin-dependent immune regulation in perforin-deficient mice. Blood. 2013;122(15):2618-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsh RA, Kim MO, Liu C, et al. An intermediate alemtuzumab schedule reduces the incidence of mixed chimerism following reduced-intensity conditioning hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Biol Blood Marrow Transplant. 2013;19(11):1625-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marsh RA, Fukuda T, Emoto C, et al. Pretransplant absolute lymphocyte counts impact the pharmacokinetics of alemtuzumab. Biol Blood Marrow Transplant. 2017;23(4):635-641. [DOI] [PubMed] [Google Scholar]
- 65.Linz U, Hupert M, Santiago-Schübel B, Wien S, Stab J, Wagner S. Transport of treosulfan and temozolomide across an in-vitro blood-brain barrier model. Anticancer Drugs. 2015;26(7):728-736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.