

Abstract
Monitoring factor VIII (FVIII) activity has traditionally been complicated by discrepancies between assays for the various sorts of FVIII molecules. The advent of novel nonfactor therapies (emicizumab, fitusiran, and anti-tissue factor pathway inhibitor antibodies) in hemophilia A poses a new level of difficulty on the laboratory monitoring of these patients. To use the correct assays and for a proper interpretation of their results, it is pertinent to understand the mode of action of these nonfactor agents. Furthermore, the biochemical consequences for the different types of activity assays (whether it be specific FVIII activity assays or global coagulation assays) should be taken into account as well. In this review, these aspects will be discussed. In addition, the use of various animal models to estimate FVIII-equivalence of the nonfactor therapies will be presented.
Introduction
During the previous 4 to 5 decades, hemophilia A treatment mostly involved replacement therapy using concentrates enriched in factor VIII (FVIII). Early management options consisted of cryoprecipitate concentrates, which over time evolved into high-purity, plasma-derived FVIII concentrates and recombinant FVIII concentrates.1,2 Laboratory monitoring of FVIII-replacement therapy is performed via FVIII-specific assays, in most cases by using activated partial thromboplastin time (aPTT)–based 1-stage clotting assays or via 2-stage chromogenic activity assays that use purified proteins.3 In the aPTT-based coagulation assay, the patient’s plasma is mixed with FVIII-deficient plasma, and via the addition of an activating reagent and Ca2+ ions, the coagulation cascade is initiated. The activating agent usually consists of a surface activator (micronized silica, ellagic acid, or kaolin), which starts the contact activation pathway.4 In the chromogenic assay, the patient’s plasma is diluted and mixed with purified factor X (FX), factor IXa (FIXa), phospholipids, thrombin, and Ca2+ ions.5 This leads to the generation of FXa, the quantity of which is analyzed by using a small synthetic substrate that is hydrolyzed by FXa.
When analyzed in the plasma of patients, the cofactor activity of plasma-derived and full-length recombinant FVIII correlates well between 1-stage and chromogenic assay systems. In contrast, differences have been detected when assessing high-purity FVIII concentrates toward plasma standards.6 To avoid these differences, 2 distinct World Health Organization (WHO)–approved standards are now available: 1 plasma standard (NIBSC-code 07/316) has been developed to assign FVIII activity values in the plasma of patients, whereas a separate standard (NIBSC-code 07/350) has been made available for the assignment of FVIII activity levels in concentrates.7-9 Importantly, clinical and laboratory experiences over the past several decades have taught us (within limits, of course) to what extent levels of FVIII measured in the activity assays correlate with the clinical phenotype of the patients.
The first issues on FVIII activity measurements arose upon the introduction of recombinant B-domainless FVIII. These concentrates were associated with an assay discrepancy, in which levels measured using a 1-stage clotting assay were 20% to 50% lower compared with the values obtained using a chromogenic assay.10-12 To alleviate this discrepancy, a product specific standard was developed.13,14 The issue of assay discrepancy between 1-stage and chromogenic assays has regained attention with the advent of modified FVIII molecules having an extended half-life. These modifications (fusion to the Fc-portion of immunoglobulin G, the attachment of polyethylene glycol, or a combination of different types of modifications) alter the physical properties of the FVIII molecule, and may therefore affect its behavior in the different assay systems. That such modifications indeed affect FVIII activity assays, has elegantly been reviewed by Kitchen and coworkers.15
We are currently experiencing revolutionary changes in the clinical management of hemophilia A, where apart from replacement-therapy using FVIII molecules or FVIII gene therapy, also so-called nonfactor therapies have become available or are in advanced clinical development. These include the bispecific antibody emicizumab, a small interfering RNA-based approach that reduces expression of antithrombin (fitusiran) and antibodies blocking the activity of tissue factor pathway inhibitor (TFPI).16 These nonfactor therapies force us into a reassessment on how and when to monitor these patients. It is relevant therefore not only to get insight into the mechanism of action of these new therapeutic agents, but also to understand how these agents perform in the biochemical assay systems that are used to monitor hemophilia A patients.
Nonfactor therapies
The molecules that qualify as nonfactor therapies for hemophilia A (emicizumab, fitusiran, and monoclonal anti-TFPI antibodies) have extensively been reviewed elsewhere,16-20 and only a brief summary will be given here.
First, emicizumab is a recombinant humanized bispecific antibody that consists of 2 different antigen-binding domains. One domain recognizes FIX/FIXa and a second domain has its epitope on FX/FXa. The mode of action of this molecule resides in bridging FIXa and FX, and by bringing enzyme and substrate in close proximity, FIXa-mediated activation of FX is increased.17,21,22 The availability of more FXa molecules will support more thrombin generation, ultimately allowing a stronger hemostatic response. Clinical trials investigating emicizumab indeed demonstrated a strong reduction in bleeding events in hemophilia A patients with and without inhibitors.23-26
A second approach, named fitusiran, is based on the reduction of antithrombin expression in the hepatocytes.27 Transcriptional silencing in eukaryotic cells was first reported in 1988, when antisense complementary DNAs were used to silence expression of the chalcone synthase gene in tobacco and petunia plants, resulting in modified flower color patterns.28,29 This approach has evolved into therapeutically applied oligonucleotides that use the natural RNA-interference pathway to inactivate the corresponding messenger RNA.27,30 In the case of fitusiran, a GalNAc-tagged oligonucleotide is used for the posttranscriptional silencing of the antithrombin gene in hepatocytes.27,30 Consequently, protein levels of antithrombin in plasma are reduced, eliminating part of the anticoagulant pathway that targets several coagulation enzymes, including thrombin, FXa, and FIXa. Reduced antithrombin levels will thus increase the overall hemostatic potential in the absence of FVIII. Indeed, clinical data that have been reported so far indicate a strong reduction in bleeding episodes in patients receiving fitusiran.31
In a third alternative approach, another member of the anticoagulant pathway is targeted (ie, TFPI).32,33 To my knowledge, 4 monoclonal antibody-based therapeutical agents have been subject to clinical evaluation: concizumab (by Novo Nordisk), marstacimab (by Pfizer), BAY-1093884 (by Bayer), and MG1113 (by Green Cross Corporation).
The anti-TFPI antibodies are designed to neutralize the inhibitory activity of TFPI toward FXa and as such also the factor VIIa/tissue factor (FVIIa/TF) complex. A prolonged functional availability of the FVIIa/TF complex will result in the production of larger quantities of FIXa and FXa, thereby stimulating thrombin generation. The clinical data that have been presented are indeed compatible with an enforced hemostatic system in patients with hemophilia A, manifested by a reduced bleeding tendency in these patients when under anti-TFPI therapy.34 Of note, as of this date (3 April 2020), clinical trials with concizumab have been paused, whereas those with BAY-1093884 have been terminated,35 both because of thrombotic events. Thrombotic events have also been observed in patients treated with emicizumab and fitusiran.23,36
Apart from their mode of action, these nonfactor therapies also modify the management of hemophilia in a more general sense (Figure 1). Classic replacement therapy using FVIII concentrates requires regular IV infusions. The frequency of these infusions varies between patients, and may be as often as 3 times per week. These IV infusions are associated with peak levels of FVIII after infusion, which decrease gradually until the next infusion. Such peak and trough levels are absent in the nonfactor therapies, which are characterized by subcutaneous administrations and stable levels of the therapeutic agent.
Figure 1.

Comparison of traditional and novel hemophilia therapies. Similarities and differences of hemophilia A treatment between FVIII replacement therapy, emicizumab, fitusiran, and anti-TFPI antibodies with regard to patient target group, mode of administration, and therapeutic levels over time.
These differences in mode of action, route of administration, and absence of peak and troughs will have a major impact on how patients receiving these nonfactor therapies are being monitored. Indeed, a series of publications have recently discussed laboratory testing in perspective of nonfactor therapies, particularly with regard to emicizumab.37-40 In this review, focus will be on the biochemical aspects when monitoring nonfactor therapies.
Laboratory monitoring of nonfactor therapies
When considering monitoring of patients receiving nonfactor therapy, the first and most important question that comes to mind is: what is it exactly that needs to be monitored? Several answers to this question are possible, and include, for instance:
monitoring levels of the therapeutic agent.
monitoring the effect of nonfactor therapies on the hemostatic potential of patients.
monitoring the FVIII-equivalence of nonfactor therapies.
Because each of these options requires a different approach and a different assay to generate the desired response, let’s address them separately.
Monitoring levels of the therapeutic agent
Prophylactic home treatment has played an important role in the emancipation of hemophilia A therapy, providing numerous advantages to the patients. In contrast to prophylactic diabetes treatment (where patients can monitor themselves by analyzing blood sugar levels), monitoring FVIII activity levels is currently not yet accessible to patients at home. Consequently, when patients return for a clinical visit, this occasion is frequently used to test for FVIII activity and/or FVIII inhibitors. With the nonfactor therapies, such monitoring will also be difficult to perform at home, at least for now. If and when levels of the therapeutic agent should be monitored will of course be decided at the discretion of the clinician (and I assume guidance will be provided by national or international expert committees). But in case this will be done, each nonfactor variant will require a different test.
To determine emicizumab levels, various options are possible. During clinical trials, an enzyme-linked immunosorbent assay (ELISA)–based assay using anti-idiotype antibodies was used for the analysis of emicizumab concentrations.41 This assay is not commercially available, but other assays can be used instead. Because emicizumab mimics FVIII cofactor function (at least to some extent), it is possible to use FVIII activity assays to monitor emicizumab levels. Unfortunately, the classic 1-stage APTT-based assay is not useful in this regard because the APTT is normalized already at subtherapeutical emicizumab concentrations (3-5 μg/mL of emicizumab normalizes the aPTT, whereas therapeutic levels are around 55 μg/mL).37,42 To overcome this limitation, 2 other activity assays could be used. A modified aPTT assay has been described, in which plasma samples are further diluted, allowing a linear relationship between emicizumab concentrations and clotting times.43,44 A second option is to use chromogenic FVIII activity assays that contain human FIXa and FX. Also, in this assay, a near-linear relationship between emicizumab concentrations and FXa generation can be obtained.37 For both assays, specific emicizumab calibrators are commercially available (r2 Diagnostics, South Bend, IN). Of note, these calibrators are not (yet) approved for clinical use in the United States. Importantly, the activity obtained using these tests provides surrogate FVIII-like activities. These values do not relate to a true FVIII equivalence.17,45 Indeed, emicizumab activity relies on the amount of FIXa that is available. These amounts are not only variable between tests from different manufacturers, but may also differ in a lot-to-lot manner in chromogenic assay kits that use the same reagents, consequently generating different FVIII-like activity values. Likewise, different activating compounds in the aPTT will generate different quantities of FIXa, thereby influencing emicizumab activity. It is further important to note that the outcome of these assays can be affected by the presence of other procoagulant molecules (eg, FVIII or activated prothrombin complex concentrates [APCC]) present in emicizumab-containing plasma samples.37,39
As for fitusiran, it will obviously be impractical to measure residual oligonucleotide levels within the hepatocytes of the patients. However, it would be possible to measure residual antithrombin levels, either via antigen assays or via thrombin- or FXa-based activity assays.46 Again, the presence of other therapeutics, like APCC, in such samples could modify the results of the activity assays.
To measure anti-TFPI antibody levels, no specific assays for the antibodies are yet available, meaning that TFPI levels should be analyzed instead. Antigen levels of TFPI can be measured in established ELISA systems. However, it is unclear whether all of the therapeutic anti-TFPI antibodies actually reduce plasma TFPI levels or whether these monoclonal antibodies interfere in ELISA assays. Alternatively, specific activity assays are available to measure residual TFPI activity, including a diluted prothrombin time–based assay and tissue factor–dependent chromogenic assays.47,48 Note should be taken that plasma contains but 10% to 30% of all TFPI,49 and these assays will therefore be unable to give a full comprehension of how much TFPI is inhibited at the vascular lining.
In conclusion, direct measurement of emicizumab, fitusiran, or anti-TFPI antibodies is complicated, but alternative, indirect activity assays could be used to monitor whether these agents are still present and/or active.
Monitoring the effect of nonfactor therapies on the hemostatic potential of patients
When analyzing the hemostatic potential in normal individuals using global coagulation tests, a large range between individuals can be observed.50 To analyze the effect of the nonfactor therapies on the hemostatic potential of a patient, it is thus important to perform such global coagulation tests before and after onset of treatment to obtain a proper impression of the changes in hemostatic potential. A diverse arsenal of global test is available, including clot-waveform analysis, thromboelastography, and thrombin generation assays.51 The focus in this section will be on the thrombin generation assay. As illustrated in Figure 2, the addition of emicizumab, nanobodies against antithrombin,52 or polyclonal antibodies against TFPI53 has a strong stimulating effect on the thrombin generation profile in FVIII-deficient plasma. Thus, the presence of the procoagulant antibody emicizumab or the functional absence of antithrombin or TFPI results in increased thrombin peaks and the endogenous thrombin potential compared with FVIII-deficient plasma alone. This is in line with an ameliorated hemostatic potential induced by these agents. Indeed, thrombin generation profiles appear to be ameliorated in the vast majority of patients that receive emicizumab, fitusiran or anti-TFPI antibodies.31,54-56 Thus, these types of assays are excellent tools to verify whether the hemostatic profile of the patient has been changed since the onset of treatment. Nevertheless, care should be taken with the interpretation of these data. First, it is yet unclear how well the individual thrombin generation potential in vitro correlates with the actual hemostatic response in a patient (ie, whether these assays accurately predict clinical outcome).57-61 Second, these global assays will not give information about the true FVIII equivalence of the agent. This is easily illustrated by using emicizumab as an example. The functioning of emicizumab is fully dependent on the amount of FIXa available during the test. Dependent on the concentrations of TF or FXIa used to initiate the reaction, the amounts of FIXa generated will be different. This may lead to more or less thrombin generated in the presence of emicizumab, and, as such, a different equivalence to FVIII. Another example concerns the functional absence of antithrombin in FVIII-deficient plasma. The initial part of a normal thrombin generation curve is dependent on FVIIa/TF-generated FIXa and FXa, as well as the temporary presence of the labile FVIIIa molecule (the activity of which disappears within a few minutes). In contrast, the second part of the curve depends for an important part on the inhibition of FIXa, FXa, and thrombin by antithrombin. Reduced antithrombin activity in FVIII-deficient plasma will thus extend the lifespan of these enzymes in the assay, which translates into an increased thrombin potential, visualized by a delayed decline of the thrombin peak (Figure 2; see also Sehgal et al27). It is possible therefore that despite a lower thrombin peak, the total amount of thrombin generated under these conditions approaches or even exceeds that of normal plasma. However, we should not forget that α2-macroglobulin-mediated inhibition of thrombin is not taken into account in this assay (α2-macroglobulin-bound thrombin hydrolyzes the small fluorogenic substrate that is used in this assay) and only a small portion of the TFPI population is present (∼10% of total TFPI in vivo). Moreover, the thrombomodulin-dependent activated protein C pathway is present to a minor extent, whereas the platelet thrombin inhibitor protease nexin-1 is completely absent. The thrombin generation assay will thus exaggerate the amount of thrombin generated under these conditions because portions of the anticoagulant pathway are not present to compensate the absence of antithrombin.
Figure 2.
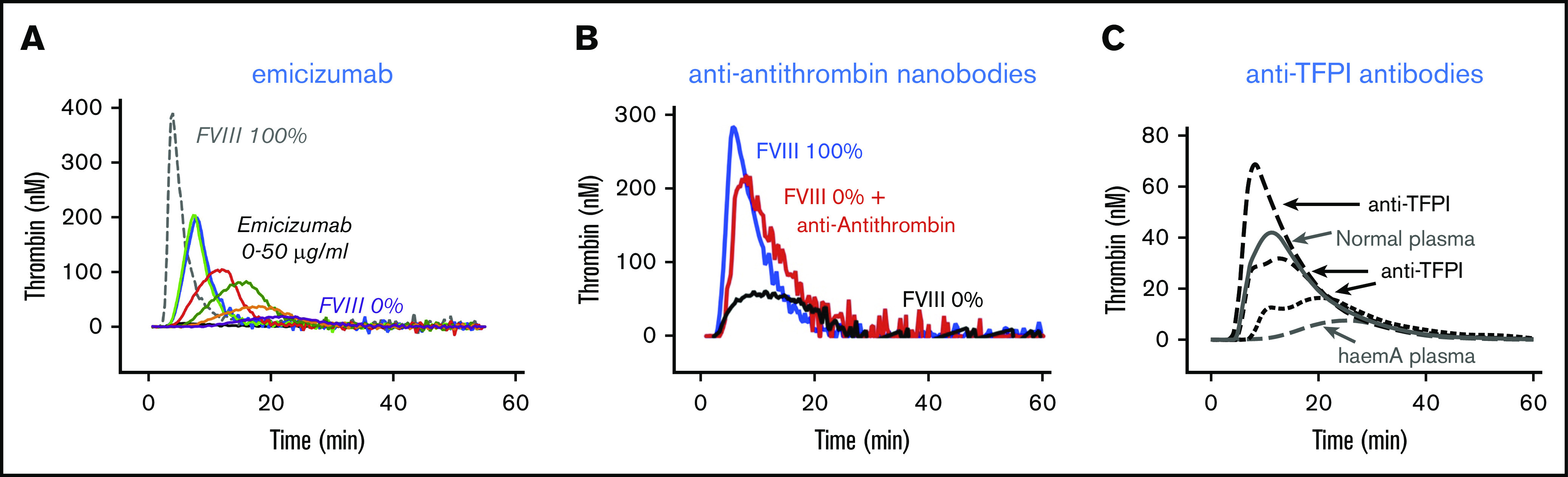
Thrombin generation in FVIII-deficient plasma. Thrombin generation in FVIII-deficient plasma in the presence of emicizumab (A), anti-antithrombin nanobodies (B), or anti-TFPI antibodies (C). In all cases, the addition of the antibodies increases thrombin generation compared with FVIII-deficient plasma alone. (A) Factor XIa-induced thrombin generation in the absence or presence of emicizumab (0-50 μg/mL; P.J.L., unpublished data). (B) Tissue factor-induced thrombin generation in the absence or presence of anti-antithrombin nanobodies.52 (C) Tissue factor-induced thrombin generation in the absence or presence of polyclonal anti-TFPI antibodies.53
In summary, global assays are an excellent tool to monitor the hemostatic potential of a patient before and after the onset of nonfactor therapies. They may even be used to analyze the effect of additive treatments, like FVIII or APCC, before they will be administered to the patient.54,62 In contrast, the global assays cannot be used to assign an FVIII equivalence to the therapeutic agent itself.
Monitoring the FVIII equivalence of nonfactor therapies
An issue that often comes up in the discussions around nonfactor therapies is: how do they compare with FVIII in terms of efficiency? The long clinical experience with FVIII replacement therapy has established what levels of FVIII are needed for certain types of activities, such as sports or surgical interventions. It would be convenient to know therefore what would be the FVIII equivalence of these nonfactor therapies to understand what level of protection will be present during this type of activities.
Again, the mode of action for each of the nonfactor approaches is fundamentally different from that of FVIII. For instance, in the FVIII-dependent FXa generation, FVIIIa is the limiting factor, and an excess of FIXa is present in the reaction. In the emicizumab-based reaction, it is FIXa that represents the limiting factor. It is thus not possible in an in vitro biochemical assay to establish a true FVIII equivalence for emicizumab because it will depend on how much FIXa is used in the assay. Perhaps a more unbiased response may be obtained by using animal models. When inflicting an injury in an animal model, there will be a natural hemostatic response, with all elements of the pro- and anticoagulant pathways present.
A first example of an animal model to obtain an FVIII equivalence for emicizumab was reported by Kitazawa and colleagues, who used a primate model for acquired hemophilia A (Figure 3).21 Primates were given an anti-FVIII antibody that specifically inhibits primate FVIII. The animals then received either emicizumab to a concentration of 6 μg/mL or porcine FVIII (which is not recognized by the anti-FVIII antibody) to a concentration of 0.01 U/mL (1%). Both treatments resulted in a similar prevention of blood loss (measured by hemoglobin levels). In contrast, porcine FVIII proved somewhat more efficient than emicizumab in reducing the bruised area in these animals.21 The authors conclude that emicizumab activity at these concentrations corresponds to about 1% FVIII activity.
Figure 3.

Animal models to estimate FVIII equivalence of nonfactor therapies. Different types of animal models have been used to estimate FVIII equivalence for nonfactor therapies. For emicizumab, a primate acquired hemophilia A model has been used21,63 and a semi-humanized mouse model for FVIII-deficiency.64 In both models, the use of an FVIII calibrator allowed to estimate the FVIII equivalence to be 10% to 20%. For fitusiran, FVIII-deficient mice displayed a strong reduction of their bleeding tendency.27 However, no FVIII calibrator curve was used to determine an FVIII equivalence. In an alternative approach using anti-antithrombin nanobodies, inhibition of antithrombin activity appeared to correspond to an FVIII equivalence of ≥20%.52 Regarding concizumab, a rabbit model for acquired hemophilia has been described.65 Also in this model, an FVIII calibrator was unavailable. Based on the observed efficiency in the cuticle bleeding model, the FVIII equivalence seems to be ≥20% FVIII.
Taking into account that the actual therapeutic dose that is now used in patients is 10-fold higher (∼55 μg/mL), it seems conceivable that the FVIII equivalence of emicizumab at these therapeutic levels will be around 10%. Indeed, in a more extensive analysis using this primate model for acquired hemophilia, the same group were able to estimate the FVIII equivalence to 10% to 20% FVIII.63
Primate studies are costly, and not accessible to all laboratories, which limits more in-depth studies to the in vivo behavior of emicizumab. Unfortunately, emicizumab is very specific to human and primate factors IX and X, complicating its assessment in more accessible animal models for hemophilia A, like mice, rats, and dogs. To overcome this limitation, we have developed an adapted mouse model (Figure 3) that combines FVIII deficiency with the presence of human factors IX and X.64 Without going into the specific details, as they will be outlined in a separate manuscript, this model allowed us to directly compare the activity of emicizumab to that of FVIII in a tail clip-bleeding model. Basically, mice were given different doses of FVIII to establish a calibrator curve, in which a specific FVIII concentration correlated with a specific amount of blood loss. We then measured blood loss in mice that were given emicizumab. The calibration curve with FVIII was subsequently used to estimate the FVIII-equivalence for emicizumab in this particular model. By doing so, we observe that the reduction in blood loss observed in the presence of therapeutic levels of emicizumab corresponds to an FVIII concentration of 9%, which is very close to the 10% estimated in the primate model.
Of course, each model has its limitations and a semihumanized mouse model is no exception. The FVIII equivalence obtained is true for this particular type of injury in this particular tissue (amputation of the terminal 3 mm of the tail tip). It is possible that in another bleeding model (eg, a tail vein transection or saphenous vein injury), the activity of emicizumab will be different. Its activity will be fully dependent on the local production of FIXa, which in turn is dependent on how much TF or FXIa will be present following the injury. It seems conceivable that this will also be true in the human setting: the severity of the injury and its location will determine how much FIXa will be generated and will be available to support emicizumab activity. Continuous clinical observation of patients receiving emicizumab will help us to further understand how efficient emicizumab is compared with FVIII in different conditions of injury.
As for fitusiran, a saphenous vein bleeding model has been used to investigate its in vivo efficacy in comparison with FVIII.27 FVIII-deficient mice were treated with fitusiran to obtain a reduction of 70% antithrombin antigen or with FVIII at a dose of 25 U/kg (which results in FVIII levels of ∼0.5 U/mL or 50%). In this model, the reduction in antithrombin proved as efficient as FVIII. However, it is unclear if lower levels of FVIII would have been sufficient to stop the bleeding in this model. Based on recent data using nanobodies that block antithrombin activity in a tail clip model using FVIII-deficient mice,52 it would be fair to assume that a reduction of antithrombin levels to 30% of normal corresponds to an FVIII-equivalence of at least 20% FVIII.
With regard to anti-TFPI antibodies, preclinical data on the analysis of concizumab (presented under its original name mAb2021) in a cuticle bleeding model in FVIII-deficient rabbits have been published.65 When given IV at a dosing range of 0.5 to 8 mg/kg 35 minutes before the induction of bleeding, a dose of 0.5 mg/kg was already sufficient to markedly reduce the bleeding tendency in this model. Moreover, all doses of ≥1 mg/kg reduced blood loss to levels observed in nonhemophilic rabbits. Unfortunately, it is not reported what were the levels of the antibody in plasma. Assuming a 90% availability and a plasma volume of 40 mL/kg in rabbits, the antibody concentration following a 1 mg/kg IV injection would be about 22 μg/mL. Noteworthy, an FVIII calibrator curve could not be used in this acquired hemophilia model, complicating the estimation of a true FVIII equivalence. However, given the strong reduction in blood loss it seems fair to assume that the FVIII equivalence is at least 20% FVIII.
Taken together, animal models provide an alternative approach to establish an FVIII equivalence for nonfactor therapies because they will generate a natural, unbiased response to an injury. By doing so, we should keep in mind that animal models are inherently hampered by species differences, which could influence the reactivity of these agents. Nevertheless, when considering the animal models currently available, it seems that emicizumab provides an FVIII equivalence of ∼10% to 20%. Unfortunately, FVIII calibrators were lacking in the reports assessing fitusiran and concizumab. Based on their capacity to fully correct the bleeding in these models, it seems fair to assume that their FVIII-equivalence is ≥20% FVIII.
Discussion
Laboratory monitoring of hemophilia A goes back a long time, and the presence of different sorts of FVIII (plasma-derived vs recombinant, plasma samples vs concentrates, B-domainless vs full-length, standard half-life vs extended half-life) has had an important impact on FVIII analysis. It is because of these complications that correct monitoring of FVIII activities are performed by specialized clinical laboratories. The advent of nonfactor therapies will add another layer of difficulty in this regard. For each patient, its treatment should be well indicated for the laboratory to perform the correct assay. In turn, clinical laboratories should be equipped with the necessary assays to be performed, whether it is needed to monitor levels of the therapeutic agent or to investigate how the patient reacts to the treatment.
As to the assays themselves, it is important to have a thorough biochemical knowledge on the properties of the novel therapies because this is pertinent for the interpretation of the obtained results. It is also relevant to have an understanding on how the novel therapies affect other clinical assays or how other types of medications affect the analysis of the nonfactor therapies, issues that have recently been discussed for emicizumab.37-39 Hemophilia treatment is entering into a new era, and its laboratory monitoring should follow in parallel.
Footnotes
Send data sharing requests to the corresponding author (peter.lenting@inserm.fr).
Authorship
Contribution: P.J.L. wrote the manuscript.
Conflict-of-interest disclosure: P.J.L. received speaker fees from Biotest, Chugai, Novo Nordisk, Roche, Sanofi, Sobi, and Takeda.
Correspondence: Peter J. Lenting, INSERM U1176, 80 Rue du General Leclerc, 94276 Le Kremlin-Bicêtre Cedex, France; e-mail: peter.lenting@inserm.fr.
References
- 1.Muczynski V, Christophe OD, Denis CV, Lenting PJ. Emerging therapeutic strategies in the treatment of hemophilia A. Semin Thromb Hemost. 2017;43(6):581-590. [DOI] [PubMed] [Google Scholar]
- 2.Sankar AD, Weyand AC, Pipe SW. The evolution of recombinant factor replacement for hemophilia. Transfus Apher Sci. 2019;58(5):596-600. [DOI] [PubMed] [Google Scholar]
- 3.Potgieter JJ, Damgaard M, Hillarp A. One-stage vs. chromogenic assays in haemophilia A. Eur J Haematol. 2015;94(suppl 77):38-44. [DOI] [PubMed] [Google Scholar]
- 4.Peyvandi F, Oldenburg J, Friedman KD. A critical appraisal of one-stage and chromogenic assays of factor VIII activity. J Thromb Haemost. 2016;14(2):248-261. [DOI] [PubMed] [Google Scholar]
- 5.Rosén S, Andersson M, Blombäck M, et al. Clinical application of a chromogenic substrate method for determination of factor VIII activity. Thromb Haemost. 1985;54(4):818-823. [PubMed] [Google Scholar]
- 6.Barrowcliffe TW. Factor VIII and Factor IX Sub-Committee. Recommendations for the assay of high-purity factor VIII concentrates. Thromb Haemost. 1993;70(5):876-877. [PubMed] [Google Scholar]
- 7.Raut S, Costanzo A, Daniels S, Heath A, Buchheit KH. Calibration of human coagulation factor VIII concentrate Ph. Eur. BRP Batch 4 for use in potency assays. Pharmeur Bio Sci Notes. 2010;2010(2):1-29. [PubMed] [Google Scholar]
- 8.Hubbard AR, Hamill M, Beeharry M, Bevan SA, Heath AB; SSC Sub-Committees on Factor VIII/Factor IX and von Willebrand factor of ISTH . Value assignment of the WHO 6th International Standard for blood coagulation factor VIII and von Willebrand factor in plasma (07/316). J Thromb Haemost. 2011;9(10):2100-2102. [DOI] [PubMed] [Google Scholar]
- 9.Raut S, Daniels S, Heath AB; SSC Sub-Committee on Factor . Value assignment of the WHO 8th International Standard for factor VIII, concentrate (07/350). J Thromb Haemost. 2012;10(6):1175-1176. [DOI] [PubMed] [Google Scholar]
- 10.Mikaelsson M, Oswaldsson U, Sandberg H. Influence of phospholipids on the assessment of factor VIII activity. Haemophilia. 1998;4(4):646-650. [DOI] [PubMed] [Google Scholar]
- 11.Hubbard AR, Weller LJ, Bevan SA. Activation profiles of factor VIII in concentrates reflect one-stage/chromogenic potency discrepancies. Br J Haematol. 2002;117(4):957-960. [DOI] [PubMed] [Google Scholar]
- 12.Morfini M, Cinotti S, Bellatreccia A, Paladino E, Gringeri A, Mannucci PM; ReFacto-AICE Study Group . A multicenter pharmacokinetic study of the B-domain deleted recombinant factor VIII concentrate using different assays and standards. J Thromb Haemost. 2003;1(11):2283-2289. [DOI] [PubMed] [Google Scholar]
- 13.Ingerslev J, Jankowski MA, Weston SB, Charles LA; ReFacto Field Study Participants . Collaborative field study on the utility of a BDD factor VIII concentrate standard in the estimation of BDDr Factor VIII:C activity in hemophilic plasma using one-stage clotting assays. J Thromb Haemost. 2004;2(4):623-628. [DOI] [PubMed] [Google Scholar]
- 14.Pouplard C, Ternisien C, Desconclois C, Lasne D, Aillaud MF, Caron C. Discrepancies between one stage assay and chromogenic substrate assay in patients treated with recombinant or plasma-derived FVIII and usefulness of a specific standard in ReFacto AF®-treated patients. Haemophilia. 2016;22(2):e101-e103. [DOI] [PubMed] [Google Scholar]
- 15.Kitchen S, Tiefenbacher S, Gosselin R. Factor activity assays for monitoring extended half-life factor VIII and factor IX replacement therapies. Semin Thromb Hemost. 2017;43(3):331-337. [DOI] [PubMed] [Google Scholar]
- 16.Weyand AC, Pipe SW. New therapies for hemophilia. Blood. 2019;133(5):389-398. [DOI] [PubMed] [Google Scholar]
- 17.Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;130(23):2463-2468. [DOI] [PubMed] [Google Scholar]
- 18.Spadarella G, Di Minno A, Milan G, et al. Paradigm shift for the treatment of hereditary haemophilia: towards precision medicine. Blood Rev. 2020;39:100618. [DOI] [PubMed] [Google Scholar]
- 19.Nogami K, Shima M. New therapies using nonfactor products for patients with hemophilia and inhibitors. Blood. 2019;133(5):399-406. [DOI] [PubMed] [Google Scholar]
- 20.Butterfield JSS, Hege KM, Herzog RW, Kaczmarek R. A molecular revolution in the treatment of hemophilia. Mol Ther. 2020;28(4):997-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570-1574. [DOI] [PubMed] [Google Scholar]
- 22.Kitazawa T, Esaki K, Tachibana T, et al. Factor VIIIa-mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117(7):1348-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809-818. [DOI] [PubMed] [Google Scholar]
- 24.Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379(9):811-822. [DOI] [PubMed] [Google Scholar]
- 25.Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study. Lancet Haematol. 2019;6(6):e295-e305. [DOI] [PubMed] [Google Scholar]
- 26.Young G, Liesner R, Chang T, et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134(24):2127-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sehgal A, Barros S, Ivanciu L, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. 2015;21(5):492-497. [DOI] [PubMed] [Google Scholar]
- 28.Van der Krol AR, Lenting PE, Veenstra J, et al. An anti-sense chalcone synthase gene in transgenic plants inhibits flower pigmentation. Nature. 1988;333(6176):866-869. [Google Scholar]
- 29.van der Krol AR, Mol JN, Stuitje AR. Antisense genes in plants: an overview. Gene. 1988;72(1-2):45-50. [DOI] [PubMed] [Google Scholar]
- 30.Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics [published correction appears in Nat Rev Drug Discov. 2020;19:290-291]. Nat Rev Drug Discov. 2019;18(6):421-446. [DOI] [PubMed] [Google Scholar]
- 31.Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377(9):819-828. [DOI] [PubMed] [Google Scholar]
- 32.Chowdary P. Anti-tissue factor pathway inhibitor (TFPI) therapy: a novel approach to the treatment of haemophilia. Int J Hematol. 2020;111(1):42-50. [DOI] [PubMed] [Google Scholar]
- 33.Peterson JA, Maroney SA, Mast AE. Targeting TFPI for hemophilia treatment. Thromb Res. 2016;141(suppl 2):S28-S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro AD, Angchaisuksiri P, Astermark J, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood. 2019;134(22):1973-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrante S, Ingham S, Kunze M, Michaels LA. Anti-TFPI antibody BAY 1093884: early termination of phase II dose escalation study due to thrombosis [abstract]. Haemophilia. 2020;26(S2):50. Abstract P099. [Google Scholar]
- 36.Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med. 2018;9:135-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adamkewicz JI, Chen DC, Paz-Priel I. Effects and interferences of emicizumab, a humanised bispecific antibody mimicking activated factor VIII cofactor function, on coagulation assays. Thromb Haemost. 2019;119(7):1084-1093. [DOI] [PubMed] [Google Scholar]
- 38.Tripodi A, Chantarangkul V, Novembrino C, Peyvandi F. Advances in the treatment of hemophilia: implications for laboratory testing. Clin Chem. 2019;65(2):254-262. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins PV, Bowyer A, Burgess C, et al. Laboratory coagulation tests and emicizumab treatment A United Kingdom Haemophilia Centre Doctors’ Organisation guideline. Haemophilia. 2020;26(1):151-155. [DOI] [PubMed] [Google Scholar]
- 40.Aleman MM, Leksa NC, Peters R, Salas J. Assay challenges (and opportunities) with non-factor VIII therapies for hemophilia A. Expert Rev Mol Diagn. 2019;19(1):1-3. [DOI] [PubMed] [Google Scholar]
- 41.Uchida N, Sambe T, Yoneyama K, et al. A first-in-human phase 1 study of ACE910, a novel factor VIII-mimetic bispecific antibody, in healthy subjects. Blood. 2016;127(13):1633-1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nogami K, Soeda T, Matsumoto T, Kawabe Y, Kitazawa T, Shima M. Routine measurements of factor VIII activity and inhibitor titer in the presence of emicizumab utilizing anti-idiotype monoclonal antibodies. J Thromb Haemost. 2018;16(7):1383-1390. [DOI] [PubMed] [Google Scholar]
- 43.Shinohara S, Saito T, Noguchi-Sasaki M, Ishiwata T, Morris M. Evaluation of emicizumab calibrator and controls with a modified one-stage FVIII assay on an automated coagulation analyzer [abstract]. Res Pract Thromb Haemost. 2019;3(suppl 1). Abstract PB1305. [Google Scholar]
- 44.Wilmot HV, Hogwood J, Williams S, et al. Laboratory measurement of emicizumab requires a product specific calibrator. Res Pract Thromb Haemost. 2019;3(suppl 1):PB1190. [Google Scholar]
- 45.Leksa NC, Aleman MM, Goodman AG, Rabinovich D, Peters R, Salas J. Intrinsic differences between FVIIIa mimetic bispecific antibodies and FVIII prevent assignment of FVIII-equivalence. J Thromb Haemost. 2019;17(7):1044-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cooper PC, Coath F, Daly ME, Makris M. The phenotypic and genetic assessment of antithrombin deficiency. Int J Lab Hematol. 2011;33(3):227-237. [DOI] [PubMed] [Google Scholar]
- 47.Dahm AE, Andersen TO, Rosendaal F, Sandset PM. A novel anticoagulant activity assay of tissue factor pathway inhibitor I (TFPI). J Thromb Haemost. 2005;3(4):651-658. [DOI] [PubMed] [Google Scholar]
- 48.Berrettini M, Malaspina M, Parise P, Lucarelli G, Kisiel W, Nenci GG. A simple chromogenic substrate assay of tissue factor pathway inhibitor activity in plasma and serum. Am J Clin Pathol. 1995;103(4):391-395. [DOI] [PubMed] [Google Scholar]
- 49.Maroney SA, Mast AE. New insights into the biology of tissue factor pathway inhibitor. J Thromb Haemost. 2015;13(suppl 1):S200-S207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calzavarini S, Brodard J, Quarroz C, et al. Thrombin generation measurement using the ST Genesia Thrombin Generation System in a cohort of healthy adults: normal values and variability. Res Pract Thromb Haemost. 2019;3(4):758-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lancé MD. A general review of major global coagulation assays: thrombelastography, thrombin generation test and clot waveform analysis. Thromb J. 2015;13(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barbon E, Ayme G, Mohamadi A, et al. Single-domain antibodies targeting antithrombin reduce bleeding in hemophilic mice with or without inhibitors. EMBO Mol Med. 2020;12(4):e11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marlu R, Polack B. Gla-domainless factor Xa: molecular bait to bypass a blocked tenase complex. Haematologica. 2012;97(8):1165-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kizilocak H, Yukhtman CL, Marquez-Casas E, Lee J, Donkin J, Young G. Management of perioperative hemostasis in a severe hemophilia A patient with inhibitors on emicizumab using global hemostasis assays. Ther Adv Hematol. 2019;10:2040620719860025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chowdary P, Lethagen S, Friedrich U, et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J Thromb Haemost. 2015;13(5):743-754. [DOI] [PubMed] [Google Scholar]
- 56.Patel-Hett S, Martin EJ, Mohammed BM, et al. Marstacimab, a tissue factor pathway inhibitor neutralizing antibody, improves coagulation parameters of ex vivo dosed haemophilic blood and plasmas. Haemophilia. 2019;25(5):797-806. [DOI] [PubMed] [Google Scholar]
- 57.Chelle P, Montmartin A, Piot M, et al. Prediction of individual factor VIII or IX level for the correction of thrombin generation in haemophilic patients. Haemophilia. 2018;24(6):995-1001. [DOI] [PubMed] [Google Scholar]
- 58.Olsson A, Hellgren M, Berntorp E, Holmström M, Baghaei F. Bleeding phenotype in carriers of haemophilia A does not correlate with thrombin generation. Haemophilia. 2015;21(1):e111-e113. [DOI] [PubMed] [Google Scholar]
- 59.Dargaud Y, Béguin S, Lienhart A, et al. Evaluation of thrombin generating capacity in plasma from patients with haemophilia A and B. Thromb Haemost. 2005;93(3):475-480. [DOI] [PubMed] [Google Scholar]
- 60.Santagostino E, Mancuso ME, Tripodi A, et al. Severe hemophilia with mild bleeding phenotype: molecular characterization and global coagulation profile. J Thromb Haemost. 2010;8(4):737-743. [DOI] [PubMed] [Google Scholar]
- 61.Young G, Sørensen B, Dargaud Y, Negrier C, Brummel-Ziedins K, Key NS. Thrombin generation and whole blood viscoelastic assays in the management of hemophilia: current state of art and future perspectives. Blood. 2013;121(11):1944-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dargaud Y, Lienhart A, Janbain M, Le Quellec S, Enjolras N, Negrier C. Use of thrombin generation assay to personalize treatment of breakthrough bleeds in a patient with hemophilia and inhibitors receiving prophylaxis with emicizumab. Haematologica. 2018;103(4):e181-e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muto A, Yoshihashi K, Takeda M, et al. Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12(2):206-213. [PubMed] [Google Scholar]
- 64.Ferrière S, Peyron I, Christophe OD, et al. A hemophilia A mouse model for the in vivo assessment of emicizumab function. Blood. In press. [DOI] [PubMed] [Google Scholar]
- 65.Hilden I, Lauritzen B, Sørensen BB, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119(24):5871-5878. [DOI] [PubMed] [Google Scholar]