

Abstract
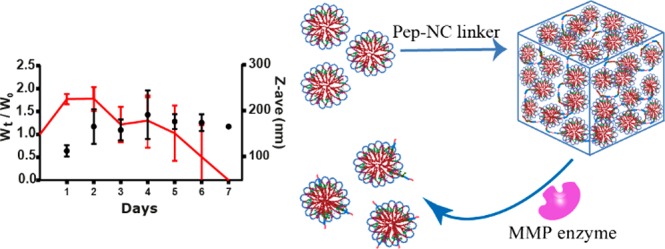
In this study, a new type of injectable hydrogel called “HyMic” that can convert into core cross-linked (CCL) micelles upon exposure to matrix metalloproteinases (MMP’s), was designed and developed for drug delivery applications. HyMic is composed of CCL micelles connected via an enzyme cleavable linker. To this end, two complementary ABA block copolymers with polyethylene glycol (PEG) as B block were synthesized using atom transfer radical polymerization (ATRP). The A blocks were composed of a random copolymer of N-isopropylacrylamide (NIPAM) and either N-(2-hydroxypropyl)methacrylamide-cysteine (HPMA-Cys) or N-(2-hydroxypropyl) methacrylamide-ethylthioglycolate succinic acid (HPMA-ETSA). Mixing the aqueous solutions of the obtained polymers and rising the temperature above the cloud point of the PNIPAM block resulted in the self-assembly of these polymers into flower-like micelles composed of a hydrophilic PEG shell and hydrophobic core. The micellar core was cross-linked by native chemical ligation between the cysteine (in HPMA-Cys) and thioester (in HPMA-ETSA) functionalities. A slight excess of thioester to cysteine groups (molar ratio 3:2) was used to allow further chemical reactions exploiting the unreacted thioester groups. The obtained micelles displayed a Z-average diameter of 80 ± 1 nm (PDI 0.1), and ζ-potential of −4.2 ± 0.4 mV and were linked using two types of pentablock copolymers of P(NIPAM-co-HPMA-Cys)-PEG-peptide-PEG-P(NIPAM-co-HPMA-Cys) (Pep-NC) to yield hydrogels. The pentablock copolymers were synthesized using a PEG-peptide-PEG ATRP macroinitiator and the peptide midblock (lysine-glycine-proline-glutamine-isoleucine-phenylalanine-glycine-glutamine-lysine (Lys-Gly-Pro-Gln-Gly-Ile-Phe-Gly-Gln-Lys)) consisted of either l- or d-amino acids (l-Pep-NC or d-Pep-NC), of which the l-amino acid sequence is a substrate for matrix metalloproteases 2 and 9 (MMPs 2 and 9). Upon mixing of the CCL micelles and the linker (l/d-Pep-NC), the cysteine functionalities of the l/d-Pep-NC reacted with remaining thioester moieties in the micellar core via native chemical ligation yielding a hydrogel within 160 min as demonstrated by rheological measurements. As anticipated, the gel cross-linked with l-Pep-NC was degraded in 7–45 days upon exposure to metalloproteases in a concentration-dependent manner, while the gel cross-linked with the d-Pep-NC remained intact even after 2 months. Dynamic light scattering analysis of the release medium revealed the presence of nanoparticles with a Z-average diameter of ∼120 nm (PDI < 0.3) and ζ-potential of ∼−3 mV, indicating release of core cross-linked micelles upon HyMic exposure to metalloproteases. An in vitro study demonstrated that the released CCL micelles were taken up by HeLa cells. Therefore, HyMic as an injectable and enzyme degradable hydrogel displaying controlled and on-demand release of CCL micelles has potential for intracellular drug delivery in tissues with upregulation of MMPs, for example, in cancer tissues.
1. Introduction
Hydrogels are three-dimensional networks of cross-linked hydrophilic polymers that can retain large amounts of waters while maintaining their structure.1,2 Hydrogels have been extensively studied for delivery of a variety of therapeutics ranging from small molecules3 to large proteins4,5 and nucleic acids.6,7 Various water-soluble therapeutics can be loaded into the hydrogel matrixes during their formation8,9 or encapsulated in carriers such as polymeric nanoparticles,10 liposomes,11,12 or micelles.13 Characteristics of the hydrogel such as pore size, swelling kinetics, and degradation mechanism play an important role in the release kinetics of loaded therapeutics.14 Regarding the release of drug-loaded nanoparticles from hydrogels, characteristics of the nanoparticles, in particular, size, charge, and stability, as well as uniform distribution into the gel matrix, affect particle release kinetics.15,16 In recent years, hydrogels that are converted into nanoparticles have gained interest for drug delivery applications. For instance, de Graaf et al. reported on the development of a drug-loaded hydrogel based on an ABA block polymer having A blocks of poly(N-isopropylacrylamide) PNIPAM and a B block of polyethylene glycol (PEG). They demonstrated that this hydrogel gradually and spontaneously converts into drug-loaded flower-like micelles.17 In other studies, micelles have been connected to each other using a linker exploiting, for example, an aldehyde and hydroxylamine reaction,18 radical polymerization,19 and metal–ligand interactions,20,21 to yield a macroscopic hydrogel structure. Although the mentioned systems showed interesting properties for drug delivery applications, they lack a triggered drug release mechanism upon disease-induced stimuli.
Hydrogels can be designed as stimuli-responsive materials that respond to signals from the surrounding environment resulting in, for example, triggered drug release.22−24 Incorporation of functional groups or enzyme responsive blocks such as trans azobenzene,25N-isopropylacrylamide (NIPAM),26 acrylic acid,27 or cleavable peptides28 in polymer structures can result in stimuli-responsive hydrogels. These materials can release the loaded therapeutics upon a trigger by, for example, light exposure, temperature, or pH changes, or the presence of enzymes upregulated in diseased tissues and organs.
Among stimuli-responsive materials, enzyme degradable hydrogels have shown to exhibit autoregulated degradation and accordingly they release their drug payload (for drug delivery purposes) or enhance cell migration (for tissue engineering purposes).29,30 For instance, Burdick et al.31 reported a matrix metalloproteinase (MMPs) responsive hydrogel loaded with a recombinant tissue inhibitor of MMPs (rTIMP-3). This polysaccharide-based hydrogel was cross-linked by MMP cleavable peptides. The authors demonstrated that the presence of MMP caused degradation of the gel and consequently release of the loaded rTIMP-3, which in turn resulted in a reduction in MMP activity in the overexpressed region.31 In another study, it was shown that MMP responsive blocks in a heparin/PEG hydrogel network in the presence of the matching enzyme enhanced cell viability and proliferation of mesenchymal stromal cells (MSC), which is important for cell-based cartilage regeneration.32
MMPs are upregulated in blood and tissues of patients with many types of human cancers.33,34 The expression of MMPs varies in different cancers as well as in different stages of the disease and can serve as a cancer biomarker.35 MMPs promote cancer progression by supporting cancer cell proliferation, migration, invasion, metastasis, and angiogenesis.36 Therefore, designing a therapeutic tool such as a hydrogel that undergoes degradation to yield nanoparticles triggered by overexpressed MMPs in pathological tissues, is of high interest for the intracellular delivery of, for example, anticancer drugs and biotherapeutics. In this contribution, an enzyme responsive hydrogel consisting of core-cross-linked (CCL) flower-like micelles named “HyMic” is developed and investigated. To construct HyMic, core cross-linked flower-like micelles based on two complementary thermosensitive ABA triblock copolymers of P(NIPAM-co-HPMA-Cys)-PEG-P(NIPAM-co-HPMA-Cys) (PNC) and P(NIPAM-co-HPMA-ETSA)-PEG-P(NIPAM-co-HPMA-ETSA) (PNE), were prepared using Native Chemical Ligation (NCL) as core-cross-linking method.37 The formed micelles were subsequently linked together using a pentablock copolymer of P(NIPAM-co-HPMA-Cys)-PEG-peptide-PEG-P(NIPAM-co-HPMA-Cys) (Pep-NC) yielding a micellar hydrogel network. The selected peptide block (Lys-Gly-Pro-Gln-Gly-Ile-Phe-Gly-Gln-Lys) is an MMP responsive sequence.38 Additionally, uptake of the released CCL micelles by HeLa cells (human epithelial cervix carcinoma cell line) was explored to investigate the possibility of intracellular drug delivery by HyMic.
2. Materials and Methods
2.1. Materials
All commercial chemicals were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands) and used as received unless indicated otherwise. N-(2-Hydroxypropyl)methacrylamide (HPMA) was synthesized by a reaction of methacryloyl chloride with 1-aminopropan-2-ol in dichloromethane according to a previously published procedure.39 Peptide grade dichloromethane (DCM), tetrahydrofuran (THF), and hexafluoro-2-propanol (HFIP) were obtained from Biosolve (Valkenswaard, The Netherlands). N-(2-Hydroxypropyl)methacrylamide-Boc-S-acetamidomethyl-l-cysteine (HPMA-Boc-Cys(Acm)) and N-(2-hydroxypropyl) methacrylamide-ethylthioglycolate succinic acid (HPMA-ETSA) were synthesized as described previously.37,40 Phosphate buffered saline 10× (PBS) pH 7.4 (1.37 M NaCl, 0.027 M KCl, and 0.119 M phosphates) BioReagents were purchased from B. Braun (Melsungen, Germany). Alexa Fluor 750 and 568 C5 maleimide dyes were obtained from Thermo Fisher Scientific (Massachusetts, U.S.). α-t-Butyloxycarbonylamino-ω-carboxy succinimidyl ester poly(ethylene glycol) (Boc-NH-PEG-NHS) (PEG-Mn 3 kDa) was purchased from Iris Biotech GMBH (Marktredwitz, Germany). Acetylated N-terminal l- and d-peptides (sequence: Lys-Gly-Pro-Gln-Gly-Ile-Phe-GLy-Gln-Lys) were purchased from GenScript (Leiden, The Netherlands). PD-10 desalting columns were purchased from GE Healthcare (Uppsala, Sweden). Dialysis tubes (molecular weight cutoff (MWCO) 3.5 and 10 kDa) were obtained from Fisher Scientific (Bleiswijk, The Netherlands). PEG standards (molecular weights ranging from 106 to 969000 Da) for GPC characterization were purchased from Agilent Technologies BV (Santa Clara, U.S.).
2.2. Polymer Synthesis and Characterization
2.2.1. Synthesis of d- or l-Pep-(NH2-PEG3k)2
d-Peptides and l-peptides (40 mg, 36 μmol; lysine-glycine-proline-glutamine-isoleucine-phenylalanine-glycine-glutamine-lysine (Lys-Gly-Pro-Gln-Gly-Ile-Phe-Gly-Gln-Lys), Figure 1) were separately dissolved in 2 mL of DMSO, followed by the addition of 60 μL of triethylamine. Subsequently, Boc-NH-PEG-NHS (221 mg, 72 μmol) was added and dissolved in the reaction mixture and allowed to react for 48 h at room temperature. The crude product, d- or l-Pep-(Boc-NH-PEG3k)2 was dialyzed against water (MWCO, 3.5 kDa) at room temperature for 2 days and subsequently lyophilized. The obtained product was analyzed by 1H NMR and GPC (NMR: Figure 2, SI-Figure 2B and GPC: Figure 3).
Figure 1.

Synthesis route for (A) d- or l-Pep-(Br-NH-PEG3k)2 ATRP macroinitiator, (B) ABA triblock copolymer of d- or l-Pep-NC containing d- or l-Pep-(PEG3k)2 as midblocks and copolymer of NIPAM and HPMA-Cys as outer-blocks.
Figure 2.

1H NMR spectra of the peptide Lys-Gly-Pro-Gln-Gly-Ile-Phe-Gly-Gln-Lys (1), peptide-PEG conjugated product with Boc protecting groups at the terminal ends (2), peptide-PEG conjugated polymers after Boc deprotection of the terminal amine groups (3), and the ATRP peptide-PEG macroinitiator (4). Deuterated DMSO was used as solvent.
Figure 3.

GPC chromatograms of starting compound (Boc-NH-PEG-NHS), peptide-conjugated products ((Boc-NH2-PEG 3 kDa)2-Pep and (NH2-PEG 3 kDa)2-Pep), ATRP peptide-PEG macroinitiator ((Br-C(CH3)2-CO-NH-PEG 3 kDa)2-Pep), and final products (d-Pep-NC and l-Pep-NC).
To remove the Boc protecting group from l/d-Pep-(Boc-NH-PEG)2, 200 mg of the polymers were dissolved in a 4 mL solution of dry DCM and trifluoroacetic acid (TFA; 1:3 v/v) and left to react for 1 h at room temperature. Subsequently, the solvents were evaporated, and the residues were dissolved in water and dialyzed against water (MWCO, 3.5 kDa) for 2 days at room temperature and then lyophilized. The obtained products were characterized by GPC (Figure 3) and 1H NMR (Figure 3, SI-Figure 2).
2.2.2. Synthesis of d- and l-Pep-(Br-C(CH3)2-CO-NH-PEG3k)2Macroinitiator for ATRP Polymerization
d- and l-Pep-(NH2-PEG3k)2 (220 mg, 30 μmol; section 2.2.1) were separately dissolved in 9 mL of dry THF, followed by the addition of 1 mL of hexafluoro-2-propanol (HFIP). Under a nitrogen atmosphere, triethylamine (20 μL) and α-bromoisobutyryl bromide (15 μL, 120 μmol; 2 equiv to the amine end groups of d- and l-Pep-(NH2-PEG3k)2) were added, and the reaction mixtures were stirred overnight at room temperature. Next, the formed ammonium bromide salts were filtered off and the solvents were evaporated under reduced pressure. The crude products were dissolved in water and the obtained solutions were dialyzed against water (MWCO, 3.5 kDa) for 2 days and subsequently lyophilized. The obtained macroinitiators were characterized by 1H NMR (Figure 2 and SI-Figures 2C and 3) and GPC (Figure 3 and SI-Figure 4).
2.2.3. Synthesis of P(NIPAM-co-HPMA-Cys)-PEG-Peptide-PEG-P(NIPAM-co-HPMA-Cys), Pep-NC
d- and l-Pep-(Br–C(CH3)2-CO-NH-PEG3k)2 macroinitiators (section 2.2.2; 60 mg, 8.0 μmol), together with CuBr (4.5 mg, 31 μmol), CuBr2 (4.7 mg, 21 μmol), NIPAM (264 mg; 2.3 mmol), and HPMA-Cys (67 mg, 0.16 mmol) were separately dissolved in a mixture of 2.0 mL of water and 0.84 mL of acetonitrile.41 The mixtures were stirred and deoxygenated by flushing with nitrogen for 15 min at room temperature, followed by 15 min in an ice bath. Polymerization started after the addition of 16 μL (0.06 mmol) of tris[2-(dimethylamino)ethyl]amine (Me6TREN) and the reaction mixtures were stirred for 2 h in an ice bath. Next, the formed polymers were diluted in water to 15 mL, then dialyzed (MWCO, 10 kDa) against water at room temperature for 1 day and subsequently lyophilized and analyzed by 1H NMR (SI-Figure 5A) and GPC (Figure 3). Finally, the Acm and Boc protecting groups of cysteine were removed as described previously.42 Briefly, the Boc-protecting group of cysteines were removed by dissolving 250 mg of the polymer in dry DCM and trifluoroacetic acid (TFA; 1:1 v/v, 10 mL) and the mixture was stirred for 1 h at room temperature in a nitrogen atmosphere. Subsequently, the solvent was evaporated under reduced pressure and the crude product was dissolved in 2 mL of DCM and subsequently precipitated in cold diethyl ether. The Acm protecting groups were removed by dissolving 250 mg of the Boc deprotected polymer in methanol (MeOH) and water (1:1 v/v, 10 mL) under a nitrogen atmosphere followed by the addition of 500 μL of HCl (1 M) and 1 mL of iodine 0.2 M in MeOH. The mixture was stirred for 1 h at room temperature, followed by the addition of 1 mL of ascorbic acid (1 M) in water to quench the excess of iodine. Subsequently, excess of tris(2-carboxyethyl)phosphine (TCEP; 250 mg) was added to reduce the formed disulfide bonds. The mixture was stirred overnight, dialyzed against water for 2 days at room temperature (MWCO, 10 kDa), and the final product was obtained after lyophilization. The obtained product was analyzed by 1H NMR (SI-Figure 5B) and GPC (SI-Figure 9). The same method was used for deprotection of (NIPAM-co-HPMA-Cys)-PEG-d-Peptide-PEG-P(NIPAM-co-HPMA-Cys) (d-Pep-NC). The obtained polymer was characterized by 1H NMR (SI-Figure 6A,B) and GPC (Figure 3, SI-Figure 9).
2.2.4. Synthesis of P(NIPAM-co-HPMA-Cys)-PEG-P(NIPAM-co-HPMA-Cys)(PNC) and P(NIPAM-co-HPMA-ETSA)-PEG-P(NIPAM-co-HPMA-ETSA) (PNE)
The polymerization solvent for the synthesis of PNC was a mixture of 2.8 mL of water and 0.9 mL of acetonitrile. For the synthesis of PNE, a mixture of 2.5 mL of water, 0.6 mL of acetonitrile, and 1.3 mL of DMSO was used. Poly(ethylene glycol) bis(2-bromoisobutyrate)37 (50 mg, 7.9 μmol), CuBr (4.5 mg, 31 μmol), CuBr2 (4.7 mg, 21 μmol), NIPAM (264 mg; 2.3 mmol), and either HPMA-ETSA (56 mg, 0.16 mmol) for the synthesis of PNE or HPMA-Boc-Cys(Acm) for the synthesis of PNC (67 mg, 0.16 mmol) were dissolved in the corresponding polymerization solvent. Next, the mixtures were deoxygenated by flushing with nitrogen for 15 min at room temperature and subsequently for 15 min upon cooling in an ice bath. To initiate the polymerization, the ligand Me6TREN (16 μL, 60 μmol) was added to the solution, and the reaction mixture was stirred for 2 and 5 h in an ice bath for PNC and PNE, respectively. The final products were diluted with 15 mL water and subsequently dialyzed (MWCO, 10 kDa) against water at room temperature for 1 day and lyophilized. The obtained polymers were characterized by 1H NMR (PNE, SI-Figure 7; protected PNC, SI-Figure 8A) and GPC (PNE, SI-Figure 10; PNC, SI-Figure 11A).
Finally, the Acm and Boc protecting groups of cysteine in protected PNC were removed as described in section 2.2.3. The obtained PNC was characterized by 1H NMR (SI-Figure 8B) and GPC (SI-Figure 11B).
2.2.5. NMR Spectroscopic Analysis
The obtained polymers were characterized by 1H NMR using a Bruker 600 UltraShield spectrometer (Billerica, Massachusetts, U.S.A.). The chemical shifts were calibrated against residual solvent peaks of CHCl3 (δ = 7.26 ppm) and DMSO (δ = 2.50 ppm).
2.2.6. Gel Permeation Chromatography (GPC) Analysis
The molecular weights and molecular weight distribution of the synthesized polymers were determined by GPC using a Waters Alliance 2414 System (Waters Corporation, Milford, MA) equipped with a refractive index detector. The separation was performed using 2 PLgel 5 μm Mixed-D columns (Polymer Laboratories, U.K.) at a temperature of 65 °C and DMF containing 10 mM LiCl at a flow of 1 mL·min–1 was used as mobile phase.43 Samples were prepared in the same solvent at a concentration of 5 mg·mL–1 and 50 μL was injected into the column for each measurement. A series of linear PEGs with narrow and defined molecular weights were used as calibration standards. Data were recorded and analyzed with Empower software v.3, 2010.
2.2.7. Determination of Cloud Points of Polymers
The cloud point (CP), defined as the onset of increasing scattering intensity, was measured using a Jasko FP-8300 spectrofluorometer (JASCO, Tokyo, Japan) at 650 nm. The polymers were dissolved at a concentration of 1 mg·mL–1 in PBS (0.13 M NaCl, 2.7 mM KCl, and 11.9 mM phosphates pH 7.4). The scattering intensity was monitored while heating the sample from 10 to 50 °C at 1 °C per minute.
2.2.8. Kinetics of Pep-NC Cleavage
To investigate the cleavage rate of the peptide in the Pep-NC polymer, 40 mg of l-Pep-NC polymer was dissolved in 8 mL of PBS (0.13 M NaCl, 2.7 mM KCl, 11.9 mM phosphates, 0.9 mM CaCl2, 0.02% NaN3, pH 7.4) at a concentration of 5 mg·mL–1 at 4 °C for 3 h and subsequently at 37 °C. Metalloprotease (type IV) collagenase from C. histolyticume (a mixture of enzymes with a molecular weight distribution 63–130 kDa) was used as a model enzyme for MMP-2 and MMP-9.44 Subsequently, polymer solutions were incubated with collagenase at a concentration of 0.5 units·mL–1 at 37 °C (this concentration is close to the total tissue concentration of MMP-1 and MMP-9 (500 ng·mL–1) reported for breast cancer).45 At several time points, 1 mL samples were withdrawn and diluted with 1 mL of an ethylenediaminetetraacetic acid (EDTA) solution (0.1 M) in PBS (0.13 M NaCl, 2.7 mM KCl, 11.9 mM phosphates, pH 7.4) and incubated for 1 h at room temperature. EDTA inhibits the enzyme activity via chelation of the metal ions (calcium and magnesium) required for catalytic activity.46,47 Next, the samples were dialyzed against water (MWCO, 10 kDa) for 2 days at 4 °C and subsequently lyophilized. d-Pep-NC at a polymer concentration of 5 mg·mL–1 was dissolved in 1 mL of PBS (0.13 M NaCl, 2.7 mM KCl, 11.9 mM phosphates, 0.9 mM CaCl2, 0.02% NaN3, pH 7.4) and incubated with collagenase at a concentration of 30 units·mL–1 for 24 h. Next, the sample was treated with 1 mL of EDTA solution (0.1 M) for 1 h and then dialyzed against water (MWCO, 10 kDa) for 2 days at 4 °C and subsequently lyophilized. The obtained polymers were dissolved at a concentration of 5 mg·mL–1 in DMF containing 10 mM LiCl, and the molecular weights were analyzed by GPC (section 2.2.6). The results are shown in Figure 4 and SI-Figure 12.
Figure 4.

(A) Formation of flower-like micelles from pentablock copolymer Pep-NC and cleavage of peptide midblock by collagenase. (B) GPC chromatograms (IR detection) of l-Pep-NC incubated with collagenase (0.5 units·mL–1) at different times at 37 °C. (C) Mn of l-Pep-NC as a function of time, as determined by GPC.
2.3. Micelle Preparation and Characterization
2.3.1. Micelle Preparation
Micelles were formed by a fast heating method as follows: PNC and PNE were dissolved separately in PBS (0.13 M NaCl, 2.7 mM KCl, and 11.9 mM phosphates, pH 7.4) at a concentration of 10 mg·mL–1 and left at 4 °C for 3 h. Subsequently, the solutions were mixed in a 2:3 (PNC:PNE) volume ratio (i.e., the molar ratio of HPMA-Cys to HPMA-ETSA was 2:3). Subsequently, the mixture was heated to 50 °C using an oil bath and stirred at this temperature overnight. The resulting micellar dispersion was dialyzed against water (MWCO, 10 kDa) for 2 days at room temperature and subsequently lyophilized. The obtained micelles were characterized by DLS before and after lyophilization (Figure 7A,C).
Figure 7.

DLS analysis of the release medium of l- and d-HyMic upon incubation with collagenase: (A) Z-average, (B) derived count rate, (C) PDI, and (D) the percentage of the released dye-conjugated micelles from HyMic (n = 3). Fluorescence imaging of (E) the release medium and (F) HyMic hydrogels incubated with collagenase at 30 units·mL–1 (incubation time for l-HyMic and d-HyMic were 2 and 30 days, respectively).
2.3.2. Preparation of Fluorescently Labeled Micelles
Micelles were formed following the same procedure as described in section 2.3.1. Then, the buffer medium was refreshed using a PD-10 desalting column equilibrated with PBS (0.13 M NaCl, 2.7 mM KCl, and 11.9 mM phosphates, pH 7.4) according to the method provided by the supplier. Subsequently, 10 μL of a 20 μg·mL–1 maleimide-Alexa fluor C5 750 or C5 568 solution in DMSO was added to 1 mL of micellar dispersion (10 mg·mL–1) and left to react at 4 °C for 2 days. Next, the labeled micelles were dialyzed against water (MWCO, 10 kDa) at room temperature for 1 day and subsequently purified three times by PD-10 columns equilibrated with deionized water and finally lyophilized.
2.3.3. Dynamic Light Scattering (DLS)
The size of the obtained micelles at a concentration of 1 mg·mL–1 in PBS (0.13 M NaCl, 2.7 mM KCl, and 11.9 mM phosphates, pH 7.4) was measured by DLS using a Zetasizer Nano S (ZEN 1600; Malvern Instruments Ltd., Malvern, U.K.) equipped with a He–Ne 4 mW, 632.8 nm laser. The measurements were carried out at a 173° angle at 37 °C controlled by the instrument. The Z-average and polydispersity index were calculated by Zetasizer software v7.13. The solvent viscosity was corrected for temperature changes by the software.
2.3.4. Zeta Potential
The obtained micelles in PBS were diluted (1:20) in HEPES (20 mM, pH 7.0) to the final concentration of 0.5 mg·mL–1 and their zeta potential was measured at 37 °C using a Zetasizer Nano Z (Malvern Instruments Ltd., Malvern, U.K.).
2.4. HyMic Preparation, Characterization, and Degradation
2.4.1. Preparation of Micellar Hydrogel (HyMic)
HyMic was prepared at a total polymer concentration of 20 wt % as follows: 6 mg of lyophilized micelles were weighed in a 1.5 mL Eppendorf vial followed by the addition of 15 μL of PBS (0.13 M NaCl, 2.7 mM KCl and 11.9 mM phosphates, pH 7.4), and hydrated for 1 h at 4 °C. In a separate vial, 15 μL of PBS was added to 1.3 mg of d- or l-Pep-NC (23 wt % of the amount of micelles) and left to dissolve for 1 h at 4 °C. Next, the d- or l-Pep-NC solution was added to the micellar dispersions and the mixtures were incubated at 4 °C for 1 h and subsequently incubated for 6 h at 37 °C for hydrogel formation.
2.4.2. Rheological Characterization
Rheological analysis of the hydrogel samples and micellar dispersions was performed on a DHR-2 rheometer (TA Instrument, New Castel, DE) using a 20 mm aluminum cone (1°) geometry equipped with a solvent trap. Time sweeps were performed for 3 h at 37 °C at a frequency of 1 Hz and 1% strain. For each measurement, 70 μL samples were used.
2.4.3. Swelling and Degradation Study
HyMic (fluorescently labeled) hydrogels with a volume of ∼30 μL were prepared as described in section 2.4.1. The obtained hydrogels were transferred into 2 mL glass vials and the gel weights were recorded (W0). The samples were then immersed into 500 μL of PBS (0.13 M NaCl, 2.7 mM KCl, 11.9 mM phosphates, 0.9 mM CaCl2, 0.02% NaN3, pH 7.4) with 0.0, 7.5, 15.0, or 30.0 units·mL–1 of collagenase and incubated at 37 °C. At regular time points, the medium was removed, and the weight of the gel was recorded (Wt), and subsequently, 500 μL of fresh medium was added and the samples were further incubated at 37 °C. The gel release medium was analyzed by DLS for size, polydispersity index (PDI), and derived count rate of the released core cross-linked micelles. In addition, the fluorescence intensity of the supernatant was analyzed as described in section 2.4.4. The swelling and degradation were recorded three times a week until complete degradation or until 2 months after which the experiment was stopped. The swelling ratio (SR = Wt/W0) is reported as the weight of the gel at a certain time point (Wt) divided by the initial gel weight (W0). The mesh sizes of the hydrogels were estimated based on the following equation:
where NA is the Avogadro constant, R is the molar gas constant, and T is temperature.
For cell internalization study, hydrogels composed of Cy5-conjugated micelles were made using the l-Pep-NC linker. These samples were incubated with collagenase at a concentration of 10 units·mL–1. The fluorescence intensity of the medium was measured in time to quantify the concentration of the released micelles. On day 21, the concentration of the released CCL micelles reached ∼8 mg·mL–1. The released micelles were used for incubation with cells, as detailed in section 2.5.
2.4.4. Fluorescence Intensity Measurement
To measure the fluorescent signals of Cy7 labeled micelles in the gel release medium, 30 μL of the release medium was transferred into a clear 384-well plate and analyzed using Odyssey infrared scanner (LI-COR Biosciences, Westburg, The Netherlands) at 700 nm. To measure the fluorescent intensity of the Cy5-conjugated micelles (released from hydrogel and used in cell study), 100 μL of release medium was transferred into a black 384-well plate and analyzed using a Jasko FP-8300 spectrofluorometer (JASCO, Tokyo, Japan). The excitation and emission wavelengths were set at 578 and 603 nm, respectively. Standard curves of the corresponding dye conjugated micelles were used for quantification of fluorescent signals.
Images of the gels and release medium were taken by a LI-COR Pearl impulse imager (LI-COR, Lincoln, Nebraska, U.S.A.).
2.5. Cellular Internalization Study
Cellular uptake of the released micelles was investigated using HeLa cells. The cells were seeded in a glass-bottomed 96 well-plate at a density of 8000 cells/well and incubated at 37 °C for 24 h. Then, the fluorescently labeled micelles either released from the gel or control micelles (freshly prepared micelles that were not converted into a gel) were added at a concentration of 400 μg·mL–1 for 1, 6, and 24 h at 37 °C. The cells were washed twice with PBS (0.13 M NaCl, 2.7 mM KCl, 11.9 mM phosphates, pH 7.4), and the plate was transferred into a Yokogawa CV7000 (Tokyo, Japan) spinning disk microscope with a 60× 1.2NA water objective.
3. Results and Discussion
3.1. Polymer Synthesis and Characterization
3.1.1. Synthesis and Characterization of Peptide-PEG ATRP Macroinitiator
Figure 1A shows the three-step synthesis route of the PEG-Pep-PEG atom transfer radical polymerization (ATRP)48 macroinitiator. The commercial Boc-NH-PEG-NHS (Figure 1, step A-1, compound 2) was characterized by GPC and 1H NMR. NMR analysis confirmed the presence of tert-butyloxycarbonyl (Boc) and succinimidyl ester (NHS) groups in compound 2 with a molar ratio of 1:1 (SI-Figure 2A). GPC characterization displayed a peak at 15.6 min corresponding to polymer with a number-average molecular weight (Mn) of 3.4 kDa in agreement with the specifications of the supplier and a shoulder at a retention time of 14.7 min (∼7% of the total peak area), corresponding with a polymer of Mn = 7.3 kDa (Figure 3). This shoulder can most likely be attributed to the presence of PEG chains of higher molecular weight possibly also derivatized with NHS and Boc functionalities. The two free amines of the N-terminus acetylated Lys-Gly-Pro-Gln-Gly-Ile-Phe-Gly-Gln-Lys were conjugated to the NHS-end group of PEG (Figure 1, step A-1). After dialysis and lyophilization, PEG-Pep-PEG (Figure 1, step A-1, compound 3) was obtained in a yield of 87%. The 1H NMR spectrum showed that the molar ratio of the phenyl group of phenylalanine in the peptide sequence and Boc group in the PEG was 1:2 (Figure 2-2 and SI-Figure 2B), demonstrating the successful synthesis of the macroinitiator. Additionally, GPC analysis showed that the retention time of compound 3 was shifted to a lower retention time representing a polymer of higher molecular weight (Mn: 7.8 kDa; Figure 3), which again confirms the formation of PEG-Pep-PEG. A small peak with a retention time of 15 min (∼7% of the total peak area) was detected, which corresponds to nonconjugated PEG (Mn: 3.4 kDa), while the peak at 13.8 min may be assigned to conjugation of peptide to the higher Mn PEG derivative present in the commercial starting compound 2. The Boc groups at both ends of the PEG-Pep-PEG were removed by trifluoroacetic acid (TFA) and the crude product was dialyzed and lyophilized to result in deprotected PEG-Pep-PEG in a yield of 79% (Figure 1, step A-2, compound 4). 1H NMR analysis of compound 4 showed that the signal corresponding to Boc at δ = 1.4 had indeed disappeared (Figure 2-3), demonstrating the quantitative removal of Boc groups. As expected, the molecular weight of the deprotected PEG-Pep-PEG (Mn: 8.1) was similar to the Boc protected PEG-Pep-PEG (Mn: 8.3 kDa) (Figure 3). It should be noted that the acetyl protecting group of the N-terminal peptide is stable under the deprotection procedure applied,49 which means that there is no risk for functionalization of this moiety by ATRP initiator. To functionalize the PEG-Pep-PEG with an ATRP initiator group at both chain ends, the free terminal amine groups were reacted with α-bromoisobutyryl bromide to result in a PEG-Pep-PEG ATRP macroinitiator with a yield of 78% (Figure 1, step A-3, compound 6). The presence of the 1H NMR signal at 1.8 ppm belonging to the methyl groups of the ATRP initiator (12 protons per polymer chain) confirmed that all chains were functionalized with an ATRP initiator (Figure 2-4; SI-Figure 2C). GPC and 1H NMR showed an Mn of 8.6 and 7.5 kDa for this macroinitiator, respectively. As a control, the same peptide sequence made of unnatural amino acids (d-amino acids) was conjugated to PEG and subsequently to the ATRP initiator. The resulting initiator was obtained in the yield of 71% and had an Mn of 7.9 kDa, as determined by GPC (SI-Figure 4). 1H NMR analysis confirmed quantitative modification of the chain ends by α-bromoisobutyryl bromide (SI-Figure 3).
3.1.2. Synthesis and Characterization of Thermosensitive ABCBA Pentablock Copolymers and ABA Triblock Copolymers
The pentablock (ABCBA) copolymers were synthesized by ATRP using the above-described PEG-Pep-PEG macroinitiator (PEG as B block and peptide as C block; section 3.1.1). The polymer structure is shown in Figure 1B and SI-Figure 1, and the polymer characteristics are summarized in Table 1. The A blocks consisting of NIPAM (N) and HPMA-Cys (C) with a feed molar ratio of 93:7 were polymerized from either the l- or d-peptide-PEG macroinitiator (l-Pep or d-Pep) using a previously established method37 (Figure 1B). In the present work, the resulting copolymers are referred to as l-Pep-NC and d-Pep-NC. Incorporation of HPMA-Cys in the thermosensitive domain provides cysteine functionalities that can be exploited for native chemical ligation (NCL).50 After polymerization, the final products were obtained after dialysis and lyophilization in high yields (82–87%).
Table 1. Characteristics of PNC and PNE ABA Triblock Copolymers Containing a PEG B-block of 6 kDa and ABCBA Pentablock Copolymers: l-Pep-NC and d-Pep-NC containing l-Pep-(PEG3k)2 or d-Pep-(PEG3k)2 as Mid BCB-blocks, Respectivelya.
| obtained
molar ratiob |
|||||||
|---|---|---|---|---|---|---|---|
| polymer | [NIPAM]:[HPMA-Boc-Cys-(Acm)] | [NIPAM]:[HPMA-ETSA] | Mnb (kDa) | Mnc (kDa) | PDIc | CP (°C) | yield (%) |
| PNC | 91:9 | 43.6 | 58.9 | 1.42 | 34.1d | 93 | |
| PNE | 92:8 | 40.1 | 64.1 | 1.78 | 29.2 | 88 | |
| l-Pep-NC | 90:10 | 44.8 | 62.5 | 1.75 | 31.8d | 87 | |
| d-Pep-NC | 91:9 | 44.2 | 64.3 | 1.72 | 31.4d | 82 | |
The outer blocks of the different polymers are composed of either NIPAM and HPMA-Boc-Cys-(Acm) (PNC, l-Pep-NC, and d-Pep-NC) or NIPAM and HPMA-ETSA (PNE). In all polymerizations, the feed molar ratio of NIPAM to either HPMA-Boc-Cys-(Acm) or HPMA-ETSA was 93:7.
Determined by 1H NMR.
Determined by GPC.
Cloud point of the deprotected polymer.
Two complementary thermosensitive triblock copolymers of P(NIPAM-co-HPMA-Cys)-PEG-P(NIPAM-co-HPMA-Cys) (PNC) and P(NIPAM-co-HPMA-ETSA)-PEG-P(NIPAM-co-HPMA-ETSA) (PNE) (SI-Figure 1) are needed for micelle formation, as reported before.37 PNC and PNE were synthesized by ATRP using a PEG ATRP macroinitiator37 (Mn: 6 kDa) and a feed molar ratio of NIPAM/HPMA-Cys or NIPAM/ETSA of 93:7. After polymerization and purification by dialysis, the final polymers (protected) PNC and PNE were obtained in yields of 93 and 88%, respectively. The Mns, as determined by GPC, were higher than Mns obtained by NMR analysis, which have been observed before for PNIPAM-based (co)polymers due to inter- and intramolecular hydrogen bonding.51,52 The PDIs of the obtained polymers (1.4–1.7) were similar to what has been reported for these types of polymers. The different reactivities of acrylamide (NIPAM) and methacrylamide (HPMA) have been shown to result in blocky structures in PNC and PNE polymers rather than a random distribution of the two types of monomers.37
The cysteine moieties in HPMA-Cys carry two protecting groups, namely, a Boc group on the terminal amine groups and an Acm group on the thiol side chain groups. Therefore, the polymers containing HPMA-Cys groups underwent deprotection of Boc groups under acidic conditions and an inert atmosphere.53 Thiol protecting groups (Acm) were removed by first oxidation by iodine under acidic conditions and were subsequently reduced to free thiols using TCEP.40,54
The synthesized PNC and PNE polymers exhibit a cloud point (CP) in aqueous solutions, attributed to the thermosensitive PNIPAM block,26 at 34.1 and 29.2 °C, respectively, which is similar to values reported before for the same polymers.37 The CPs of the Pep-NC polymers were very similar (31.8 and 31.4 °C for l- and d-Pep-NC, respectively) and slightly lower than the CP of PNC (34.1 °C), likely because the peptide sequence in Pep-NCs contains several hydrophobic amino acid residues55 (Table 1).
3.1.3. Enzymatic Cleavage of the Thermosensitive ABCBA Pentablock Polymers (Pep-NC)
The pentablock copolymer, Pep-NC, forms flower-like micelles in PBS with a Z-average of 87 ± 1 nm (PDI 0.1) at temperatures above the lower critical solution temperature (LCST ∼ 31 °C, Table 1; Figure 4A). To study the accessibility of the peptide block in the polymer backbone for metalloproteases, d/l-Pep-NC polymers were incubated with metalloprotease type IV collagenase (a model for MMP types 2 and 9) above the LCST. Analysis of the obtained polymers by GPC showed that the molecular weight of d-Pep-NC did not change after incubation with collagenase while that of l-Pep-NC reduced to half its original molecular weight under the same conditions (SI-Figure 12). These results demonstrate that the peptide sequence in l-Pep-NC in the micelles is indeed accessible for collagenase. As expected, cleavage of the peptide block in d-Pep-NC by the enzyme did not occur.
To investigate the kinetics of peptide cleavage of l-Pep-NC, the polymer was incubated with and without collagenase at 37 °C and thus above the LCST of the polymer in PBS and samples were collected at different time points and analyzed by GPC (Figure 4B,C). No change in the molecular weight was observed in the absence of the enzyme even after 24 h of incubation (SI-Figure 12). In contrast, in the presence of collagenase, a gradual increase in the retention time as a function of incubation time was observed. The change in the retention time (from 11.9 to 12.7 min) corresponds to a decrease in Mn from 64 to 35 kDa indicating cleavage of the peptide block within 160 min. The cleavage of the peptide at 37 °C (above LCST of the Pep-NC) demonstrates the accessibility of the peptide block for the enzyme, even though other reported PNIPAM-peptide conjugates have shown the opposite.56 The cleavage of the peptide in the l-Pep-NC pentablock copolymer can be explained by the presence of PEG as flanking blocks that force the peptide blocks to be exposed in the loops of hydrophilic PEG shells of the flower-like micelles formed above the LCST (Figure 4A).
3.2. Preparation of Micellar Hydrogel (HyMic), Characterization, and Degradation
3.2.1. Preparation of Micellar Hydrogel and Characterization
Preparation of HyMic was performed in two steps. In the first step, PNC and PNE polymer solutions were mixed followed by increasing the temperature above LCST of the polymers, which resulted in self-assembly of the polymers into flower-like micelles. The micellar core was cross-linked by native chemical ligation of the cysteine and thioester functionalities present in PNC (HPMA-Cys) and PNE (HPMA-ETSA), respectively.37 The obtained micelles displayed a Z-average of 80 ± 1 nm (PDI 0.09) and ζ-potential of −4.2 ± 0.4 at 37 °C. Subsequently, the prepared micelles were purified by dialysis against water and then lyophilized without cryo-protectant. The slight increase in their size after lyophilization of micelles from 80 ± 1 to 93 ± 2 nm (PDI: 0.1) indicates that aggregation occurred to a limited degree. The excess of PNE for the preparation of micelles resulted in reactive thioester functionalities in the micellar core, which can be used for bridging the micelles using Pep-NC linkers via native chemical ligation to yield a hydrogel network. In the second step, Pep-NC having free cysteine moieties was added to the CCL micelle dispersion with free thioester functionalities in the micellar cores at a temperature below the LCST of PNIPAM. At this temperature, Pep-NC and the polymeric chains in the core of the CCL micelles are swollen and consequently, Pep-NC molecules can diffuse into the hydrated core of the CCL micelles. The close proximity of thioester functionalities in the core of micelles to the cysteine functionalities in the Pep-NC linker facilitates native chemical ligation and triggers gel formation. Subsequent incubation of the CCL micelle-Pep-NC mixture above the LCST of the PNIPAM blocks resulted in a hydrogel, while the sample composed of only CCL micelles displayed a low viscous dispersion (Figure 5A). The storage modulus of sample composed of only micelles remained constant (around 2 Pa) during the entire experiment (SI-Figure 13). In more detail, the CCL micelle-Pep-NC mixture displayed a G′ of 600 Pa shortly after heating to 37 °C, which increased up to 1300 Pa after 160 min. A decrease in tan δ from 0.20 to 0.06 confirmed formation of a chemically cross-linked hydrogel. The relatively high G′ (and low tan δ) value of the CCL micelle-Pep-NC mixture at the start of the measurement can likely be attributed to cross-linking of the micelles that occurred during sample preparation and before the start of the measurement. Although the G′ is relatively high (600 Pa), 150 μL of the CCL micelle-Pep-NC mixture was easily passed through a needle (gauge 23) at 37 °C; therefore, it can be considered as an injectable material at body temperature. This is in line with what has been shown before by Van Tomme et al. that hydrogels with G′ < 4000 Pa are suitable for injection.57 These results demonstrate the formation of a cross-linked network of micelles, which is further abbreviated as “HyMic” (Figure 5A). The overall gel concentration can be adjusted according to the final application and the required gel stiffness. Stable gels can be formed at polymer concentrations above 12% and at least up to 30%. The gel stiffness can be increased by increasing the total polymer concentration. However, the ratio between the cysteine functionalities in the linker and thioester functionalities in the micellar core should be considered for optimal cross-linking.
Figure 5.

Formation of HyMic (A) lyophilized core cross-linked flower-like micelles were dispersed in PBS and subsequently mixed with l-Pep-NC to form a hydrogel. (B) Storage modulus (G′) and tan δ as a function of time for micelles after mixing with l-Pep-NC at 37 °C.
3.2.2. Enzymatic Degradation of HyMic
As shown in section 3.1.3, the peptide midblock in the l-Pep-NC polymer can be cleaved by collagenase. To investigate the accessibility of l-Pep-NC in the gel structure and consequently enzyme responsivity of HyMic, the gel composed of CCL micelles and the l-Pep-NC (l-HyMic) was incubated with collagenase at different concentrations at 37 °C (Figure 6A). HyMic composed of CCL micelles and d-Pep-NC (d-HyMic) was only treated with the highest concentration of collagenase used for l-HyMic treatment (30 units·mL–1).
Figure 6.

(A) Enzymatic degradation of l-HyMic at different concentrations of collagenase. (B) Degradation time as a function of enzyme concentration at 37 °C and pH 7.4. Degradation time is reported as the recorded time for full degradation of a gel (n = 3).
Both types of gels displayed a maximum swelling ratio of 2.0 and no gel erosion was observed in the absence of the enzyme. l-HyMic exhibited complete degradation in 7 days at an enzyme concentration of 30 units·mL–1, while d-HyMic remained intact even after 2 months at the same enzyme concentration (Figure 6A), which demonstrates that degradation is indeed triggered by cleavage of l-Pep-NC linker. Interestingly, the degradation rate of l-HyMic was significantly slower than of l-Pep-NC (7 days vs 160 min (Figure 4)), even though the substrate to enzyme ratio was 5× lower (see section 3.1.3). This slower degradation of l-HyMic could be attributed to the limited accessibility of collagenase to the peptide block in the hydrogel network. The gel degradation as a function of enzyme concentration (Figure 6) can reveal insights into the degradation mechanism. The observed degradation times were 45, 21, and 7 days in the presence of 7.5, 15.0, and 30.0 units of enzyme per mL, respectively. The samples incubated with 30 units·mL–1 exhibited a weight loss after reaching maximum swelling, while the gels incubated with 15 and 7.5 units·mL–1 of collagenase showed a constant gel weight for about 10 and 25 days, respectively (Figure 6A). The plateau value in hydrogel weight can be ascribed to absorption of water due to a decreasing cross-link density of the hydrogel network, which is compensated by shedded particles. At the decaying point of the graph (Figure 6A; around days 10 and 25 for gels incubated with 15 and 7.5 units·mL–1 of collagenase, respectively), the gel network became very weak, resulting in rapid disintegration of the gels. Figure 6B shows that the degradation time of the gels decreased with increasing enzyme concentration. It is known that, for surface erosion, the degradation rate is not affected by enzyme concentration above a certain concentration due to saturation of the surface with enzyme molecules. On the other hand, for bulk degradation the degradation rate increases with increasing enzyme concentration.58,59 Estimation of hydrogel mesh size (ζ) based on rubber elasticity theory60,61 (equation in section 2.4.3) showed a mesh size of ∼7 nm for the gel with a G′ of 1300 Pa. This means that collagenase with a molecular weight of 63–130 kDa62,63 (Rh ∼ 3.5–4.5 nm)64 can penetrate into the hydrogel network and initiate bulk degradation. Therefore, the observed degradation is very likely due to combination of bulk degradation and surface erosion.
The release medium of the l-HyMic and d-HyMic hydrogels incubated with 30 units·mL–1 of collagenase was refreshed daily and analyzed using DLS. Additionally, the fluorescence intensity of the release medium was measured to determine the concentration of the released dye-conjugated CCL micelles (Figure 7). The d-HyMic release medium only exhibited a detectable signal for the derived count rate on the first day, which was much lower than the recorded value for l-HyMic (1800 vs 48000). From day 2 on, no signal above background was recorded, and thus, no nanoparticles were present in the l-HyMic release medium (Figure 7B). The released particles at day one exhibited a Z-average of ∼100 nm and PDI of 0.1, suggesting the release of intact CCL micelles (Figure 7A,C). Measuring the concentration of the released dye-conjugated CCL micelles using fluorescence showed a 10% release of the CCL micelles on the first day (Figure 7D). Taken together, during the first day, CCL micelles were released from the d-HyMic that were not connected to the gel network. Clearly, collagenase is unable to cleave d-Pep-NC to result in shedding of CCL micelles over time.
In contrast to d-HyMic, the release medium of l-HyMic exhibited high values for the derived count rate in the first days, which decreased over time (Figure 7B). The released particles displayed a size of ∼110 (PDI ∼ 0.2), which raised to ∼200 nm (PDI ∼ 0.3) from day 2 on. The high derived count rate values in the first days can be explained by the release of CCL micelles on the surface, which may have a low number of connections to the micellar network. The latter can also explain the similar size and PDI of the released particles compared with lyophilized micelles that were used for l-HyMic formation (Figure 7A). The bigger size and PDI after the first day indicate the release of nanosized gel fragments composed of more than one CCL micelle (Figure 7A). Measuring the fluorescence intensity of the released dye-conjugated CCL micelles showed that approximately 50% of the CCL micelles were released upon gel exposure to 30 unit·mL–1 collagenase at day one and the CCL micelle release was completed after 7 days. This is in line with the high value of the derived count rated on the first day (Figure 7B), low swelling ratio, and immediate weight loss of this gel (Figure 6A). Fluorescence imaging of l-HyMic and d-HyMic hydrogels and their release medium was performed after 2 and 30 days incubation with collagenase (Figure 7E,F). As expected, a strong fluorescent signal was detected in the release medium of l-HyMic, while the signal in d-HyMic release medium was below the detection limit. Interestingly, fluorescence imaging showed a fluffy structure for the l-HyMic gel, while the d-HyMic displayed a dense structure even after a one month incubation with collagenase. This fluffy swollen structure is most likely due to cleavage of Pep-NC linkers inside the gel network due to diffusion of the enzyme in the hydrogel matrix, as mentioned above. This observation supports that degradation of the hydrogel in the presence of collagenase is due to a combination of surface erosion and bulk degradation. Such a micellar hydrogel could be injected in tumor tissues. The upregulation of MMPs in tumor tissues would then trigger the gel degradation, resulting in the release of micelles. When micelles are loaded with anticancer therapeutics, the internalization of the released micelles in cancer cells can cause cytotoxicity and, consequently, tumor regression.
3.3. Cellular Internalization of CCL Micelles by HeLa Cells
The suitability of the released CCL upon enzymatic degradation of l-HyMic for intracellular drug delivery was investigated using HeLa cells. To this end, dye-conjugated CCL micelles were formulated into HyMic using l-Pep-NC and subsequently incubated with collagenase. The concentration of the released CCL micelles was monitored by measuring the dye concentration in the release medium. The released CCL micelles obtained after 21 days of incubation with collagenase displayed a Z-average of 120 ± 2 nm (PDI 0.2) and ζ-potential of −2.7 ± 0.0 at 37 °C. The confocal images (Figure 8) showed punctate fluorescence, confirming the internalization of both control micelles and released CCL micelles after 24 h incubation with the cells. The uptake of released CCL micelles from l-HyMic upon its enzymatic degradation shows the potential of enzymatic cleavable l-HyMic for intracellular drug delivery.
Figure 8.

Internalization of freshly prepared micelles and micelles released from the l-HyMic hydrogel upon enzymatic degradation. Laser confocal scanning microscopy of HeLa cells incubated for 24 h with (A) freshly prepared fluorescently labeled CCL micelles at a concentration of 400 μg·mL–1 and (B) CCL micelles released from the gel at a concentration of 400 μg·mL–1. Micelles are visualized with maleimide-Alexa fluor C5 568 in red.
4. Conclusion
This paper describes the design and synthesis of an enzyme responsive hydrogel (HyMic) consisting of CCL flower-like micelles and an enzyme responsive linker (Pep-NC). The complete degradation of HyMic in the presence of collagenase in a concentration dependent manner shows the programmability of this hydrogel. Upon enzymatic degradation, HyMic is converted into CCL micelles that can be taken up by HeLa cells. These results demonstrate the great potential of HyMic for sustained release of CCL micelles for intracellular drug delivery in tissues with upregulation of MMP, for example, cancer tissue. The introduced micellar hydrogel technology can be easily used for the development of other types of enzyme responsive micellar hydrogels. To this end, the peptide block in the linker can be substituted by a peptide that matches the specificity of the desired enzyme. Moreover, micelles can be decorated with targeting ligand to improve their cellular uptake upon release from the hydrogel.
Acknowledgments
The Netherlands Organization for Scientific Research (NWO/Aspasia 015.009.038 and NWO/VIDI 13457) is acknowledged for funding.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.9b01675.
Overview of PNC, PNE, Pep-NC, core cross-linked flower-like micelle, and HyMic structures (SI-Figure 1), 1H NMR spectrum of Boc-NH-PEG-NHS (SI-Figure 2A), l-Pep-PEG conjugate (SI-Figure 2B), l-Pep-PEG ATRP macroinitiator (SI-Figure 2C), 1H NMR spectrum of d-Pep-PEG (SI-Figure 3), GPC chromatogram of d-Pep-PEG (SI-Figure 4), 1H NMR spectrum of protected l-Pep-NC (SI-Figure 5A) and l-Pep-NC (SI-Figure 5B), 1H NMR spectrum of protected d-Pep-NC (SI-Figure 6A) and d-Pep-NC (SI-Figure 6B), 1H NMR of PNE (SI-Figure 7), 1H NMR of protected PNC (SI-Figure 8A) and PNC (SI-Figure 8B), GPC chromatograms of l-Pep-NC and d-Pep-NC (SI-Figure 9), GPC chromatogram of PNE (SI-Figure 10), GPC chromatograms of protected PNC and PNC (SI-Figure 11A,B), GPC chromatogram of l- and d-Pep-NC before and after enzymatic treatment (SI-Figure 12), storage modulus (G′), and tan δ as a function of time for micelles at 37 °C (SI-Figure 13) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hoffman A. S. Hydrogels for biomedical applications. Adv. Drug Delivery Rev. 2012, 64, 18–23. 10.1016/j.addr.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Buwalda S. J.; Boere K. W. M.; Dijkstra P. J.; Feijen J.; Vermonden T.; Hennink W. E. Hydrogels in a historical perspective: From simple networks to smart materials. J. Controlled Release 2014, 190, 254–273. 10.1016/j.jconrel.2014.03.052. [DOI] [PubMed] [Google Scholar]
- Hoare T. R.; Kohane D. S. hydrogels in drug delivery:progress and challenges. Polymer 2008, 49, 1993–2007. 10.1016/j.polymer.2008.01.027. [DOI] [Google Scholar]
- Vermonden T.; Censi R.; Hennink W. E. Hydrogels for protein delivery. Chem. Rev. 2012, 112 (5), 2853–2888. 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- Peppas N. A.; Wood K. M.; Blanchette J. O. Hydrogels for oral delivery of therapeutic proteins. Expert Opin. Biol. Ther. 2004, 4 (6), 881–887. 10.1517/14712598.4.6.881. [DOI] [PubMed] [Google Scholar]
- Fliervoet L. A. L.; Engbersen J. F. J.; Schiffelers R. M.; Hennink W. E.; Vermonden T. Polymers and hydrogels for local nucleic acid delivery. J. Mater. Chem. B 2018, 6 (36), 5651–5670. 10.1039/C8TB01795F. [DOI] [PubMed] [Google Scholar]
- Dosio F.; Arpicco S.; Stella B.; Fattal E. Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv. Drug Delivery Rev. 2016, 97, 204–236. 10.1016/j.addr.2015.11.011. [DOI] [PubMed] [Google Scholar]
- Buwalda S. J.; Vermonden T.; Hennink W. E. Hydrogels for Therapeutic Delivery: Current Developments and Future Directions. Biomacromolecules 2017, 18 (2), 316–330. 10.1021/acs.biomac.6b01604. [DOI] [PubMed] [Google Scholar]
- McKenzie M.; Betts D.; Suh A.; Bui K.; Kim D. L.; Cho H. Hydrogel-Based Drug Delivery Systems for Poorly Water-Soluble Drugs. Molecules 2015, 20 (11), 20397–20408. 10.3390/molecules201119705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou M.; Li X.; Dai M.; Gong C.; Wang X.; Xie Y.; Deng H.; Chen L.; Zhao X.; Qian Z.; Wei Y. A novel injectable local hydrophobic drug delivery system: Biodegradable nanoparticles in thermo-sensitive hydrogel. Int. J. Pharm. 2008, 359 (1), 228–233. 10.1016/j.ijpharm.2008.03.023. [DOI] [PubMed] [Google Scholar]
- O’Neill H. S.; Herron C. C.; Hastings C. L.; Deckers R.; Lopez Noriega A.; Kelly H. M.; Hennink W. E.; McDonnell C. O.; O’Brien F. J.; Ruiz-Hernández E.; Duffy G. P. A stimuli responsive liposome loaded hydrogel provides flexible on-demand release of therapeutic agents. Acta Biomater. 2017, 48, 110–119. 10.1016/j.actbio.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Stenekes R. J. H.; Loebis A. E.; Fernandes C. M.; Crommelin D. J. A.; Hennink W. E. Controlled Release of Liposomes from Biodegradable Dextran Microspheres: A Novel Delivery Concept. Pharm. Res. 2000, 17 (6), 664–669. 10.1023/A:1007526114744. [DOI] [PubMed] [Google Scholar]
- Wei L.; Cai C.; Lin J.; Chen T. Dual-drug delivery system based on hydrogel/micelle composites. Biomaterials 2009, 30 (13), 2606–2613. 10.1016/j.biomaterials.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Li J.; Mooney D. J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eljarrat-Binstock E.; Orucov F.; Aldouby Y.; Frucht-Pery J.; Domb A. J. Charged nanoparticles delivery to the eye using hydrogel iontophoresis. J. Controlled Release 2008, 126 (2), 156–161. 10.1016/j.jconrel.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Henke M.; Brandl F.; Goepferich A. M.; Tessmar J. K. Size-dependent release of fluorescent macromolecules and nanoparticles from radically cross-linked hydrogels. Eur. J. Pharm. Biopharm. 2010, 74 (2), 184–192. 10.1016/j.ejpb.2009.08.011. [DOI] [PubMed] [Google Scholar]
- de Graaf A. J.; Azevedo Prospero dos Santos I. I.; Pieters E. H.; Rijkers D. T.; van Nostrum C. F.; Vermonden T.; Kok R. J.; Hennink W. E.; Mastrobattista E. A micelle-shedding thermosensitive hydrogel as sustained release formulation. J. Controlled Release 2012, 162 (3), 582–590. 10.1016/j.jconrel.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Qiu S.; Zhuang J.; Jin S.; Yang N.-L. Nitrocatecholic copolymers – synthesis and their remarkable binding affinity. Chem. Commun. 2019, 55 (72), 10748–10751. 10.1039/C9CC04425F. [DOI] [PubMed] [Google Scholar]
- Xiao L.; Zhu J.; Londono J. D.; Pochan D. J.; Jia X. Mechano-responsive hydrogels crosslinked by block copolymer micelles. Soft Matter 2012, 8 (40), 10233–10237. 10.1039/c2sm26566d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoorchian A.; Simon J. R.; Bharti B.; Han W.; Zhao X.; Chilkoti A.; López G. P. Bioinspired Reversibly Cross-linked Hydrogels Comprising Polypeptide Micelles Exhibit Enhanced Mechanical Properties. Adv. Funct. Mater. 2015, 25 (21), 3122–3130. 10.1002/adfm.201500699. [DOI] [Google Scholar]
- Guillet P.; Mugemana C.; Stadler F. J.; Schubert U. S.; Fustin C.-A.; Bailly C.; Gohy J.-F. Connecting micelles by metallo-supramolecular interactions: towards stimuli responsive hierarchical materials. Soft Matter 2009, 5 (18), 3409–3411. 10.1039/b910325b. [DOI] [Google Scholar]
- Hoffman A. S. Stimuli-responsive polymers: Biomedical applications and challenges for clinical translation. Adv. Drug Delivery Rev. 2013, 65 (1), 10–16. 10.1016/j.addr.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Soppimath K. S.; Aminabhavi T. M.; Dave A. M.; Kumbar S. G.; Rudzinski W. E. Stimulus-Responsive “Smart” Hydrogels as Novel Drug Delivery Systems. Drug Dev. Ind. Pharm. 2002, 28 (8), 957–974. 10.1081/DDC-120006428. [DOI] [PubMed] [Google Scholar]
- Dadsetan M.; Liu Z.; Pumberger M.; Giraldo C. V.; Ruesink T.; Lu L.; Yaszemski M. J. A stimuli-responsive hydrogel for doxorubicin delivery. Biomaterials 2010, 31 (31), 8051–8062. 10.1016/j.biomaterials.2010.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K.; Tomatsu I.; Kros A. Light controlled protein release from a supramolecular hydrogel. Chem. Commun. 2010, 46 (23), 4094–4096. 10.1039/c002565h. [DOI] [PubMed] [Google Scholar]
- Najafi M.; Hebels E.; Hennink W. E.; Vermonden T.. Poly(N-isopropylacrylamide): Physicochemical Properties and Biomedical Applications: Chemistry, Properties and Applications. In Temperature-Responsive Polymers: Chemistry, Properties, and Applications; Khutoryanskiy V. V., Georgiou K. T., Eds.; John Wiley & Sons, 2018; pp 1–34. [Google Scholar]
- Qiu Y.; Park K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Delivery Rev. 2001, 53 (3), 321–339. 10.1016/S0169-409X(01)00203-4. [DOI] [PubMed] [Google Scholar]
- West J. L.; Hubbell J. A. Polymeric Biomaterials with Degradation Sites for Proteases Involved in Cell Migration. Macromolecules 1999, 32 (1), 241–244. 10.1021/ma981296k. [DOI] [Google Scholar]
- Chandrawati R. Enzyme-responsive polymer hydrogels for therapeutic delivery. Exp. Biol. Med. 2016, 241 (9), 972–979. 10.1177/1535370216647186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi H. W.; Hafeez S.; van Blitterswijk C. A.; Moroni L.; Baker M. B. Hydrogels that listen to cells: a review of cell-responsive strategies in biomaterial design for tissue regeneration. Mater. Horiz. 2017, 4 (6), 1020–1040. 10.1039/C7MH00373K. [DOI] [Google Scholar]
- Purcell B. P.; Lobb D.; Charati M. B.; Dorsey S. M.; Wade R. J.; Zellars K. N.; Doviak H.; Pettaway S.; Logdon C. B.; Shuman J. A.; Freels P. D.; Gorman J. H. III; Gorman R. C.; Spinale F. G.; Burdick J. A. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat. Mater. 2014, 13, 653–661. 10.1038/nmat3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse E.; Freudenberg U.; Niemietz T.; Greth C.; Weisser M.; Hagmann S.; Binner M.; Werner C.; Richter W. Peptide-functionalized starPEG/heparin hydrogels direct mitogenicity, cell morphology and cartilage matrix distribution in vitro and in vivo. J. Tissue Eng. Regener. Med. 2018, 12 (1), 229–239. 10.1002/term.2404. [DOI] [PubMed] [Google Scholar]
- Westermarck J.; Kähäri V.-M. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999, 13 (8), 781–792. 10.1096/fasebj.13.8.781. [DOI] [PubMed] [Google Scholar]
- Foda H. D.; Zucker S. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discovery Today 2001, 6 (9), 478–482. 10.1016/S1359-6446(01)01752-4. [DOI] [PubMed] [Google Scholar]
- Roy R.; Yang J.; Moses M. A. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J. Clin. Oncol. 2009, 27 (31), 5287–5297. 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M.; Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2 (3), 161–174. 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Najafi M.; Kordalivand N.; Moradi M. A.; van den Dikkenberg J.; Fokkink R.; Friedrich H.; Sommerdijk N.; Hembury M.; Vermonden T. Native Chemical Ligation for Cross-Linking of Flower-Like Micelles. Biomacromolecules 2018, 19 (9), 3766–3775. 10.1021/acs.biomac.8b00908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzel-Arnett S.; Sang Q. X.; Moore W. G. I.; Navre M.; Birkedal-Hansen H.; Van Wart H. E. Comparative sequence specificities of human 72- and 92-kDa gelatinases (type IV collagenases) and PUMP (matrilysin). Biochemistry 1993, 32 (25), 6427–6432. 10.1021/bi00076a016. [DOI] [PubMed] [Google Scholar]
- Ulbrich K.; Šubr V.; Strohalm J.; Plocová D.; Jelínková M.; Říhová B. Polymeric drugs based on conjugates of synthetic and natural macromolecules: I. Synthesis and physico-chemical characterisation. J. Controlled Release 2000, 64 (1), 63–79. 10.1016/S0168-3659(99)00141-8. [DOI] [PubMed] [Google Scholar]
- Boere K. W. M.; Soliman B. G.; Rijkers D. T. S.; Hennink W. E.; Vermonden T. Thermoresponsive Injectable Hydrogels Cross-Linked by Native Chemical Ligation. Macromolecules 2014, 47 (7), 2430–2438. 10.1021/ma5000927. [DOI] [Google Scholar]
- de Graaf A. J.; Mastrobattista E.; Vermonden T.; van Nostrum C. F.; Rijkers D. T. S.; Liskamp R. M. J.; Hennink W. E. Thermosensitive Peptide-Hybrid ABC Block Copolymers Obtained by ATRP: Synthesis, Self-Assembly, and Enzymatic Degradation. Macromolecules 2012, 45 (2), 842–851. 10.1021/ma2024667. [DOI] [Google Scholar]
- Boere K. W.; van den Dikkenberg J.; Gao Y.; Visser J.; Hennink W. E.; Vermonden T. Thermogelling and Chemoselectively Cross-Linked Hydrogels with Controlled Mechanical Properties and Degradation Behavior. Biomacromolecules 2015, 16 (9), 2840–2851. 10.1021/acs.biomac.5b00802. [DOI] [PubMed] [Google Scholar]
- Soga O.; van Nostrum C. F.; Ramzi A.; Visser T.; Soulimani F.; Frederik P. M.; Bomans P. H. H.; Hennink W. E. Physicochemical Characterization of Degradable Thermosensitive Polymeric Micelles. Langmuir 2004, 20 (21), 9388–9395. 10.1021/la048354h. [DOI] [PubMed] [Google Scholar]
- Tsurkan M. V.; Chwalek K.; Levental K. R.; Freudenberg U.; Werner C. Modular StarPEG-Heparin Gels with Bifunctional Peptide Linkers. Macromol. Rapid Commun. 2010, 31 (17), 1529–1533. 10.1002/marc.201000155. [DOI] [PubMed] [Google Scholar]
- Wu Z.-S.; Wu Q.; Yang J.-H.; Wang H.-Q.; Ding X.-D.; Yang F.; Xu X.-C. Prognostic significance of MMP-9 and TIMP-1 serum and tissue expression in breast cancer. Int. J. Cancer 2008, 122 (9), 2050–2056. 10.1002/ijc.23337. [DOI] [PubMed] [Google Scholar]
- Ando H.; Twining S. S.; Yue B. Y.; Zhou X.; Fini M. E.; Kaiya T.; Higginbotham E. J.; Sugar J. MMPs and proteinase inhibitors in the human aqueous humor. Invest. Ophthalmol. Visual Sci. 1993, 34 (13), 3541–3548. [PubMed] [Google Scholar]
- Thompson J. M.; Agee K.; Sidow S. J.; McNally K.; Lindsey K.; Borke J.; Elsalanty M.; Tay F. R.; Pashley D. H. Inhibition of Endogenous Dentin Matrix Metalloproteinases by Ethylenediaminetetraacetic Acid. J. Endod. 2012, 38 (1), 62–65. 10.1016/j.joen.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyjaszewski K.; Xia J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101 (9), 2921–2990. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- Isidro-Llobet A.; Álvarez M.; Albericio F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109 (6), 2455–2504. 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. Synthesis of proteins by native chemical ligation. Science 1994, 266 (5186), 776–779. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- Smithenry D. W.; Kang M.-S.; Gupta V. K. Telechelic Poly(N-isopropylacrylamide): Polymerization and Chain Aggregation in Solution. Macromolecules 2001, 34 (24), 8503–8511. 10.1021/ma010769b. [DOI] [Google Scholar]
- Ganachaud F.; Monteiro M. J.; Gilbert R. G.; Dourges M.-A.; Thang S. H.; Rizzardo E. Molecular Weight Characterization of Poly(N-isopropylacrylamide) Prepared by Living Free-Radical Polymerization. Macromolecules 2000, 33 (18), 6738–6745. 10.1021/ma0003102. [DOI] [Google Scholar]
- Blondelle S. E.; Houghten R. A. Comparison of 55% TFA/CH2Cl2 and 100% TFA for Boc group removal during solid-phase peptide synthesis. Int. J. Pept. Protein Res. 1993, 41 (6), 522–527. 10.1111/j.1399-3011.1993.tb00473.x. [DOI] [PubMed] [Google Scholar]
- Kamber B.; Hartmann A.; Eisler K.; Riniker B.; Rink H.; Sieber P.; Rittel W. The Synthesis of Cystine Peptides by Iodine Oxidation of S-Trityl-cysteine and S-Acetamidomethyl-cysteine Peptides. Helv. Chim. Acta 1980, 63 (4), 899–915. 10.1002/hlca.19800630418. [DOI] [Google Scholar]
- Trzebicka B.; Robak B.; Trzcinska R.; Szweda D.; Suder P.; Silberring J.; Dworak A. Thermosensitive PNIPAM-peptide conjugate – Synthesis and aggregation. Eur. Polym. J. 2013, 49 (2), 499–509. 10.1016/j.eurpolymj.2012.11.005. [DOI] [Google Scholar]
- Molawi K.; Studer A. Reversible switching of substrate activity of poly-N-isopropylacrylamide peptide conjugates. Chem. Commun. 2007, (48), 5173–5175. 10.1039/b713083j. [DOI] [PubMed] [Google Scholar]
- Van Tomme S. R.; van Nostrum C. F.; Dijkstra M.; De Smedt S. C.; Hennink W. E. Effect of particle size and charge on the network properties of microsphere-based hydrogels. Eur. J. Pharm. Biopharm. 2008, 70 (2), 522–530. 10.1016/j.ejpb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Tsuji H.; Ishida T. Poly(L-lactide). X. Enhanced surface hydrophilicity and chain-scission mechanisms of poly(L-lactide) film in enzymatic, alkaline, and phosphate-buffered solutions. J. Appl. Polym. Sci. 2003, 87 (10), 1628–1633. 10.1002/app.11605. [DOI] [Google Scholar]
- Wachiralarpphaithoon C.; Iwasaki Y.; Akiyoshi K. Enzyme-degradable phosphorylcholine porous hydrogels cross-linked with polyphosphoesters for cell matrices. Biomaterials 2007, 28 (6), 984–993. 10.1016/j.biomaterials.2006.10.024. [DOI] [PubMed] [Google Scholar]
- Rubinstein M.; Colby R. H.. Polymer Physics; Oxford University Press: Oxford; New York, 2003. [Google Scholar]
- Atallah P.; Schirmer L.; Tsurkan M.; Putra Limasale Y. D.; Zimmermann R.; Werner C.; Freudenberg U. In situ-forming, cell-instructive hydrogels based on glycosaminoglycans with varied sulfation patterns. Biomaterials 2018, 181, 227–239. 10.1016/j.biomaterials.2018.07.056. [DOI] [PubMed] [Google Scholar]
- Bond M. D.; Van Wart H. E. Purification and separation of individual collagenases of Clostridium histolyticum using red dye ligand chromatography. Biochemistry 1984, 23 (13), 3077–3085. 10.1021/bi00308a035. [DOI] [PubMed] [Google Scholar]
- Matsushita O.; Jung C.-M.; Katayama S.; Minami J.; Takahashi Y.; Okabe A. Gene Duplication and Multiplicity of Collagenases in Clostridium histolyticum. J. Bacteriol. 1999, 181 (3), 923–933. 10.1128/JB.181.3.923-933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson H. P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 2009, 11, 32–51. 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.