

Key Points
We have developed a new anti-human C6 mAb that could have therapeutic value.
This mAb inhibits MAC formation by blocking both free C6 and C6 in the C5b6 complex.
Abstract
Membrane attack complexes (MACs; C5b-9) assembled after complement activation can directly injure self-tissues, leading to various diseases. Eculizumab, a monoclonal antibody (mAb) against complement component C5, is being used in the clinic to treat diseases in which MAC-mediated tissue damage is a primary cause. However, C5 is not a selective target for MAC assembly inhibition, and some patients respond incompletely or not at all to the eculizumab treatment. Therefore, C6, the next essential component in the terminal pathway of complement activation, may be an alternative target for the selective inhibition of MAC formation. Surprisingly, few reports describe a functional blockade of C6 using a specific mAb. Here, we report the development of an anti-human C6 mAb (clone 1C9) that recognizes C6 both in free circulation and within C5b6 complexes. This mAb blocked C7 binding to C5b6 complexes and consequently inhibited MAC formation and protected affected paroxysmal nocturnal hemoglobinuria patient red blood cells from MAC-mediated damage in vitro. In addition, this mAb cross-reacts with rhesus monkey but not mouse complement C6. Finally, 1C9 significantly reduced human complement–mediated intravascular hemolysis in vivo in a mouse model. These results suggest that the anti-C6 mAb holds promise as a new therapeutic agent that selectively targets MAC for many complement-mediated pathological conditions.
Visual Abstract

Introduction
Complement is a key component of the innate immune system and a critical aspect of infection control.1 Under normal conditions, most complement components exist in the blood as zymogens. However, in response to an invasive pathogen or certain other circumstances, complement is activated through 1 of 3 major pathways: the classical, alternative, or lectin pathway. The cascades of all 3 pathways merge at the terminal pathway, in which C5 is cleaved to release C5a, a potent anaphylatoxin that recruits and activates many cells, especially leukocytes, to promote local inflammation. Simultaneously, C5b, the larger product of C5 cleavage, binds to a single C6 unit in the surrounding milieu to form the C5b6 complex, which then binds sequentially to C7, C8, and multiple copies of C9 and thus assembles a membrane attack complex (MAC) on the target cell membrane.2 The MAC (C5b-9) forms a functional pore on the cell membrane via a ring-like structure and thus directly injures or even kills the target cell (eg, pathogen) via osmotic damage or lysis. However, under certain conditions, excessive MAC formation on self-cells can damage host tissues, leading to diseases such as paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and myasthenia gravis.3 Therefore, the inhibition of MAC formation is a valid approach to the treatment of diseases in which the MAC plays an integral role in pathogenesis.
To date, eculizumab, an anti-C5 monoclonal antibody (mAb), has been used successfully to treat many complement-mediated diseases and is officially approved as treatment for PNH,4 aHUS,5 myasthenia gravis,6 and neuromyelitis optica spectrum disorders.7 Mechanistically, eculizumab binds to quiescent C5, which is present in peripheral blood at an approximate concentration of 75 μg/mL, and thus prevents its activation and subsequent MAC formation. However, the effects of a C5 blockage are not limited to the inhibition of MAC formation; rather, C5a production is also suppressed,8 with potentially beneficial or detrimental effects on patients, because C5a-initiated inflammation could battle infection or worsen disease. In addition to the relatively high blood concentration of C5, this complement component also has a rapid turnover rate. Consequently, large quantities of eculizumab (as high as 1 g per patient per dose) are required to inhibit MAC formation in patients. Additionally, clinical observations have revealed that some patients are incompletely or not at all responsive to eculizumab treatment because of residual C5 activity9 or certain polymorphisms within the C5 gene.10 Therefore, a reagent that selectively inhibits MAC formation by targeting another component within the terminal pathway may provide a reasonable alternative or supplement to eculizumab and other C5-targeted reagents currently under extensive clinical development.
After C5, C6 is the next essential component in the MAC assembly process. This component is present in the blood at an approximate concentration of 45 to 60 μg/mL.11 Theoretically, C6 inhibition should prevent MAC formation without affecting C5 activation or C5a release. Therefore, this component should be another attractive target in the treatment of MAC-mediated diseases. Here, we describe the development of mAbs against this component of the terminal complement pathway. We identified a resulting anti-C6 mAb, clone 1C9, that exhibited potent inhibitory activity against MAC-mediated cell damage (ie, hemolysis). We further characterized this anti-C6 mAb using an enzyme-linked immunosorbent assay (ELISA) and surface plasmon resonance (SPR) analysis and determined that it binds to C6 both in the free circulation and within the C5b6 complex to prevent and inhibit MAC formation. We also evaluated the ability of this anti-C6 mAb to inhibit MAC-mediated hemolysis both in vitro and in vivo. Taken together, our results indicate that this newly developed anti-C6 mAb demonstrates promise as a potential therapeutic agent for diseases in which MACs are integrally involved in the pathogenesis.
Methods
Complement reagents
All complement-related reagents, including purified human C5b6, complement components C6 and C7, pooled normal human serum (NHS), and C6- or C7-depleted human serum, were purchased from Complement Technology, Inc. (Tylor, TX). Rhesus monkey sera were provided by the School of Veterinary Medicine, University of California Davis (Davis, CA).
Development of anti-C6 hybridomas
Wild-type (WT) C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) were immunized with purified human C6 protein (Complement Technology, Inc.). Hybridomas were developed according to an established protocol.12 Clone 1C9 was identified by conventional ELISA screening of the supernatants of resultant hybridoma cultures. All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of Cleveland Clinic.
Determination of anti-C6 mAb binding to free C6 and C6 in C5b6 complexes by ELISA
ELISA plate wells were coated with 50 µL of purified C5b6 complexes, C6, or C5 in phosphate-buffered saline (PBS; 1 nM) and incubated overnight at 4°C. The plate was then washed and blocked with 1 mg/mL of bovine serum albumin in PBS for 1 hour at room temperature. Next, different dilutions of the anti-C6 mAb (1 mg/mL) were added to each well and incubated at room temperature for another 2 hours. After washing, a horseradish peroxidase–conjugated goat anti-mouse immunoglobulin G (IgG) antibody was added, and the plate was incubated for 1 hour before development.
Determination of the affinity of 1C9 for C6 or C5b6 by SPR
SPR studies were performed using a BIAcore T200 device (GE Healthcare, Marlborough, MA) at 25°C. The binding assays were conducted in PBSP+ buffer, which contained 20 mM of phosphate (pH, 7.4), 137 mM of sodium chloride (NaCl), and 2.7 mM of potassium chloride, plus 0.05% surfactant P20. Next, mAb clone 1C9 at a concentration of 50 μg/mL in 10 mM of sodium acetate (pH, 4.5) was injected at a rate of 10 µL per minute for 15 seconds and amine coupled to S-series CM5 sensor chips (BIAcore) using a standard amine coupling kit (BIAcore). The final density of 1C9 reached 350 RU. Twofold series of C6 dilutions ranging from 1.95 to 125 nM were then injected at a rate of 30 µL per minute over surfaces on which 1C9 had been immobilized. Each data point was repeated twice, and double referencing was applied to all detected traces using BIAevaluation software (version 6). Briefly, the data were referenced by subtracting an unmodified surface to correct the binding responses for the bulk refractive index change. Next, the response from an average of blank injections was subtracted from the data values to remove any systematic artifacts. Same concentration range was applied in the binding assay of C5b6 and 1C9, except a lower density of 1C9 (40RU) was immobilized on the surface, considering the larger size of C5b6 compared with C6.
In vitro classical pathway complement-mediated hemolytic assay using normal human or rhesus monkey sera
Sheep red blood cells (RBCs; Hemostat Laboratories, Dixon, CA) were initially incubated at 37°C for 30 minutes with a 1:800 dilution of rabbit anti-sheep RBC serum (MP Biomedicals, Santa Ana, CA) in gelatin veronal buffer (GVB) with EDTA (10 mM of barbital, 145 mM of NaCl, 0.1% gelatin, and 10 mM of EDTA; pH, 7.2) for sensitization. Approximately 5 × 106 sensitized sheep RBCs (EshA) were incubated with 1% sera in the presence or absence of different concentrations of purified 1C9 IgG in GVB containing Mg++ and Ca++ (GVB++; 10 mM of barbital, 145 mM of NaCl, 0.5 mM of magnesium chloride, 0.15 mM of calcium chloride, and 0.1% gelatin; pH, 7.2 ± 0.15; Boston BioProducts, Ashland, MA) at 37°C. For negative controls, 5 mM of EDTA was added to the tubes to inhibit complement activity.
After a 5-minute incubation, the EshA were centrifuged, and the supernatants were collected. The optical density was measured at 414 nm (OD414), and the following equation was used to calculate the percentage of hemolysis: hemolysis (%) = ([A − B]/[C − B]) × 100%. Here, A, B, and C represent the OD readings of the sample with serum, the sample with serum and EDTA, and the maximum hemolysis induced by water, respectively.
In vitro alternative pathway complement-mediated hemolytic assay using normal mouse sera
Rabbit RBCs (Erabb; Hemostat Laboratories) were incubated with GVB-Mg-EGTA (5 mM of barbital, 145 mM of NaCl, 0.5 mM of magnesium chloride, 10 mM of EGTA, and 0.1% gelatin; pH, 7.2; Boston BioProducts) containing 10% freshly collected sera from WT C57BL/6 mice. The percentage of hemolysis was calculated using the equation described in the previous section.
Modified Ham’s test using PNH RBCs
A whole-blood sample was obtained from a deidentified PNH patient with consent following a Cleveland Clinic Institutional Review Board–approved protocol. The RBCs were washed and resuspended in GVB-Mg-EGTA buffer at a concentration of 2 × 108/mL. Then, the RBCs were incubated with 50% acidified NHS (pH, 6.4) in GVB-Mg-EGTA buffer at 37°C for 15 minutes in the presence of different concentrations of 1C9 (0-1500 nM) or 10 mM of EDTA (control). After incubation, the RBCs were washed again, stained with an APC-conjugated anti-human CD59 mAb (Biolgend, CA), and analyzed by flow cytometry.
Determination of the mechanism by which the anti-C6 mAb inhibits MAC formation
To determine whether our anti-C6 mAb would inhibit MAC formation by binding to free C6, we preincubated 0.1 μg of C6 with 0.2 μg/mL of mAb clone 1C9 or the same concentration of control mouse IgGs for 10 minutes at room temperature. We then combined this mixture with EshA and 1% C6-depleted sera. After a 10-minute incubation at 37°C for 10 minutes, the supernatants were collected, and hemolysis was quantitated by measuring the OD414.
To determine whether our anti-C6 mAb could inhibit MAC formation by preventing the binding of C7 to an assembled C5b6 complex, EshA were incubated with 1% C7-depleted sera at 37°C for 20 minutes to allow the formation of C5b6 on EshA. Next, anti-C6 mAb 1C9 or the same concentration of control mouse IgGs and 2 μg of purified C7 were added to the reactions. After a 20-minute incubation, the supernatants were collected, and hemolysis was quantitated by measuring the OD414.
Cross-reactivity assays
To determine the potential cross-reactivity of clone 1C9 with monkey C6, which would have implications for future clinical development, we added 1C9 to sera from different individual rhesus monkeys and performed the above-described EshA-based hemolytic assay. To determine the potential cross-reactivity of 1C9 to mouse C6, 1C9 was added to sera from different individual WT C57BL/6 mice, and the above-described Erabb-based alternative pathway hemolytic assay was performed to account for the weak hemolytic activity associated with the mouse classical complement pathway.
In vivo intravascular hemolytic studies
We induced complement-mediated intravascular hemolysis by injecting NHS IV into WT mice according to a previously published protocol.13 Briefly, each WT C57BL/6 mouse (6-8 week male and female mice with body weights ranging from 18-28 g) was weighed and injected IV with NHS and 1C9 or the same concentration of control mouse IgG via the tail vein. The required amount of NHS per mouse was determined by calculating 1.5 µL/g body weight, and 4 μg of 1C9 or mouse IgGs were used per each µL of NHS, with a total volume of 200 µL per mouse in sterile PBS. Ten minutes after injection, blood samples were collected via retroorbital bleeding into tubes containing 10 µL of 0.5-M EDTA in each tube to stop complement activation and prevent coagulation. After centrifugation, plasma samples were collected, and the extent of NHS-induced hemolysis in each was measured by quantitating the OD414 as a spectrophotometric measure of hemoglobin release.
Results
Development of a mouse anti-human C6 mAb
After immunizing WT mice with purified human C6 proteins, we developed hybridomas according to a conventional protocol14 after confirming high titers of anti-C6 IgGs in sera from the immunized mice (data not shown). ELISA screening of the culture supernatants from ∼600 hybridomas identified 20 clones that yielded positive signals at least fivefold above those of the negative controls (culture media alone). We then subjected these culture supernatants to a rapid EshA-based hemolytic assay and evaluated the subsequent presence of RBC pellets. Only 1 mAb, clone 1C9, seemed to block the function of C6, as indicated by the presence of an RBC pellet after incubation and centrifugation (data not shown).
Functional assay to detect the anti-C6 mAb 1C9-mediated inhibition of MAC-mediated hemolysis
Encouraged by the positive results of the initial screening experiments, we expanded the 1C9 clone and purified the IgGs from bulk culture supernatants by affinity chromatography using a protein G column and performed a complement-mediated hemolytic assay using EshA and NHS in the presence of 0 to 30 nM of 1C9 to test the efficacy of 1C9 against MAC-mediated RBC damage. The results demonstrated that 1C9 suppresses MAC-mediated hemolysis in a concentration-dependent manner (Figure 1).
Figure 1.

The anti-human C6 mAb 1C9 inhibits complement-mediated hemolysis in a dose-dependent manner. Different concentrations of 1C9 (0-30 nM) were assessed in a conventional complement-mediated hemolytic assay containing sensitized sheep RBCs and 1% normal human serum in GVB containing Ca++ and Mg++. Data represent mean ± standard deviation. Representative results from 3 independent experiments.
Determination of 1C9 binding to free C6 and C6 within C5b6 complexes by ELISA
Potentially, 1C9 could inhibit MAC formation by binding to free C6 and/or C6 within C5b6 complexes. To elucidate the mechanism by which 1C9 inhibits MAC formation, we performed an ELISA to examine the binding of 1C9 to purified C6, C5b6 complexes, and C5 protein (control). Notably, 1C9 (1 mg/mL) detected both free C6 and C6 within C5b6 complexes at dilution factors as low as 1:27 000 (Figure 2). As expected, no cross-reaction with C5, the other component of the C5b6 complex, was observed, even at the highest tested concentration, thus confirming the specific binding of 1C9 to C6 both in free circulation and within the C5b6 complex.
Figure 2.

The mAb 1C9 binds to both free C6 and C6 within C5b6 complexes. An ELISA plate was coated with 1 nM of purified C6, C5b6, or C5 and then incubated with different dilutions of 1C9 (maximum, 1 μg/mL). After washing and development, the binding of 1C9 in each well was measured by reading the optical density at 450 nm (OD450). Negative controls (Ctrl) were wells to which the highest concentration of mouse IgG (1 μg/mL) was added. Data represent mean ± standard deviation in triplicate wells. Representative results from 2 independent experiments.
Determination of the affinity of 1C9 for free C6 or C6 within the C5b6 complex by SPR
Next, we quantitated the affinity of 1C9 for free C6 or C6 within the C5b6 complex via SPR by fixing the mAb on a sensor chip and running purified C6 or C5b6 complexes in the fluid phase. The binding data revealed that C6 and 1C9 were globally fitted to a 1:1 Langmuir binding model. The on-rate of binding (Ka) for free C6 was 1.36 × 105 ± 94 M−1s−1s-1, and its off-rate (Kd) was 2.82 × 10−4 ± 1.3 × 10−6 s−1-1, which yielded a KD of 2.07 ± 0.14 nM (Figure 3A). Meanwhile, the on-rate of binding (Ka) for C5b6 was 1.82 × 105 ± 340 1s-M−1s−1, and its off-rate (Kd) was 3.47 × 10−4 ± 3.5 × 10−6 ss-−1, which yielded a KD of 1.91 ± 1.03 nM (Figure 3B).
Figure 3.
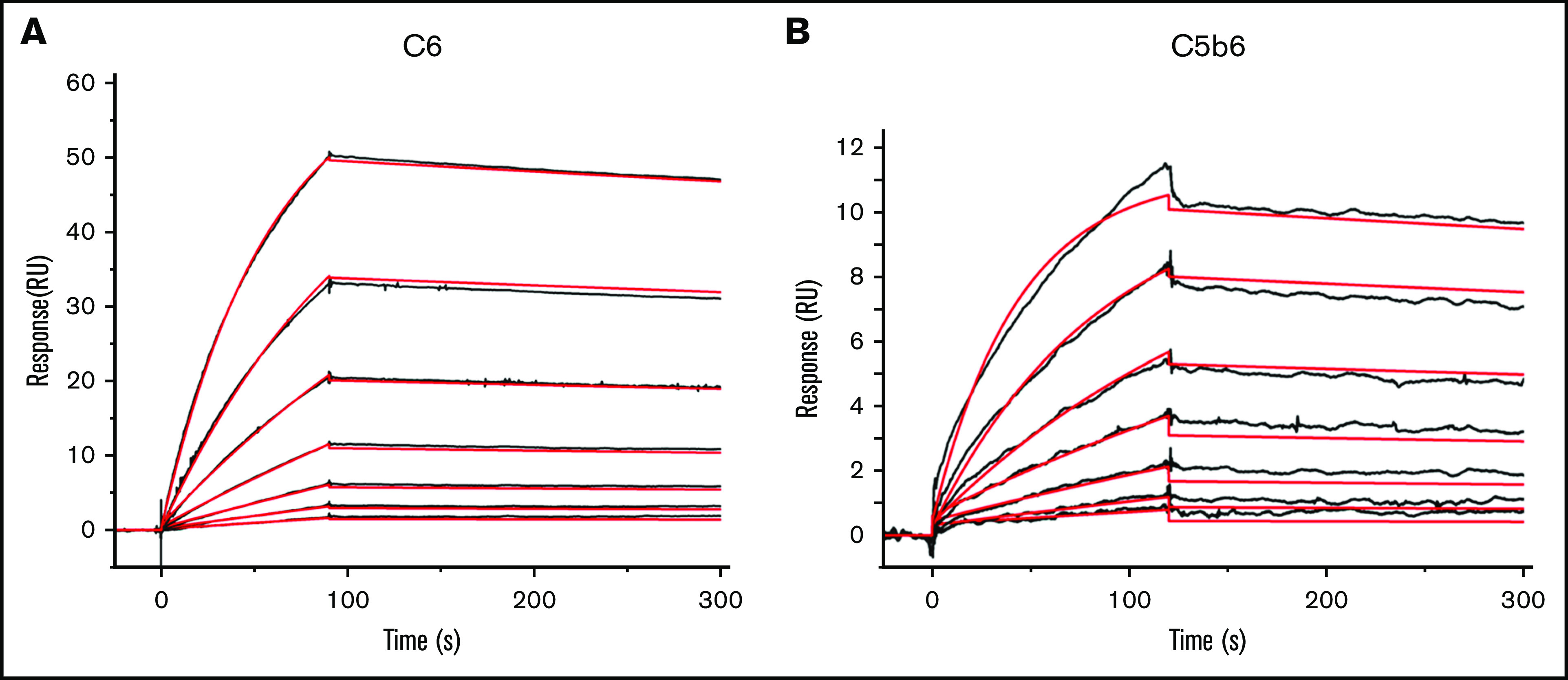
The mAb 1C9 binds to C6 in free circulation and within the C5b6 complex with high affinity. SPR sensorgram shows the binding kinetics for C6 (A) and C5b6 (B) with immobilized 1C9 antibody. Data are shown as black lines, and the best fits of the data to a 1:1 binding model (antigen-binding fragment) are shown in red.
Mechanisms by which 1C9 inhibits MAC formation
The ability of 1C9 to bind both free and complex-bound C6 suggests that this mAb could inhibit MAC formation by blocking the binding of free C6 to C5b and/or by preventing the binding of C7 to the C5b6 complex. To determine the mechanism by which 1C9 inhibits MAC formation, we performed functional hemolytic assays using C6- or C7-depleted sera in the presence of 1C9 together with purified C6 or C7 proteins. The addition of C6 incubated with control IgG to the C6-depleted sera resulted in massive hemolysis, whereas the addition of C6 incubated with 1C9 did not (Figure 4A), suggesting that binding free C6 and thus preventing complex formation with C5b is a potential underlying mechanism of 1C9. To determine whether 1C9 bound to C6 within the C5b6 complexes blocks the incorporation of C7 for the MAC assembly, we performed additional experiments in which EshA were preincubated with C7-depleted sera to enable C5b6 complex formation before adding purified C7; the addition of 1C9, but not control IgG, inhibited hemolysis (Figure 4B). These results confirmed that 1C9 does prevent the binding of C7 to C5b6 complexes.
Figure 4.

Mechanistic studies of how the mAb 1C9 inhibits MAC formation. (A) 1C9 or mouse IgG (mIgG) was preincubated with purified C6 and then mixed with EshA and 1% C6-depleted sera. After incubation, the inhibition of hemolysis was measured. (B) EshA were first incubated with 1% C7-depleted sera and then mixed with 1C9 or mIgG, followed by the addition of purified C7 protein. After incubation, the inhibition of hemolysis was measured. Data represent mean ± standard deviation. Representative results from 3 independent experiments. *P < .05 by Student t test.
The anti-C6 mAb protects affected PNH RBCs from MAC-mediated destruction
To determine whether 1C9 could protect affected PNH RBCs from MAC-mediated damage, we performed a modified Ham’s test15,16 by incubating RBCs from a PNH patient with 50% acidified NHS in the presence of different concentrations of 1C9 ranging from 0 to 1500 nM. By identifying and quantitating the affected PNH RBCs using CD59 as the marker following common clinical practice,16 we found that consistent with previous reports,17,18 CD59− PNH RBCs were selectively destroyed by complement in this assay. We also found that 1C9 protected the CD59− PNH RBCs from MAC-mediated destruction in this assay in a concentration-dependent manner (Figure 5A)
Figure 5.
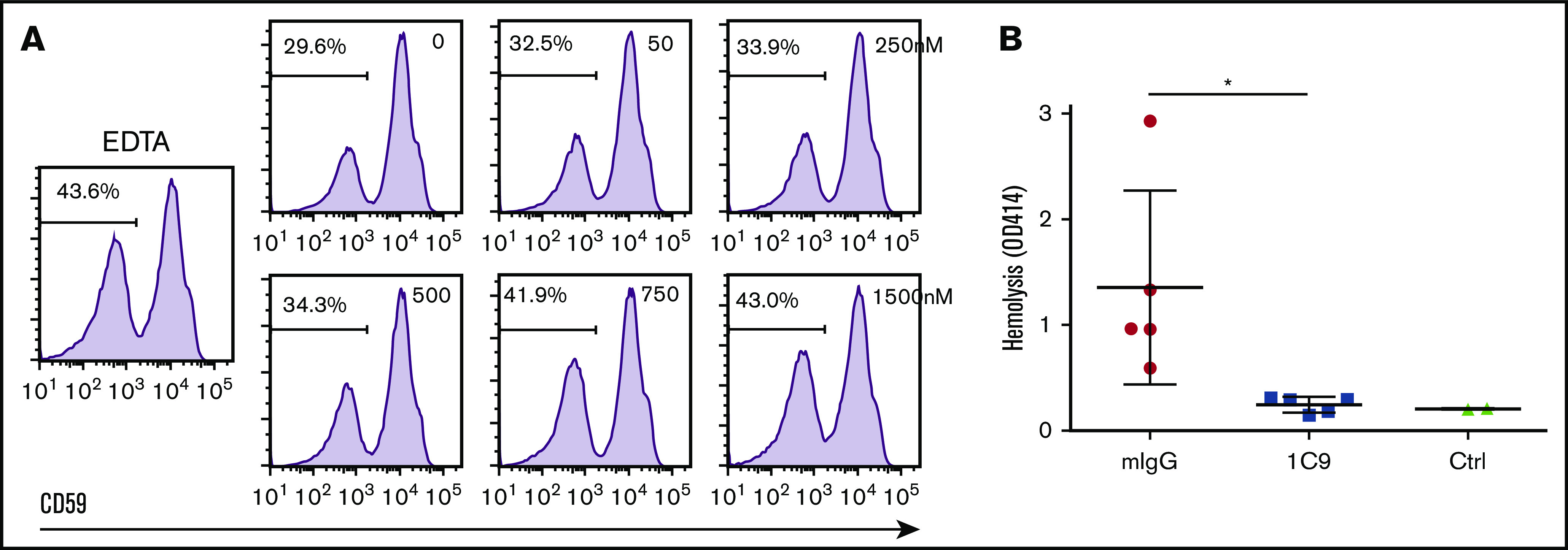
The mAb 1C9 protects PNH RBCs in vitro and prevents complement-mediated intravascular hemolysis in vivo in a mouse model. (A) RBCs from a PNH patient were incubated with 50% acidified NHS in the presence of 0 to 1500 nM of 1C9 or 5 mM of EDTA (control [Ctrl]). After incubation, surviving PNH RBCs (CD59−) were quantitated by flow cytometry. Representative results from 2 repetitive experiments. (B) NHS was mixed with 1C9 or mouse IgG (mIgG) and then immediately injected IV into WT C57BL/6 mice via the tail vein. After 10 minutes, blood samples were collected in EDTA-containing tubes via terminal retroorbital bleeding. In vivo intravascular hemolysis was measured in the plasma by reading the optical density at 450 nm (OD450). Each dot presents 1 mouse. Ctrl were naïve C57BL/7 mice. *P < .05.
The anti-C6 mAb 1C9 prevents human complement–mediated hemolysis in vivo
Taken together, the in vitro assay results suggest that the anti-C6 mAb 1C9 may be useful in preventing MAC-mediated tissue damage. To validate these in vitro data in an in vivo model, we used a previously established mouse model of human complement–mediated intravascular hemolysis wherein NHS is injected into mice to induce erythrocyte lysis via the activation of human complement and thus MAC formation, as described in Methods. Whereas control IgG-treated mice exhibited massive intravascular hemolysis after NHS injection, consistent with previous reports, anti-C6 IgG-treated mice exhibited significantly reduced and even completely suppressed complement-mediated hemolysis (Figure 5B). These results demonstrate the efficacy of 1C9 as an inhibitor of MAC-mediated hemolysis in vivo.
Examination of cross-reactivity of the anti-human C6 mAb to rhesus monkey and mouse C6
C6 proteins share a relatively high level of homology among various species. Because mice and rhesus monkeys are commonly used as in vivo animal models before human clinical trials, we examined the ability of 1C9 to inhibit hemolysis mediated by rhesus monkey and mouse complements. Notably, 10 nM of 1C9 significantly inhibited the hemolysis induced by rhesus monkey complement (Figure 6A). However, this mAb had no effect on mouse complement–mediated hemolysis, even at a concentration of 10 μM (Figure 6B). These results suggest that this mAb recognizes and blocks monkey C6 but does not cross-react with mouse C6.
Figure 6.

The mAb 1C9 inhibits hemolysis mediated by activated complement from rhesus monkeys but not from mice. (A) Ten nM of 1C9 or mouse IgG (mIgG) was incubated with EshA and 1% sera from 9 individual rhesus monkeys. Hemolysis was measured by reading the optical density at 450 nm (OD450). (B) Ten μM of 1C9 or mIgG was incubated with Erabb and 10% sera from 5 individual C57BL/6 mice. Hemolysis was measured by reading the OD414. *P < .05.
Discussion
In this project, we used conventional hybridoma technology to develop a panel of anti-human C6 mAbs and identified 1, clone 1C9, as a functional inhibitor of C6. Particularly, we determined that 1C9 binds to free C6 or C6 within the C5b6 complexes at a 1:1 ratio with a KD of 2.07 and 1.91 nM, respectively, and that it recognizes both free and complex-bound C6. Using C6- and C7-depleted sera and purified C6 and C7 proteins, we confirmed that 1C9 inhibits MAC formation by blocking the binding of C7 to existing C5b6 complexes. We further discovered that this anti-C6 mAb cross-reacts with and therefore inhibits rhesus monkey, but not mouse, C6 in functional assays. More importantly, this anti-human C6 mAb 1C9 protects affected PNH RBCs from MAC-mediated destruction in vitro and prevents human complement–mediated intravascular hemolysis in vivo in a mouse model. In summary, we have developed a new anti-human C6 mAb clone 1C9 that inhibits the formation of MAC.
Excessive complement activation plays an integral role in many diseases, and MAC formation consequent to complement activation is a primary mechanism by which activated complement causes pathological injury to self-tissues.19 Accordingly, inhibition of any stage of the complement activation cascade could suppress MAC formation and then ameliorate the severity of associated diseases. Many small- and large-molecule inhibitors targeting different complement components and different activation cascade steps are undergoing extensive drug development. These potential drugs include inhibitors of factor D,20 factor B,21 C1,22,23 and MASP-2,24 and both animal studies and clinical trials have yielded encouraging treatment efficacy data, showing that inhibiting MAC formation is a valid therapeutic approach.25
All complement activation pathways share a terminal pathway that ends with MAC assembly. Therefore, at least in theory, all components of this terminal pathway are valid and logical targets for the treatment of many complement-mediated diseases. Although these components are present at similar concentrations in the blood (C5, ∼75 μg/mL; C6, ∼45 μg/mL; C7, ∼56 μg/mL; C8, 55 μg/mL; C9, ∼60 μg/mL),26 so far only C5 has been targeted clinically via eculizumab and its related second-generation product, ravulizumab.27 Other C5 inhibitors, such as zilucoplan, are under extensive clinical development and have yielded positive treatment results in clinical trials.25
Potentially, C6 may be an equally effective or even superior inhibitory target within the terminal complement pathway. Studies of rats carrying a spontaneous C6 mutation that leads to C6 deficiency28 revealed that in the absence of C6 activity, many models of human diseases, including intravascular hemolysis, sepsis, nephritis, thrombotic microangiopathy, transplant rejection, and neurodegenerative diseases, demonstrated attenuation. In addition, rabbits with C6 deficiency resulting from spontaneous C6 gene mutations exhibit marked protection against membranous nephropathy, a disease in which the glomeruli are damaged by MAC.29 Detailed mechanistic studies19,30 have established that the half-life of C5b after C5 activation and cleavage is very short (∼2 minutes), unless the subunit can bind to C6 to form the stable C5b6 complex. However, C5b6 cannot attach to a cell membrane before binding to C7, which exposes a membrane-binding region that extends into the bilipid layer. The subsequent binding of C8 enables the complex to extend farther into the cell membrane and to bind multiple units of C9, thus completing the formation of MAC and enabling cell damage. Accordingly, a blockade of free C6 should prevent its binding to C5b and therefore the initiation of the terminal pathway, whereas a blockade of C6 within the C5b6 complex should inhibit sequential C7 binding to abolish the formation of MAC. Our mAb 1C9 binds to C6 both in the free circulation and in the C5b6 complex, suggesting that 1C9 acts via both mechanisms, which might account for its high level of efficacy in terms of preventing MAC-mediated cell damage (eg, hemolysis). However, even though our mechanistic in vitro studies using C7-depleted sera and purified C7 protein confirmed that 1C9 blocks C7 binding to C5b6, the experiments using C6-depleted sera and purified C6 protein could not definitely prove that 1C9 blocks free C6 binding to C5b because of its observed activity in blocking C7 binding downstream of the terminal pathway.
Surprisingly, there have been few published reports describing the development of C6-targeted mAbs for potential therapeutic use, in contrast to the extensive development of C5-targeted reagents. In a study published almost 3 decades ago, newly generated anti-human C6 mAbs barely inhibited complement-mediated hemolysis, whereas their developed anti-C5 mAbs were potent inhibitors of MAC-mediated hemolysis.31 Similarly, only 1 of 20 C6-reactive mAbs identified in our study inhibited complement-mediated hemolysis in our initial functional screening assays, even though all displayed high affinities for C6 in our ELISAs (data not shown). These results suggest that most immune-dominant epitopes in human C6 generally elicit the production of mAb binding, with no inhibitory ability. However, we note that an anti-human C6 mAb developed in rats was described in a patent application published in 2018 (US20180015162A1), although no related literature is available. This rat anti-human C6 mAb efficiently inhibited MAC-mediated formation and hemolysis and reduced axon and myelin destruction in a rat model of sciatic nerve injury. A humanized form of the antibody is apparently under further development as a new reagent for the treatment of MAC-mediated neurological diseases. However, detailed underlying mechanisms by which this mAb works remain unclear.
The mouse anti-human C6 mAb 1C9 binds to C6 both in free circulation and within the C5b6 complex as shown by both ELISA and SPR analyses. Interestingly, even though the ELISA results suggest that 1C9 might have a slightly higher affinity against free C6 than C6 within the C5b6 complex, the SPR results showed that the affinities are comparable (2.07 vs 1.91 nM). This apparent discrepancy could be due to the different experimental conditions in these assays. For example, in the ELISA, C5b6 complexes were coated on a solid phase (the plate surface), which might lead to conformation changes, whereas in the SPR experiments, they were in the fluid phase, which might help to maintain their correct conformation.
The mAb 1C9 inhibits MAC-mediated hemolysis, possibly because of the simultaneous binding of both free C6 in the blood and C6 within C5b6 complexes. As described above, either mechanism would block the terminal pathway of complement activation and thus MAC formation, and the concurrent use of both mechanisms might yield a synergistic effect, resulting in the more efficient reduction of MAC formation and attenuation of MAC-mediated tissue damage. In PNH patients, affected RBCs (CD59−) differentiated from hematopoietic stem cells carrying the mutant PIGA gene are more susceptible to MAC-mediated destruction than the unaffected RBCs (CD59+) from the normal hematopoietic stem cells.32 Under certain conditions, including infections and the drop of pH in the blood, complement activation occurs and thus leads to MAC-mediated intravascular hemolysis of the affected RBCs in PNH patients.33-35 In a modified Ham’s test in which PNH RBCs were selectively destroyed by MAC under an acidic condition, we found that 1C9 protected the CD59− RBCs from complement-mediated destruction in a concentration-dependent manner. These data, together with results from other in vitro and in vivo studies, suggest that this newly developed mAb, 1C9, exhibits potential as another inhibitor of MAC formation and is therefore a new candidate for further development as a potential therapeutic agent for diseases in which MAC is integrally involved in the pathogenesis. Our observation of cross-reactivity with rhesus monkey C6 suggests that this mAb can be tested directly in nonhuman primates during future clinical development endeavors. Furthermore, we also note that 1C9 did not completely abolish hemolysis in our assays, even when excess molar concentrations were used, suggesting that C6 residual activity might exist, similar to the reported C5 residual activity when eculizumab is used.9
In this work, we used a convenient mouse model of human complement–mediated intravascular hemolysis, which has been used to test the inhibitory capacities of novel reagents against human complement–mediated cell damage in vivo.13 In these mice, after NHS injection, mouse intravascular hemolysis mediated by activated human complement can be easily quantitated by measuring released hemoglobin in the plasma. Using this model, we and others demonstrated previously that complement inhibitors such as FUT-17513 and SSL-736 significantly reduced intravascular hemolysis, a common feature in patients with PNH, cold agglutinin disease, and aHUS. We found that when blood was collected 10 minutes after NHS injection to measure intravascular hemolysis, it showed that 1C9 administration significantly prevented or even abolished the human complement–mediated RBC destruction. However, in our pilot treatment experiments to titrate the optimal blood collection time points for hemolytic measurements, we found that significant hemolysis had occurred in all mice at 1 hour after NHS injection, regardless of whether the mice had been simultaneously treated with our anti-C6 mAb (data not shown). Given the hemolytic inhibitory activity of this mAb displayed in vitro in the presence of human NHS, we hypothesized that mouse C6 may have a reduced but functional ability to form a human-mouse hybrid MAC in the absence of human C6. Indeed, the injection of an equivalent amount of C6-depleted human sera into a WT mouse similarly induced massive hemolysis at only 30 minutes postadministration (supplemental Figure 2). These observations are consistent with our previous finding that mouse C5 could supplement human C5 during the process of MAC formation, because the injection of C5-depleted human sera into a WT mouse induced hemolysis, which was reduced by the further inhibition of mouse C5.37 These results suggest that this simple model should be used with caution when testing the efficacy of human complement–targeted reagents because of the potential interference from mouse complement counterparts. In addition, the precise initiation and regulatory mechanisms underlying the intravascular hemolysis in this xenogeneic model could be different from those in patients. Nevertheless, the ability of 1C9 to significantly prevent intravascular hemolysis during the acute phase in this mouse model provides in vivo evidence supporting the further development of this mAb as a new agent for MAC-mediated intravascular hemolysis.
In summary, we have developed and characterized a new mouse anti-human C6 mAb (clone 1C9). This mAb binds to both free C6 and C6 within the C5b6 complex to inhibit MAC formation. Thus, 1C9 reduces MAC-mediated hemolysis both in vitro and in vivo. In addition, this mouse anti-human C6 mAb also cross-reacts with rhesus monkey C6 and inhibits rhesus monkey MAC–mediated hemolysis. All these features warrant the further development of this novel mAb as a new agent for the treatment of diseases in which MAC is integrally involved in the pathogenesis.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Jarek Maciejewski at Cleveland Clinic for providing the blood sample from a deidentified PNH patient. The authors also thank Sara Thomasy, School of Veterinary Medicine, University of California Davis, for providing the rhesus monkey sera.
This project was supported in part by National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases grants DK103581 (F.L.) and EY031087 (F.L.). J.X. is supported in part by NIH, National Cancer Institute grant K08 CA228039.
Footnotes
Send data sharing requests via e-mail to the corresponding author, Feng Lin (linf2@ccf.org).
Authorship
Contribution: K.L., L.Z., M.K., M.Y., Y.C., E.P., and M.H. performed the experiments and analyzed the data; J.X. and C.T. discussed the results and edited the manuscript; and F.L. designed the experiments, analyzed the data, and wrote the manuscript, together with K.L.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Feng Lin, Department of Immunity and Inflammation, Lerner Research Institute, Cleveland Clinic, 2111 E 96th St, Cleveland, OH 44106; e-mail: linf2@ccf.org.
References
- 1.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Müller-Eberhard HJ. Transmembrane channel-formation by five complement proteins. Biochem Soc Symp. 1985;50:235-246. [PubMed] [Google Scholar]
- 3.Wong EKS, Kavanagh D. Diseases of complement dysregulation-an overview. Semin Immunopathol. 2018;40(1):49-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria [published correction appears in Nat Biotechnol. 2007;25(12):1488]. Nat Biotechnol. 2007;25(11):1256-1264. [DOI] [PubMed] [Google Scholar]
- 5.Westra D, Wetzels JF, Volokhina EB, van den Heuvel LP, van de Kar NC. A new era in the diagnosis and treatment of atypical haemolytic uraemic syndrome. Neth J Med. 2012;70(3):121-129. [PubMed] [Google Scholar]
- 6.Howard JF Jr., Utsugisawa K, Benatar M, et al. ; REGAIN Study Group . Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16(12):976-986. [DOI] [PubMed] [Google Scholar]
- 7.Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(7):614-625. [DOI] [PubMed] [Google Scholar]
- 8.Thomas TC, Rollins SA, Rother RP, et al. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996;33(17-18):1389-1401. [DOI] [PubMed] [Google Scholar]
- 9.Harder MJ, Kuhn N, Schrezenmeier H, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129(8):970-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura J, Yamamoto M, Hayashi S, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370(7):632-639. [DOI] [PubMed] [Google Scholar]
- 11.Würzner R, Orren A, Potter P, et al. Functionally active complement proteins C6 and C7 detected in C6- and C7-deficient individuals. Clin Exp Immunol. 1991;83(3):430-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C. Hybridoma technology for the generation of monoclonal antibodies. Methods Mol Biol. 2012;901:117-135. [DOI] [PubMed] [Google Scholar]
- 13.Ino Y, Sato T, Suzuki S, Iwaki M, Yoshikawa T. Inhibitory effects of FUT-175, a new synthetic protease inhibitor, on intravascular hemolysis by human serum in mice. Int J Immunopharmacol. 1987;9(5):533-537. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama WM. Production of monoclonal antibodies. Curr Protoc Cytom. 2006;3(Appendix):3J. [DOI] [PubMed] [Google Scholar]
- 15.Preis M, Lowrey CH. Laboratory tests for paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2014;89(3):339-341. [DOI] [PubMed] [Google Scholar]
- 16.Sutherland DR, Kuek N, Davidson J, et al. Diagnosing PNH with FLAER and multiparameter flow cytometry. Cytometry B Clin Cytom. 2007;72(3):167-177. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira VP, Pangburn MK. Factor H mediated cell surface protection from complement is critical for the survival of PNH erythrocytes. Blood. 2007;110(6):2190-2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maibaum J, Liao SM, Vulpetti A, et al. Small-molecule factor D inhibitors targeting the alternative complement pathway. Nat Chem Biol. 2016;12(12):1105-1110. [DOI] [PubMed] [Google Scholar]
- 19.Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology. 2016;221(6):747-751. [DOI] [PubMed] [Google Scholar]
- 20.Wiles JA, Galvan MD, Podos SD, Geffner M, Huang M. Discovery and development of the oral complement factor D inhibitor ACH-4471 [published online ahead of print 1 October 2019]. Curr Med Chem. doi:10.2174/0929867326666191001130342. [DOI] [PubMed] [Google Scholar]
- 21.Schubart A, Anderson K, Mainolfi N, et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc Natl Acad Sci USA. 2019;116(16):7926-7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szilágyi K, Hajdú I, Flachner B, et al. Design and selection of novel C1s inhibitors by in silico and in vitro approaches. Molecules. 2019;24(20):E3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang E, Vo A, Choi J, et al. Three-year outcomes of a randomized, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Clin J Am Soc Nephrol. 2020;15(1):109-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwaeble WJ, Lynch NJ, Clark JE, et al. Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2011;108(18):7523-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. 2018;14(1):26-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barnum SR, Schein TN, eds.. The Complement FactsBook, 2nd ed. London, United Kingdom: Elsevier/Academic Press; 2018: [Google Scholar]
- 27.Stern RM, Connell NT. Ravulizumab: a novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Ther Adv Hematol. 2019;10:2040620719874728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leenaerts PL, Stad RK, Hall BM, Van Damme BJ, Vanrenterghem Y, Daha MR. Hereditary C6 deficiency in a strain of PVG/c rats. Clin Exp Immunol. 1994;97(3):478-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Groggel GC, Salant DJ, Darby C, Rennke HG, Couser WG. Role of terminal complement pathway in the heterologous phase of antiglomerular basement membrane nephritis. Kidney Int. 1985;27(4):643-651. [DOI] [PubMed] [Google Scholar]
- 30.Morgan BP, Boyd C, Bubeck D. Molecular cell biology of complement membrane attack. Semin Cell Dev Biol. 2017;72:124-132. [DOI] [PubMed] [Google Scholar]
- 31.Würzner R, Schulze M, Happe L, et al. Inhibition of terminal complement complex formation and cell lysis by monoclonal antibodies. Complement Inflamm. 1991;8(5-6):328-340. [DOI] [PubMed] [Google Scholar]
- 32.Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017;3:17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomita M. Biochemical background of paroxysmal nocturnal hemoglobinuria. Biochim Biophys Acta. 1999;1455(2-3):269-286. [DOI] [PubMed] [Google Scholar]
- 34.Crosby WH. Paroxysmal nocturnal hemoglobinuria: relation of the clinical manifestations to underlying pathogenic mechanisms. Blood. 1953;8(9):769-812. [PubMed] [Google Scholar]
- 35.Parker CJ. Historical aspects of paroxysmal nocturnal haemoglobinuria: “defining the disease”. Br J Haematol. 2002;117(1):3-22. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Clow F, Fraser JD, Lin F. Therapeutic potential of staphylococcal superantigen-like protein 7 for complement-mediated hemolysis. J Mol Med (Berl). 2018;96(9):965-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Qiu W, Crooke S, et al. Development of autologous C5 vaccine nanoparticles to reduce intravascular hemolysis in vivo. ACS Chem Biol. 2017;12(2):539-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.