

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited, progressive nephropathy accounting for 4–10% of end stage renal disease worldwide. PKD1 and PKD2 are the most common disease loci, but even accounting for other genetic causes, about 7% of families remain unresolved. Typically, these unsolved cases have relatively mild kidney disease and often have a negative family history. Mosaicism, due to de novo mutation in the early embryo, has rarely been identified by conventional genetic analysis of ADPKD families. Here we screened for mosaicism by employing two next generation sequencing screens, specific analysis of PKD1 and PKD2 employing long-range polymerase chain reaction, or targeted capture of cystogenes. We characterized mosaicism in 20 ADPKD families; the pathogenic variant was transmitted to the next generation in five families and sporadic in 15. The mosaic pathogenic variant was newly discovered by next generation sequencing in 13 families, and these methods precisely quantified the level of mosaicism in all. All of the mosaic cases had PKD1 mutations, 14 were deletions or insertions, and 16 occurred in females. Analysis of kidney size and function showed the mosaic cases had milder disease than a control PKD1 population, but only a few had clearly asymmetric disease. Thus, in a typical ADPKD population, readily detectable mosaicism by next generation sequencing accounts for about 1% of cases, and about 10% of genetically unresolved cases with an uncertain family history. Hence, identification of mosaicism is important to fully characterize ADPKD populations and provides informed prognostic information.
Keywords: ADPKD, PKD1, Mosaicism, Genotype/Phenotype Correlations, Mutations, Diagnostics, Prognostics
Graphical Abstract
Introduction
Tuberous sclerosis (TSC) is an example of a dominant disease where de novo mutations are common, accounting for 60–70% of cases1. These new mutations usually happen in germ cells but can occur postzygotically, resulting in mosaicism where the subject is composed of cells with and without the mutation. Mosaic pathogenic variants can be difficult to detect by Sanger sequencing (SS), and when targeted capture next generation sequencing (tNGS) was applied to the 10–15% of TSC patents with no (Sanger) mutation identified (NMI), 49% were found to be mosaics2. Hence, mosaicism is a diagnostic challenge but its identification is important to fully characterize a population for family planning purposes, and to determine likely disease severity within a family.
Population studies indicate that 10–20% of autosomal dominant polycystic kidney disease (ADPKD) families can be traced to a de novo mutation within living generations3, 4. ADPKD is a late-onset, systemic, inherited disorder with a worldwide prevalence of ~1 in 10005. It is characterized by the development and growth of kidney cysts, resulting in end stage renal disease (ESRD) in ~50% of individuals by 60y6. The major ADPKD genes are PKD1 (~78% of pedigrees) and PKD2 (~15%); PKD1 is associated with more severe disease, and PKD1 truncating (PKD1T) alleles are more severe than nontruncating (PKD1NT)7–9. A small proportion of NMI cases have mutations to other genes, e.g. GANAB, DNAJB11, or HNF1B, but ~7% remain unresolved10–13. Mutation screening of PKD1 is complicated by the presence of six pseudogenes, traditionally requiring a long-range PCR (LR-PCR) approach for SS14. More recently, LR-NGS and specifically designed tNGS approaches have been shown to successfully screen this locus15, 16, 17, 18.
Mosaicism is a possible explanation for NMI cases since they often have a negative/indeterminate family history, typically milder disease, and sometimes an asymmetric/unilateral renal disease presentation; five mosaic ADPKD families have been described4, 9, 19. One with gonadal and somatic mosaicism (mixed) illustrated the complexity of identifying a potential living related donor by linkage analysis alone20. In a second, an affected father with two ADPKD daughters had the familial large deletion in just ~15% of cells21. Other examples showed milder than expected disease for a PKD1T pathogenic variant in a mosaic mother22, LR-NGS analysis detected low level mosaicism (3–10%) in different cell types23, and quantitative PCR and cloning detected mosaicism in a patient with asymmetric disease4. Importantly, all described cases were identified because an offspring had fully penetrant disease.
Here, we employed two NGS approaches to detect and characterize mosaicism in 20 ADPKD pedigrees.
Results
Next generation sequencing analysis to detect mosaicism
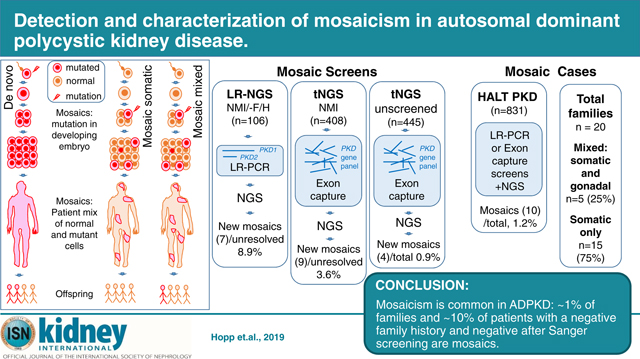
To detect mosaicism, two NGS methods were employed, a modified LR-NGS screen of just PKD1 and PKD215 and a tNGS panel of 65 or 137 cystogenes (Figure 1, Table S1)12, with DNA isolated from blood cells, mimicking a clinical diagnostic setting. The LR-NGS screen included 106 ADPKD families with a known/suspected negative family history and NMI from SS of PKD1 and PKD2. Four suspected mosaics detected by family/conventional analysis were controls (Figure 1). The tNGS screening included two parts, 408 SS PKD1/PKD2 NMI ADPKD-like families12 and 445 unscreened ADPKD-like families (Figure 1). Previous analysis showed good coverage of the duplicated PKD1 gene using this tNGS approach12. Both screening methods have the advantage over whole exome sequencing (WES) of high average sequence read depth (LR-NGS PKD1=7317, PKD2=10988; 65-Gene-tNGS PKD1=839; PKD2=475; 137-Gene-tNGS PKD1=619; PKD2=779; Table S2), allowing detection of pathogenic variants found in just a small percentage of reads.
Figure 1. Diagram showing the study design and detection rates for the different groups involved in the screen for mosaicism.

ADPKD patients were included in the study from those collected for mutation screening at the Mayo Translational PKD Center (MTPC), ADPKD Modifier patients, and PKD1 and PKD2 SS NMI HALT PKD patients. The analysis for mosaic cases was performed in three screens, LR-NGS of PKD1 and PKD2 SS NMI ADPKD patients with a proven or suspected negative family history (-ve F/H), a tNGS screen of PKD1 and PKD2 SS NMI ADPKD patients, and a tNGS screen of previously unscreened patients. The number of total families included in each screen, and their origin is indicated at the top portion of the figure. The detection rate breakdown of each screen is indicated in boxes and includes number of families resolved with typical heterozygous variants (non mosaics resolved), number of families remaining unresolved, and the number of families with detected mosaic variants. The numbers in the circles represents families shared among the LR-NGS and tNGS approach. The number of newly detected mosaics as a proportion of the unresolved plus mosaics (New mosaics/residual) is also calculated. Approximately 1% of not families (HALT PKD (1.2%) and tNGS (Unscreened; 0.9%)) were mosaics. In addition, newly resolved mosaics represented 8.9% of the unresolved plus mosaic families from the LR-NGS screen (-ve F/H and SS PKD1/PKD2 NMI) and 3.6% from the tNGS (SS PKD1/PKD2 NMI) screen. The number of unresolved families from the tNGS (unscreened) analysis is greater than expected from a typical ADPKD population, which is likely because of the large number of very mild and atypical patient screened from the MTPC population. *For the non-mosaic patients resolved from the LR-NGS screen, results from other genes detected by tNGS are also included.
Two families illustrated the need for caution in defining mosaic cases and the value of employing NGS plus Sanger methodologies. In pedigree M796, SS of individual II-2 suggested a missense pathogenic variant at a low level but subsequent tNGS analysis showed that 581/1130 reads (51%) had the pathogenic variant (Figure S1A–C). Follow up SS consistently showed low level signal of the pathogenic variant (Figure S1B). These SS findings are likely explained by allele dropout due to a polymorphism under the LR or exon-specific PCR primer. Family P1317 showed the opposite scenario where tNGS identified a 26 bp duplication in just 7.9% of reads in II-1 (Figure S1D,E). However, SS showed the duplication to be equally represented (Figure S1F), and subsequently the patient was found to have a positive family history; highlighting, that using tNGS, larger duplications may be captured inefficiently
Definitions and details of the detected mosaic cases
To be certain that the described cases are genuine mosaics, we required the pathogenic variant to be consistently detected at a reduced level by NGS and SS or also detected by allele specific-PCR (AS-PCR). To avoid calling cases with apparent small reductions in read representation that may be false mosaics, or false positives, the mutant allele needed to be present in 2–25% of reads (4–50% of the expected level of the pathogenic allele); two exceptions (Pedigrees 390010 and 790057) are explained below. Tables 1 and S3 describe and evaluate the detected mosaic pathogenic variants to PKD1 and provide clinical information of the 20 resolved families. All families have a negative or equivocal family history, with details and diagnostic information summarized in Table 2.
Table 1.
Clinical presentation and genetic details of PKD1 families with mosaicism
| Image Analysis | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % reads +ve | Kidney | Liver | ||||||||||||||
| eGFR | HTN | Vol (ht) | ||||||||||||||
| Pedigree | Pathogenic variant | Subject | LR-NGS | tNGS | Sex | age | age (y) | Age (y) | Cystic description | RK | LK | TKV | MIC | Fig | Cystic Description | Vol (ht) |
| Variant transmitted | ||||||||||||||||
| 590013 | c.935_937delinsA | II-2 | NS | 17.3 | F | 63/56y | 43 | 52 | Multiple bilateral, large LK | 249 | 639 | 888 | 1C | 1D | Some small | 1013 |
| p.(Ala312fs) | III-1 | Offspring | M | 67/29y | 18 | 25 | Multiple bilateral | 326 | 396 | 722 | 1E | 1E | Few very small | 828 | ||
| 690020 | c.l2440_12443dup | II-2 | 9.1 | NS | F | 57/67y | 61 | 61 | Many large bilateral (US) | 14.2^ | 16.6^ | NA | ND | 5.3 cm cyst | NA | |
| p.(Phe4149fs) | III-1 | Offspring | F | 109/43y | 41 | 42 | Multiple bilateral | 402 | 424 | 826 | 1C | S2A | Few small | 947 | ||
| 870348 | c.2548_2557del | II-2 | NS | 8.2 | M | 87/47y | 36 | 47 | Multiple bilateral | 244 | 351 | 595 | 1B | 1J | Few small | 696 |
| p.(Asp850fs) | III-1 | Offspring | M | Diagnosed at 26y but no further information available | ||||||||||||
| 590046 | c.8970_8972del | II-4 | 7.0 | NS | F | 91/56y | ′22 | 53 | Multiple tiny, 1 large LK | 223 | 325 | 548 | 2A | 1M | One large cyst | 1529 |
| p.(Tyr2991del) | III-2 | Offspring | M | 127/22y | NA | 19 | Few bilateral | 120 | 114 | 234 | 1B | 1N | No cysts | 951 | ||
| III-4 | Offspring | M | NA | NA | 11 | Multiple bilateral (US) | NA | ND | NA | |||||||
| 390010 | c.H654_11683del | II-2 | 0.1 | ND | F | 74/52y | ′51 | 48 | Few, 1 large RK & LK | 113 | 144 | 257 | 1A | 1Q | Few tiny | 777 |
| p.(Val3885_Ser3894del) | III-1 | Offspring | M | 82/38y | 20 | 35 | Multiple bilateral | 373 | 436 | 809 | 1D | 1R | Multiple small | 1054 | ||
| Single affected individual | ||||||||||||||||
| 790057 | c. 1386–34_1606+26del282bp, p.(Ser463fs) | II-2 | NI | 37.7 | F | 82/37 | Y/29 | 31 | Multiple bilateral | 303 | 348 | 651 | 1C | 2D | Few large | 1382 |
| 590039 | c.215+8043_1850–141del11.7kb, p.(Leu72fs) | II-4 | NI | 22.7, 25.5^ | F | ES/55y 28/53y |
Y/39 | 49 | Multiple large bilateral | 785 | 1073 | 1858 | 1D | 2H | Severe PLD# | 4228 |
| M484 | c.10922dupC, p.(Arg3642fs) | II-8 | 20.6 | 20.1 | F | 88/47y | Y/44 | 46 | Many large bilateral | 400 | 573 | 973 | 1C | 2K | Severe PLD | 3334 |
| M375 | c.11379delG, p.(Thr3794fs) | II-3 | 19.1 | NS | F | 113/34y | N/34 | 37 | Multiple small | 97 | 136 | 233 | 1A | 2N | Few small | 908 |
| 290001 | c.4520G>A, p.(Trp1507*) | II-1 | 17.5 | 20.6 | F | 41/66y | Y/42 | 42 | Multiple bilateral (US) | NA | ND | NA | ||||
| 870005 | c.844_845dupGG, p.(Pro283fs) | II-2 | NS | 14.8 | F | 72/54y | Y/43 | 49 | Many large bilateral | 400 | 431 | 831 | 1C | 3C | Moderate PLD | NA |
| M646 | c.8426C>T, p.(Pro2809Leu) | II-3 | NS | 13.4 | M | 44/60y | Y/<54 | 54 | Bilateral, several large LK | 206 | 282 | 487 | 1B | 3F | None | 1191 |
| 870452 | c.11157–2A>G, p.(Arg3719?) | II-2 | NS | 13.4 | F | 77/57y | Y/45 | 52 | Multiple bilateral | 527 | 735 | 1262 | 1C | −70 small | NA | |
| M1312 | c.5352_5353insTG, p.(Asn1785*) | II-2 | NS | 12.2 | F | 125/35y | N/36 | 36 | Multiple, 1 large RK | 280 | 256 | 536 | 1C | 3K | Few tiny | 536 |
| 290034 | c.3685delG, p.(Val1229fs) | II-3 | 12.3 | 12.0 | M | 34/68y | Y/52 | 67 | Bilateral renal enlargement | 1506 | 1179 | 2685 | 1C | NA | NA | |
| M1327 | c.10373_10386del14, p.(Pro3458fs) | II-2 | NS | 9.0 | M | 78/48y | Y/40 | 46 | Multiple cysts, few large | 186 | 198 | 384 | 1B | 4C | Few small | 876 |
| M855 | c.74_75delGCinsT, p.(Gly25fs) | II-4 | 8.2 | NS | F | 76/47y | N/47 | 39 | Multiple small RK, few LK | 230 | 103 | 333 | 1B | 4F | Multiple small | 847 |
| 290084 | c.1722G>T+, p.(Glu574?) | II-7 | 6.0 | 7.3 | F | 74/53y | Y/44 | 50 | Many large bilateral | 439 | 359 | 798 | 1C | 4I | Severe PLD | 4173 |
| M174 | c.12682C>T, p.(Arg4228*) | II-1 | 7.1 | 3.5 | F | ES/83y 16/83y |
Y/64 | 82 | Few large bilateral | 512 | 742 | 1254 | 1B | 4L | Multiple small | 649 |
| 290114 | c.9185_9201+7del24, p.(Val3062fs) | II-1 | 4.1 | 1.3 | F | 70/40y | Y/34 | 38 | Multiple small, 3 large LK | 278 | 164 | 442 | 1B | 4O | Few small | 1299 |
Vol (ht), height adjusted kidney or liver volume, RK, right kidney; LK, left kidney; TKV, total kidney volume; +ve, positive; y, years; F, female; M, male; HTN, hypertension (Y, yes; N, No); MIC, Mayo Imaging Class; Fig, Figure within this manuscript; NS, not screened, ND, not detected; NI, not informative; ES, ESRD,
, kidney length;
, value before liver resection;
, predicted to disrupt splicing
Table 2.
Diagnostic and family history details of PKD1 patients with mosaicism
| Proband Diagnosis | Family History | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pedigree | Subject | Age (y) | Why | Initial Molecular | Confirmed Molecular | Father | Mother | Sibs | Children | Comments |
| Variant transmitted | ||||||||||
| 590013 | II-2 | 28 | Pregnancy, US | FH/ Sanger | tNGS | 80y†, PC, 79y BRC | 87, no PKD Dx | 2, no PKD Dx | 1 +ve, HALTA, III-1 | Father had some cysts |
| 690020 | II-2 | 61 | severe HTN, US | FH/Sanger | LR-NGS | >90y, no PKD Dx | ~40y LTFU, no PKD Dx | 2, no PKD Dx | 1 +ve, HALTA, III-1 2GD, +ve US 19y & <19y |
Father reported kidney & AAA problems |
| 870348 | II-2 | 44 | HTN, enlarged kidneys | tNGS | Sanger/AS-PCR | No PKD Dx | No PKD Dx | 2, no PKD Dx | 1 +ve 26y, 1 untested | |
| 590046 | II-4 | 46 | NA | FH/Sanger | LR-NGS | −ve US 78y | −ve US 76y | 2 −ve US, 50, 37y, 2† other causes | 2 +ve 16, 11y, 1 –ve US 10y, 1 RF untested | Brother† encephalopathy, sister MRKH, III-4, PH |
| 390010 | II-2 | 39 | Pain, US | FH/LR-NGS | AS-PCR | 76y† cardiac, no PKD Dx | 80y† No PKD Dx | 2, no PKD Dx, 1† accidental | 1 +ve, HALT A, 1, BRC, LTFU, | |
| Single affected individual | ||||||||||
| 790057 | II-2 | 30 | HTN, MR | MLPA | tNGS | no PKD Dx | no PKD Dx, stones | 3, no PKD Dx | 2, NT PKD | |
| 590039 | II-4 | 32 | Pain, imaging | MLPA | tNGS/CNV | 68y† no PKD Dx | 74y, no PKD Dx | 4, no PKD Dx, 1 sister liver cysts | 1, no PKD Dx | Distant relatives with PKD? |
| M484 | II-8 | 37 | Fullness, possible mass, imaging | 2x NGS | Sanger | 50y† EtOH, no PKD Dx | no PKD Dx | 6, no PKD Dx | 2, no PKD Dx | |
| M375 | II-3 | 26 | CT, Pain | Sanger | LR-NGS | †MI, no PKD Dx | 2 liver cysts | 3, no PKD Dx | None | |
| 290001 | II-1 | 42 | US | 2x NGS | Sanger | No PKD Dx | no PKD Dx | None | 2 −ve US, 20y, 14y, 1 untested | |
| 870005 | II-2 | 43 | Pain, imaging | tNGS | Sanger | †melanoma, no PKD Dx | no PKD Dx | 1, no PKD Dx | 3, untested | |
| M646 | II-3 | 53 | HTN, US | tNGS | Sanger | no PKD Dx | no PKD Dx | 4, no PKD Dx | 1, no PKD Dx | |
| 870452 | II-2 | 45 | HTN, flank pain, imaging | tNGS | Sanger | no PKD Dx, 76y | no PKD Dx, 75y | 1, no PKD Dx, 57y | 1 −ve US, 2 untested | Grandfather ESRD 75y |
| M1312 | II-2 | 25 | Pregnancy US | tNGS | AS-PCR | −ve US | −ve US | 1, −ve US | 4 untested | 6mm kidney stone |
| 290034 | II-3 | 56 | Hematuria, imaging | 2x NGS | Sanger | 77y†, no PKD Dx | no PKD Dx | 2, no PKD Dx | 1 no PKD, 1 untested | |
| M1327 | II-2 | 46 | Cyst hemorrhage | tNGS | Sanger | −ve US, 70s, MND | −ve US, 70s, MND | 2, no PKD Dx | 3 untested | |
| M855 | II-4 | 38 | Pain, US | Sanger | LR-NGS | −ve US, 72y | −ve US, 68y | 3, no PKD Dx | 3 no PKD Dx | |
| 290084 | II-7 | 44 | HTN, CT | 2x NGS | AS-PCR | no PKD Dx, 83y | no PKD Dx, 77y | 6, no PKD Dx | 2 no PKD Dx | |
| M174 | II-1 | 64 | Fullness, CT | 2x NGS | Sanger | 78y†, no PKD Dx | 94y†, no PKD Dx | 2, no PKD Dx | 1 adopted | |
| 290114 | II-1 | 35 | Stones, HTN, MR | 2x NGS | Sanger | no PKD Dx | no PKD Dx | None | 3 no PKD Dx | |
US, ultrasound; HTN, hypertension; FH, family history; 2x NGS, both the LR- and tNGS methods; PC, pancreatic cancer;
, died;
BRC, bilateral renal cysts; EtOH, alcoholic; MI, myocardial infarction; MND, mutation not detected; LTFU, lost to follow up; RF, reflux; AAA, abdominal aortic aneurysm, MRKH, Mayer Rokitansky, Küster Hauser syndrome; PH, prenatal hydrocephalous; y, years; GD, granddaughter; CT, computer tomography; MR, magnetic resonance; Dx, diagnosis
Families with a transmitted PKD1 mosaic variant (mixed)
In five families, the mosaicism was somatic but the pathogenic variant was also germinal, since the mutation was transmitted to the next generation. Mosaicism was suspected from SS of Family 590013 since the frameshifting change detected in the son (III-1) was only apparent at a low level in the affected mother (II-2; Figure 2A,B). tNGS analysis confirmed the mutant allele in just 157/906 reads (17.3%) compared to 702/1415 (49.6%) in III-1 (Figure 2C). Asymmetric disease was seen in II-2, with a few large cysts in the left kidney, in contrast to multiple small cysts detected in III-1 (Figure 2D,E). II-2 had normal renal function at 56y.
Figure 2. Pedigree, imaging and sequencing data of mosaic families segregating the pathogenic variant to the next generation.

(A) Pedigree 590013 showing ADPKD in II-2 and III-1, with I-1 found to have a few, likely simple, cysts at 79y. (B) SS showing the indel (red line) and reduced peak height of the frameshifted sequence (red) in II-2 compared to III-1. (C) tNGS showing the CCG deletion and T insertion in a reduced number of reads in II-2 compared to III-1. (D) MRI of II-2 at 47y showing asymmetric disease with just a few large cysts in the left kidney compared to a more even distribution in III-1 (E). (F) Pedigree 690020 with ADPKD in three generations. (G) SS of II-2 showing the frameshifted sequence following the 4bp duplication at a low level, reflecting mosaicism. (H) Pedigree of family 870348 with ADPKD in II-2 and III-1. (I) SS of II-2 (reverse strand) showing frameshifted sequence due to a 10bp deletion at a very low level, which is confirmed by AS-PCR (Figure S2B). (J) MRI showing robust PKD in II-2. (K) Pedigree 590046 showing the mosaic case (II-4) with two affected children (III-2 and III-4). (L) The inframe codon deletion is seen at only a low level in II-4 compared to III-2. (M) MRI of II-4 showing very mild kidney and liver cystic disease. (N) Contrast (C) enhanced CT of III-2 at 19y showing a few kidney cysts. (O) Pedigree 390010 shows ADPKD in the II-2 and III-1. (P) SS of the AS-PCR from a normal individual (N), the son (III-1) and the mosaic mother (II-2) shows the 30bp deletion in the son, but due to the low-level mosaicism, the AS-PCR is not completely specific in II-2, hence the doublet sequence. (Q) CT of II-2 shows just a couple of cysts, in contrast to the typical PKD shown by MRI in III-1 (R). Pedigree: red shaded, mosaic; gray, equivocal or unknown; white, ADPKD negative. The percentage of the observed versus the expected level of the pathogenic allele (PAL) is indicated next to each radiological image.
In Family 690020, a PKD1T pathogenic variant was readily detected by SS in III-1, but only found in the mother (II-2) after focused analysis (Figure 2F,G). LR-NGS identified the change in II-2 in 589/6476 reads (9.1%). II-2’s ultrasound (US) imaging showed moderately enlarged kidneys at 61y, while III-1 had significantly enlarged kidneys (Mayo Imaging Class; MIC-1C8; Figure S2A); II-2 has just mild renal impairment at 67y.
Individual 870348 II-2 was initially screened by tNGS and found to have a deletion in 515/6247 reads (8.2%; Figure 2H). The deletion was confirmed at a low level by SS and AS-PCR (Figures 2I, S2B). II-2 has multiple bilateral cysts (MIC-1B), with normal renal function at 47y (Figure 2J). One son, III-1, was diagnosed at 26y.
Initial SS of 590046 II-4 did not identify a likely mutation; however, SS of the son (III-2) detected a single codon deletion (Table S3). LR-NGS of II-2 showed that 899/12858 reads (7.0%) had the deletion, which was confirmed by close inspection of the Sanger sequence (Figure 2K,L). II-4 had an atypical radiological presentation (MIC-2A) with one large kidney and liver cyst and normal renal function at 56y (Figure 2M). Two sons were affected, and mild cystic disease (MIC-1B) was characterized in III-2 at 19y (Figure 2N).
SS of 390010 III-1 identified a large inframe deletion (Table S3), but it was not detected in his affected mother (II-2) (Figure 2O). LR-NGS also did not readily detect the variant in II-2 but careful analysis showed that 6/4246 reads (0.1%) contained the deletion. SS of AS-PCR showed the deleted sequence in III-1 and a mixture of the normal and deleted sequence in II-2, reflecting an enrichment of the rare deleted sequence (Figure 2P). Imaging of II-2 at 48y showed very mild cystic disease (MIC-1A) and normal renal function at 52y, while the disease in III-1 was more typical for an PKD1 fully penetrant pathogenic variant (MIC-1D; Figure 2Q,R).
Families with a mosaic variant in a single individual (somatic)
Fifteen families had a somatic mosaic PKD1 pathogenic variant that was not shown to be transmitted. 790057 II-2 was suspected of a whole exon deletion from multiplex ligation-dependent probe amplification (MLPA), but the level of signal suggested mosaicism (Figure 3A,B). Reanalysis by LR-NGS confirmed the deletion and SS of a breakpoint fragment defined it (Figures 3C, S2C). Analysis of read depth within (4731) and flanking the deletion (7810) indicated that 37.7% reads had the deletion; a margin, along with the MLPA data, consistent with mosaicism. The disease of II-2 was moderately severe (MIC-1C; Figure 3D), but with normal renal function at 31y.
Figure 3. Pedigree, imaging and sequencing data of mosaic families not segregating the pathogenic variant.

(I). (A) Pedigree 790057 showing the mosaic mother (II-2) and her untested offspring. (B) Screening by MLPA found a possible mosaic deletion of ex7 that was confirmed by log2 copy number variant (CNV) analysis of the LR-NGS (C). (D) MRI of II-2 shows typical ADPKD at 31y. (E) Pedigree 590039 shows the mosaic subject (II-4) as the only affected. (F) A suspected mosaic PKD1 deletion was detected by MLPA and confirmed by log2 CNV analysis (Figure S2D). (G) Amplification and SS (Figure S2E) of a specific breakpoint fragment defined the deletion. (H) MRI of II-4 shows significant kidney disease at 49y (prior to ESRD) and severe PLD (after partial liver resection). (I) Pedigree M484 showing two untested daughters of the mosaic case (II-8). (J) Sanger confirmation of the mosaic single nucleotide duplication in II-8. (K) Non-contrast (NC) enhanced CT of II-8 at 46y shows significant PKD and severe PLD. (L) Pedigree M375 shows just one affected subject (II-3). (M) Sanger sequence shows II-3 is mosaic for a single nucleotide deletion. (N) CT of II-3 at 37y shows very mild kidney disease. (O) Pedigree 290001 shows the mosaic subject (II-1) and three children without PKD or untested. (P) Sanger sequence of II-1 confirms mosaicism of a nonsense change. Pedigree: red shaded, mosaic; gray, equivocal or unknown; white, ADPKD negative. The percentage of the observed versus the expected level of the PAL is indicated next to each radiological image.
In 590039 II-4, MLPA detected an apparently mosaic multiexon deletion (Figure 3E,F). The deletion was not found by the LR-NGS assay since it spanned the end of a LR-PCR product (Table S4), but the deletion was detected by tNGS 49/167 reads (22.7%) from blood derived DNA; 25.5% in PLD tissue (Figure S2D). Analysis of breakpoint reads and amplifying across the deletion defined the pathogenic variant (Figure 3G, S2E). The PKD in II-4 was severe (MIC-1D), resulting in ESRD at 55y, and she also had severe PLD that required partial liver resection (Figure 3H).
M484 II-8 was negative from SS but a single nucleotide duplication was detected by both LR-NGS, 1152/5207 reads (20.6%) and tNGS, 518/2578 reads (20.1%), and confirmed by re-SS (Figure 3I,J). II-8 had quite severe kidney disease (MIC-1C), but normal renal function at 47y, and severe PLD requiring cyst aspiration (Figure 3K).
A possible mosaic frameshifting deletion was detected by SS in M375 II-3 that was confirmed and quantified in 930/4858 reads (19.1%) by LR-NGS (Figure 3L,M). The ADPKD was mild (MIC-1B) with normal renal function at 34y (Figure 3N).
290001 II-1 was negative from SS but a nonsense pathogenic variant was detected by the LR-NGS in 1296/7347 reads (17.5%) and tNGS in 1045/5061 reads (20.6%), and seen at a low level after re-SS (Figure 3O,P). US identified multiple bilateral cysts and the patient had Stage 3b CKD at 66y.
Initial SS of 870005 II-2 was negative, but tNGS identified a frameshifting pathogenic variant in 84/566 reads (14.8%), which was subsequently seen at a low level in the SS by visual inspection (Figure 4A,B). This patient had moderately severe PKD (MIC-1C), moderate PLD, but normal renal function at 54y (Figure 4C).
Figure 4. Pedigree, imaging and sequencing data of mosaic families not segregating the pathogenic variant.

(II). (A) Pedigree 870005 where the mosaic subject (II-2) has three untested children. (B) SS confirmation in II-2 of mosaicism of the GG duplication (reverse strand shown). (C) MRI of II-2 at 49y shows multiple large cysts. (D) Pedigree M646 where the mosaic proband (II-3) has an untested son. (E) SS confirms mosaicism for a previously described missense substitution in II-3. (F) CT imaging of II-3 at 54y shows bilateral disease with large cysts in the left kidney. (G) Pedigree 870452 showing three offspring of the mosaic case II-2. (H) SS of II-2 confirms mosaicism for a typical splicing variant. (I) Pedigree M1312 shows mosaicism in II-2 with four untested children. (J) AS-PCR shows a specific fragment just in II-2 but not the normal (N) control, that was confirmed to have the TG insertion by SS of the product (Figure S2F). (K) CT of II-2 at 36y shows bilateral cysts with several large cysts in the right kidney. (L) Pedigree 290034 indicates mosaicism in II-3 and two children either negative or untested. (M) Sanger sequence of II-3 confirms mosaicism of a G deletion. Pedigree: red shaded, mosaic; gray, equivocal or unknown; white, ADPKD negative. The percentage of the observed versus the expected level of the PAL is indicated next to each radiological image.
SS was negative in M646 II-3 but tNGS identified the missense variant p.Pro2809Leu in 454/3392 reads (13.4%), that was subsequently seen at a low level by SS (Figure 4D,E). p.Pro2809Leu is a non-conservative substitution of a residue invariant in orthologs to fish, is not listed in gnomAD24, and has been described as a likely pathogenic variant (Table S3)14. II-3 has relatively small, asymmetric kidneys (MIC-1B; Figure 4F) and has Stage 3b CKD at 60y.
870452 II-1 was only screened by tNGS that detected a typical splicing change in 162/1208 reads (13.4%), that was confirmed by SS (Figure 4G,H). The patient had normal renal function at 57y.
M1312 II-2 was screened initially by tNGS that revealed a frameshifting insertion in 199/1637 reads (12.2%). This insertion was not detected by SS but AS-PCR amplified only from the patient’s DNA, and SS confirmed the insertion in the AS-PCR product (Figure 4I,J, S2F). Imaging showed multiple kidney cysts, including a large one, but renal function was normal at 36y (Figure 4K).
SS of 290034 II-3 was negative but analysis by both LR-NGS, 786/6407 reads (12.3%) and tNGS, 523/4352 reads (12.0%), detected a mosaic frameshifting deletion that was subsequently confirmed by close examination of the SS (Figure 4L,M). II-3 has moderate kidney disease (MIC-1C and Stage 3b CKD at 68y).
In patient M1327 II-2, no mutation was detected by clinical SS but tNGS detected a frameshifting deletion in 134/1489 reads (9.0%), that was confirmed by repeat SS; the parents were negative for the pathogenic variant (Figure 5A,B). II-2 had relatively mild PKD, MIC-1B, and normal renal function at 48y (Figure 5C).
Figure 5. Pedigree, imaging and sequencing data of mosaic families not segregating the pathogenic variant.

(III). (A) Pedigree M1327 shows the mosaic proband (II-2) has untested twin girls and a son. (B) SS of II-2 confirms mosaicism for a 14bp deletion. (C) Just a few bilateral cysts are detected in II-2 by MRI at 46y. (D) The mosaic proband in M855 (II-4) has 3 untested children. (E) SS of II-4 confirms mosaicism of a deletion/insertion. (F) CT of II-4 at 39y show just a few small cysts in the kidney and liver. (G) Pedigree 290084 shows two offspring of the mosaic subject (II-7) either negative for PKD or untested. (H) AS-PCR shows the mutant allele just in II-7 but not a control (N), and sequencing confirms the substitution (Figure S2G). (I) MRI of II-7 show moderate kidney disease and severe PLD. (J) The mosaic proband (II-1) in M174 is the only affected member. (K) AS-PCR shows that II-1 has p.Arg4228* that is also found in a positive control (P) but not an individual without this change (N), and SS shows the substitution at a very low level (Figure S2H). (L) CT of II-1 at 82y, shortly before ESRD, shows a few large cysts in both kidneys. (M) Pedigree 290114 shows the mosaic individual (II-1) has three untested children. (N) SS of II-1 shows mosaicism for a 24bp, splice site spanning, deletion. (O) II-1 has mild cystic disease with a few moderately sized cysts. Pedigree: red shaded, mosaic; gray, equivocal or unknown; white, ADPKD negative. The percentage of the observed versus the expected level of the PAL is indicated next to each radiological image.
A pathogenic variant was suspected but undefined by SS in M855 II-4 and then characterized by LR-NGS as a deletion/insertion in 725/8889 reads (8.2%; Figure 5D,E). The kidney phenotype was mild cystic disease (MIC-1B) with normal renal function at 47y (Figure 5F).
No mutation was detected in 290084 II-7 by initial SS but a conservative missense change (p.Glu574Asp) was found by both LR-NGS, 423/7044 reads (6.0%) and tNGS, 279/3540 reads (7.3%), and confirmed by AS-PCR (Figure 5G,H, S2G). The substitution, c.1722G>T, changes the last nucleotide of exon 8 and is predicted to result in loss of the IVS8 donor site (Figure S2G legend and Table S3). The patient has normal renal function at 53y but relatively large kidneys (MIC-1C) and severe PLD (Figure 5I).
Initial SS of M174 II-1 was negative but a nonsense variant was detected by LR-NGS, 427/6018 reads (7.1%) and tNGS, 40/1159 reads (3.5%), and confirmed by AS-PCR and SS (Figure 5J,K, S2H). The patient has rather few, larger cysts in each kidney and reached ESRD at 83y (Figure 5L).
290114 II1 was NMI from initial SS but the LR-NGS, 304/7506 reads (4.1%), and tNGS, 46/3490 reads (1.3%), identified a deletion extending over a splice junction. This was subsequently confirmed by re-SS (Figure 5M,N). This patient has relatively mild cystic disease (MIC-1B) and normal renal function at 40y (Figure 5O).
Phenotypic features of the mosaic individuals
We compared the eGFR and height adjusted total kidney volume (htTKV) for the mosaic cases to a non-mosaic Mayo PKD1 population with similar mutation types; truncating or strongly predicted nontruncating mutations (Mutation Strength Group [MSG] 1 and 2)9. This analysis showed the mosaic group had a greater eGFR of 30.2 mL/min/1.73 m2 (p<0.001) and a htTKV 32.5% (p=0.021) smaller than the controls, but there was little correlation between the level of the mosaic mutant allele and either phenotype (Figure 6A,B).
Figure 6. Regression comparison of eGFR and htTKV in mosaic subjects compared to PKD1 controls.

The control populations (blue) are PKD1 patients with truncating (MSG1) or strongly predicted non-truncating changes (MSG2)9 from a MTPC cohort without known mosaicism; n=550 for eGFR and 403 for htTKV. The mosaic population is indicated in red and the percentage level of the mosaic allele compared to a fully penetrant allele is listed for each case. (A) The renal function analysis shows that the mosaic population (red) has a significantly higher eGFR than the controls (blue). (B) Analysis of renal structure shows that the mosaic population has a significantly lower htTKV than the controls. The htTKV is plotted on a log2 scale. No correlation was observed between the level of mosaicism and disease severity (eGFR or htTKV).
Discussion
We describe here two NGS methods to robustly detect mosaic pathogenic variants (~4–50% expected allele level) in the PKD1 or PKD2 gene, including in the duplicated region of PKD1. Unlike previous descriptions of ADPKD mosaicism, 15 pathogenic variants were detected when just one family member was affected. Where both methods were employed (6 cases), there was good agreement in the mutant allele level, which was more quantifiable and reliable than from SS. Both methods have good read depth over most parts of the screened genes, although read depth was lower for both in very GC-rich regions (Table S2). However, the much easier application of tNGS versus LR-PCR means it is likely to be more widely employed15, 17, 18. The greater read depth, plus better coverage of PKD1, shows the value of a targeted approaches for screening (especially for mosaicism) of this gene compared to WES17, 18, 25. Here we did not attempt to identify patients with possible high-level mosaicism (50–90% expected allele level), since further study is needed to reliably differentiate mosaicism from de novo germline mutations.
Detecting mosaicism is important as it can genetically resolve SS NMI cases (13 families in this study). Also, it provides important prognostic information; on average the kidney disease in mosaics is milder than expected for the mutation type, with important implications for the expected renal disease severity in offspring compared to the parent. However, the precise level of mosaicism in blood cell DNA did not strongly correlate with disease severity, presumably reflecting that the level of the mutant allele in the kidney that can diverge considerably from that in blood cells. Interestingly, three mosaics had severe PLD, consistent with previous data that ADPKD genotype is less important for the development of liver versus kidney disease26. In the one case where severe PLD tissue was assayed, the level of the mutant allele was only slightly higher than in the blood cell DNA.
In transmitted cases, knowledge that the disease is likely to be more severe in the offspring is important. In the families studied here, the mutant allele was transmitted in 25% of cases. There is likely a positive bias in detecting cases with a fully penetrant offspring, but many families only had unscreened, young, or no offspring and so the transmission rate may be underestimated. Nevertheless, it seems likely that >50% of mosaic cases are sporadic and are not transmitted. In males, fuller analysis of germ cells will determine the level of mixed rather than strictly somatic mosaics and the level of germline mosaicism. Overall, 16/20 mosaic individuals were female, suggesting an enrichment but this did not reach statistical significance compared to the female enriched control population (p=0.11).
Mosaicism is often considered an explanation for unilateral and severely asymmetric cases that usually have a negative family history. Indeed, we did identify six cases with asymmetric or lopsided disease. However, over the whole cohort this was not a consistent feature, and 26 fully unilateral or markedly asymmetry cases remained unresolved after these NGS-based screens. This is likely because the mosaics identified by screening blood cell DNA have broad penetrance of the mutant allele in the body’s organs. Screening DNA isolated from other tissues, such as cells isolated from urine, buccal cells, hair roots, skin fibroblasts and sperm, plus methods to detect pathogenic variants at <2%, may solve these cases. Although not a significant problem in our study, the degree to which the duplicated PKD1 gene will complicate detecting very low-level mosaicism remains to be determined.
All of the detected mosaic cases were PKD1. An enrichment for PKD1 would be expected as it is the most common ADPKD gene (~78% of cases) and a larger mutational target (~13kb coding region compared to ~3kb for PKD2), and mosaic PKD2 subjects may have a very mild phenotype and so not be recognized as PKD. But it does raise the question again whether PKD1 is unusually prone to de novo mutations27. Most mutations (15/20) were deletions or insertions (~39% of germline PKD1 mutations), 14 of which were frameshifting, including six deletions or insertions >10bp, two >250bp. It has been previously described that mosaicism of larger rearrangements is more common than for base-pair changes28, but it is not clear if the predominance of indels here is just due to ease of detection.
The HALT PKD cohort is a typical ADPKD population where patients were hypertensive at baseline and recruited patients had either normal renal function (15–49y) or an eGFR 25–60ml/m/1.732 (18–64y). Notably, there was no kidney size requirement for recruitment and the finding of five different genes in this population show its diversity9, 11, 12; 29. All HALT PKD patients with DNA available were screened by SS9 and unresolved and suspected mosaic cases rescreened by NGS (LR and/or tNGS). Overall, 10/831 HALT PKD families (1.2%) were shown to have ADPKD originating from a mosaic case (Figure 1). A similar level (0.9%) was found from the newly screened patients employing tNGS.
In the LR-NGS cohort, where all patients had a negative or equivocal family history and NMI from SS, seven cases were newly resolved as mosaic (Figure 1). Removing the patients resolved in other ways (fully penetrant PKD1/PKD2 pathogenic variants or other disease genes), the mosaics were equivalent to 8.9% of the residual families (Figure 1). Therefore, in a typical ADPKD population, ~1% of families, and ~10% of NMI families, without a clear family history, are readily resolvable mosaics. Including very low (<4% expected level) and high-level (>50%) mosaics, and analyzing other DNA sources, the total number of mosaics would likely be even higher, although, the representation of mosaics in a population selected for rapidly progressive disease would likely be lower30, 31.
Methods
Study participants and clinical analysis
The participants were recruited through different ADPKD cohorts: the HALT-PKD clinical trial (n=10 positive cases: 590013, 690020, 590046, 390010, 790057, 590039, 290001, 290034, 290084, 290114)9, 32, 33, the ADPKD Modifier Study (n=3: 870348, 870005, 870452), and the Mayo Clinic Translational PKD Center (MTPC; n=7: M484, M375, M646, M1312, M1327, M855, M174) (Figure 1). The relevant Institutional Review Boards or ethics committees approved all studies, and participants gave informed consent. Clinical and imaging data were obtained by review of clinical and study records. Kidney function was calculated from clinical serum creatinine measurements with the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula34. Blood samples for standard DNA isolation were collected from the probands and all available family members.
NGS of PKD1/PKD2 LR-PCR amplicons (LR-NGS)
A total of 110 ADPKD patients were screened that had a negative/equivocal family history and were PKD1/PKD2 NMI by SS (Figure 1). Four of the selected patients were suspected mosaic cases based on either prior SS or family data. The samples were analyzed employing a modification of the LR-PCR and NGS approach15, which is described in detail in the Supplemental Methods and Table S4. For the bioinformatics analysis, FASTQ files were aligned to the hg19 reference genome using bwa-mem (VN:V7.10) with default options. Variant calling was performed using the GATK (VN:3.6) Haplotype Caller. Generated vcf files were prioritized for variants fulfilling the following criteria: Genotype Quality ≥20; Read Depth ≥20; Alternate Allele Frequency ≥3%; Forward/Reverse Read Balance ≥0.25; ExAc/ESP6500/1000Genome Allele Frequency ≤0.1%; Frequency Count within screened patient population <4; Exonic +/−15bp; Non-synonymous, dbNSFP Evaluation ≥ 2 as damaging (Lrt, MetaLr, PolyPhen2, Provean, SIFT). BAM files of variants of interest were reviewed and designated as possible mosaics if the alternate allele was present in 2–35% of reads.
Targeted Next-Generation Sequencing (tNGS)
Samples were run on a custom Agilent SureSelect gene panel containing the coding regions ±50bp of either 6512 or 137 genes (Table S1). Library preparation, sequencing, and sequence alignment was performed as described12. The SNP and Variation Suite (SVS, Golden Helix) was used for small nucleotide variant mining, utilizing the following filtering thresholds: variant locus read-depth (DP) ≥10 and quality (GQ) ≥20; GnomAD, MAF ≤1.0%; removal of non-coding variants >15bp from the splice site. The remaining variants were individually evaluated for pathogenicity based on: inclusion in the online ADPKD database (pkdb.pkdcure.org); or predicted loss of function, or SIFT score ≤0.10 and alignGVGD class ≥C35, and not present in an orthologous sequence; or predicted to alter splicing by Berkeley Drosophila Genome Project (BDGP) and Human Splicing Finder 3.0. BAM files of variants of interest were reviewed and designated as possible mosaics if the alternate allele was present in 1–35% of reads. Large CNVs were assessed by calculating the LOG2 ratio of actual read-depth over expected read-depth for a given locus. Variants with LOG2 ratios between 0.5 and 0.1 OR −0.2 and −0.9 were considered mosaic candidates. BAM files were reviewed to identify exact breakpoints of the CNV and the final level of mosaicism was calculated based on the change of read-depth at the 5’ end of the rearrangement.
Confirmation of variants by SS, MLPA or AS-PCR
All changes were confirmed by SS for PKD1 as previously described14. When family samples were available, segregation analysis of the variant of interest was performed. MLPA was performed employing the MRC Holland kits. For AS-PCR, allele specific primers were designed for mosaic variants which were not or poorly detected by SS. Either the forward or reverse primer was designed specifically to match the pathogenic variant (Table S5), with a second mismatch often introduced35, and used to PCR amplify the mosaic variant using standard methods.
Analysis of Phenotypic Endpoints of the Mosaic Population
The most recent eGFR and htTKV available on mosaic cases and PKD1 controls (MSG 1 & 2) was employed for the regression analysis to correlate the age and phenotypic endpoints.
Supplementary Material
Acknowledgements
We thank the families and coordinators for involvement in the study and Andrew Metzger and Timothy Kline (Mayo Clinic). The study was supported by: NIDDK grant DK058816 (PCH), the Mayo Clinic Robert M. and Billie Kelley Pirnie Translational Polycystic Kidney Disease Center (DK090728; VET), an American Society of Nephrology (ASN) Foundation Ben J Lipps Fellowship (KH), ASN Kidney Research Fellowship (EC-LG), the Fulbright Association and the Foundation Monaham (EC-LG), the Zell Family Foundation and Robert M. and Billie Kelley Pirnie. The HALT-PKD studies were supported by NIDDK cooperative agreements (DK062410, DK062408, DK062402, DK082230, DK062411, and DK062401) and National Center for Research Resources General Clinical Research Centers (RR000039, RR000585, RR000054, RR000051, RR023940, RR001032) and National Center for Advancing Translational Sciences Clinical and Translational Science Awards (RR025008, TR000454, RR024150, TR00135, RR025752, TR001064, RR025780, TR001082, RR025758, TR001102, RR033179, TR000001). The ADPKD Modifier Study is supported by NIDDK grant DK079856.
Other HALT PKD and/or ADPKD Modifier Investigators are also thanked: Yu A, Winklhofer FT (Kansas Medical Center), Abebe KZ, Patterson CG (University of Pittsburgh), Schrier RW, Brosnahan GM (University of Colorado Denver), Miskulin DC (Tufts University), Braun WE (Cleveland Clinic), Czarnecki PG (Brigham and Women’s Hospital), Chebib FT, Hogan MC (Mayo Clinic), Mrug M (University of Alabama at Birmingham), Pei, Y (University of Toronto), Sandford, R (University of Cambridge), Rennert H (The Rogosin Institute, New York), Le Meur Y (Université de Brest), Watnick, T (University of Maryland), Peter DJM (Leiden University Medical Center), Gansevoort RT (University Medical Center Groningen), Demoulin N (Université Catholique de Louvain), Devuyst O (University Hospital of Zürich).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
All the authors declared no competing interests.
Supplementary Material
Supplementary Methods:
LR-NGS Methods.
Supplementary Figures:
Figure S1, Analysis of two false positive “mosaic” PKD1 families.
Figure S2, Details of mosaic positive PKD1 families.
Supplementary Tables:
Table S1. Genes included on the 137 gene panel.
Table S2: Mean read counts for the exonic regions of PKD1 and PKD2 by the 65 and 137 gene panel.
Table S3. Details of the PKD1 pathogenic variants detected in the 20 mosaic families.
Table S4. Details of primers used for amplification of PKD1 and PKD2 for the LR-NGS protocol.
Table S5. Primers and conditions for AS-PCR.
References
- 1.Sampson JR, Scahill SJ, Stephenson JB, et al. Genetic aspects of tuberous sclerosis in the west of Scotland. J Med Genet 1989; 26: 28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tyburczy ME, Dies KA, Glass J, et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet 2015; 11: e1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossetti S, Strmecki L, Gamble V, et al. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet 2001; 68: 46–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iliuta IA, Kalatharan V, Wang K, et al. Polycystic Kidney Disease without an Apparent Family History. J Am Soc Nephrol 2017; 28: 2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007; 369: 1287–1301. [DOI] [PubMed] [Google Scholar]
- 6.Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41: 1311–1319. [DOI] [PubMed] [Google Scholar]
- 7.Cornec-Le Gall E, Audrezet MP, Chen JM, et al. Type of PKD1 Mutation Influences Renal Outcome in ADPKD. J Am Soc Nephrol 2013; 24: 1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol 2015; 26: 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heyer CM, Sundsbak JL, Abebe KZ, et al. Predicted Mutation Strength of Nontruncating PKD1 Mutations Aids Genotype-Phenotype Correlations in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol 2016; 27: 2872–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clissold RL, Hamilton AJ, Hattersley AT, et al. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol 2015; 11: 102–112. [DOI] [PubMed] [Google Scholar]
- 11.Porath B, Gainullin VG, Cornec-Le Gall E, et al. Mutations in GANAB, Encoding the Glucosidase IIalpha Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am J Hum Genet 2016; 98: 1193–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornec-Le Gall E, Olson RJ, Besse W, et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am J Hum Genet 2018; 102: 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornec-Le Gall E, Torres VE, Harris PC. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol 2018; 29: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18: 2143–2160. [DOI] [PubMed] [Google Scholar]
- 15.Rossetti S, Hopp K, Sikkink RA, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol 2012; 23: 915–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan AY, Michaeel A, Liu G, et al. Molecular diagnosis of autosomal dominant polycystic kidney disease using next-generation sequencing. J Mol Diagn 2014; 16: 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trujillano D, Bullich G, Ossowski S, et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol Genet Genomic Med 2014; 2: 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenberger T, Decker C, Hiersche M, et al. An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease. PLoS One 2015; 10: e0116680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braun WE, Abebe KZ, Brosnahan G, et al. ADPKD Progression in Patients With No Apparent Family History and No Mutation Detected by Sanger Sequencing. Am J Kidney Dis 2018; 71: 294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Connor A, Lunt PW, Dolling C, et al. Mosaicism in autosomal dominant polycystic kidney disease revealed by genetic testing to enable living related renal transplantation. Am J Transplant 2008; 8: 232–237. [DOI] [PubMed] [Google Scholar]
- 21.Consugar MB, Wong WC, Lundquist PA, et al. Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int 2008; 74: 1468–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiterova J, Stekrova J, Merta M, et al. Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol 2013; 14: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan AY, Blumenfeld J, Michaeel A, et al. Autosomal dominant polycystic kidney disease caused by somatic and germline mosaicism. Clin Genet 2015; 87: 373–377. [DOI] [PubMed] [Google Scholar]
- 24.Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-offunction intolerance across human protein-coding genes BioRxiv 2019. [Google Scholar]
- 25.Tan AY, Zhang T, Michaeel A, et al. Somatic Mutations in Renal Cyst Epithelium in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol 2018; 29: 2139–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chebib FT, Jung Y, Heyer CM, et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2016; 31: 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu G, Myers S, Chen X, et al. Replication fork stalling and checkpoint activation by a PKD1 locus mirror repeat polypurine-polypyrimidine (Pu-Py) tract. J Biol Chem 2012; 287: 33412–33423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet 2007; 121: 389–400. [DOI] [PubMed] [Google Scholar]
- 29.Cornec-Le Gall E, Chebib FT, Madsen CD, et al. The Value of Genetic Testing in Polycystic Kidney Diseases Illustrated by a Family With PKD2 and COL4A1 Mutations. Am J Kidney Dis 2018; 72: 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367: 2407–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cornec-Le Gall E, Blais J, Irazabal MV, et al. Can we further enrich ADPKD clinical trials for rapidly progressive patients? Application of the PROPKD score in the TEMPO trial. Nephrol Dial Transpl 2018; 33: 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371: 2255–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371: 2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newton CR, Graham A, Heptinstall LE, et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res 1989; 17: 2503–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.