

Summary
Redox balance is essential for normal brain, hence dis-coordinated oxidative reactions leading to neuronal death, including programs of regulated death, are commonly viewed as an inevitable pathogenic penalty for acute neuro-injury and neurodegenerative diseases. Ferroptosis is one of these programs triggered by dyshomeostasis of three metabolic pillars—iron, thiols and polyunsaturated phospholipids. This review is focused on: i) lipid peroxidation (LPO) as the major instrument of cell demise, ii) iron as its catalytic mechanism, and iii) thiols as regulators of pro-ferroptotic signals, hydroperoxy-lipids. Given the central role of LPO, we discuss the engagement of selective and specific enzymatic pathways vs. random free radical chemical reactions in the context of the phospholipid substrates, their biosynthesis, intracellular location and related oxygenating machinery as participants in ferroptotic cascades. These concepts are discussed in lieu of emerging neuro-therapeutic approaches controlling intracellular production of pro-ferroptotic phospholipid signals and their non-cell-autonomous spreading leading to ferroptosis-associated necroinflammation.
Keywords: regulated cell death, neurodegeneration, cerebral ischemia, traumatic brain injury, cerebral hemorrhage, redox lipidomics, phospholipid, glutathione peroxidase 4, lipoxygenase
Graphical Abstract
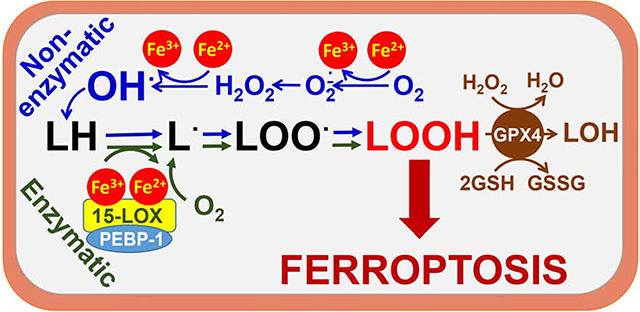
Bayır et al., review ferroptotic death resulting from lipid peroxidation triggered by failure to coordinate iron, lipid and thiol metabolism. Despite several deciphered genetic/biochemical mechanisms, involvement of free radical chemistry is conceivable. Appreciation of selectivity and specificity of these contributory pathways will strongly affect development of anti- and pro-ferroptotic therapies.
Achieving life is not the equivalent of avoiding death.
-Ayn Rand
Introduction
The compatibility and even the necessity of death for the continuation of harmonized life has been allegorically emphasized by many famous poets and artists. Surprisingly, the necessity of death as a physiological mechanism has not been commonly accepted until recently, in spite of the fact that it has been recognized as an evolutionary conserved factor maintaining the health of cells, organismal populations and communities. The requirement of cell demise has become obvious with the discovery of several genetically predetermined programs of regulated death. Conceptualized and described less than a decade ago, ferroptosis is one of these programs that engages three major metabolic pathways operated by thiols, lipid peroxidation, and iron (Dixon et al., 2012a). In the absence of characteristic morphological features of ferroptotic death as well as readily detectable biomarkers, there is a commonly accepted functional definition of ferroptosis as an iron-dependent lipid peroxidation-driven and glutathione peroxidase 4 (GPX4)-inhibitable process leading to cell death preventable by select anti-ferroptotic compounds (most commonly, ferrostatin-1 [Fer-1]) (Dixon et al., 2012b; Gaschler et al., 2018; Skouta et al., 2014; Yang et al., 2014b; Yang and Stockwell, 2016).
In spite of the dramatic outcome of this program—cell death—the biological role of ferroptosis is not completely understood (Stockwell et al., 2017), although several speculations have been made. According to one school of thought, ferroptotic death is a penalty imposed on a particular cell within a multicellular community that exhibits severe and irreparable dis-coordinated systems of redox regulation. This leads to the excessive accumulation of pro-oxidant metabolites that may represent a threat for the healthy environment of the community (Kagan et al., 2019). This may manifest in several ways; for example, excessive production of lipid mediators, acting as opposing or controversial signals, may disrupt essential biological functions (Fig. 1). To prevent this confusion and equivocal signaling, the program of ferroptotic elimination is triggered. This program operates with the same major components of the protein machinery involved in the generation of lipid mediators but with slightly modified substrates and products of the enzymatic process. As an example, the dis-coordinated production of the canonic lipid mediators (e.g., eicosanoids) catalyzed by one or several representatives of the lipoxygenase (LOX) family (5-, 8-, 12-, 15-LOX) leads to the assembly of 15-LOX and phosphatidylethanolamine binding protein 1 (PEBP1) into 15-LOX/PEBP1 enzymatic complexes. These complexes generate death signals by changing the LOX substrate selectivity (arachidonic acid [AA] esterified to phosphatidylethanolamine [PE] instead of free AA) and product specificity (15-hydroperoxy AA-containing PE instead of oxygenated forms of AA) causing cellular demise (Fig. 1) (Kagan et al., 2017; Wenzel et al., 2017).
Fig. 1. Lipoxygenases (LOXs) catalyze the formation of essential lipid mediators with pro- and anti-inflammatory propensities.
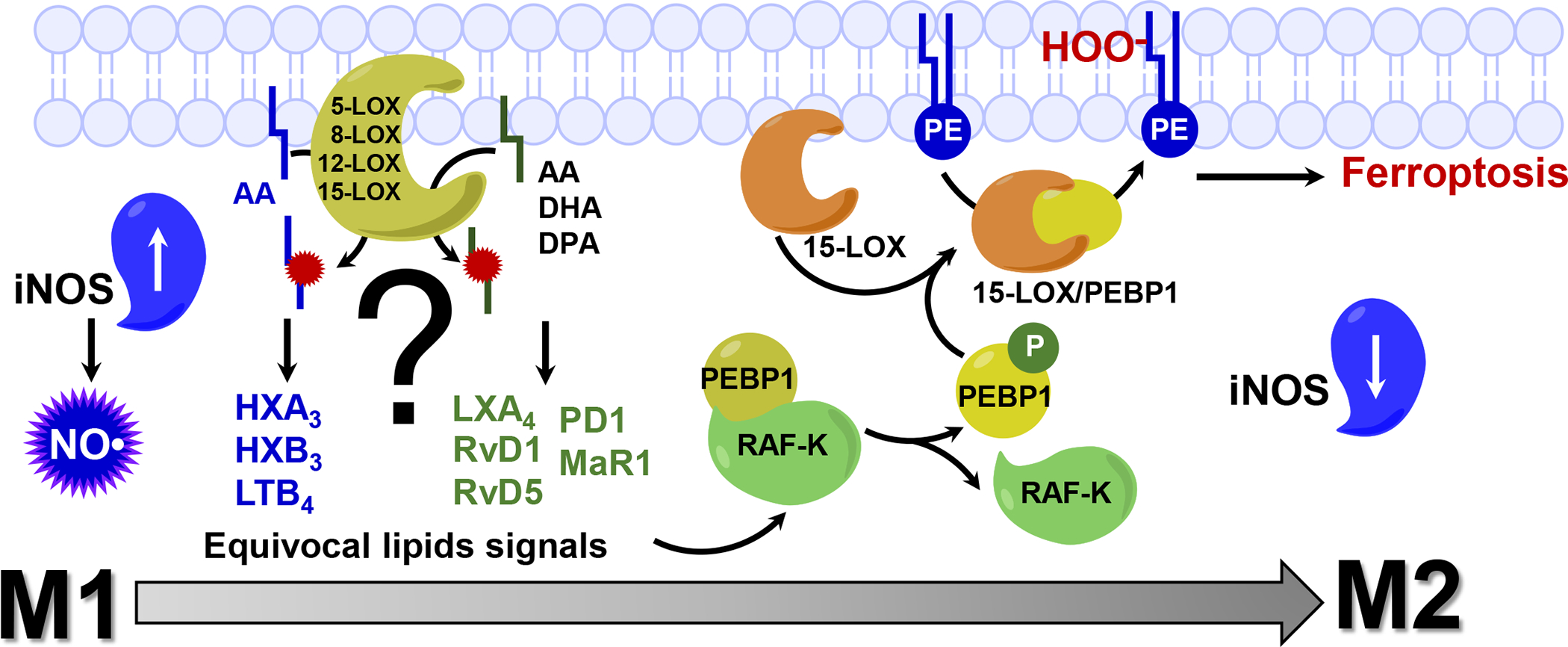
In cells, essential signaling molecules, lipid mediators, are formed by lipoxygenases (LOX) from free PUFA (w-6 and w-3 series). These oxidation products coordinate many metabolic processes and cell responses, including inflammation. 12-LOX and 5-LOX are involved in the generation of pro-inflammatory mediator leukotriene B4 (LTB4) from arachidonic acid (AA). The first step in biosynthesis of hepoxilins A3 and B3 (HXA3 and HXB3) is catalyzed by 12-LOX. 15-LOX is implicated in biosynthesis of anti-inflammatory lipid mediators such as docosahexaenoic acid (DHA) derived resolvins (RvD1 and RVD5), docosapentaenoic acid (DPA) derived protectin D1, AA-derived lipoxins (LXA3). Maresin R1 (MaR1), a DHA-derived lipid mediator generated by 12-LOX, stimulates switch of macrophages from M1 to M2 type. Dysregulated production of equivocal lipid signal by LOXs may lead to the triggering of a specialized program of cell death, ferroptosis, in dis-coordinated cells aberrantly producing confusing signals. Dis-regulation of lipid mediators biosynthesis leads to the assembly and engagement of 15-LOX/PEBP1 enzymatic complexes that start producing pro-ferroptotic death signals. PEBP1 (Phosphatidylethanolamine Binding Protein 1), a scaffold protein, an inhibitor of protein kinase (RAF-K), complexes with 15-LOX, and changes the substrate selectivity of the dioxygenase (AA-PE instead of free AA) and catalyzes a specific oxygenation generating 15-HOO-AA-PE as a ferroptotic cell death signal.
Another example may be related to the regulation of inflammation. Cells of the innate immune system—neutrophils, macrophages, microglia—may be present in several polarization states with substantially different phenotypic manifestations, including different sensitivities to ferroptosis (Kapralov et al., 2020). Interestingly, this may be used as a means of fine-tuning of the inflammatory response. Indeed, professional phagocytes activated to the pro-inflammatory M1 state with a high level of inducible nitric oxide synthase (iNOS)/NO• expression display a remarkably high resistance to ferroptosis, whereas alternatively activated pro-resolving M2 cells are highly vulnerable to ferroptotic elimination (Fig. 1). Thus, the numbers and ratio of M1 cells (which generate pro-inflammatory regulators such as cytokines and lipid mediators) to M2 cells (which are a source of anti-inflammatory regulators) may act as a “rheostat” controlling the production of pro-inflammatory vs pro-resolving signals (Dalli et al., 2013). The distinctive sensitivity or tolerance of these cells to ferroptosis may command the context-dependent adaptive changes of the innate immune system to support more aggressive pro-inflammatory vs more pro-resolving anti-inflammatory milieus. Of course, other examples of the functional role(s) of ferroptosis in different types of cells will emerge as our understanding of its role and mechanisms advances. The above examples, however, raise an interesting general question: do differential sensitivity and execution of ferroptosis in specific cell types of a given community represent a mechanism of death, or rather do they represent a reprograming on the level of a cell population? From this point of view, the engagement of the three pillars—iron, thiols, and lipid peroxidation—in the cascade of reprograming events utilizes the major intracellular redox pathways.
The pathophysiological relevance of ferroptosis compared to necrosis is likely due to the “regulated” nature of the ferroptotic program with specific biochemical and genetic mechanisms that can be targeted therapeutically. The pathophysiological relevance of ferroptosis compared to apoptosis is likely defined by the differences in the inflammatory responses associated with these death pathways. Apoptosis is considered strongly anti-inflammatory due to preservation of the plasma membrane integrity along with the hydrolytic “digestion” of the intracellular contents, and generation of apoptotic bodies engulfed by macrophages. Ferroptosis is regarded as one of the pro-inflammatory (necro-inflammatory) regulated cell death pathways characterized by plasma membrane rupture and release of intracellular contents as damage associated molecular patterns (Tonnus et al., 2019).
IRON IN FERROPTOSIS
The chemically reactive, kinetically labile iron pool is directly coordinated by glutathione
Specific mechanisms of ferroptosis are continuing to emerge; however, the thiol-driven regulation seems to be better understood than the roles of iron and lipid peroxidation pathways. In fact, the role of the two major components of the thiol system—GPX4 and reduced glutathione (GSH)—are well-characterized (Forcina and Dixon, 2019; Friedmann Angeli et al., 2014a; Seibt et al., 2019; Sun et al., 2018b). In contrast, specific mechanisms of lipid peroxidation and redox activity of iron in ferroptosis remain less explored in spite of their definitively established importance as discussed below.
The species of iron thought to directly participate in ferroptosis are likely represented by two pools of iron: i) catalytic centers of non-heme iron proteins, e. g. LOX (discussed below), and ii) ferrous iron of the cytosolic labile iron pool (LIP). The LIP is largely defined by what it is not. Most of the iron in mammalian cells (>90%) is in the form of cofactors (heme, iron-sulphur clusters, mono- and diiron centers) and storage pools (ferritin), with the amount of iron as cofactors staying relatively constant over time and the amount of iron in ferritin varying widely (Chakrabarti et al., 2015). The “labile iron pool” is the iron that remains. It is the chemically reactive and kinetically labile pool that can be transferred to mitochondria and used for heme and iron-sulfur cluster synthesis. It can be transferred directly to the active sites of non-heme iron enzymes in the cytosol or to ferritin for storage. It can bind to iron-sensing proteins to affect their activity or stability. Based on the reduction potential of the cytosol, Williams in 1982 suggested that the low-molecular-weight pool of iron in cells is almost exclusively ferrous [Fe(II)] (Williams, 1982). Subsequent experimental evidence confirmed that the LIP is Fe(II) (Kozlov et al., 1992; Yegorov et al., 1993) and is present in concentrations between 0.5 and 5 μM (Esposito et al., 2002; Ma et al., 2006; Petrat et al., 2002). However, under physiological conditions Fe(II) is highly reactive and can catalyze the conversion of endogenously produced hydrogen peroxide to highly reactive intermediate species (Galaris et al., 2019), such as hydroxyl radical (HO•) or a high-valence oxo-ferryl species via the Fenton reaction (Merkofer et al., 2006; Yamamoto et al., 2012). Both of these are predicted to attack and oxidatively damage multiple cellular components, especially lipids containing polyunsaturated fatty acyl chains (PUFAs). It is also possible that iron centers of LOXs catalyze the formation of the primary enzymatic products of lipid peroxidation, hydroperoxy-lipids (L-OOH), while Fe(II) from the LIP participates in the secondary reactions of L-OOH decomposition to produce oxidatively-truncated electrophilic products of lipid peroxidation (see below) (Stoyanovsky et al., 2019). This chemical reactivity of Fe(II) in the LIP is mitigated through coordination by a complex buffer composed of small molecules and iron chaperone proteins.
Many small molecule ligands have been proposed to coordinate cytosolic LIP. Iron (II) exhibits relatively strong interactions with free thiols, including cysteine and GSH. Although Fe(II) binds cysteine, the amino acid is not present in the cytosol at sufficiently high levels to support complex formation. Systematic in vitro evaluation suggests that only GSH exists in the cytosol at the necessary concentrations (2–10 mM) to form complexes with Fe(II) (Hider and Kong, 2013; Hider and Kong, 2011). It is proposed that Fe(II) forms a 1:1 complex with GSH, where the free thiol directly coordinates the iron. Iron speciation plots covering the physiological concentration ranges of GSH (1–10 mM) and iron (0.5–5 μM) indicate that >95% of the cytosolic LIP is predicted to be a Fe(II)-GSH complex. Moreover, a recent empiric study confirms the formation of Fe-GSH complexes in cells (Patel et al., 2019). These quantitative estimates also indicate that only a relatively small fraction of intracellular GSH (~0.1mol%) is engaged in the LIP function in many types of cells, including CNS cells, particularly neurons (Sun et al., 2006).
Iron-glutathione complexes of the labile iron pool are coordinated by PCBP1
The poly(rC)-binding protein (PCBP) family plays a key role in cellular Fe(II) distribution (Philpott and Jadhav, 2019) (Fig. 2). PCBPs are multifunctional adaptor proteins that bind single-stranded nucleic acids, proteins, and iron, affecting the fate of each ligand. PCBP1 and PCBP2 are iron chaperones: they bind iron and can receive or transfer it to other proteins via metal-mediated protein-protein interactions. In cells, PCBPs function by chaperoning iron to/from: transport proteins (divalent metal transporter [DMT1], ferroportin1), storage sites (ferritin), and iron-containing proteins (prolylhydroxylase [PHD], deoxyhypusine hydroxylase [DOHH], hemeoxygenase-1 [HO1], BolA2). In vitro studies using isothermal titration calorimetry indicate both PCBP1 and PCBP2 can coordinate three Fe (II) ions per protomer with low μM affinity (Leidgens et al., 2013; Shi et al., 2008). More recent work demonstrates that GSH is required for the formation of stable iron complexes on PCBP1, both in cells and in model (bio)chemical systems (Patel et al., 2019). PCBPs are structurally comprised of three hnRNP-K-homology (KH) domains, which are ancient, conserved RNA-binding modules (Makeyev and Liebhaber, 2002). The third KH domain of PCBP1 coordinates iron via conserved cysteine and glutamate residues and a molecule of noncovalently bound GSH, indicating that the preferred ligand for PCBP1 is an Fe-GSH complex (Patel et al., 2019). These studies also showed that GSH binding can allosterically stabilize the Fe-PCBP1 complex to facilitate iron transfer to client proteins. In vitro analysis also indicated that PCBP1 can coordinate Fe-GSH molecules with affinities in the sub-μM range, which suggests that much of the cytosolic iron pool is in the form of PCBP1-Fe-GSH complexes.
Fig. 2. Iron chaperone limits the chemical reactivity of cytosolic labile iron pool.
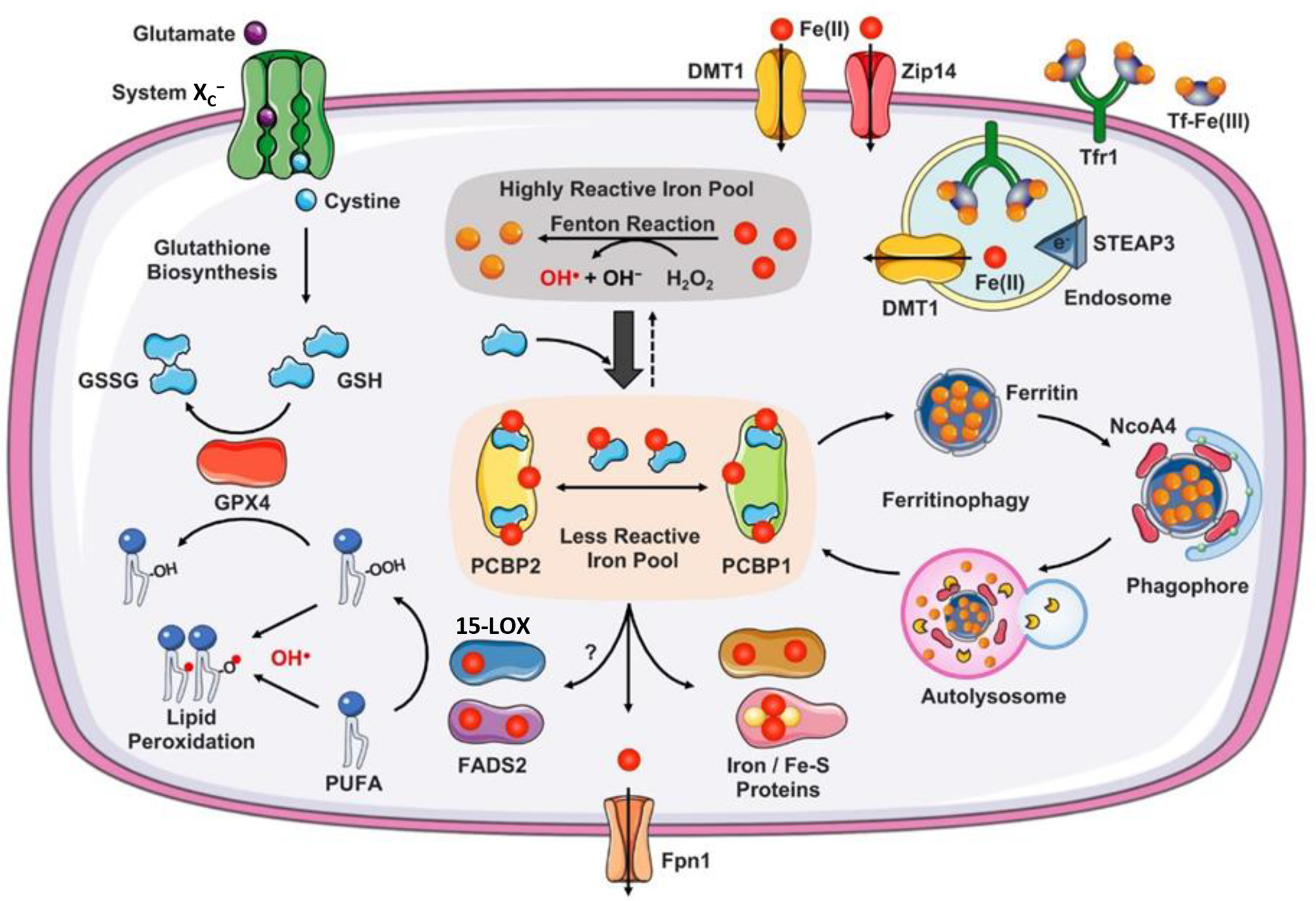
Non-transferrin-bound iron is reduced to Fe(II) by ferrireductases at the plasma membrane surface and transported into the cytosol via divalent metal transporter, DMT1, or Zip14. Circulating iron in the form of Fe(III)2-transferrin (Tf) binds to transferrin receptor 1 (Tfr1) on the plasma membrane to form Tf-Tfr1 complex, which is endocytosed. At the low pH of the endocytic vesicle, Fe(III) is released from Tf and ferrireductases (e.g., STEAP3) reduce Fe(III) to Fe(II), which is translocated into the cytosol by DMT1. If it remains free, Fe(II) reacts with hydrogen peroxide (H2O2) to produce cytotoxic hydroxyl radicals (OH•) via the Fenton reaction. Cytosolic Fe(II) is coordinated by reduced glutathione (GSH) in the cytosolic labile iron pool. PCBP1 and PCBP2 are iron chaperones that play integral roles in intracellular iron trafficking. Structurally PCBPs contain three tandem K-homology domains (KH1–3) that can bind GSH coordinated Fe(II) with high affinity in 1:1 ratio. Under physiological conditions a significant proportion (>90%) of the labile iron pool may be coordinated by PCBPs. PCBP2 binds iron loaded DMT1 to facilitate iron influx, binds heme degrading heme oxygenase to facilitate iron redistribution, and binds Ferroportin (Fpn1) to facilitate iron efflux. PCBP1 (to a lesser extent, PCBP2) binds and delivers iron to ferritin and mono/di-nuclear iron proteins and Fe-S carrier proteins. Ferritin binds to Nuclear Receptor Coactivator 4 (NcoA4) to undergo lysosomal degradation, process referred to as ferritinophagy. Ferritin iron released in the lysosome can be transferred back to the cytosol or transported to mitochondria. PCBPs may also metalate di-iron containing fatty acid desaturases (e.g. FADS2) and mono-iron containing lipoxygenases (e.g. 15-LOX), which are integral in lipid metabolism. FADS2 contributes to the formation of polyunsaturated fatty acids (PUFAs) and 15-LOX catalyzes the dioxygenation of PUFAs to generate phospholipid hydroperoxide (PL-OOH). Both PUFA and PL-OOH are the targets of the peroxidation reactions associated with ferroptosis. System XC− imports cystine, which is reduced to cysteine and used to synthesize GSH. Glutathione peroxidase 4 (GPX4) uses GSH to eliminate lipid peroxides formed in phospholipids containing PUFAs. Arrows indicate promotion; broad arrow indicate higher extent; dotted arrow indicates lesser extent.
PCBP1 limits the chemical reactivity of the cytosolic labile iron pool
Studies demonstrating the coordination of Fe-GSH complexes by PCBP1 suggest that PCBP1 exerts control over the chemical reactivity as well as the trafficking of the labile iron pool. In humans, PCBP1 is uniformly present in many tissues and its expression in different areas of the brain is high (Uhlén et al., 2015) Mice lacking PCBP1 in hepatocytes spontaneously develop steatohepatitis, with accumulation of di- and triglycerides and cholesteryl esters in the liver (Protchenko, 2019). This accumulation of lipid appears to be driven by increased production of reactive oxygen species (ROS) in the presence of “unchaperoned” iron. Shifting these animals to diets with very low amounts of iron blocks the development of steatosis and the accumulation of oxidized lipids in the liver. Thus, PCBP1 is required in to limit the chemical reactivity of iron.
The iron chaperone activities of PCBP1 sequester labile iron in ferritin
PCBP1 may also limit the chemical reactivity of the cytosolic LIP by keeping the pool small. PCBP1 was initially identified as an iron chaperone for ferritin (Shi et al., 2008). Ferritin is the major site of iron storage in mammalian cells and is a hollow sphere, composed of 24 subunits of H- and L-chains, that can sequester large amounts of iron as ferric oxyhydroxides. In the brain, all four major cell types (neurons, glia, microglia, and oligodendrocytes) contain ferritin, with glia being the most iron-rich cells (Reinert et al., 2019). PCBP1 can deliver iron to ferritin via a direct, protein-protein interaction that results in iron transfer to the mineral core, likely via pores formed at subunit interfaces. Iron sequestered in ferritin is relatively inert, but cells can mobilize the iron stored in ferritin by directing the oligomer to the lysosome for degradation (Philpott et al., 2017). This process occurs through a specific form of autophagy, referred to as ferritinophagy, which is mediated by the ferritin-specific, autophagic cargo receptor, nuclear receptor coactivator 4 (NCOA4) (Dowdle et al., 2014; Mancias et al., 2015; Mancias et al., 2014). Under conditions of iron deficiency, NCOA4 can also directly bind to ferritin and mediate its incorporation into the autophagosome. Once delivered to the autolysosome, iron liberated from degraded ferritin can be transferred back to the cytosol or, in the case of erythroid precursors, to the mitochondria for heme synthesis.
Cell-based in vitro studies and murine studies of erythropoietic precursors and hepatocytes confirm that efficient storage of iron in ferritin is dependent on PCBP1. In hepatocytes, PCBP1 is needed to maintain intracellular levels of iron and ferritin (Protchenko, 2019). In erythroid precursors, iron trafficking through ferritin is critical for efficient utilization of iron for heme synthesis in the mitochondria (Ryu et al., 2017). Similarly, NCOA4-mediated turnover of ferritin is also required for optimal iron utilization in erythroid precursors. Because the PCBP1- and NCOA4-mediated flux of iron through ferritin is associated with the mobilization of potentially large amounts of stored iron, these processes may have an impact on ferroptosis in some settings. Empiric data to support or refute this hypothesis are scant, and further studies are needed. PCBP1 has been found in neurons, and it has been established that its knockdown or overexpression affect transcriptional and translational regulation of several signaling pathways, cell cycling and apoptosis (Huo et al., 2012) as well as neuronal development (Vidaki et al., 2017). However, the neuronal role of PCBP1 in regulation of iron homeostasis has yet to be established and may represent the subject of future studies. Similarly, additional studies are necessary to clarify the expression, regulation, and function of NCOA4, as well as it is role in ferroptotic cell death in the CNS (Quiles Del Rey and Mancias, 2019).
PCBP1 may deliver iron to client proteins involved in ferroptosis
Client proteins that may receive their iron cofactors from PCBP1 are key mediators of the ferroptotic process. The iron chaperone activities of PCBP1 include the delivery of ferrous ions to mononuclear and dinuclear iron centers. The PHDs that regulate hypoxia-inducible factor (HIF) 1-α contain mononuclear iron centers. PCBP1 can directly interact with PHD2 to transfer iron to its active site. Cells lacking PCBP1 exhibit reduced PHD activity due to loss of the iron cofactor (Nandal et al., 2011). Similarly, PCBP1 can activate the diiron center of DOHH, a monooxygenase that catalyzes the hydroxylation step in the formation of hypusine, a modified amino acid in eIF5a. DOHH and PHD2 are structurally unrelated and yet PCBP1 binds both and the PCBP1-mediated interaction with DOHH is dependent on iron (Frey et al., 2014). Other mononuclear and dinuclear iron centers may also depend on PCBP1 for full metalation. Two classes of enzymes containing nonheme iron centers are integral in lipid metabolism: fatty acid desaturases, which contain a diiron center (Lee et al., 2016), and LOXs, which contain a monoiron center (Boyington et al., 1993). The fatty acid desaturases contribute to the formation of PUFA-containing lipids, which are the targets of the peroxidation reactions associated with ferroptosis. While some PUFAs originate from dietary sources (e.g. Ω-3 FAs, linoleic and linolenic acids), endogenous desaturases contribute to the formation of arachidonoyl- and adrenoyl-PE (AA- and AdA-PE) that are implicated in ferroptosis. LOX, especially the 15-LOX, may directly catalyze the hydroperoxidation of AA- or AdA-containing lipids (Stoyanovsky et al., 2019). Hydroperoxides of AA (HOO-AA) are further metabolized to form signaling molecules (Kuhn et al., 2015), while hydroperoxides of AA-PE (HOO-AA-PE) are proposed to trigger the ferroptotic response. Although PCBP1 has not been directly demonstrated to activate these non-heme enzymes, overall reductions in the non-heme iron content of cells lacking PCBP1 suggest they could be affected. Furthermore, removal of iron from the recombinant human 5-LOX not only altered the catalytic activity of the enzyme, but also impaired its membrane-binding. In cells exposed to increasing amounts of iron, a redistribution of the cytosolic 5-LOX to the nuclear fraction was observed (Dufrusine et al., 2019).
Management of iron through PCBP2
Although PCBP1 and PCBP2 have very similar structures and an 80% identical amino acid sequence, they are independently required for murine embryonic development and have non-overlapping functions in cells. The iron chaperone activities of PCBP2 have been most closely associated with the transfer of iron between transporters or enzymes at cellular membranes. Cell-based models suggest that iron import through DMT1 and export through ferroportin are assisted by PCBP2 and that the iron released through the activity of heme oxygenases at the endoplasmic reticulum is captured by PCBP2 (Yanatori et al., 2016; Yanatori et al., 2017; Yanatori et al., 2014). The chaperone activities of PCBP2 may not be active in all cell types, as HEK cells depleted of PCBP2 exhibit levels of intracellular iron similar to PCBP2-expressing cells (Frey et al., 2014) and developing erythrocytes depleted of PCBP2 also exhibit wild-type levels of iron uptake (Ryu et al., 2017). Similar to PCBP1, PCBP2 is also markedly expressed in different types of neuronal cells where it fulfills different regulatory and signaling functions (Xu et al., 2009; Zhu et al., 2002). It was shown that PCBP2 expression increases in neurons and astrocytes after an acute CNS insult (Mao et al., 2016). PCBP2 deficiency enhances glutamate-induced neuronal death while promoting cell cycle arrest in astrocytes. However, its direct relevance to control of iron metabolism and homeostasis in neurons and astrocytes needs further studies.
Mammalian tissues that specialize in iron handling, e.g. the intestinal epithelium, the liver, and the erythron, may exhibit differing management of the cytosolic LIP than cells with more limited metabolic requirements for iron, e.g. neurons or other terminally-differentiated cells. These potential differences in the LIP may render cells more or less susceptible to iron-mediated ROS damage and ferroptotic cell death. Recent studies indicate that GSH has an integral, direct role in the management of the cytosolic LIP that goes beyond its role in supporting glutathione peroxidases (GPXs) or general redox buffering. Further studies will clarify the roles of iron chaperones in the management of the LIP and protection against ferroptosis.
Pathogenic dysregulation of iron in the brain possibly related to ferroptosis.
Increasingly, dysregulation of iron handling and increased pool of the labile redox active iron is viewed as a possible pathogenic mechanism of neurodegeneration related to ferroptosis. In the majority of these cases, the specific mechanisms responsible for the mishandling of iron are not firmly identified, yet positive protective (therapeutic) effects of iron chelators or Fer-1 are considered as strong evidence supporting the role of ferroptosis. Examples of this type are numerous but a direct mechanistic understanding of how iron dyshomeostasis translates into specific disease pathogenesis is still missing in most cases. Among several recent examples, neuroferritinopathies associated with alterations in the L-ferritin gene that increase the cytosolic pool of labile iron (Cozzi et al., 2019) cause the formation of ferritin aggregates, oxidative damage, and the onset of a senescence phenotype, which is particularly severe in neurons. In this spontaneous senescent neuroferritinopathy-based model, cells are susceptible to ferroptotic death (Cozzi et al., 2019). Another pathological example involving a possible ferroptotic mechanism is intracerebral hemorrhage (ICH), whereby neurological deficits, memory impairment, and brain atrophy were reduced by Fer-1 treatment (Chen et al., 2019). Temporal lobe epilepsy following kainic acid (KA) in rats demonstrated sensitivity to the Fer-1 treatment that prevented the initiation and progression of ferroptosis in the hippocampus and rescued cognitive function in KA-induced TLE in rats (Ye et al., 2019). Friedreich’s ataxia is a progressive neurodegenerative disorder characterized by ataxia and sensory loss associated with by GAA repeat expansions in the first introns of both alleles of the FXN gene. Its pathology leads to the decreased expression of the encoded protein, frataxin, which is required for iron-sulfur-cluster biosynthesis in mitochondria (Santos et al., 2010). It has been demonstrated that frataxin deficiency triggers mitochondrial dysfunction and iron accumulation as well as increased oxidative stress, and these typical manifestations were preventable by inhibitors of ferroptosis (Cotticelli et al., 2019). Based on the sensitivity of the spinal cord trauma to iron chelators (e.g. deferoxamine) or ferroptosis inhibitors (eg, SRS 16–86), the pathogenic engagement of ferroptosis has been proposed (Yao et al., 2019; Zhang et al., 2019a). Based on partially understood crosstalk mechanisms of proteins pathologically associated with neurodegeneration, such as α-synuclein, tau, and amyloid precursor protein with iron homeostatic proteins, conclusions have been made on the participatory role of ferroptosis in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (Masaldan et al., 2019). While the above-mentioned and other similar cases certainly deserve further thorough research attention, it is difficult to definitively conclude that ferroptosis is the leading cause of their pathogenic mechanisms. As will be discussed in the next section, a highly selective and specific enzymatically catalyzed phospholipid peroxidation by non-heme iron-proteins (LOXs) rather than disorganized iron homeostasis, is required for the fulfillment of the ferroptotic death program. Therefore, focused studies of iron chaperones participating in the delivery of iron to LOXs may be particularly important.
LIPID PEROXIDATION IN FERROPTOSIS
In spite of the unambiguous leading role of lipid peroxidation as the major instrument of ferroptosis, its mechanisms, and the role of iron in its initiation and regulation are poorly understood (Dixon et al., 2012b; Doll and Conrad, 2017; Latunde-Dada, 2017). In fact, whether the lipid peroxidation process has a strictly controlled enzymatic nature or develops as a poorly controlled free radical reaction still remains the subject of active discussions (Stoyanovsky et al., 2019; Zilka et al., 2017a). To a large extent, this is due to the lack of adequate methodology for studying ferroptotic lipid peroxidation, which led researchers to use simple and readily available semi-quantitative and nonspecific protocols rather than precise and accurate measurements and quantitative characterizations.
Adequacy of the analytical tools and methods to assess lipid peroxidation and its substrates.
The cell lipidome includes tens of thousands of individual species of lipids that vary markedly in their abundance, intracellular distribution, physical organization, and functions (Braverman and Moser, 2012; Lordan et al., 2017; Mejia and Hatch, 2016). A significant part of lipidome is represented by molecular species of lipids containing readily oxidizable PUFA residues (Guichardant et al., 2011). The oxidation process—enzymatic or non-enzymatic—begins with the abstraction of a bis-allylic hydrogen, rearrangement of the resonance radical structure, addition of the molecular oxygen leading to the peroxyl radical, and the formation of the primary molecular product, L-OOH (Fig. 3). A relatively low dissociation energy of the O-O bond (Porter et al., 1995; Pratt et al., 2011) leads to the L-OOH cleavage yielding a number of secondary oxidation products, frequently with a shortened carbon chain and an oxygen-containing functionality with highly electrophilic propensities (Fig. S1). The most commonly formed electrophiles include epoxy, oxo-, or aldehydic groups that may be located at different sites of the hydrocarbon chain (Fig. S1). The electrophilic group may be formed on either truncated phospholipid or on the leaving shorter PUFA fragment (Fig. S1).
Fig. 3. Principle schemas of the reaction mechanism and specific features of enzymatic vs random non-enzymatic peroxidation of PUFA phospholipids.
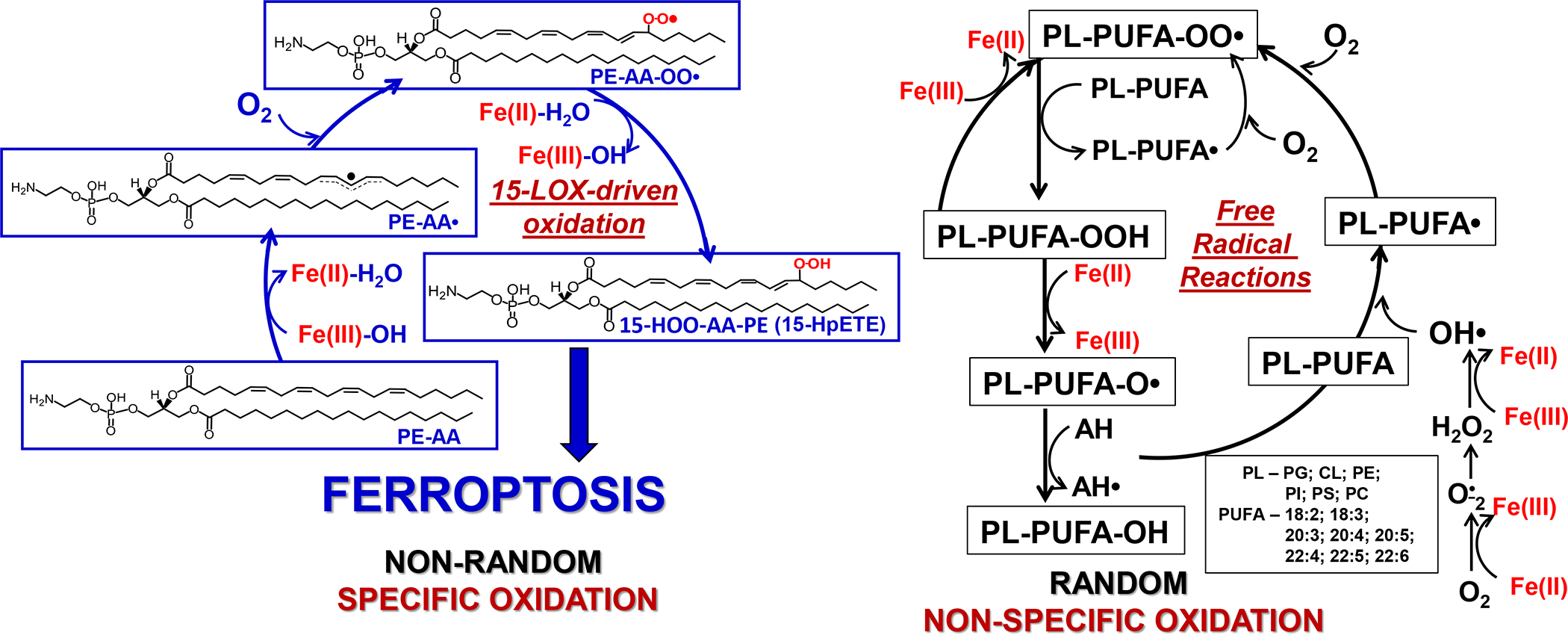
Both enzymatic - selective (left panel) and non-enzymatic - non-selective (right panel) lipid peroxidation begins with the abstraction of bis-allylic hydrogen, rearrangement of the resonance radical structure, addition of the molecular oxygen leading to the generation of peroxyl radical and the formation of the primary molecular product, hydroperoxy-lipid. During random oxidation all PUFA containing phospholipids undergo oxidation whereby the rates are proportional to the number of readily abstractable bis-allylic H, resulting in the accumulation of a highly diversifies pattern of oxidation products with the dominance of oxygenated PUFA-PLs with 6, 5, 4, 3, and 2 double bonds. LOX-driven oxidation results in the preferential generation of specific AA-PE oxidation yielding 15-HOO-AA-PE. The selectivity and specificity of the reaction is due to the organization of the catalytic site in 15-LOX/PEBP1 complex (Wenzel et al., 2017) resulting in highly selective, site-specific and stereo-selective product- 15-HOO-AA-PE. The substrate radical rearrangement is accompanied by the addition of molecular oxygen delivered via a special channel in the protein. Non-enzymatic lipid peroxidation proceeds by a free radical chain reaction. This process is not specific and non-selective.
Given the huge diversification of the lipidome and a large number of possible oxidation products, full characterization of the oxidation process and its products represents a daunting task (Tyurina et al., 2019). Assuming that one or more of these diversified lipid peroxidation products may act as direct executioners of ferroptotic death, ideally all of these oxidation products should be characterized using contemporary high resolution liquid chromatography with tandem mass spectrometry (LC-MS/MS) platforms (Kagan et al., 2017; Tyurina et al., 2019; Wenzel et al., 2017). Because redox lipidomics is still in the initial stages of its development, this ideal analytical approach is not always possible, and “much easier” simplified protocols, frequently inadequate, are commonly utilized as surrogate measures of lipid peroxidation in ferroptosis (Friedmann Angeli et al., 2014a).
Leaving aside an almost “ancient” protocol with thiobarbituric reactive substances (TBARS or malondialdehyde assay) that gives nonspecific positive responses with many compounds that have aldehyde-containing functionalities (Kohn and Liversedge, 1944) and thus proven to be completely non-applicable for any metabolic system (cells, tissues of model organisms) (Janero, 1990), there are only three major approaches currently used to assess lipid peroxidation in ferroptosis research: i) fluorescence assay of BODIPY (also called Lipid ROS); ii) linoleate-based Click-iT® LAA (linoleamide alkyne) lipid peroxidation assay, and iii) Liperfluo fluorogenic assay of hydro-peroxy-containing groups (Fig. 4). All three of these protocols suffer from substantial deficiencies. The BODIPY® 581/591 undecanoic acid assay demonstrates a strong correlation with the fluorescence activation due to oxidation of the BODIPY® 581/591 fragment of the probe (MacDonald et al., 2007). The oxidation is catalyzed by some kind of pro-oxidant activity towards a non-lipid substrate clearly unrelated to characterization of lipid peroxidation. The second protocol, which relates to linoleic acid-dependent click-chemistry, still does not represent a lipid peroxidation assay. Among these three approaches, the LiperFluo technique is the most closely connected with the mechanisms of ferroptosis dependent on GPX4. Indeed, the fluorogenic response from the probe emerges as a result of reduction of lipid hydroperoxides by a tri-phenylphosphine moiety of the probe in a way similar to GPX4 (Fig. 4) (Kagan et al., 2017). Thus, the fluorescence response detects lipid hydroperoxides. The deficiency of this protocol, however, is that it does not discriminate between many types of lipid hydroperoxides—PUFA-OOH, phospholipid-OOH (PL-OOH), cholesterol-OOH. Most notably, in ferroptosis-resistant Acsl4-deficient Pfa1 fibroblasts, the LiperFluo response was higher than in the ferroptosis-prone WT cells (Kagan et al., 2017).
Fig. 4. Most commonly used assays for indirect assessments of different “peroxidation/peroxidase activities associated with the execution of ferroptosis. Note that only Liperfluo assay detects hydroperoxyl-lipids.
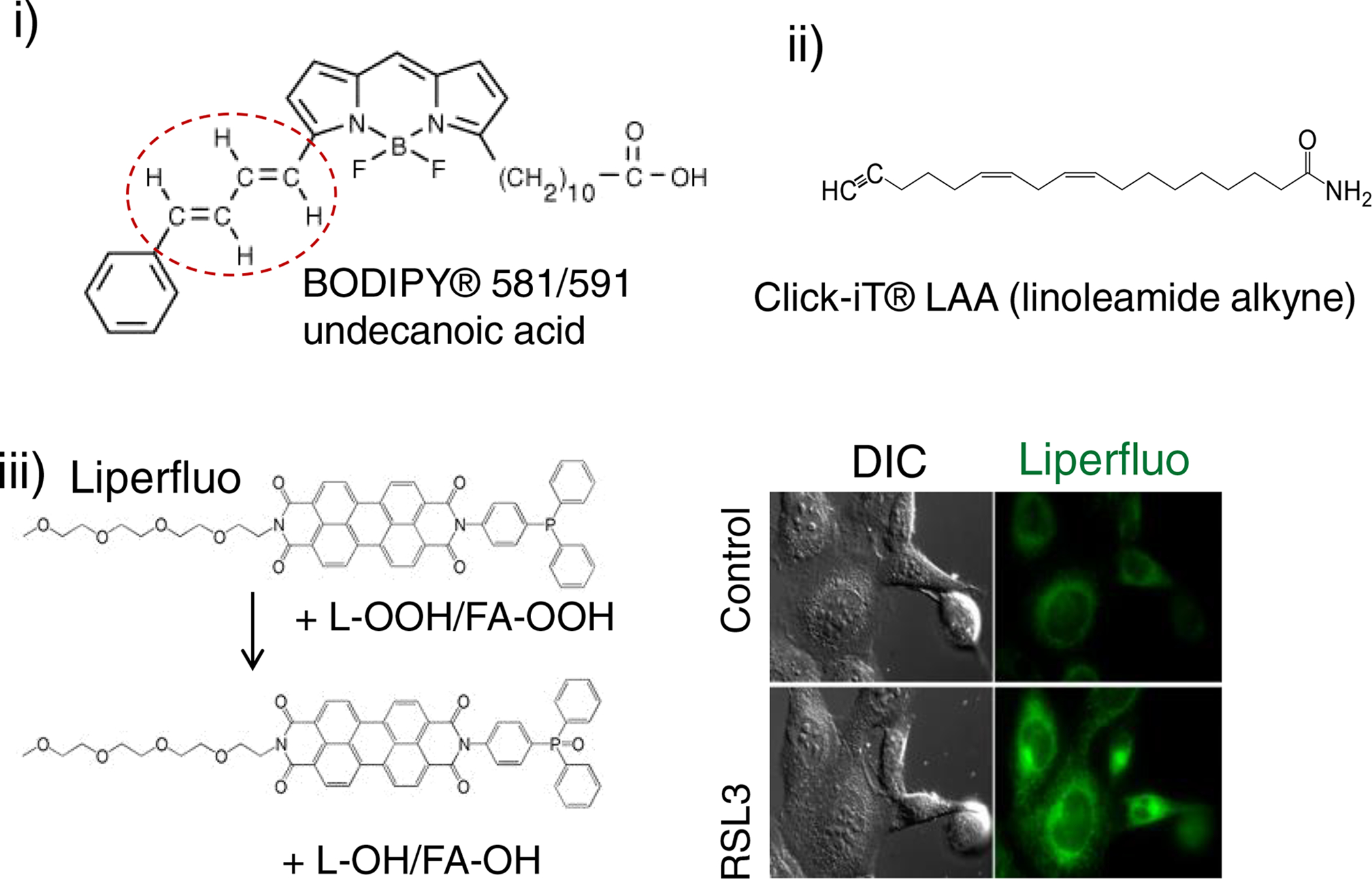
i) BODIPY® 581/591 detects “peroxidase activity” resulting in changed fluorescence characteristics of the non-lipidic BODIPY chromophore. Oxidation of butadiene portion (circled in red) of the BODIPY® 581/591 results in a shift of fluorescence emission peak from 590 nm to 510 nm.
ii) linoleate-based Click-iT® LAA (linoleamide alkyne) assay. This assay is designed to detect lipid-peroxidation derived modification of protein in fixed cells. Incorporated into cellular membranes Click-iT® LAA can undergo lipid peroxidation resulting in production of 9- and 13-hydroperoxy linoleic acid that further decomposes to unsaturated aldehydes which can modify proteins. These modified proteins are detected by Click-iT® chemistry.
iii) Liperfluo fluorogenic assay of hydroperoxy-containing groups. Liperfluo, a perylene derivative containing oligooxyethylene, detects L-OOH. LiperFluo can react with L-OOH and its fluorescence reliably reports intracellular sites of L-OOH accumulation by a fluorescence microscopy. Both free PUFA-OOH and PUFA-OOH esterified into phospholipids display high reactivity toward LiperFluo. The results in the robust fluorescence response of oxidatively modified LiperFluo in cells exposed to pro-ferroptotic stimuli) are shown on the right panel. Chemically, Liperfluo reduces L-OOH to the respective alcohols similarly to the reaction catalyzed by GPX4.
Given the huge diversity of (phospho)lipid peroxidation products detectable by Liquid chromatography mass spectrometry (LC-MS) protocols in control cells and tissues, and the enhanced lipid peroxidation response upon triggering ferroptotic death, it is becoming increasingly clear that high resolution LC-MS currently represents the only adequate platform for identification and quantification of ferroptosis-associated lipid peroxidation. Studies of this kind can help in resolving one of the major conundrums of ferroptosis: enzymatic or non-enzymatic free radical lipid peroxidation process. Fig. 5 demonstrates a typical schema of global redox lipidomics analysis of oxygenated lipids and products of their oxidative truncation including major classes of phospholipids and neutral lipids.
Fig. 5. A schema explaining the major stages of LC-MS based non-targeted Global (Redox) Lipidomics Analysis (GRLA).
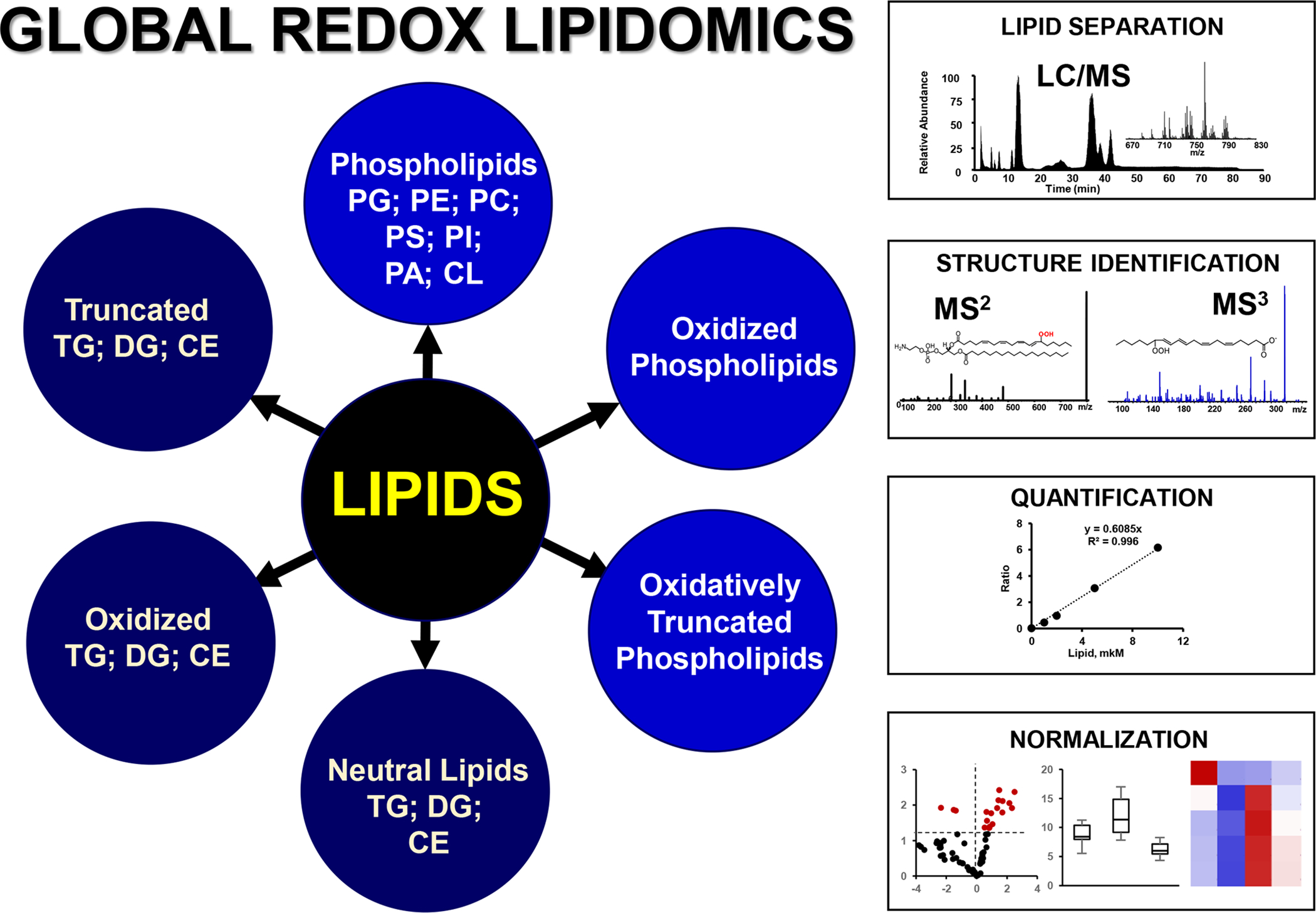
GRLA includes analysis of phospholipids and neutral lipids including their oxidized and oxidatively truncated species and consists of several steps. The first step is separation of lipids by liquid chromatography and their detection by mass spectrometry. Normal and reverse phase chromatographic protocols are used for separation of lipids based on their polarity and hydrophobicity. Electrospray ionization (ESI) is widely used for the detection of lipids and their oxidatively modified species. Second step is the identification of lipids and their oxidation products. This is achieved by using ion trap or high resolution mass spectrometry (MS) orbitrap instrumentations with unlimited fragmentation capacity. Step three includes quantitative analysis and normalization of results. LC-MS and LC-MS/MS can be set up for quantitative analysis using internal standards and calibration curves established with reference standards.
The high resolution of LC-MS/MS analysis yielding detailed information about the lipid oxidation products and numerous signals is also its “curse”: which of these products are predictive biomarkers of ferroptosis? Answering this question usually relies on the comparative analysis of cells exposed to pro-ferroptotic stimulation (e.g., by a GPX4 inhibitor, RSL3, or system XC− inhibitor, erastin) in the presence and absence of a typical inhibitor of ferroptosis (e.g. Fer-1). The signals from oxidized lipids disappearing/decreasing upon treatment with Fer-1 represent predictive biomarkers of ferroptosis.
Enzymatic vs non-enzymatic lipid peroxidation: the role of LOXs, PEBP1, selectivity/specificity, and adduct formation.
The general reaction schemas of enzymatic and non-enzymatic lipid peroxidation are similar as they include the catalytic abstraction of a bis-allylic hydrogen from the oxidizable substrate molecule (Fig. 3). For non-enzymatic peroxidation of PUFA, the reaction rates are proportional to the number of methylene-interrupted double bonds (i.e. the number of bis-allylic hydrogens) (Yin et al., 2011). Therefore, hexaenoyl- and pentaenoyl-phospholipids should be preferred oxidation substrates with relatively low selectivity with regards to the nature of the polar head of the phospholipids (Yin et al., 2011). Further, the non-enzymatic mechanism should not lead to positional specificity of the oxidation products in the PUFA residue. In contrast to this random profile of free radical peroxidation, enzymatic lipid peroxidation by di-oxygenases (e.g. cyclooxygenases [COXs] and LOXs) is highly substrate-selective and product-specific due to strictly coordinated juxtapositioning of the PUFA residues at the catalytic site (Kuhn et al., 2015). These substantial differences suggest that LC-MS/MS characterization of the reaction substrates and products may be diagnostic in establishing the enzymatic vs non-enzymatic nature of the peroxidation process.
This background information may be useful for the critical analysis of the known facts related to ferroptotic lipid peroxidation (Fig. 67). Very early studies established the essentiality of Acyl-CoA Synthetase Long Chain Family Member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) for the effective execution of ferroptosis (Doll et al., 2017; Kagan et al., 2017). Given that these two proteins are necessary for the activation of PUFA, particularly AA, to yield CoA-forms utilized for the esterification of free PUFA and re-acylation of lyso-phospholipids, respectively, it has been concluded that PUFA-phospholipids, rather than free PUFA were utilized as peroxidation substrates (Kagan et al., 2017). Subsequent LC-MS/MS work indicated that AA-PE are the preferred peroxidation substrates and HOO-AA-PE are the most common products (Kagan et al., 2017).
With regards to the positional specificity, there is evidence, albeit less unequivocal, that 15-HOO-AA-PE is the likely oxidation product (Anthonymuthu et al., 2018). The fact that COXs oxidize free rather than esterified peroxidized PUFA has led to the conclusion that LOXs may be the likely candidate enzymatic catalysts of ferroptotic peroxidation (Kagan et al., 2017). Of several mammalian LOXs, two isoforms of 15-LOX are known to be capable of attacking and peroxidizing membrane phospholipids (Chaitidis et al., 1998). This chain of logic put forward 15-LOX as the catalytic mechanism of ferroptotic AA-PE peroxidation (Kagan et al., 2017). While this conclusion was in line with the initial work on the involvement of LOXs in non-apoptotic cell death (Seiler et al., 2008a), subsequent studies with genetic deletion of different LOXs produced less definitive results (Friedmann Angeli et al., 2014a), still implicating some LOX isoforms in the ferroptotic peroxidation (Friedmann Angeli et al., 2014a). These studies might also be indicative of the requirement of additional regulatory factors controlling the phospholipid peroxidation process. Indeed, it has been demonstrated that the formation of the complex of 15-LOX with a scaffold protein, PEBP1, was required for the effective generation of pro-ferroptotic 15-HOO-AA-PE in several types of mammalian cells and tissues in normal and disease conditions (e.g. brain trauma, asthma, acute kidney injury) (Wenzel et al., 2017). Further analysis also demonstrated that the 15-LOX/PEBP1 complex, rather than 15-LOX alone, was important for the selective and specific peroxidation of AA-PE and the formation of 15-HOO-AAPE. LC-MS/MS analysis confirmed this remarkable catalytic selectivity and specificity of the 15-LOX/PEBP1 complex (Anthonymuthu et al., 2018) (Fig. 6) and demonstrated that AA-PE—out of hundreds of molecular species of alternative oxidizable PUFA-phospholipids—was the predominantly peroxidized phospholipid. Furthermore, even among dozens of oxidizable PUFA-PE-species, AA-PE was identified as the preferred peroxidation substrate and oxidized AA-PE as the peroxidation product (Fig. 6). While the initial studies (Kagan et al., 2017) indicated that the major locations of the generators of HOO-AA-PE are predominantly associated with ER sites, further studies are necessary to explore the involvement of AA-PE pools of mitochondriaassociated membranes and mitochondria.
Fig. 6. Schematic representation of the major stages of lipid redox metabolism leading to the formation of HOO-AA-PE as a pro-ferroptotic signal.
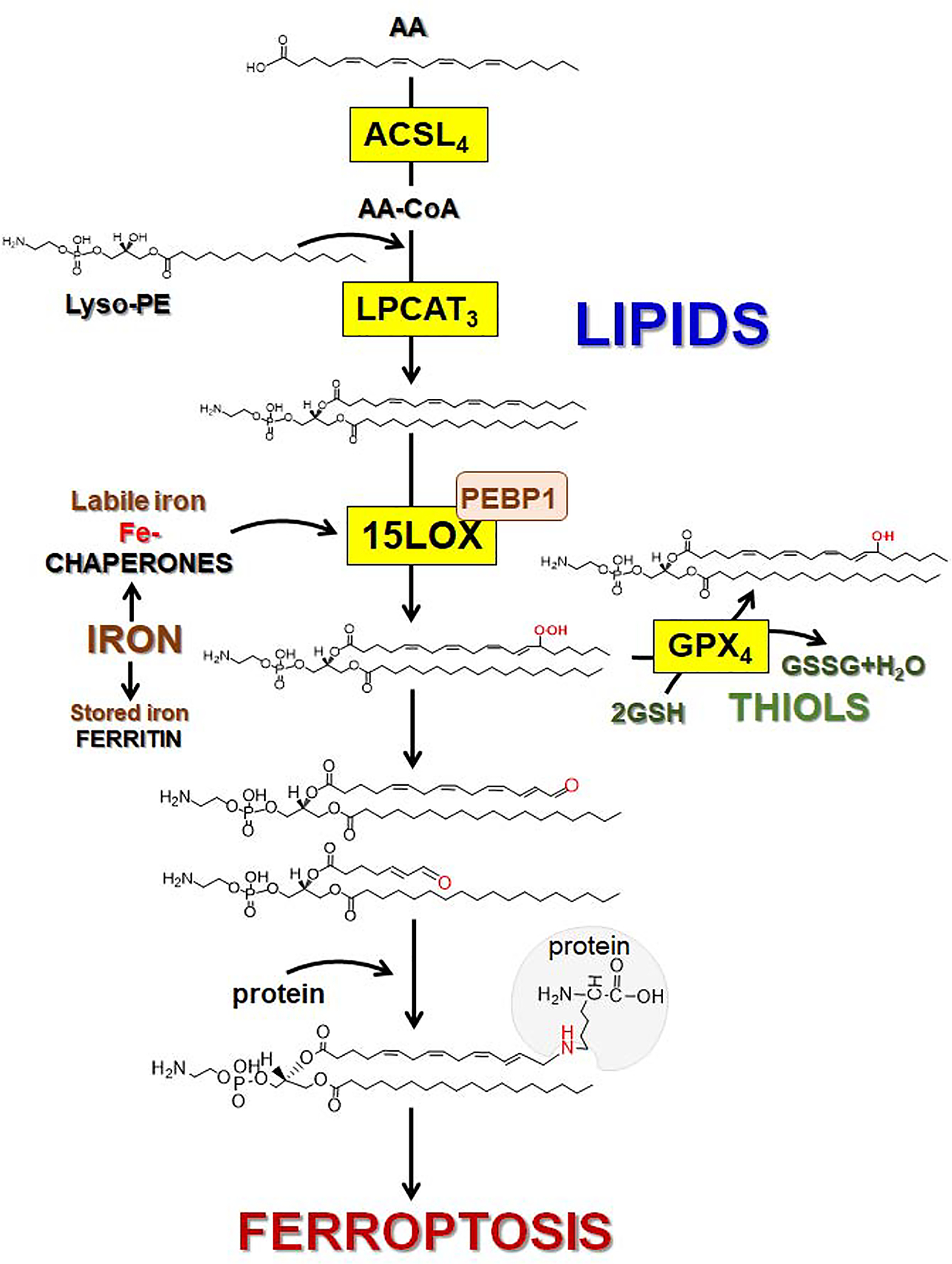
Esterification of arachidonic acid (AA) into phosphataidylethanolamine (PE) requires acyl-CoA synthase 4 (ACSL4) catalyzed formation of AA-CoA which is used for the AA esterification into lyso-PE driven by lysophosphatidyl-choline acyltransferase 3 (LPCAT3) to yield AA-PE. Assembly and engagement of 15-LOX/PEBP1 enzymatic complexes changes 15-LOX substrate selectivity from AA to AA-PE thus facilitating the generation of specific product (15-HOO-AA-PE). Under ferroptotic conditions, the activity of GPX4 is inhibited, thus HOO-AA-PE cannot be reduced to HO-AA-PE. In contrast oxidatively-truncated electrophilic products of HOO-AA-PE with shortened side chains and electrophilic oxygen functionalities are formed. These oxidatively-truncated PE products form adducts by attacking nucleophilic sites in target proteins thus leading to the yet to be identified gateway ferroptotic complexes.
Interestingly, the 15-LOX gene and its protein product, pLoxA have also been found in a prokaryotic organism, a pathogenic Gram-negative bacterium Pseudomonas aeruginosa, which does not contain its own PUFA peroxidation substrates but uses the protein for the pro-ferroptotic attack of the host AA-PE phospholipids (Fig. S2) (Dar et al., 2018). This work not only confirmed that the 15-LOX-driven PE-peroxidation represents an evolutionary conserved and ancient effective mechanism of cell death, but it also confirmed the sufficiency of 15-LOX as a mechanism of ferroptotic death (Dar et al., 2018). Assuming that 15-LOX/PEBP1 is an important catalyst of pro-ferroptotic phospholipid peroxidation, further attempts have been made to explore the possible mechanisms of its regulation, possibly duplicating the central anti-ferroptotic role of GPX4. Several regulatory mechanisms targeting different stages of the process leading to the generation and accumulation of the pro-ferroptotic signals have been identified. One of them is based on the iNOS/NO•-driven suppression of 15-LOX/PEBP1 catalytic activity preventing the formation of the death signal (Kapralov et al., 2019). Interestingly, this regulatory mechanism may act independently of the GPX4/GSH system. Another regulatory system, acting independently of the canonical thiol-driven pathway, includes the ferroptosis suppressor protein 1 (FSP1) that blocks and/or attenuates the production of ferroptotic signals via controlling the reduction of a non-mitochondrial CoQ (Bersuker et al., 2019; Doll et al., 2019). Upon recruitment of FSP1 to the plasma membrane, its oxidoreductase activity reduces CoQ and suppresses the accumulation of L-OOH.
In spite of this significant support for the enzymatic mechanism of the production of the pro-ferroptotic death signal, there are two groups of facts contending the enzymatic nature of lipid peroxidation: i) high effectiveness of a number of free radical scavengers of different nature—aromatic amines, phenolic compounds, thiols—in preventing/suppressing ferroptosis and ii) the lack of correlation between the high anti-ferroptotic activity and the ability to inhibit LOXs. These important arguments deserve further focused research attention and necessitate testing of these compounds as regulators not only of LOX alone but also of 15-LOX/PEBP1 complexes. Such studies have not been reported so far. Their meaningfulness, however, should be considered in view of the possible design of highly specific anti-ferroptotic remedies suppressing the activity of 15-LOX/PEBP1 complexes without affecting the physiologically important reactions by LOXs catalyzing the production of numerous lipid mediators.
While the enzymatic nature of the HOO-AA-PE production seems highly likely, there is yet another possibility for the essential participation of free radical scavengers in the regulation of ferroptosis. This relates to the poorly explored second stage of the lipid peroxidation process and formation of the oxidatively truncated electrophilic species that interact with nucleophilic sites of yet to be identified proteins and may serve as the proximate executioners of ferroptosis. This difficult task may be significantly simplified assuming that these secondary products are generated from a limited number of PL-OOH.
Clearly definitive decoding of the mechanism of lipid peroxidation—enzymatic vs nonenzymatic—is not only an academic matter as a considerable number of injuries and disease conditions include ferroptotic cell death as one of the important pathogenic mechanisms. Hence, therapeutic and prevention strategies should rely on strong and unequivocal evidence.
Prerequisites and specific features of pro-ferroptotic lipid peroxidation in the brain.
The content of PUFA glycerophospholipids, particularly AA (C20:4)- and docosahexaenoic acid (DHA, C22:6)-phospholipids, is high (~60%) in the brain (Chen et al., 2008; Choi et al., 2018; Sun et al., 2018a). Among all phospholipids, PE (18:0/20:4) and PE (18:0/22:6) are the most abundant species (Choi et al., 2018). Given that AA-PE is essential for the production of ferroptotic death signals, this means that AA-PE is readily available and does not represent the limiting factor for the 15-LOX/PEBP1 catalyzed generation of HOO-AA-PE. Within the brain tissue, the amount of PUFA containing phospholipids differs depending on the cell type and developmental stage. For example astrocytes have relatively lower content of PUFA containing phosholipids vs. neurons, and premature brain has less PUFA containing phospholipids vs. adult brain (Martinez and Mougan, 1998; Tyurina et al., 2014). Thus, it is possible that ferroptosis sensitivity could depend on age and cell type even if the ferroptotic insult is similar in magnitude. However, this does not take into consideration the other key components of the ferroptotic program: 15-LOX and GSH. Notably, contemporary MS imaging, particularly the protocol of secondary ion MS utilizing gas cluster ion beams (Tian et al., 2019), detects AA-PE species in different subcellular localizations of brain cells (e.g. neurons, astrocytes) (Fig. S3), thus offering remarkable opportunities for identifying the major sites of catalytic initiation of the ferroptotic process.
While most LOXs usually oxygenate free PUFA, some of isoforms (e.g. 15-LOX) can directly attack PUFA (including AA) esterified into phospholipids, particularly PE. Canonically, the AA released from phospholipids in phospholipase A2-driven reactions is metabolized by COXs or LOXs to form lipid mediators (Mouchlis and Dennis, 2019; Shearer and Walker, 2018). Among the three forms of LOXs (5-LOX, 12-LOX, and 15-LOX) in the brain, 5-LOX is localized in the cytosol in neuronal cells and involved in generation of 5-HOO-AA, 5-HOO-AdA, and pro-inflammatory lipid mediators, leukotriens (Radmark et al., 2015). 12-LOX catalyzes oxygenation of AA to generate 12-HpETE and 12-HOO-AA (Li et al., 1997a). 15-LOX is the predominant isoform in the mammalian brain (Shalini et al., 2018). Normally, the level of the enzyme is low, but its expression increases many-fold in an array of diverse pathologies, including in response to acute brain injury (Kagan et al., 2017; Tyurina et al., 2019; Wenzel et al., 2017). 15-LOX can utilize AA and DHA as its substrates to generate anti-inflammatory lipid mediators, including AA-derived lipoxin A4 and DHA-derived resolvins (RvD1, RvD5) and neuroprotectins (Protectin D1) (Dalli et al., 2013; Green et al., 2018; Shalini et al., 2018). In addition, esterified oxygenated PUFA can act as signals for triggering different death pathways in cells (Kagan et al., 2017; Kagan et al., 2005) and elimination of dead cells (Tyurin et al., 2014). Importantly, 15-LOX is implicated in the generation of HOO-AA and HOO-AdA molecular species of PE recognized as ferroptotic cell death signals (Kagan et al., 2017; Wenzel et al., 2017). PEBP1 is abundantly expressed in the brain (Burgula et al., 2010) (Wang et al., 2017) specifically in neurons (Hellmann et al., 2009; Liu et al., 2019), and its role and contribution to different types of neuronal injury are subjects of active investigation (Jung et al., 2018; Kim et al., 2019). Recent studies established that 15-LOX/PEBP1 complexes are engaged in the mechanisms of ferroptotic neuronal death after traumatic brain injury (TBI) (Kenny et al., 2019; Stockwell et al., 2017; Wenzel et al., 2017).
GSH AND GPX4 IN FERROPTOSIS
Ferroptosis was originally described as a non-apoptotic form of cell death induced by the inhibition of cystine transportation by system XC− that can be rescued by Fer-1 or by iron chelators, such as deferoxamine (Dixon et al., 2012a). Since then, perturbations of GSH and GPX4, either through depletion of GSH or by inhibition of GPX4 activity, have been used as the major ferroptosis inducing method (Dixon and Stockwell, 2019). Although cell death caused by inhibition of GSH synthesis (Zaman et al., 1999) and GPX4 inactivation (Seiler et al., 2008b) has been known long before, the relationships between GSH, GPX4, iron, and lipid peroxidation have been clearly conceptualized in the context of ferroptosis.
GSH Synthesis
The thiol-containing tripeptide GSH (γ-L-glutamyl-L-cysteinylglycine) is the major component of the cellular antioxidant defense system against oxidative stress including lipid hydroperoxides (Meister and Anderson, 1983). GSH is the most abundant small molecule antioxidant present in cells. The concentration of GSH can vary in different cell types with the highest level reaching up to 12 mM in liver cells (Cooper, 1997). GSH is synthesized from its constituent amino acids—glutamic acid, cysteine, and glycine—through a two-step pathway. The initial step is the joining of glutamate and cysteine to produce the dipeptide γ-GluCys. Subsequently, γ-GluCys is combined with glycine to produce GSH. The first and rate-limiting step of GSH synthesis is catalyzed by glutamate cysteine ligase (GCL, previously called glutamylcysteine synthetase). Mechanistically, the gamma-carboxyl group of the amino acid L-Glu is activated by ATP-assisted phosphorylation followed by the reaction with an amino group of L-Cys (Meister, 1974). GCL is a dimeric protein comprised of a heavy, catalytic subunit and a light, modifier subunit, each of which is encoded by separate genes. Synthesis of GSH is precisely regulated by alteration of GCL activity through feedback inhibition by GSH, availability of L-Cys, and transcriptional and post-transcriptional regulation (Hibi et al., 2004). Buthionine sulfoximine (BSO) is a classical GCL inhibitor shown to induce ferroptosis (Griffith, 1982; Nishizawa et al., 2018; Yang et al., 2014a). Conversion of γ-GluCys to GSH is catalyzed by glutathione synthetase (GS). GS is comprised of two subunits and like GCL, uses ATP as a co-substrate (Oppenheimer et al., 1979). Though GS is not the rate-limiting enzyme during normal GSH synthesis, it could play a decisive role during GSH synthesis especially during pathological conditions (Lu, 2009a). Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcriptional regulator of both the GLC and GS shown to be involved in the execution of ferroptosis (Abdalkader et al., 2018).
System XC−
GSH synthesis is limited by the availability of one of its constituting amino acids, L-Cys (Shih et al., 2006). Cysteine is normally derived from two pathways: i) transmethylation and transsulfuration of the essential amino acid methionine (Tarver and Schmtdt, 1939) and ii) reduction of cellular cystine. Normally, cysteine is transported as its oxidized dimer, cystine, at the extracellular space (Kranich et al., 1998) (Bannai and Tateishi, 1986). Cystine is imported into the cells via an exchange for glutamic acid through system XC−, a cystine-glutamate exchanger. System XC− is a member of the heteromeric amino acid transporter (HAT) family, which consists of heterodimers of solute carrier 3 (SLC3) and solute carrier 7 (SLC7) family proteins (Broer and Wagner, 2002). Likewise, system XC− is composed of SLC3A2 (4F2hc, heavy chain) and SLC7A11 (xCT). The heavy chain is responsible for the amino acid transport while the xCT unit is required for anchoring the complex at the cell surface. Once transported, cystine is immediately converted into cysteine by the intracellular GSH pool or through thioredoxin reductase (Mandal et al., 2010). Because of instantaneous depletion of intracellular cystine combined with the high cellular glutamate concentration, system XC− predominantly acts as cystine importer and glutamate exporter (Bannai, 1986). Extracellular glutamate is a competitive inhibitor of cellular cystine uptake (Makowske and Christensen, 1982). System XC− is constitutively expressed; however, its activity can be regulated at the transcriptional and post-transcriptional levels (Lewerenz et al., 2012). System XC− is the most extensively studied axis in ferroptosis execution, and multiple system XC− inhibitors such as erastin, imidazole-keto-erastin, sulfasalazine, and glutamate have been shown to activate ferroptosis (Stockwell et al., 2017; Zhang et al., 2019b). Mechanistically erastin and sulfasalazine promote the phosphorylation of BECN1 through AMPKα and subsequent formation of the BECN1-SLC7A11 complex, which inhibits the cystine uptake (Song et al., 2018). System XC− regulates two important molecules implicated in neuronal cell death, extracellular glutamate and intracellular GSH. Extracellular glutamate can also overactivate ionotropic glutamate receptors such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), KA, and N-methyl-D-aspartic acid (NMDA) types (Lodge, 2009; Soria et al., 2014), leading to excitoxic neuronal death (Sheldon and Robinson, 2007). This makes system XC− one of the major therapeutic targets in neuronal disorders. Expression of system XC− is also regulated by the transcription factor Nrf2, making Nrf2 the major transcriptional regulator in modulating GSH levels (Harvey et al., 2009). Mice with deleted system XC− are fully viable, suggesting alternative pathways in GSH synthesis. However, the development of ferroptosis in such a system has not yet been studied (Sato et al., 2005).
Role of mitochondria in GSH synthesis.
The synthesis of GSH occurs exclusively in the cytosol of the cells with subsequent distribution to subcellular organelles (Griffith and Meister, 1985). Since mitochondria are responsible for the majority of ROS production in cells, a significant amount of GSH is transported into the mitochondria. Two inner mitochondrial membrane anion transporters, the dicarboxylate carrier (DIC, Slc25a10) and the 2-oxoglutarate carrier (OGC, Slc25a11) transport GSH into the mitochondria (Azzi et al., 1967; Lash, 2006; Meijer et al., 1972). Mitochondria receive 10–15 % of the total cellular GSH that equalizes the mitochondrial GSH concentration to the cytosolic GSH concentration (Griffith and Meister, 1985). In turn, mitochondria play a vital role in the synthesis and regulation of GSH. More specifically, the availability of glutamate is tightly controlled by mitochondrial metabolism. Three of the major glutamic acid metabolic enzymes, glutaminase (GLS), glutamate dehydrogenase (GDH), and aspartate aminotransferase, are housed in the mitochondria (Newsholme et al., 2005). GLS converts glutamine into glutamate through a deamination reaction. Mitochondrial glutamate is converted into the tricarboxylic acid (TCA) cycle cycle intermediate alpha-ketoglutarate (α-KG) either by glutamate dehydrogenase (GDH) or by the alanine or aspartate transaminases (TAs) (Watford, 2000). Both GDH and TAs are considered as cataplerotic enzymes forming products necessary for removing TCA cycle intermediates (Owen et al., 2002). These pathways are involved in the precise regulation of glutamate in the cytosol which in turn regulates the cystine intake by system XC− and subsequent GSH synthesis (Lu, 2000).
Function and metabolism of GSH.
Cellular GSH is required for several vital functions such as: 1) modulating cellular processes such as protein s-glutathionylation (Dalle-Donne et al., 2009), DNA synthesis (Suthanthiran et al., 1990), and immune response (Furukawa et al., 1987), 2) supplying cysteine for protein synthesis (Meister and Anderson, 1983), 3) metal ion homeostasis (Jozefczak et al., 2012), 4) maintaining the redox status of the proteins, and 5) providing an antioxidant defense (Lu, 2009b). GSH is also a critical component used to assemble iron-sulfur clusters in the cytosol and the mitochondria. Assembling Fe-S clusters is a vital function of mitochondria. Furthermore, GSH is the major small molecule that coordinates iron in the cytosol and is necessary for its coordination by iron chaperones (Patel, et al, 2019). The antioxidant capacity of GSH is useful in detoxifying xenobiotics and in the removal of ROS (Forman et al., 2009). Removal of ROS by GSH is achieved by non-enzymatic and enzymatic ways. In the non-enzymatic process, GSH reacts with hydroxyl radical, nitric oxide, and superoxide radicals (Dringen, 2000). However, this reaction generates a strong pro-oxidant thiyl radical. Hence, the protective non-enzymatic role of GSH as a radical scavenger may not be commonly realized in biological systems (Sagristá et al., 2002; Sturgeon et al., 1998).
GSH is an obligatory co-substrate of the non-heme enzymes, GPXs, which are involved in the reduction of peroxides to water or corresponding alcohols. Eight families of human GPXs have been identified (Brigelius-Flohé and Maiorino, 2013). GPX1–4 and 6 are selenoenzymes containing a selenocysteine at the catalytic center, whereas GPX5, 7, and 8 contain cysteine instead of selenocysteine. Hydrogen peroxide is the major oxidizing substrate for GPX1, 3, 7, and 8. Small molecular organic hydroperoxides including soluble free fatty acid peroxides are also reduced by GPX1 and 3. GPX4 is involved in reducing peroxides at the cellular membranes e.g. hydroperoxides of fatty acid residues in phospholipid peroxides and cholesterol hydroperoxides (Brigelius-Flohé and Maiorino, 2013). During the reduction of peroxides, GSH is converted into its oxidized dimeric form, GSSG, which can be recycled back into GSH using electrons transferred from reduced form of nicotinamide adenine dinucleotide phosphate by glutathione reductase (GR) (Carlberg and Mannervik, 1985). Thus, the availability of reduced GSH is regulated by NAD(P)H supply. The major metabolic pathways contributing to NAD(P)H production are glucose-6-phosphate dehydrogenase and 6-phosphoglucanate dehydrogenase, enzymes of pentose-phosphate pathway (PPP) (Lunt and Vander Heiden, 2011; Xiao et al., 2018; Yang and Sauve, 2016). Besides PPP, glyceraldehyde phosphate dehydrogenase of glycolysis (Lunt and Vander Heiden, 2011), isocitrate dehydrogenase, and malate dehydrogenase (Lunt and Vander Heiden, 2011; Yang and Vousden, 2016) of TCA cycle are the enzymes involved in the synthesis of NADPH. The levels of GSH/GSSG are closely associated with NADPH/NADP+ concentration. Numerous studies have shown involvement of metabolism in the regulation of ferroptosis. Inhibition of PPP enzymes ameliorate resistance to ferroptosis induced by erastin (Dixon et al., 2012a). Blockage of glycolysis by 2-deoxy-d-glucose substantially inhibits lipid peroxidation and ferroptosis, reportedly via AMPK pathway (Lee et al., 2020). Cellular GSH is also controlled by thioredoxin superfamily proteins such as thioredoxins, peroxiredoxins, and glutaredoxins (Xiao and Loscalzo, 2019). A high content of thioredoxin superfamily proteins is characteristic of cells of the central nervous system (Aon-Bertolino et al., 2011; Silva-Adaya et al., 2014), and different redoxins provide proper redox-regulations during neuronal differentiation and maturation processes (Olguín-Albuerne and Morán, 2018). Specifically, peroxiredoxin 6, one of the six peroxiredoxins which is involved in reduction of phospholipid hydroperoxides to phospholipid alcohols is protective in non-neuronal cells against ferroptosis (Lu et al., 2019)
While much of the GSH is utilized inside the cell, GSH is also transported across the plasma membrane for maintaining extracellular and inter-organ GSH homeostasis. Seven ATP-binding cassette transporter (ABC) family proteins—multidrug resistance-associated protein (MRP) 1–5, cystic fibrosis transmembrane conductance regulator, ATP Binding Cassette Subfamily G Member 2, and two organic-anion-transporting polypeptide (OATP) family members (OATP1, OATP2)—are involved in the export of cellular GSH (Fig. 7). Three solute carrier proteins, OAT1, OAT3, and Na+/Ca2+-Exchange Protein 3 (NaC3), are shown to be involved in the import of GSH to the cytoplasm (Bachhawat et al., 2013).
Figure 7.
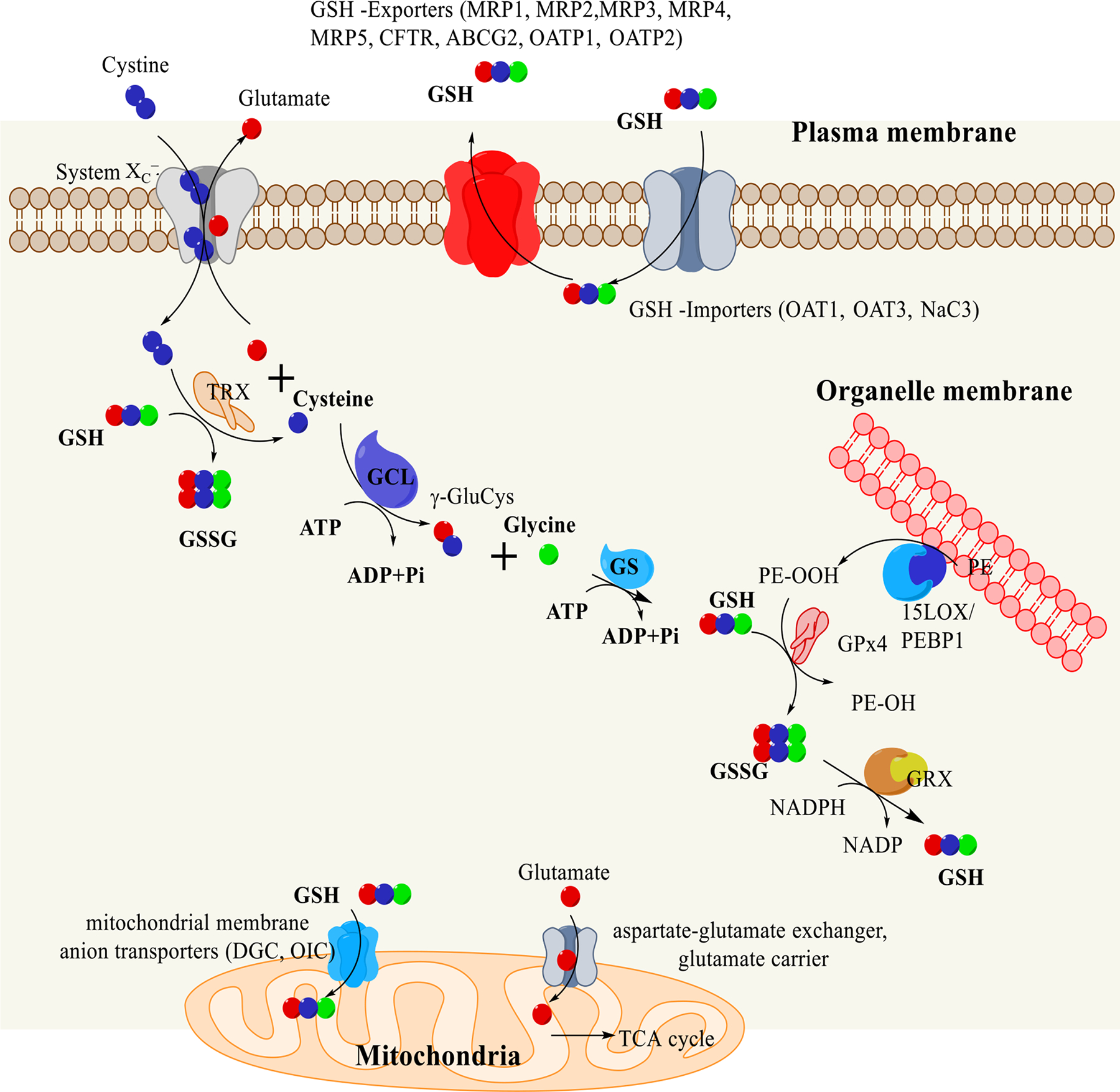
Metabolism of GSH. Cystine is imported into the cells with an expense of glutamate by system Xc-. Glutamate levels in the cytoplasm dictates the cystine transport. Cytosolic glutamate level is controlled by its transport to mitochondria through two transporters aspartate glutamate exchanger and glutamate carrier. Mitochondrial glutamate is used in the TCA cycle after its conversion to α-ketoglutarate. Transported cystine is converted to cysteine by thioredoxin (TRX) or by GSH. Cysteine is then converted to γ-glutamylcysteine (γ-GluCys) by glutamate cysteine ligase (GCS). GSH is synthesized by the addition of a glycine group to γ-GluCys by glutathione synthetase (GS). 10–15% of the GSH are transported to the mitochondria by dicarboxylate carrier (DIC) and the 2-oxoglutarate carrier (OGC). GSH is also transported across various transporters. PE-OOH produced from the organelle membrane is reduced to PE-OH by glutathione peroxidase4 (GPX4) using GSH. During the reduction process of PE-OOH to PE-OH, GSH is oxidized to GSSG, which are then converted into GSH by glutathione reductase (GRX).
GPX4
Among various GPXs, only GPX4 is known to reduce phospholipid hydroperoxides (Thomas et al., 1990; Ursini et al., 1982). GPX4 exists in 4 different isoforms, 3 of which have their specific subcellular locations: cytosolic GPX4, mitochondrial GPX4, and nuclear GPX4. A DHA-inducible splice variant of GPX4 identified in murine hippocampal HT22 cells is the fourth isoform of GPX4. Cytosolic GPX4 is the ubiquitously expressed predominant form of GPX4. Mitochondrial GPX4 is essential for survival and apoptotic resistance (Liang et al., 2009). GPX4 is a monomeric N-glycosylated protein (Wang et al., 2013) containing four α-helixes and 7 β-strands that form a thioredoxin motif (Scheerer et al., 2007). Like other GPXs, GPX4 follows a two-stage ping-pong mechanism to reduce peroxide. The first step involves the reduction of the PL-OOH to an alcohol with spontaneous oxidation of selenium. This step changes the position of GPX4 on the membrane making the selenium oxide accessible to the soluble GSH. Subsequently, selenium oxide is reduced by GSH in the second step (Brigelius-Flohé and Maiorino, 2013) forming GSSG and active GPX4 (Cozza et al., 2017).
GPX4 is a unique component of the essential antioxidant enzymes responsible for maintaining redox homeostasis in the cells. As a result, embryonic inactivation of GPX4 is lethal in mice (Ingold et al., 2015). However, adult mice with conditional GPX4 knockout die from acute renal injury (Friedmann Angeli et al., 2014c; Imai and Nakagawa, 2003; Yant et al., 2003). GPX4 is ubiquitously expressed in the human body, albeit with varying expression levels. Testis has the highest level of GPX4 expression, while brain has a low to moderate GPX4 expression. However, GPX4 is one of the most abundant constitutively expressed selenoproteins in the brain (Pitts et al., 2014). Depletion of neuronal GPX4 is lethal that can be prevented by either by α-tocopherol, 12/15-LOX inhibitors, or siRNA-mediated apoptosis-inducing factor silencing (Seiler et al., 2008b). GPX4 is one of the major regulators in the execution of ferroptosis.
THERAPEUTIC APPROACHES TARGETING INHIBITION OF FERROPTOSIS IN NEURONAL DISEASES
As the execution of ferroptosis engages three major metabolic redox pathways, it may be subject to multi-faceted regulation. This multiplicity of possible targets also represents a practical difficulty: which of the participating processes should be targeted? Another dilemma is that there are many already known inhibitors of ferroptosis—do we need to seek out new ones? The involvement of the multiple important pathways is a blessing because of the many choices of targets it offers, but it is also a curse because many essential pathways may be affected thus causing undesirable side effects. Using inhibitors which limit the availability and delivery of iron to its protein clients might lead to multiple manifestations of iron insufficiency. Iron is an essential cellular nutrient that is involved in vital cellular activities, such as oxygen transport, energy metabolism, and DNA synthesis (Lu, 2000). In fact, iron deficiency-related anemia is one of the major global health problems (Camaschella, 2015). Similarly, targeting the GSH/GPX4 axis of ferroptosis may interfere with numerous essential functions of thiols in regulating the cellular redox homeostasis. While the control of lipid peroxidation may represent the most attractive strategy, this approach may also cause nonspecific side effects. For example, baicalein is an excellent pan-inhibitor of LOX, and its application will likely affect multiple important pathways for biosynthesis of lipid mediators, making the prediction of the outcomes difficult. Even targeting one of the LOXs, 15-LOX, may have multiple consequences and side effects due to the important roles of 15-LOX in several biosynthetic reactions, producing pro- and anti-inflammatory lipid mediators, eicosdanoids and docosanoids (Dennis and Norris, 2015). These considerations lead to the conclusion that selective inhibitors targeting specific pro-ferroptotic reactions of 15-LOX/PEBP1 complexes may be the optimized solution. In the following section, we will present the ideas and data on the possible control and regulation of ferroptotic pathways with regard to the development of new therapeutic approaches.
GSH replenishment.
GSH/GPX4 acts as a negative regulator of ferroptosis; increased levels/activity of GSH/GPX4 suppresses ferroptosis, whereas a decrease in iron and lipid peroxidation is necessary for ferroptotic inhibition. Increasing GSH levels as a therapeutic strategy had already been attempted in many neurological diseases such as Alzheimer’s and Parkinson’s diseases (Cacciatore et al., 2012; Mischley et al., 2016). N-acetyl cysteine (NAC) (Bavarsad Shahripour et al., 2014), liposome encapsulated GSH (Cacciatore et al., 2012), and glutathione esters (Anderson et al., 1985) are some of the compounds used for replenishing GSH levels. Few studies however investigated the effects of GSH replenishment in the context of ferroptosis (Karuppagounder et al., 2018). The cysteine pro-drug, NAC, has attracted attention due to its clinical use as an FDA approved medication for acetaminophen-induced liver injury (Smilkstein et al., 1988). In an elegant study, Ratan and his group showed that hemin (heme oxidation product)-induced ferroptosis in primary neurons can be attenuated by NAC even 12h after the exposure (Karuppagounder et al., 2018). The mechanism of protection was, in a large part, due to increased intracellular GSH levels enabling the GSH utilizing enzyme, GSH-S-transferases, to detoxify 5-LOX-mediated lipid-derived protein oxidation (Karuppagounder et al., 2018). Interestingly while NAC was successful in attenuating collagenase-induced hemorrhagic stroke in mice, it was ineffective in rats due to drug tolerability issues since large doses of NAC (millimolar concentrations) were required to attenuate ferroptosis both in vivo and in vitro (with hemin as the hemorrhagic stroke model in vitro in primary neurons). Thus, translating these studies to humans might be challenging. NAC is known to have poor brain exposure when the blood brain barrier (BBB) is intact (Samuni et al., 2013). This might be less of a problem in disease states such as TBI or hemorrhagic stroke where the BBB is temporarily disrupted. However, even in these situations BBB integrity recovers with time (Kushi et al., 1994; Stevens et al., 1999). NAC’s poor brain exposure is not only due to its low BBB permeability but also active transport of this drug out of CNS via transporters (Hagos et al., 2017; Samuni et al., 2013) A recent phase-I study showed that brain exposure of NAC can be increased in patients when NAC is combined with an inhibitor of the transporters, probenecid (Clark et al., 2017). Probenecid has been shown to prevent active efflux of GSH and its conjugates by inhibiting MRP transporters (Versantvoort et al., 1995). In addition, co-administration of probenecid reduces the clearance of NAC and increases both plasma and brain levels of NAC by inhibiting OAT1- and OAT3-mediated uptake of NAC into renal tubule (Hagos et al., 2017). In line with a synergistic effect of probenecid with NAC, the combination rescues HT22 neuronal cells from glutamate-induced ferroptosis while neither NAC nor probenecid was effective by itself. Expectedly, however, neither NAC nor the combination of NAC and probenecid has a beneficial effect when GPX4 is inhibited by RSL3 (unpublished observations) indicating the critical role that GPX4 plays in neuronal health. If GPX4 is inhibited, the high levels of GSH do not protect the cells from ferroptotic insult.
This brings us to potential strategies of regulating GPX4 function i.e. reducing PL-OOH to corresponding alcohols. Although its effect in ferroptotic death has not been explored, GPX-mimic Ebselen has been shown to ameliorate neuronal injury in both acute and chronic CNS diseases (Maiorino et al., 1992; Schewe, 1995). Another strategy could be augmenting neuronal expression of GPX4 as it was shown by Alim et al., via injection of the AAV8 vector encoding GPX4 under the control of Synapsin 1 promoter into the brain parenchyma (Alim et al., 2019). However, this strategy requires pretreatment prior to CNS insult and would have limited clinical translational potential. GPX4 requires selenium in the form of selenocysteine for its activity (Ingold et al., 2018). While substitution of selenocysteine with cysteine rescues embryonic lethality in GPX4-deficient mice, GABAergic parvalbumin interneurons do not develop in the absence of selenocysteine deficient GPX4 leading to seizures (Ingold et al., 2018), indicating an essential role for selenium in CNS function. In addition to its roles in normal development, pharmacological supplementation of selenium has been shown to improve outcome after acute brain injury (Alim et al., 2019; Reisinger et al., 2009; Savaskan et al., 2003). However, a narrow therapeutic index and the requirement of intracerebroventricular delivery to be effective in CNS insults (Alim et al., 2019) make selenium administration a challenging therapy. Overcoming these shortcomings, a peptide containing selenium, TatSelPep, was recently shown to be effective in attenuating neurological dysfunction after hemorrhagic and ischemic stroke in mice (Alim et al., 2019). The mechanism was found to be transcriptional upregulation of GPX4 and inhibition of ferroptosis as well as excitotoxic and endoplasmic stress-mediated cell death.
GPX4 deficiency leads to early embryonic lethality (Friedmann Angeli et al., 2014b), which can be prevented by a vitamin E enriched diet containing α-tocopheryl acetate (Carlson et al., 2016). Conditional ablation of GPX4 in neurons results in rapid motor neuron degeneration due to ferroptosis and paralysis in adult mice. Supplementation with vitamin-E delayed the onset of paralysis and death induced by GPX4 ablation (Chen et al., 2015). Conversely, when mice with conditional deletion of GPX4 in forebrain neurons were fed a diet deficient in Vitamin E, hippocampal neurodegeneration and deficits in spatial learning and memory were accelerated (Hambright et al., 2017). These studies point to the important role of vitamin-E in the detoxification of lipid peroxyl radicals. During this process, the resultant tocopherol radical is reduced back to tocopherol by ascorbic acid and dihydrolipoic acid via the vitamin E recycling process (Kagan et al., 1992). Vitamin E derivatives—tocopherol and to a greater extent tocotrienols—are known to inhibit LOX activity (Khanna et al., 2003). The mechanism of inhibition is likely via specific liganding of the LOX catalytic site outcompeting binding and oxygenation of free or PE-esterified AA and AdA (Kagan et al., 2017). An endogenous metabolite of vitamin E, α-tocopherol hydroquinone inhibits ferroptosis via reducing active Fe(III) to an inactive Fe(II) at the active site of 15-LOX (Hinman et al., 2018). There have been numerous experimental and clinical studies evaluating the potential beneficial effects of vitaminE for prophylaxis or treatment of both acute and chronic CNS disorders (Anthonymuthu et al., 2016; Lee and Ulatowski, 2019). Although most studies evaluated tocopherols in this respect, the neuroprotective potential of tocotrienols may be greater than tocopherols (Patel et al., 2011).
Recent studies identified the flavoprotein, apoptosis-inducing factor mitochondria-associated 2 as a GPX4-independent controlling mechanism of ferroptosis in cancer cells (Bersuker et al., 2019; Doll et al., 2019). Another potential substitute for GPX4 function is the thioredoxin system. Thioredoxins contain a di-thiol disulfide in their active site. Their primary function is the reduction of oxidized cysteine residues and the cleavage of disulfide bonds (Arner and Holmgren, 2000). Inhibitors of the thioredoxin system lead to ferroptosis (Llabani et al., 2019). While harnessing the power of thioredoxin in therapy might be more challenging, another reducing cascade that involves dihydrolipoic acid is amenable to therapeutic targeting. In line with this, α-lipoic acid has been shown to improve functional outcomes in experimental and clinical studies of acute and chronic neurological diseases (Packer et al., 1997; Prehn et al., 1992). Although the exact mechanisms of how lipoic acid might inhibit ferroptosis are unknown, its interactions with lipoamide dehydrogenase in α-ketoglutarate complex could be critical in the reduction of PL-OOH (Bunik, 2019).
Radical trapping antioxidants
Lipid peroxidation is central to ferroptosis. Regulating ferroptosis is accomplished by controlling either the production or accumulation of select PL-OOH. Exogenous strategies targeting PL-OOH may be divided into two groups: LOX inhibitors and lipophilic antioxidants. Between these two, the latter—lipophilic antioxidants—demonstrated the greatest potential in experimental studies. It should also be mentioned that free radical scavengers may act via their interactions with radical intermediates of the 15-LOX-catalyzed enzymatic reaction (Kagan et al., 2017).
Liproxstatin (Lip-1) and Fer-1 were the first small molecule anti-ferroptotic agents characterized. Both of these lipophilic compounds were identified based on their low nanomolar anti-ferroptotic potency using a high-throughput screening approach (Dixon et al., 2012c; Friedmann Angeli et al., 2014b). They were found to suppress the accumulation of PL-OOH (Skouta et al., 2014; Wenzel et al., 2017) without altering the execution of other regulated death pathways (apoptosis, necroptosis). The precise mechanisms and their contributions to the anti-ferroptotic action remain debatable. First, both Fer-1 and Lip-1 are capable of inhibiting LOX activity. However, this effect is weak and requires micromolar concentrations to be observed (Zilka et al., 2017b), contrasted with the nanomolar anti-ferroptotic efficacy observed in cells. This may be interpreted in terms of a relatively low significance of the LOX inhibition in the overall protective mechanism. Assuming, however, that not only 15-LOX alone but rather the 15-LOX/PEBP1 complex act as the catalysts of AA-PE peroxidation, detailed studies of possible effects on the catalytic complex should be conducted to realistically evaluate possible contribution of the enzymatic 15-LOX/PEBP1 in the total anti-ferroptotic effects of Fer-1 and Lip-1. Alternatively, Fer-1 and Lip-1 may serve as radical-trapping antioxidants (RTAs), scavenging peroxyl radicals and preventing the non-enzymatic propagation of the radical chain reactions in membrane PUFA lipids. Though naturally occurring lipophilic RTA, α-tocopherol (vitamin E), is a potent ferroptosis inhibitor, its non-metabolizable isobaric analog, α-tocopherol hydroquinone did not inhibit ferroptosis despite its RTA capacity (Hinman et al., 2018). The anti-ferroptotic activity of Lip-1 and Fer-1 may lie in their active groups’ reduced H-bond donor capacity, which may improve the availability of these key active groups to react with membranous PL-OOH rather than off-target H-bond donors (Zilka et al., 2017b). As these studies primarily measured oxidation of non-physiologic reporter molecules (e.g. C11-BODIPY) in artificial liposomes, further validation is required to clarify the RTA mechanism in Fer-1- and Lip-1-mediated cytoprotection. Despite these uncertainties in precise mechanisms of their action, Lip-1 and Fer-1 have already displayed great promise for clinical translation across an array of acute and chronic neuropathologies. Intraventricular Fer-1 following TBI attenuated neuronal degeneration, reduced lesion volume, and improved long-term motor and cognitive function (Xie et al., 2019). Similarly, Fer-1 or Lip-1 improved histologic and neurofunctional outcomes in ICH models (Li et al., 2017). This benefit was correlated with lower levels of 4-HNE, a toxic electrophilic breakdown product of PL-OOH (Shoeb et al., 2014). Further, ferroptosis has been implicated in several chronic neurodegenerative conditions (Angeli et al., 2017; Cardoso et al., 2017). Lipid peroxidation is increased in Alzheimer’s disease (Praticò et al., 2004; Yang et al., 2010), Huntington’s disease, and Parkinson’s disease (PD) (Li et al., 1997b). Increased LOX activity is compounded by dysfunctional GSH metabolism (Saharan and Mandal, 2014; Smeyne and Smeyne, 2013) that is permissive to PL-OOH accumulation and death. Ultimately, the combination of increased LOX activity and diminished detoxification capacity primes neurons for ferroptotic death. Fer-1 completely ameliorated injury observed in rat corticostriatal brain slices expressing a pathogenic huntingtin exon 1 repeat fragment (mN90Q73) (Skouta et al., 2014). Similarly, anti-ferroptotics were effective at reducing dopaminergic neuron loss and improving behavioral outcomes in models of MPTP-induced PD (Do Van et al., 2016). These studies support the engagement of ferroptosis as a pathogenic mechanism in an array of neurological diseases, as well as the efficacy of anti-ferroptotic lipophilic antioxidants.
LOX inhibitors:
Among the five mammalian LOX enzymes (Kuhn et al., 2015), 5-LOX, 12-LOX, and 15-LOX have been implicated in ferroptosis execution in different cell types (Anthonymuthu et al., 2018; Chu et al., 2019; Karuppagounder et al., 2018; Liu et al., 2015). Baicalein, a pan-LOX inhibitor, and more specific LOX inhibitors such as the 5-LOX inhibitor, Zileuton, (Liu et al., 2015) have been shown to inhibit ferroptosis in vitro. In the case of acute TBI (Farias et al., 2011; Kenny et al., 2019), ICH (Poloyac et al., 2005), and cerebral ischemic-reperfusion (van Leyen et al., 2006; Yigitkanli et al., 2017), LOX expression and activity increased significantly in hours to days post-injury. The timeline of LOX expression and activity correlated with ferroptotic PL-OOH accumulation and offers a good therapeutic window. Furthermore, baicalein has been shown to attenuate ferroptotic PE-OOH accumulation, neuronal death, and the resultant cognitive deficits after TBI (Farias et al., 2011; Kenny et al., 2019).
LOXs are involved in the production of octanoids, eicosanoids, and docosanoids (Kuhn et al., 2015). These lipid mediators generated by LOX activity have important regulatory roles in inflammation and vascular tone (Dennis and Norris, 2015). Thus, their inhibition can also lead to detrimental effects depending on the timing and duration of activation and their enzymatic products. Identification of PEBP1 as a switch for the substrate specificity of 15-LOX from free to esterified into AA-PE and AdA-PE allows specific targeting of PE-OOH generation without interfering with the production of octanoids, eicosanoids, and docosanoids. Such compounds would not inhibit oxygenation of free arachidonic acid but inhibit PE-esterified arachidonic acid and inhibit ferroptotic death. As key sites that enable the adaptation of 15-LOX to specific substrate binding and allosteric activity upon binding with PEBP1 emerge (Mikulska-Ruminska et al., 2019), small molecule inhibitors of this interaction could be potential therapies targeting ferroptosis in the future.
ACSL4 and LPCAT3 inhibitors
Pharmacological inhibition of ACSL4 using thiazolidinediones (a class of peroxisome proliferator-activated receptor gamma [PPARγ] agonists) namely rosiglitazone, pioglitazone, and troglitazone, has been shown to ameliorate ferroptotic tissue injury in murine models of acute renal failure and intestinal ischemia/reperfusion (Doll et al., 2017; Li et al., 2019; Moerke et al., 2019). While PPARγ inhibitors have been studied in CNS disorders (Galimberti and Scarpini, 2017), few studies to date have evaluated the role of ACSL4 and LPCAT3 in neuronal ferroptotic cell death and potential therapies targeting these pathways in CNS diseases. It was shown that after TBI, ACSL4 expression is increased in conjunction with elevated levels of 15-LOX2 protein amount and activity (Kenny et al., 2019). ACSL4 inhibitor Triacsin C and LPCAT3 inhibitor CI-976 (but not Thimerosal) attenuated glutamate and mechanical stretch-induced neuronal death with similar efficacy to Fer-1 or Lip-1 in hippocampal neuronal cells. However, in vivo applications of these compounds were challenging. Intracerebroventricular administration of Triacsin C was required to attenuate TBI-induced hippocampal neuronal death, making clinical translation difficult (unpublished results). Therapeutic targeting of ACSL4 and LPCAT3 pathways as inhibitors of ferroptosis could be problematic since other ACSLs and acyl transferases can compensate for the lack of ACSL4 and LPCAT3 (Rossi Sebastiano and Konstantinidou, 2019), in addition to issues with specificity, solubility and blood brain permeability of small molecule inhibitors (Hishikawa et al., 2014; Moerke et al., 2019) (Gelderblom et al., 2001).
CONCLUSIONS
While regulated mechanisms of cell death and reprograming have been identified decades ago, deciphering their precise molecular mechanisms is still an ongoing process (Ellis and Horvitz, 1986). In addition to apoptosis with its clearly expressed anti-inflammatory propensities, several necrotic pro-inflammatory cell death programs have been identified and represent the objects of active and focused research (Linkermann et al., 2014). Ferroptosis is one of the latest additions to the list of these necrotic programs of cell death (Linkermann et al., 2014). Significant knowledge has been accumulated with regard to the engagement of three metabolic redox pillars—iron, thiols, and polyunsaturated phospholipids—in the trigger and propagation of the ferroptotic cell demise. Yet, many features and precise mechanisms of the formation of the proximate ferroptotic signals and their gateway interactions culminating in the cell death still remain elusive. As a significant part of these unresolved issues are related to the lack of adequate methodologies in accurate and sensitive analytical approaches to the analysis of lipid peroxidation products, the work in this direction is urgently required. The exact location of the intracellular pro-ferroptotic initiation centers and the spreading mechanisms of the death signals within the cell and beyond its confinements to generate pro-ferroptotic death clusters in a cell non-autonomous fashion are still missing pieces of information. Similarly, the specific role of mitochondria—clearly involved in the overall ferroptotic process—still awaits more rigorous research attention. On the positive side, extensive genetic and biochemical studies have already revealed several important metabolic networks actively participating in the regulation of the ferroptotic process (Stockwell et al., 2017; Zhang et al., 2019b). The initially claimed unique role of GPX4/GSH as the sole regulator of ferroptosis has been revised to include a number of several independent regulatory mechanisms such as FSP1 and iNOS/NO• (Bersuker et al., 2019; Doll et al., 2019; Kapralov et al., 2020). One of the “hot-spots” of the discussion on the lipid peroxidation mechanisms is its enzymatic vs non-enzymatic free radical chain reaction (Conrad and Pratt, 2019; Kagan et al., 2019; Stoyanovsky et al., 2019). While mechanistic reactive intermediates of these peroxidation reactions may be similar, the central issues of selectivity, specificity, and regulation will strongly affect the development of new generations of anti- and pro-ferroptotic small molecules as therapeutic modalities. The exponential kinetics of research in the field of ferroptosis is a warranty not only for the upcoming fundamental mechanistic discoveries but also in the applied research that will bring new effective and clinically significant anti-ferroptotic agents.
Supplementary Material
Fig. S1. A schema illustrating the formation of diversified peroxidation products from phospholipids with polyunsaturated fatty acids (PUFA-PL).
PUFA-PL can undergo enzymatic (e.g., catalyzed by LOXs) or non-enzymatic (e.g., catalyzed by transition metals) peroxidation. The major types of peroxidation products include: hydroperoxy-lipids generated as the primary molecular products. The latter can subsequently undergo reduction to hydroxy-lipids or conversion into epoxy-, oxo-, or oxidatively-truncated electrophilic products with shortened side chains.
Fig. S2. A Gram-negative bacterium, Pseudomonas aeruginosa, (biofilm form) expresses a specialized 15LOX - pLoxA - and releases using extracellular membrane vesicles. The vesicles, containing many proteins including pLOX, are taken-up by the host epithelial cells where pLoxA can selectively and specifically oxidize membrane phospholipids, particularly arachidonoyl-phosphatidylethanolamine (AA-PE) to produce pro-ferroptotic 15-HOO-AA-PE thus triggering ferroptotic death of the airways epithelium.
Figure S3. Secondary ion mass spectrometry (SIMS) utilizing gas cluster ion beams, detects AA-PE species in different subcellular localizations of neurons and astrocytes in brain tissue. Superimposition of immunofluorescence confocal images with SIMS images of coronal rat brain. A) NeuN (green, neurons) and GFAP (red, astrocytes) signals are marked with arrows and their thresholded overlays were used to define cell body boundaries (scale bar =10 μm). B) The PE(36:4p) SIMS signal (m/z 722.5) from the same region is rendered as a pseudocolored heatmapped panel and is intensity-scaled relative to the field of view. The thresholded NeuN and GFAP signals have been overlaid on the SIMS image. C) PE(36:4p) signal is cropped to the NeuN and GFAP thresholds. D) PE(38:4) m/z 766.5, and, E) PE(40:6) m/z 790.5 signals are also cropped to the NeuN and GFAP thresholds.
Reproduced with permission from Tian et al., 2019
Acknowledgments
This work was supported by NIH (U19AI068021, HL114453, NS076511, NS061817, GM113908, CA165065).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References:
- Abdalkader M, Lampinen R, Kanninen KM, Malm TM, and Liddell JR (2018). Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front Neurosci 12, 466–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ, et al. (2019). Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 177, 1262–1279 e1225. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Powrie F, Puri RN, and Meister A (1985). Glutathione monoethyl ester: preparation, uptake by tissues, and conversion to glutathione. Archives of biochemistry and biophysics 239, 538–548. [DOI] [PubMed] [Google Scholar]
- Angeli JPF, Shah R, Pratt DA, and Conrad M (2017). Ferroptosis Inhibition: Mechanisms and Opportunities. Trends in Pharmacological Sciences 38, 489–498. [DOI] [PubMed] [Google Scholar]
- Anthonymuthu TS, Kenny EM, and Bayir H (2016). Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res 1640, 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthonymuthu TS, Kenny EM, Shrivastava I, Tyurina YY, Hier ZE, Ting HC, Dar HH, Tyurin VA, Nesterova A, Amoscato AA, et al. (2018). Empowerment of 15-Lipoxygenase Catalytic Competence in Selective Oxidation of Membrane ETE-PE to Ferroptotic Death Signals, HpETE-PE. J Am Chem Soc 140, 17835–17839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon-Bertolino ML, Romero JI, Galeano P, Holubiec M, Badorrey MS, Saraceno GE, Hanschmann E-M, Lillig CH, and Capani F (2011). Thioredoxin and glutaredoxin system proteins—immunolocalization in the rat central nervous system. Biochimica et Biophysica Acta (BBA)-General Subjects 1810, 93–110. [DOI] [PubMed] [Google Scholar]
- Arner ES, and Holmgren A (2000). Physiological functions of thioredoxin and thioredoxin reductase. European journal of biochemistry 267, 6102–6109. [DOI] [PubMed] [Google Scholar]
- Azzi A, Chappell J, and Robinson B (1967). Penetration of the mitochondrial membrane by glutamate and aspartate. Biochemical and biophysical research communications 29, 148–152. [DOI] [PubMed] [Google Scholar]
- Bachhawat AK, Thakur A, Kaur J, and Zulkifli M (2013). Glutathione transporters. Biochimica et Biophysica Acta (BBA) - General Subjects 1830, 3154–3164. [DOI] [PubMed] [Google Scholar]
- Bannai S (1986). Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem 261, 2256–2263. [PubMed] [Google Scholar]
- Bannai S, and Tateishi N (1986). Role of membrane transport in metabolism and function of glutathione in mammals. Journal of Membrane Biology 89, 1–8. [DOI] [PubMed] [Google Scholar]
- Bavarsad Shahripour R, Harrigan MR, and Alexandrov AV (2014). N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav 4, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks J, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyington JC, Gaffney BJ, and Amzel LM (1993). The three-dimensional structure of an arachidonic acid 15-lipoxygenase. Science 260, 1482–1486. [DOI] [PubMed] [Google Scholar]
- Braverman NE, and Moser AB (2012). Functions of plasmalogen lipids in health and disease. Biochimica et biophysica acta 1822, 1442–1452. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohé R, and Maiorino M (2013). Glutathione peroxidases. Biochimica et Biophysica Acta (BBA) - General Subjects 1830, 3289–3303. [DOI] [PubMed] [Google Scholar]
- Broer S, and Wagner CA (2002). Structure-function relationships of heterodimeric amino acid transporters. Cell Biochem Biophys 36, 155–168. [DOI] [PubMed] [Google Scholar]
- Bunik VI (2019). Redox-Driven Signaling: 2-Oxo Acid Dehydrogenase Complexes as Sensors and Transmitters of Metabolic Imbalance. Antioxid Redox Signal 30, 1911–1947. [DOI] [PubMed] [Google Scholar]
- Burgula S, Medisetty R, Jammulamadaka N, Musturi S, Ilavazhagan G, and Singh SS (2010). Downregulation of PEBP1 in rat brain cortex in hypoxia. Journal of molecular neuroscience 41, 36–47. [DOI] [PubMed] [Google Scholar]
- Cacciatore I, Baldassarre L, Fornasari E, Mollica A, and Pinnen F (2012). Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxidative medicine and cellular longevity 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camaschella C (2015). Iron-deficiency anemia. New England journal of medicine 372, 1832–1843. [DOI] [PubMed] [Google Scholar]
- Cardoso BR, Hare DJ, Bush AI, and Roberts BR (2017). Glutathione peroxidase 4: a new player in neurodegeneration? Molecular Psychiatry 22, 328–335. [DOI] [PubMed] [Google Scholar]
- Carlberg I, and Mannervik B (1985). [59] Glutathione reductase. In Methods in enzymology (Elsevier), pp. 484–490. [DOI] [PubMed] [Google Scholar]
- Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL, and Conrad M (2016). Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox biology 9, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitidis P, Schewe T, Sutherland M, Kuhn H, and Nigam S (1998). 15-Lipoxygenation of phospholipids may precede the sn-2 cleavage by phospholipases A2: reaction specificities of secretory and cytosolic phospholipases A2 towards native and 15-lipoxygenated arachidonoyl phospholipids. FEBS Lett 434, 437–441. [DOI] [PubMed] [Google Scholar]
- Chakrabarti M, Cockrell AL, Park J, McCormick SP, Lindahl LS, and Lindahl PA (2015). Speciation of iron in mouse liver during development, iron deficiency, IRP2 deletion and inflammatory hepatitis. Metallomics 7, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Chen Z, Liu M, Gao X, Cheng Y, Wei Y, Wu Z, Cui D, and Shang H (2019). Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Research Bulletin 153, 122–132. [DOI] [PubMed] [Google Scholar]
- Chen CT, Green JT, Orr SK, and Bazinet RP (2008). Regulation of brain polyunsaturated fatty acid uptake and turnover. Prostaglandins Leukot Essent Fatty Acids 79, 85–91. [DOI] [PubMed] [Google Scholar]
- Chen L, Hambright WS, Na R, and Ran Q (2015). Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem 290, 28097–28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Yin T, Shinozaki K, Lampe JW, Stevens JF, Becker LB, and Kim J (2018). Comprehensive analysis of phospholipids in the brain, heart, kidney, and liver: brain phospholipids are least enriched with polyunsaturated fatty acids. Mol Cell Biochem 442, 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, and Gu W (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nature cell biology 21, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RSB, Empey PE, Bayir H, Rosario BL, Poloyac SM, Kochanek PM, Nolin TD, Au AK, Horvat CM, Wisniewski SR, et al. (2017). Phase I randomized clinical trial of N-acetylcysteine in combination with an adjuvant probenecid for treatment of severe traumatic brain injury in children. PLoS One 12, e0180280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, and Pratt DA (2019). The chemical basis of ferroptosis. Nat Chem Biol 15, 1137–1147. [DOI] [PubMed] [Google Scholar]
- Cooper AJ (1997). Glutathione in the brain: disorders of glutathione metabolism. The molecular and genetic basis of neurological disease, 1245–1245. [Google Scholar]
- Cotticelli MG, Xia S, Lin D, Lee T, Terrab L, Wipf P, Huryn DM, and Wilson RB (2019). Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J Pharmacol Exp Ther 369, 47–54. [DOI] [PubMed] [Google Scholar]
- Cozza G, Rossetto M, Bosello-Travain V, Maiorino M, Roveri A, Toppo S, Zaccarin M, Zennaro L, and Ursini F (2017). Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic Biol Med 112, 1–11. [DOI] [PubMed] [Google Scholar]
- Cozzi A, Orellana DI, Santambrogio P, Rubio A, Cancellieri C, Giannelli S, Ripamonti M, Taverna S, Di Lullo G, Rovida E, et al. (2019). Stem Cell Modeling of Neuroferritinopathy Reveals Iron as a Determinant of Senescence and Ferroptosis during Neuronal Aging. Stem Cell Reports 13, 832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo G, Giustarini D, and Milzani A (2009). Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends in biochemical sciences 34, 85–96. [DOI] [PubMed] [Google Scholar]
- Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggstrom JZ, Petasis NA, and Serhan CN (2013). The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J 27, 2573–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar HH, Tyurina YY, Mikulska-Ruminska K, Shrivastava I, Ting HC, Tyurin VA, Krieger J, St Croix CM, Watkins S, Bayir E, et al. (2018). Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest 128, 4639–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EA, and Norris PC (2015). Eicosanoid storm in infection and inflammation. Nature reviews Immunology 15, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, and Yang WS (2012a). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012b). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012c). Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, and Stockwell BR (2019). The hallmarks of ferroptosis. Annual Review of Cancer Biology 3, 35–54. [Google Scholar]
- Do Van B, Gouel F, Jonneaux A, Timmerman K, Gelé P, Pétrault M, Bastide M, Laloux C, Moreau C, Bordet R, et al. (2016). Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiology of Disease 94, 169–178. [DOI] [PubMed] [Google Scholar]
- Doll S, and Conrad M (2017). Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 69, 423–434. [DOI] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel C, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, et al. (2014). Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16, 1069–1079. [DOI] [PubMed] [Google Scholar]
- Dringen R (2000). Metabolism and functions of glutathione in brain. Progress in Neurobiology 62, 649–671. [DOI] [PubMed] [Google Scholar]
- Dufrusine B, Di Francesco A, Oddi S, Scipioni L, Angelucci CB, D’Addario C, Serafini M, Häfner A-K, Steinhilber D, and Maccarrone M (2019). Iron-dependent trafficking of 5-lipoxygenase and impact on human macrophage activation. Frontiers in Immunology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis HM, and Horvitz HR (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829. [DOI] [PubMed] [Google Scholar]
- Esposito BP, Epsztejn S, Breuer W, and Cabantchik ZI (2002). A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal Biochem 304, 1–18. [DOI] [PubMed] [Google Scholar]
- Farias SE, Heidenreich KA, Wohlauer MV, Murphy RC, and Moore EE (2011). Lipid Mediators in Cerebral Spinal Fluid of Traumatic Brain Injured Patients. Journal of Trauma and Acute Care Surgery 71, 1211–1218. [DOI] [PubMed] [Google Scholar]
- Forcina GC, and Dixon SJ (2019). GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 19, e1800311. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Zhang H, and Rinna A (2009). Glutathione: overview of its protective roles, measurement, and biosynthesis. Molecular aspects of medicine 30, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, Ghosh MC, Lee J, Rouault TA, Park MH, et al. (2014). Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc Natl Acad Sci U S A 111, 8031–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014a). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature cell biology 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014b). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature Cell Biology 16, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014c). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature Cell Biology 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Meydani S, and Blumberg J (1987). Reversal of age-associated decline in immune responsiveness by dietary glutathione supplementation in mice. Mechanisms of ageing and development 38, 107–117. [DOI] [PubMed] [Google Scholar]
- Galaris D, Barbouti A, and Pantopoulos K (2019). Iron homeostasis and oxidative stress: An intimate relationship. Biochim Biophys Acta Mol Cell Res 1866, 118535. [DOI] [PubMed] [Google Scholar]
- Galimberti D, and Scarpini E (2017). Pioglitazone for the treatment of Alzheimer’s disease. Expert opinion on investigational drugs 26, 97–101. [DOI] [PubMed] [Google Scholar]
- Gaschler MM, Hu F, Feng H, Linkermann A, Min W, and Stockwell BR (2018). Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem Biol 13, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom H, Verweij J, Nooter K, and Sparreboom A (2001). Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer 37, 1590–1598. [DOI] [PubMed] [Google Scholar]
- Green AR, Freedman C, Tena J, Tourdot BE, Liu B, Holinstat M, and Holman TR (2018). 5 S,15 S-Dihydroperoxyeicosatetraenoic Acid (5,15-diHpETE) as a Lipoxin Intermediate: Reactivity and Kinetics with Human Leukocyte 5-Lipoxygenase, Platelet 12-Lipoxygenase, and Reticulocyte 15-Lipoxygenase-1. Biochemistry 57, 6726–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith O (1982). Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. Journal of Biological Chemistry 257, 13704–13712. [PubMed] [Google Scholar]
- Griffith OW, and Meister A (1985). Origin and turnover of mitochondrial glutathione. Proceedings of the National Academy of Sciences 82, 4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichardant M, Chen P, Liu M, Calzada C, Colas R, Vericel E, and Lagarde M (2011). Functional lipidomics of oxidized products from polyunsaturated fatty acids. Chem Phys Lipids 164, 544–548. [DOI] [PubMed] [Google Scholar]
- Hagos FT, Daood MJ, Ocque JA, Nolin TD, Bayir H, Poloyac SM, Kochanek PM, Clark RS, and Empey PE (2017). Probenecid, an organic anion transporter 1 and 3 inhibitor, increases plasma and brain exposure of N-acetylcysteine. Xenobiotica; the fate of foreign compounds in biological systems 47, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambright WS, Fonseca RS, Chen L, Na R, and Ran Q (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox biology 12, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CJ, Thimmulappa RK, Singh A, Blake DJ, Ling G, Wakabayashi N, Fujii J, Myers A, and Biswal S (2009). Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free radical biology & medicine 46, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann J, Rommelspacher H, and Wernicke C (2009). Long-term ethanol exposure impairs neuronal differentiation of human neuroblastoma cells involving neurotrophin-mediated intracellular signaling and in particular protein kinase C. Alcoholism: Clinical and Experimental Research 33, 538–550. [DOI] [PubMed] [Google Scholar]
- Hibi T, Nii H, Nakatsu T, Kimura A, Kato H, Hiratake J, and Oda J.i. (2004). Crystal structure of γ-glutamylcysteine synthetase: insights into the mechanism of catalysis by a key enzyme for glutathione homeostasis. Proceedings of the National Academy of Sciences 101, 15052–15057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hider RC, and Kong X (2013). Iron speciation in the cytosol: an overview. Dalton Trans 42, 3220–3229. [DOI] [PubMed] [Google Scholar]
- Hider RC, and Kong XL (2011). Glutathione: a key component of the cytoplasmic labile iron pool. Biometals 24, 1179–1187. [DOI] [PubMed] [Google Scholar]
- Hinman A, Holst CR, Latham JC, Bruegger JJ, Ulas G, McCusker KP, Amagata A, Davis D, Hoff KG, Kahn-Kirby AH, et al. (2018). Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLOS ONE 13, e0201369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishikawa D, Hashidate T, Shimizu T, and Shindou H (2014). Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res 55, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L-R, Liang J-T, Zou J-H, Wang L-Y, Li Q, and Wang X-M (2012). Possible novel roles of poly (rC)-binding protein 1 in SH-SY5Y neurocytes: an analysis using a dynamic Bayesian network. Neuroscience bulletin 28, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, and Nakagawa Y (2003). Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 34, 145–169. [DOI] [PubMed] [Google Scholar]
- Ingold I, Aichler M, Yefremova E, Roveri A, Buday K, Doll S, Tasdemir A, Hoffard N, Wurst W, and Walch A (2015). Expression of a catalytically inactive mutant form of glutathione peroxidase 4 (Gpx4) confers a dominant-negative effect in male fertility. Journal of Biological Chemistry 290, 14668–14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421. [DOI] [PubMed] [Google Scholar]
- Janero DR (1990). Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med 9, 515–540. [DOI] [PubMed] [Google Scholar]
- Jozefczak M, Remans T, Vangronsveld J, and Cuypers A (2012). Glutathione is a key player in metal-induced oxidative stress defenses. Int J Mol Sci 13, 3145–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HY, Cho SB, Kim W, Yoo DY, Won M-H, Choi G-M, Cho T-G, Kim DW, Hwang IK, and Choi SY (2018). Phosphatidylethanolamine-binding protein 1 protects CA1 neurons against ischemic damage via ERK-CREB signaling in Mongolian gerbils. Neurochemistry international 118, 265–274. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Serbinova EA, Forte T, Scita G, and Packer L (1992). Recycling of vitamin E in human low density lipoproteins. J Lipid Res 33, 385–397. [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1, 223–232. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurina YY, W.Y. S, Vlasova II, Dar H, Tyurin VA, Amoscato AA, Mallampalli R, van der Wel PC, He RR, et al. (2019). Redox Phospholipidomics of Enzymatically Generated Oxygenated Phospholipids as Specific Signals of Programmed Cell Death. Free Radic Biol Med in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, St Croix CM, Mikulska-Ruminska K, Liu B, Shrivastava IH, et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol 16, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, Wilkinson CM, Nadeau CA, Kumar A, Perry S, et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol 84, 854–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny EM, Fidan E, Yang Q, Anthonymuthu TS, New LA, Meyer EA, Wang H, Kochanek PM, Dixon CE, Kagan VE, et al. (2019). Ferroptosis Contributes to Neuronal Death and Functional Outcome After Traumatic Brain Injury. Crit Care Med 47, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, and Sen CK (2003). Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem 278, 43508–43515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Cho SB, Jung HY, Yoo DY, Oh JK, Choi G-M, Cho T-G, Kim DW, Hwang IK, and Choi SY (2019). Phosphatidylethanolamine-Binding Protein 1 Ameliorates Ischemia-Induced Inflammation and Neuronal Damage in the Rabbit Spinal Cord. Cells 8, 1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn HI, and Liversedge M (1944). On a new aerobic metabolite whose production by brain is inhibited by apomorphine, emetine, er- gotamine, epinephrine, and menadione. J Pharmacol Exp Ther 82, 292–300. [Google Scholar]
- Kozlov AV, Yegorov D, Vladimirov YA, and Azizova OA (1992). Intracellular free iron in liver tissue and liver homogenate: studies with electron paramagnetic resonance on the formation of paramagnetic complexes with desferal and nitric oxide. Free Radic Biol Med 13, 9–16. [DOI] [PubMed] [Google Scholar]
- Kranich O, Dringen R, Sandberg M, and Hamprecht B (1998). Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: preference for cystine. Glia 22, 11–18. [PubMed] [Google Scholar]
- Kuhn H, Banthiya S, and van Leyen K (2015). Mammalian lipoxygenases and their biological relevance. Biochimica et biophysica acta 1851, 308–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushi H, Katayama Y, Shibuya T, Tsubokawa T, and Kuroha T (1994). Gadolinium DTPA-enhanced magnetic resonance imaging of cerebral contusions. Acta neurochirurgica Supplementum 60, 472–474. [DOI] [PubMed] [Google Scholar]
- Lash LH (2006). Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chemico-biological interactions 163, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latunde-Dada GO (2017). Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj 1861, 1893–1900. [DOI] [PubMed] [Google Scholar]
- Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, and Steinberg GR (2020). Energy-stress-mediated AMPK activation inhibits ferroptosis. Nature Cell Biology 22, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Lee H, Kang S, and Park WJ (2016). Fatty Acid Desaturases, Polyunsaturated Fatty Acid Regulation, and Biotechnological Advances. Nutrients 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, and Ulatowski LM (2019). Vitamin E: Mechanism of transport and regulation in the CNS. IUBMB life 71, 424–429. [DOI] [PubMed] [Google Scholar]
- Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, Subramanian P, Hsu E, Natarajan N, Nandal A, et al. (2013). Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem 288, 17791–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Maher P, and Methner A (2012). Regulation of xCT expression and system $$ x_{\text{c}}^{-} $$ function in neuronal cells. Amino Acids 42, 171–179. [DOI] [PubMed] [Google Scholar]
- Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2, e90777–e90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, Liu D, Zhang F, Ning S, and Yao J (2019). Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death & Differentiation, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, and Schubert D (1997a). A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19, 453–463. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, and Schubert D (1997b). A Role for 12-lipoxygenase in Nerve Cell Death Caused by Glutathione Depletion. Neuron 19, 453–463. [DOI] [PubMed] [Google Scholar]
- Liang H, Yoo S-E, Na R, Walter CA, Richardson A, and Ran Q (2009). Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. The Journal of biological chemistry 284, 30836–30844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Stockwell BR, Krautwald S, and Anders HJ (2014). Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nature reviews Immunology 14, 759–767. [DOI] [PubMed] [Google Scholar]
- Liu X, Bao Y, Meng W, Yang P, An Y, Ma J, Tang Y, Liu Z, Lu Y, and Zhou J (2019). TROY interacts with RKIP to promote glioma development. Oncogene 38, 1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang W, Li Y, Xiao Y, Cheng J, and Jia J (2015). The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biological & pharmaceutical bulletin 38, 1234–1239. [DOI] [PubMed] [Google Scholar]
- Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E, and Hergenrother PJ (2019). Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nature chemistry 11, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge D (2009). The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclature. Neuropharmacology 56, 6–21. [DOI] [PubMed] [Google Scholar]
- Lordan R, Tsoupras A, and Zabetakis I (2017). Phospholipids of Animal and Marine Origin: Structure, Function, and Anti-Inflammatory Properties. Molecules 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Chen X, Hong Y, Zhu H, He Q, Yang B, Ying M, and Cao J (2019). Identification of PRDX6 as a Regulator of Ferroptosis. Acta Pharmacol Sin 40, 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC (2000). Regulation of glutathione synthesis. Curr Top Cell Regul 36, 95–116. [DOI] [PubMed] [Google Scholar]
- Lu SC (2009a). Regulation of glutathione synthesis. Mol Aspects Med 30, 42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC (2009b). Regulation of glutathione synthesis. Molecular Aspects of Medicine 30, 42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annual review of cell and developmental biology 27, 441–464. [DOI] [PubMed] [Google Scholar]
- Ma Y, de Groot H, Liu Z, Hider RC, and Petrat F (2006). Chelation and determination of labile iron in primary hepatocytes by pyridinone fluorescent probes. Biochem J 395, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ML, Murray IV, and Axelsen PH (2007). Mass spectrometric analysis demonstrates that BODIPY 581/591 C11 overestimates and inhibits oxidative lipid damage. Free Radic Biol Med 42, 1392–1397. [DOI] [PubMed] [Google Scholar]
- Maiorino M, Roveri A, and Ursini F (1992). Antioxidant effect of Ebselen (PZ 51): peroxidase mimetic activity on phospholipid and cholesterol hydroperoxides vs free radical scavenger activity. Arch Biochem Biophys 295, 404–409. [DOI] [PubMed] [Google Scholar]
- Makeyev AV, and Liebhaber SA (2002). The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. Rna 8, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowske M, and Christensen HN (1982). Contrasts in transport systems for anionic amino acids in hepatocytes and a hepatoma cell line HTC. Journal of Biological Chemistry 257, 5663–5670. [PubMed] [Google Scholar]
- Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC, and Harper JW (2015). Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Seiler A, Perisic T, Kölle P, Canak AB, Förster H, Weiss N, Kremmer E, Lieberman MW, and Bannai S (2010). System xc− and thioredoxin reductase 1 cooperaYvely rescue glutathione deficiency. Journal of Biological Chemistry 285, 22244–22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Liu J, Chen C, Zhang W, Qian R, Chen X, Lu H, Ge J, Zhao C, Zhang D, et al. (2016). PCBP2 Modulates Neural Apoptosis and Astrocyte Proliferation After Spinal Cord Injury. Neurochem Res 41, 2401–2414. [DOI] [PubMed] [Google Scholar]
- Martinez M, and Mougan I (1998). Fatty acid composition of human brain phospholipids during normal development. Journal of Neurochemistry 71, 2528–2533. [DOI] [PubMed] [Google Scholar]
- Masaldan S, Bush AI, Devos D, Rolland AS, and Moreau C (2019). Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med 133, 221–233. [DOI] [PubMed] [Google Scholar]
- Meijer A, Brouwer A, Reijngoud D, Hoek J, and Tager J (1972). Transport of glutamate in rat-liver mitochondria. Biochimica et Biophysica Acta (BBA)-Bioenergetics 283, 421–429. [DOI] [PubMed] [Google Scholar]
- Meister A (1974). Theγ-Glutamyl Cycle: Diseases Associated with Specific Enzyme Deficiencies. Annals of Internal Medicine 81, 247–253. [DOI] [PubMed] [Google Scholar]
- Meister A, and Anderson ME (1983). Glutathione. Annual review of biochemistry 52, 711–760. [DOI] [PubMed] [Google Scholar]
- Mejia EM, and Hatch GM (2016). Mitochondrial phospholipids: role in mitochondrial function. J Bioenerg Biomembr 48, 99–112. [DOI] [PubMed] [Google Scholar]
- Merkofer M, Kissner R, Hider RC, Brunk UT, and Koppenol WH (2006). Fenton chemistry and iron chelation under physiologically relevant conditions: Electrochemistry and kinetics. Chem Res Toxicol 19, 1263–1269. [DOI] [PubMed] [Google Scholar]
- Mikulska-Ruminska K, Shrivastava I, Krieger J, Zhang S, Li H, Bayir H, Wenzel SE, VanDemark AP, Kagan VE, and Bahar I (2019). Characterization of Differential Dynamics, Specificity, and Allostery of Lipoxygenase Family Members. Journal of chemical information and modeling 59, 2496–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischley LK, Conley KE, Shankland EG, Kavanagh TJ, Rosenfeld ME, Duda JE, White CC, Wilbur TK, De La Torre PU, and Padowski JM (2016). Central nervous system uptake of intranasal glutathione in Parkinson’s disease. npj Parkinson’s Disease 2, 16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerke C, Theilig F, Kunzendorf U, and Krautwald S (2019). ACSL4 as the First Reliable Biomarker of Ferroptosis Under Pathophysiological Conditions In Ferroptosis in Health and Disease, Tang D, ed. (Cham: Springer International Publishing; ), pp. 111–123. [Google Scholar]
- Mouchlis VD, and Dennis EA (2019). Phospholipase A2 catalysis and lipid mediator lipidomics. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, Bruick RK, and Philpott CC (2011). Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab 14, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsholme P, Brennan L, Rubi B, and Maechler P (2005). New insights into amino acid metabolism, β-cell function and diabetes. Clinical science 108, 185–194. [DOI] [PubMed] [Google Scholar]
- Nishizawa S, Araki H, Ishikawa Y, Kitazawa S, Hata A, Soga T, and Hara T (2018). Low tumor glutathione level as a sensitivity marker for glutamate-cysteine ligase inhibitors. Oncol Lett 15, 8735–8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olguín-Albuerne M, and Morán J (2018). Redox signaling mechanisms in nervous system development. Antioxidants & redox signaling 28, 1603–1625. [DOI] [PubMed] [Google Scholar]
- Oppenheimer L, Wellner VP, Griffith OW, and Meister A (1979). Glutathione synthetase. Purification from rat kidney and mapping of the substrate binding sites. J Biol Chem 254, 5184–5190. [PubMed] [Google Scholar]
- Owen OE, Kalhan SC, and Hanson RW (2002). The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. Journal of Biological Chemistry 277, 30409–30412. [DOI] [PubMed] [Google Scholar]
- Packer L, Tritschler HJ, and Wessel K (1997). Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic Biol Med 22, 359–378. [DOI] [PubMed] [Google Scholar]
- Patel SJ, Frey AG, Palenchar DJ, Achar S, Bullough KZ, Vashisht A, Wohlschlegel JA, and Philpott CC (2019). A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat Chem Biol 15, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrat F, de Groot H, Sustmann R, and Rauen U (2002). The chelatable iron pool in living cells: a methodically defined quantity. Biol Chem 383, 489–502. [DOI] [PubMed] [Google Scholar]
- Philpott CC, and Jadhav S (2019). The ins and outs of iron: Escorting iron through the mammalian cytosol. Free Radic Biol Med 133, 112–117. [DOI] [PubMed] [Google Scholar]
- Philpott CC, Ryu MS, Frey A, and Patel S (2017). Cytosolic iron chaperones: Proteins delivering iron cofactors in the cytosol of mammalian cells. J Biol Chem 292, 12764–12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts MW, Byrns CN, Ogawa-Wong AN, Kremer P, and Berry MJ (2014). Selenoproteins in Nervous System Development and Function. Biological Trace Element Research 161, 231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloyac SM, Reynolds RB, Yonas H, and Kerr ME (2005). Identification and quantification of the hydroxyeicosatetraenoic acids, 20-HETE and 12-HETE, in the cerebrospinal fluid after subarachnoid hemorrhage. J Neurosci Methods 144, 257–263. [DOI] [PubMed] [Google Scholar]
- Porter NA, Caldwell SE, and Mills KA (1995). Mechanisms of free radical oxidation of unsaturated lipids. Lipids 30, 277–290. [DOI] [PubMed] [Google Scholar]
- Praticò D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, and Lee VMY (2004). 12/15-lipoxygenase is increased in Alzheimer’s disease: possible involvement in brain oxidative stress. Am J Pathol 164, 1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt DA, Tallman KA, and Porter NA (2011). Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Accounts of chemical research 44, 458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehn JH, Karkoutly C, Nuglisch J, Peruche B, and Krieglstein J (1992). Dihydrolipoate reduces neuronal injury after cerebral ischemia. J Cereb Blood Flow Metab 12, 78–87. [DOI] [PubMed] [Google Scholar]
- Protchenko O (2019). Unchaperoned iron in hepatocytes causes lipid peroxidation and fatty liver disease.
- Quiles Del Rey M, and Mancias JD (2019). NCOA4-mediated ferritinophagy: a potential link to neurodegeneration. Front Neurosci 13, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radmark O, Werz O, Steinhilber D, and Samuelsson B (2015). 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochimica et biophysica acta 1851, 331–339. [DOI] [PubMed] [Google Scholar]
- Reinert A, Morawski M, Seeger J, Arendt T, and Reinert T (2019). Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neuroscience 20, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisinger J, Hollinger K, Lang W, Steiner C, Winter T, Winter A, Mori M, Lindorfer A, Kiblbock D, and Siostrzonek P (2009). Does early administration of selenium improve neurological outcome after cardiac arrest? The American journal of emergency medicine 27, 176–181. [DOI] [PubMed] [Google Scholar]
- Rossi Sebastiano M, and Konstantinidou G (2019). Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, and Philpott CC (2017). PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J Clin Invest 127, 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagristá ML, GarcÍa AF, De Madariaga MA, and Mora M (2002). Antioxidant and pro-oxidant effect of the thiolic compounds N-acetyl-L-cysteine and glutathione against free radical-induced lipid peroxidation. Free radical research 36, 329–340. [DOI] [PubMed] [Google Scholar]
- Saharan S, and Mandal PK (2014). The emerging role of glutathione in Alzheimer’s disease. J Alzheimers Dis 40, 519–529. [DOI] [PubMed] [Google Scholar]
- Samuni Y, Goldstein S, Dean OM, and Berk M (2013). The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta 1830, 4117–4129. [DOI] [PubMed] [Google Scholar]
- Santos R, Lefevre S, Sliwa D, Seguin A, Camadro J-M, and Lesuisse E (2010). Friedreich ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid Redox Signal 13, 651–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K. i., and Moriguchi T (2005). Redox imbalance in cystine/glutamate transporter-deficient mice. Journal of Biological Chemistry 280, 37423–37429. [DOI] [PubMed] [Google Scholar]
- Savaskan NE, Brauer AU, Kuhbacher M, Eyupoglu IY, Kyriakopoulos A, Ninnemann O, Behne D, and Nitsch R (2003). Selenium deficiency increases susceptibility to glutamate-induced excitotoxicity. FASEB J 17, 112–114. [DOI] [PubMed] [Google Scholar]
- Scheerer P, Borchert A, Krauss N, Wessner H, Gerth C, Hohne W, and Kuhn H (2007). Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry 46, 9041–9049. [DOI] [PubMed] [Google Scholar]
- Schewe T (1995). Molecular actions of ebselen--an antiinflammatory antioxidant. General pharmacology 26, 1153–1169. [DOI] [PubMed] [Google Scholar]
- Seibt TM, Proneth B, and Conrad M (2019). Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 133, 144–152. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. (2008a). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, et al. (2008b). Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metabolism 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Shalini SM, Ho CF, Ng YK, Tong JX, Ong ES, Herr DR, Dawe GS, and Ong WY (2018). Distribution of Alox15 in the Rat Brain and Its Role in Prefrontal Cortical Resolvin D1 Formation and Spatial Working Memory. Mol Neurobiol 55, 1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer GC, and Walker RE (2018). An overview of the biologic effects of omega-6 oxylipins in humans. Prostaglandins Leukot Essent Fatty Acids 137, 26–38. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, and Robinson MB (2007). The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51, 333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Bencze KZ, Stemmler TL, and Philpott CC (2008). A cytosolic iron chaperone that delivers iron to ferritin. Science 320, 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, and Murphy TH (2006). Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. Journal of Neuroscience 26, 10514–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoeb M, Ansari NH, Srivastava SK, and Ramana KV (2014). 4-hydroxynonenal in the pathogenesis and progression of human diseases. Current medicinal chemistry 21, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Adaya D, Gonsebatt ME, and Guevara J (2014). Thioredoxin system regulation in the central nervous system: experimental models and clinical evidence. Oxidative medicine and cellular longevity 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136, 4551–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne M, and Smeyne RJ (2013). Glutathione metabolism and Parkinson’s disease. Free radical biology & medicine 62, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilkstein MJ, Knapp GL, Kulig KW, and Rumack BH (1988). Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 319, 1557–1562. [DOI] [PubMed] [Google Scholar]
- Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al. (2018). AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc- Activity. Current Biology 28, 2388–2399.e2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria FN, Pérez-Samartín A, Martin A, Gona KB, Llop J, Szczupak B, Chara JC, Matute C, and Domercq M (2014). Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. The Journal of Clinical Investigation 124, 3645–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens H, Jansen HM, De Reuck J, Lemmerling M, Strijckmans K, Goethals P, Lemahieu I, de Jong BM, Willemsen AT, and Korf J (1999). 55Cobalt (Co) as a PET-tracer in stroke, compared with blood flow, oxygen metabolism, blood volume and gadolinium-MRI. Journal of the neurological sciences 171, 11–18. [DOI] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanovsky DA, Tyurina YY, Shrivastava I, Bahar I, Tyurin VA, Protchenko O, Jadhav S, Bolevich SB, Kozlov AV, Vladimirov YA, et al. (2019). Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic Biol Med 133, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon BE, Sipe HJ, Barr DP, Corbett JT, Martinez JG, and Mason RP (1998). The fate of the oxidizing tyrosyl radical in the presence of glutathione and ascorbate implications for the radical sink hypothesis. Journal of Biological Chemistry 273, 30116–30121. [DOI] [PubMed] [Google Scholar]
- Sun GY, Simonyi A, Fritsche KL, Chuang DY, Hannink M, Gu Z, Greenlief CM, Yao JK, Lee JC, and Beversdorf DQ (2018a). Docosahexaenoic acid (DHA): An essential nutrient and a nutraceutical for brain health and diseases. Prostaglandins Leukot Essent Fatty Acids 136, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Shih AY, Johannssen HC, Erb H, Li P, and Murphy TH (2006). Two-photon imaging of glutathione levels in intact brain indicates enhanced redox buffering in developing neurons and cells at the cerebrospinal fluid and blood-brain interface. Journal of Biological Chemistry 281, 17420–17431. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zheng Y, Wang C, and Liu Y (2018b). Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis 9, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthanthiran M, Anderson ME, Sharma VK, and Meister A (1990). Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proceedings of the National Academy of Sciences 87, 3343–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarver H, and Schmtdt C (1939). The conversion of methionine to cystine: experiments with radioactive sulfur. Journal of Biological Chemistry 130, 67–80. [Google Scholar]
- Thomas JP, Geiger PG, Maiorino M, Ursini F, and Girotti AW (1990). Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochimica et Biophysica Acta (BBA)-Lipids and Lipid Metabolism 1045, 252–260. [DOI] [PubMed] [Google Scholar]
- Tian H, Sparvero LJ, Blenkinsopp P, Amoscato AA, Watkins SC, Bayir H, Kagan VE, and Winograd N (2019). Secondary-Ion Mass Spectrometry Images Cardiolipins and Phosphatidylethanolamines at the Subcellular Level. Angewandte Chemie 58, 3156–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnus W, Gembardt F, Latk M, Parmentier S, Hugo C, Bornstein SR, and Linkermann A (2019). The clinical relevance of necroinflammation-highlighting the importance of acute kidney injury and the adrenal glands. Cell Death and Differentiation 26, 68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, Kapralov AA, Macphee CH, and Kagan VE (2014). Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ 21, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW, et al. (2014). A mitochondrial pathway for biosynthesis of lipid mediators. Nature chemistry 6, 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurina YY, Tyurin VA, Anthonymuthu T, Amoscato AA, Sparvero LJ, Nesterova AM, Baynard ML, Sun W, He R, Khaitovich P, et al. (2019). “Redox lipidomics technology: Looking for a needle in a haystack”. Chem Phys Lipids 221, 93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Ursini F, Maiorino M, Valente M, Ferri L, and Gregolin C (1982). Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochimica et Biophysica Acta (BBA)-Lipids and Lipid Metabolism 710, 197–211. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Kim HY, Lee SR, Jin G, Arai K, and Lo EH (2006). Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke 37, 3014–3018. [DOI] [PubMed] [Google Scholar]
- Versantvoort CH, Bagrij T, Wright KA, and Twentyman PR (1995). On the relationship between the probenecid-sensitive transport of daunorubicin or calcein and the glutathione status of cells overexpressing the multidrug resistance-associated protein (MRP). International journal of cancer 63, 855–862. [DOI] [PubMed] [Google Scholar]
- Vidaki M, Drees F, Saxena T, Lanslots E, Taliaferro MJ, Tatarakis A, Burge CB, Wang ET, and Gertler FB (2017). A requirement for Mena, an actin regulator, in local mRNA translation in developing neurons. Neuron 95, 608–622. e605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wu Y, Zhou T, Guo Y, Zheng B, Wang J, Bi Y, Liu F, Zhou Z, Guo X, et al. (2013). Mapping of the N-Linked Glycoproteome of Human Spermatozoa. Journal of Proteome Research 12, 5750–5759. [DOI] [PubMed] [Google Scholar]
- Wang Z, Bu J, Yao X, Liu C, Shen H, Li X, Li H, and Chen G (2017). Phosphorylation at S153 as a functional switch of phosphatidylethanolamine binding protein 1 in cerebral ischemia-reperfusion injury in rats. Frontiers in molecular neuroscience 10, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watford M (2000). Glutamine and Glutamate Metabolism across the Liver Sinusoid. The Journal of Nutrition 130, 983S–987S. [DOI] [PubMed] [Google Scholar]
- Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, et al. (2017). PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 171, 628–641 e626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RJ (1982). Free manganese (II) and iron (II) cations can act as intracellular cell controls. FEBS Lett 140, 3–10. [DOI] [PubMed] [Google Scholar]
- Xiao W, and Loscalzo J (2019). Metabolic responses to reductive stress. Antioxidants & Redox Signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Wang R-S, Handy DE, and Loscalzo J (2018). NAD (H) and NADP (H) redox couples and cellular energy metabolism. Antioxidants & redox signaling 28, 251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF, Gao GY, and Jiang JY (2019). Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci Ther 25, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Sterling CR, and Tank AW (2009). cAMP-mediated stimulation of tyrosine hydroxylase mRNA translation is mediated by polypyrimidine-rich sequences within its 3′-untranslated region and poly (C)-binding protein 2. Molecular pharmacology 76, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Koga N, and Nagaoka M (2012). Ferryl-oxo species produced from Fenton’s reagent via a two-step pathway: minimum free-energy path analysis. J Phys Chem B 116, 14178–14182. [DOI] [PubMed] [Google Scholar]
- Yanatori I, Richardson DR, Imada K, and Kishi F (2016). Iron Export through the Transporter Ferroportin 1 Is Modulated by the Iron Chaperone PCBP2. J Biol Chem 291, 17303–17318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanatori I, Richardson DR, Toyokuni S, and Kishi F (2017). The iron chaperone poly(rC)-binding protein 2 forms a metabolon with the heme oxygenase 1/cytochrome P450 reductase complex for heme catabolism and iron transfer. J Biol Chem 292, 13205–13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanatori I, Yasui Y, Tabuchi M, and Kishi F (2014). Chaperone protein involved in transmembrane transport of iron. Biochem J 462, 25–37. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhuo J-M, Chu J, Chinnici C, and Praticò D (2010). Amelioration of the Alzheimer’s Disease Phenotype by Absence of 12/15-Lipoxygenase. Biological Psychiatry 68, 922–929. [DOI] [PubMed] [Google Scholar]
- Yang M, and Vousden KH (2016). Serine and one-carbon metabolism in cancer. Nature Reviews Cancer 16, 650–662. [DOI] [PubMed] [Google Scholar]
- Yang Wan S., SriRamaratnam R, Welsch Matthew E., Shimada K, Skouta R, Viswanathan Vasanthi S., Cheah Jaime H., Clemons Paul A., Shamji Alykhan F., Clish Clary B., et al. (2014a). Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014b). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2016). Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 26, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, and Sauve AA (2016). NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1864, 1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, and Prolla TA (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Biology and Medicine 34, 496–502. [DOI] [PubMed] [Google Scholar]
- Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, Li B, Fan BY, Wang X, Li WX, et al. (2019). Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural regeneration research 14, 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Zeng C, Dong L, Wu Y, Huang Q, and Wu Y (2019). Inhibition of ferroptosis processes ameliorates cognitive impairment in kainic acid-induced temporal lobe epilepsy in rats. American journal of translational research 11, 875. [PMC free article] [PubMed] [Google Scholar]
- Yegorov D, Kozlov AV, Azizova OA, and Vladimirov YA (1993). Simultaneous determination of Fe(III) and Fe(II) in water solutions and tissue homogenates using desferal and 1,10-phenanthroline. Free Radic Biol Med 15, 565–574. [DOI] [PubMed] [Google Scholar]
- Yigitkanli K, Zheng Y, Pekcec A, Lo EH, and van Leyen K (2017). Increased 12/15-Lipoxygenase Leads to Widespread Brain Injury Following Global Cerebral Ischemia. Translational stroke research 8, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Xu L, and Porter NA (2011). Free radical lipid peroxidation: mechanisms and analysis. Chemical reviews 111, 5944–5972. [DOI] [PubMed] [Google Scholar]
- Zaman K, Ryu H, Hall D, O’Donovan K, Lin K-I, Miller MP, Marquis JC, Baraban JM, Semenza GL, and Ratan RR (1999). Protection from Oxidative Stress-Induced Apoptosis in Cortical Neuronal Cultures by Iron Chelators Is Associated with Enhanced DNA Binding of Hypoxia-Inducible Factor-1 and ATF-1/CREB and Increased Expression of Glycolytic Enzymes, p21waf1/cip1, and Erythropoietin. The Journal of Neuroscience 19, 9821–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sun C, Zhao C, Hao J, Zhang Y, Fan B, Li B, Duan H, Liu C, Kong X, et al. (2019a). Ferroptosis inhibitor SRS 16–86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res 1706, 48–57. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, and Stockwell BR (2019b). Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell chemical biology 26, 623–633. e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Sun Y, Mao X, Jin K, and Greenberg D (2002). Expression of poly (C)-binding proteins is differentially regulated by hypoxia and ischemia in cortical neurons. Neuroscience 110, 191–198. [DOI] [PubMed] [Google Scholar]
- Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, and Pratt DA (2017a). On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci 3, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. A schema illustrating the formation of diversified peroxidation products from phospholipids with polyunsaturated fatty acids (PUFA-PL).
PUFA-PL can undergo enzymatic (e.g., catalyzed by LOXs) or non-enzymatic (e.g., catalyzed by transition metals) peroxidation. The major types of peroxidation products include: hydroperoxy-lipids generated as the primary molecular products. The latter can subsequently undergo reduction to hydroxy-lipids or conversion into epoxy-, oxo-, or oxidatively-truncated electrophilic products with shortened side chains.
Fig. S2. A Gram-negative bacterium, Pseudomonas aeruginosa, (biofilm form) expresses a specialized 15LOX - pLoxA - and releases using extracellular membrane vesicles. The vesicles, containing many proteins including pLOX, are taken-up by the host epithelial cells where pLoxA can selectively and specifically oxidize membrane phospholipids, particularly arachidonoyl-phosphatidylethanolamine (AA-PE) to produce pro-ferroptotic 15-HOO-AA-PE thus triggering ferroptotic death of the airways epithelium.
Figure S3. Secondary ion mass spectrometry (SIMS) utilizing gas cluster ion beams, detects AA-PE species in different subcellular localizations of neurons and astrocytes in brain tissue. Superimposition of immunofluorescence confocal images with SIMS images of coronal rat brain. A) NeuN (green, neurons) and GFAP (red, astrocytes) signals are marked with arrows and their thresholded overlays were used to define cell body boundaries (scale bar =10 μm). B) The PE(36:4p) SIMS signal (m/z 722.5) from the same region is rendered as a pseudocolored heatmapped panel and is intensity-scaled relative to the field of view. The thresholded NeuN and GFAP signals have been overlaid on the SIMS image. C) PE(36:4p) signal is cropped to the NeuN and GFAP thresholds. D) PE(38:4) m/z 766.5, and, E) PE(40:6) m/z 790.5 signals are also cropped to the NeuN and GFAP thresholds.
Reproduced with permission from Tian et al., 2019