

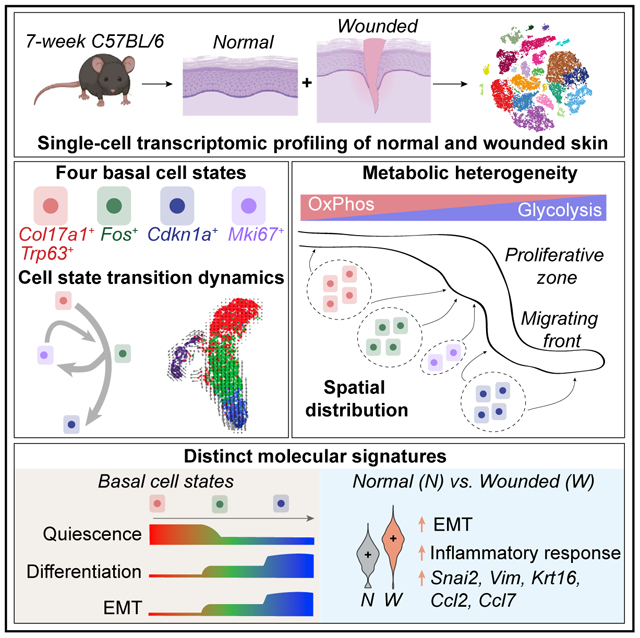
SUMMARY
Our knowledge of transcriptional heterogeneities in epithelial stem and progenitor cell compartments is limited. Epidermal basal cells sustain cutaneous tissue maintenance and drive wound healing. Previous studies have probed basal cell heterogeneity in stem and progenitor potential, but a comprehensive dissection of basal cell dynamics during differentiation is lacking. Using single-cell RNA sequencing coupled with RNAScope and fluorescence lifetime imaging, we identify three non-proliferative and one proliferative basal cell state in homeostatic skin that differ in metabolic preference and become spatially partitioned during wound re-epithelialization. Pseudotemporal trajectory and RNA velocity analyses predict a quasi-linear differentiation hierarchy where basal cells progress from Col17a1Hi/Trp63Hi state to early-response state, proliferate at the juncture of these two states, or become growth arrested before differentiating into spinous cells. Wound healing induces plasticity manifested by dynamic basal-spinous interconversions at multiple basal transcriptional states. Our study provides a systematic view of epidermal cellular dynamics, supporting a revised “hierarchical-lineage” model of homeostasis.
Graphical Abstract
In Brief
Haensel et al. performed a comprehensive dissection of the cellular makeup of skin during homeostasis and wound healing and the molecular heterogeneity and cellular dynamics within its stem-cell-containing epidermal basal layer. Their work provides insights and stimulates further investigation into the mechanism of skin maintenance and repair.
INTRODUCTION
Epithelial tissue maintenance is driven by resident stem cells, the proliferation and differentiation dynamics of which need to be tailored to the tissue’s homeostatic and regenerative needs. However, our understanding of tissue-specific cellular dynamics in vivo at single-cell and tissue scales is often very limited. The self-renewing skin epidermis represents an outstanding model to study the precise sequence of events that underlie the commitment and differentiation of epithelial stem cells toward highly specialized terminal states with important biological functions.
Within the adult mouse interfollicular epidermis, stem and progenitor cells residing in the basal layer undergo self-renewing or differentiative cell divisions to maintain a proper pool of basal cells and to generate post-mitotic differentiating (spinous and granular) cells in the suprabasal layers that ultimately form the stratum corneum—an outer permeability barrier that protects an organism from dehydration, infection, and a myriad of other harmful insults (Gonzales and Fuchs, 2017). Cumulative evidence supports multiple possible mechanisms of epidermal homeostasis: (1) a single, equipotent population of progenitor cells stochastically choosing between self-renewal and differentiation; (2) a hierarchical lineage of relatively quiescent stem cells giving rise to faster cycling, and committed progenitor cells that then exit the cell cycle and terminally differentiate; and (3) two spatially segregated populations of stem cells that divide at different rates and adopt distinct lineage trajectories (Gonzales and Fuchs, 2017; Mascré et al., 2012; Rompolas et al., 2016; Sada et al., 2016). The different criteria used for stem and progenitor fate assignment, such as molecular differentiation markers, basal layer residence status, and assumptions about stem cell division or clonal-growth kinetics, may account for the differences in data interpretation leading to these seemingly diverse models (Gonzales and Fuchs, 2017). Moreover, the observed epidermal stem cell heterogeneity in mouse back skin may reflect different cellular states of a single differentiation program (Rognoni and Watt, 2018). Clearly, single-cell resolution data are needed to provide a comprehensive picture of basal cell heterogeneity and cellular states during epidermal lineage differentiation.
Upon cutaneous wounding, the skin must alter its cellular dynamics to facilitate efficient healing for timely restoration of the protective barrier. Wound healing represents a highly regulated process composed of several distinct but overlapping stages (inflammation, re-epithelialization, and resolution) that involve the coordinated activities of epidermal, dermal, immune, and endothelial cells (Gurtner et al., 2008). Re-epithelialization is driven by spatially patterned migration and proliferation of epidermal cells at the wound periphery, as well as migration and dedifferentiation and reprogramming of hair follicle (HF) and sebaceous gland epithelial cells (Haensel and Dai, 2018; Park et al., 2017; Rognoni and Watt, 2018). What and how epidermal cells migrate during wound re-epithelialization has been a subject of debate, with two different models proposed: (1) basal cells first migrate into the wound bed and unidirectionally convert into suprabasal cells, and (2) wound peripheral epidermal cells crawl or “leapfrog” over one another such that suprabasal cells migrate in and become basal cells (Rittié, 2016; Rognoni and Watt, 2018). Recent live-cell imaging and lineage tracing studies have defined distinct zones of epidermal cellular activities in the wound area: a migratory zone next to the wound margin where both basal and suprabasal cells move toward the wound center; an intermediate, mixed zone of coordinated migration and proliferation; and a hyperproliferative zone furthest away from the wound margin (Aragona et al., 2017; Park et al., 2017). Precisely how many distinct transcriptional states exist for wound epidermal cells and whether these states correlate with or differ from their homeostatic counterparts, particularly within the basal layer, remain to be elucidated.
In this work, we performed single-cell RNA sequencing (scRNA-seq) of cells from normal or wounded (WO) mouse skin, and identified four distinct basal cell states in normal skin that alter gene expression during wound healing. Using multiplexed RNA in situ detection (RNAScope) and fluorescence lifetime imaging microscopy (FLIM), we spatially mapped the scRNA-seq-revealed molecular and metabolic heterogeneities onto the intact normal and WO skin tissue. Pseudotemporal trajectory and RNA velocity analyses placed the different basal cell states temporally onto a differentiation hierarchy and revealed enhanced cell fate and state fluidity during wound healing. Overall, our study provides a comprehensive single-cell perspective of epidermal cellular dynamics and transitional states during normal homeostasis and repair.
RESULTS
scRNA-Seq Reveals Global Changes in Skin Cellular Makeup during Wound Healing
To systematically examine major cell type and cell state differences between homeostasis and repair, we performed scRNA-seq on samples isolated from unwounded (UW) and WO mouse back skin (Figure 1A). The wound samples were taken at 4 days after the introduction of 6-mm wounds, corresponding to a stage of active re-epithelialization (Figures 1B and S1A). After quality control (Figure S1B; STAR Methods), we obtained 10,615 (from two UW biological replicates) and 16,164 (from three WO biological replicates) cells for downstream analyses. By combining all five samples using Seurat (Butler et al., 2018), we identified three major cell types: epithelial (high Krt14 or Krt1 expression), fibroblast (high Col1a2 expression), and immune (high Ptprc expression) (Figures 1C, 1D, S1C, and S1D). The average percentages of immune cells and, to a lesser extent, fibroblasts were increased, and the average percentage of epithelial cells decreased, in the WO replicates compared to UW replicates (Figures 1E and S1E).
Figure 1. scRNA-Seq Analysis of All Cells in the UW and WO Skin.

(A) Schematic diagram detailing the single-cell isolation and live-cell selection strategy.
(B) H&E analysis of WO skin showing a region equivalent to those used for scRNA-Seq. The blue dashed line at the top indicates a representative 10-mm region used for single-cell suspension. Enlarged image of the boxed area in top panel is shown at the bottom to highlight the wound migrating front and proliferative zone. Red dashed line points to the wound margin.
(C) t-Distributed Stochastic Neighbor Embedding (tSNE) plot for all five samples with the major cell type populations highlighted, and their relevant percentage per total number (26,779) of all cells indicated in parenthesis. The two small clusters in gray (labeled as “other”) are endothelial cells and skeletal muscle cells (see Figure S1C).
(D) Cells are colored by replicate identity in the tSNE plot.
(E) Bar graph representing major cell type populations. Chi-square test was used to determine the statistical significance of differences in the relative proportion of each cell type between UW and WO samples. ***p < 0.0005.
(F) tSNE plot for the two aggregated UW replicate datasets. The percentage of cells present in each cluster per total number (10,615) of cells under analysis is indicated. Markers associated with the indicated cell types are listed in Figure S2B and Table S1A.
(G) tSNE plot for the three aggregated WO replicate datasets (total 16,164 cells). Markers associated with the indicated cell types are listed in Figure S2C and Table S1B.
(H) Feature plots showing expression of the indicated genes in the UW replicates in (F). Normalized expression levels for each cell are color-coded and overlaid onto the t-SNE plot.
(I) Feature plots in the WO replicates in (G).
We also analyzed the UW and WO sample types separately (STAR Methods). Known cell type markers (Han et al., 2018; Joost et al., 2016, 2018; Jaitin et al., 2014) were used to determine cluster identity (Figures 1F, 1G, and S2A-S2C; Tables S1A and S1B). For UW and WO samples, we observed 15 and 14 cell clusters, respectively (Figures 1F and 1G). Feature plots of key cell type markers revealed population-level changes in epidermal basal (Krt14+) and spinous (Krt1+) cells, HF-associated cells (Krt17+), immune cells (Ptprc+), and fibroblasts (Col1a2+) (Figures 1H and 1I). Several new cell types, including macrophages, dendritic cells (DCs; which also includes the Cd207+ Langerhans cells), myofibroblasts, and endothelial cells, were either expanded or only detectable in the WO skin (Figure S2D). The dramatically increased presence of macrophages and myofibroblasts in WO skin was confirmed using immunofluorescence with antibodies for F4/80 and smooth muscle actin (SMA), respectively (Figure S3). Greater presence and contributions of distinct immune cell types from the WO sample was also evident when individual cell types were identified in our UW-WO combined dataset from above (Figures S2E and S2F; Table S2). Moreover, the percentage of each cell type in the combined analysis is quantitively similar to that in the separate analysis (Figures S2D and S2F), indicating the robustness of the findings.
Epithelial cells of the interfollicular epidermis, HF, and sebaceous gland drive wound re-epithelialization. Subclustering analysis of these cells revealed comparable numbers of clusters in UW and WO samples, namely, four basal cell subclusters, two spinous cell subclusters, and four HF subclusters with distinct markers (Figures 2A, 2B, and 2D-2G; Tables S3A and S3B), which are generally consistent with the reported scRNA-seq data using Fludigm C1 platform (Joost et al., 2016). Compared to the Joost et al. (2016) dataset, our dataset does not contain a minor loricrin+ granular cell population, likely due to difference in cell harvest procedure, but it features a distinct proliferative basal cell subcluster in both UW and WO skin (Figures 2A, 2B, 2D, and 2E). Overall, the relative proportions of the various epithelial cell types did not dramatically change between UW and WO skin (Figure 2C).
Figure 2. scRNA-Seq Analysis Reveals Mild Changes in Epithelial Cellular Makeup during Wound Healing.

(A) tSNE plot for all epithelial cells from UW skin with cell each type indicated. The percentage of cells in each cluster per total number (7,099) of cells under analysis is indicated in parenthesis.
(B) tSNE plot for all epithelial cells (4,021) from WO skin.
(C) Bar graph representing the major epithelial cell type populations in the UW and WO samples. Chi-square test was performed (*p < 0.05; ***p < 0.0005).
(D) Feature plots highlighting the expression of key genes in UW epithelial cells.
(E) Feature plots highlighting the expression of key genes in WO epithelial cells.
(F) Heatmap for the top 10 genes enriched in each cluster from the UW skin. Top two marker genes for each cluster are colored to match cluster identity, and additional genes used in the final annotations are colored in black. All marker genes are listed in Table S3A.
(G) Heatmap for the top 10 genes enriched in each cluster from the WO skin. All marker genes are listed in Table S3B.
Collectively, these data provide a general overview of the major changes in cellular heterogeneity from homeostasis to a wound healing state, supporting current knowledge obtained using traditional methods (Gurtner et al., 2008; Shaw and Martin, 2009).
Wound Epidermal Basal Cells Upregulate Inflammation- and Migration-Related Gene Expression
Next, we compared UW and WO epidermal basal cells by subseting out the Krt14+ but Krt17−CD34− epithelial subsets (Figures 2D and 2E). We also excluded the proliferative Krt14+ basal cell subcluster from this analysis to avoid cell cycle gene expression overshadowing other molecular differences (Figure S4A). A total of 53 and 99 genes were particularly enriched in UW and WO basal cells, respectively (Table S4). Interestingly, the expression levels of such enriched genes were not uniform across all single cells of each condition, with some UW basal cells displaying a WO-like signature but not vice versa (Figure 3A).
Figure 3. Gene Expression Differences between Epidermal Basal Cells of the UW and WO Skin.

(A) Heatmap showing the top 10 markers for basal cells from the UW and WO samples. All the identified markers are listed in Table S4.
(B) Expression of select genes in UW and WO basal cells. p values are from two-sided Wilcoxon rank-sum tests.
(C) GO analysis of the identified markers (listed in Table S4) of UW and WO basal cells using GO (left) and Hallmark (right; defined by fewer genes) gene sets.
(D) Gene scoring analysis using the indicated molecular signatures. p values are from two-sided Wilcoxon rank-sum tests.
Genes upregulated in WO basal cells included inflammatory genes Cxcl2, Ccl2, and Ccl7; epithelial-to-mesenchymal transition (EMT)-related genes Snai2 and Vim; and positive-control Krt16 (Wawersik et al., 2001; Figure 3B). Gene Ontology (GO) analysis of all differentially expressed genes revealed enrichment of inflammation (e.g., transforming growth factor α [TNF-α] signaling and interferon gamma response) and EMT signatures in WO basal cells (Figure 3C). Genes associated with wound healing, EMT, or upregulated in the leading edge of wound neo-epidermis (i.e., α5 integrin-expressing cells) (Aragona et al., 2017) showed upregulation in WO basal cells compared to UW counterparts (Figure 3D). Moreover, UW basal cells encompassed two distinct subsets scoring low and high for TNF-α signaling and hypoxia, whereas WO basal cells scored uniformly higher for the same gene sets (Figure 3D). Together, these data demonstrate that epidermal basal cells may exist in distinct inflammation-low and inflammation-high states during homeostasis and that they dramatically upregulate inflammatory and migratory gene expression during wound healing.
Three Distinct Non-proliferative (NP) Basal Cell States Exist in UW Skin
We next zoomed in on the NP UW basal cells to dissect their heterogeneity. Three distinct subclusters were observed (Figures 4A and 4B; Table S5): (1) a Col17a1Hi subcluster with top markers such as Col17a1, a gene enriched in epidermal stem cells, and Trp63, a gene enriched in quiescent bulge HF stem cells (Bu-HFSCs) compared with activated stem and progenitor cells (Lien et al., 2011); (2) an early-response (ER) subcluster enriched for immediate early genes and genes associated with activated Bu-HFSCs relative to quiescent Bu-HFSCs or with known function in regulating proliferation, such as Fos, Jun, and Id1 (Andrianne et al., 2017; Briso et al., 2013; Florin et al., 2006; Herschman, 1991; Lien et al., 2011; Rotzer et al., 2006; Zenz and Wagner, 2006; Zhu et al., 2008); and (3) a growth-arrested (GA) subcluster enriched for genes with known functions in promoting cell cycle arrest, such as Cdkn1a, Irf6, Ovol1, and Sfn (Hammond et al., 2012; Ingraham et al., 2006; Nair et al., 2006; Topley et al., 1999; Figures 4C, S4B, and S4C; Table S5). Binarizing the expression of select marker genes for Col17a1Hi, ER, and GA subclusters revealed little overlap in cells that most abundantly express Col17a1 (or Trp63) or Cdkn1a, whereas stronger overlap was seen for Id1. For example, at least 42% of basal cells uniquely express appreciable levels (i.e., >0.75 quantile) of Col17a1 (Col17a1Hi subcluster) and 10% of basal cells uniquely express appreciable levels of Cdkn1a (GA subcluster) (Figure S4D). Furthermore, GA- and Col17a1Hi-enriched genes presented a trend of inverse correlation among the three subclusters (Figure 4C). Together, our data support the existence of three major, distinct transcriptional states in NP UW basal cells.
Figure 4. scRNA-Seq and RNAScope Data Revealing Heterogeneity within UW Epidermal Basal Cells.

(A) tSNE plot for NP basal cells from two UW replicates. The percentage of each subpopulation per total number (2,838) of basal cells was indicated.
(B) Heatmap of top 10 markers for each subcluster in (A). Genes used to annotate cell identity are indicated by the corresponding colors. A complete list of marker genes is provided in Table S5.
(C) Violin plots showing expression of key marker genes. p values are from two-sided Wilcoxon rank-sum tests.
(D) Gene scoring analysis using the indicated molecular signatures. HFSCs and spinous cells from the UW sample were used as positive controls. p values are from two-sided Wilcoxon rank-sum tests.
(E) RNAScope data showing spatial distribution of Cdkn1a and Krt14 transcripts and K14 protein in UW skin. Shown are both low (left)- and high (middle, right)-magnification images. Red and white arrows indicate Cdkn1a+ and Cdkn1a− basal cells, respectively. DAPI stains the nuclei. Scale bars: 50 μm in low-magnification image; 10 μm in high-magnification images.
(F) Spatial distribution of Cdkn1a, Trp63, and Id1 transcripts and K14 protein in UW skin. Red, white, and yellow arrows indicate Id1+, Trp63+, and Cdkn1a+ basal cells, respectively. Scale bars represent the same as in (E).
(G) Quantification of fluorescence intensity (represented by a color-coded dot) for Cdkn1a and Krt14 transcripts and K14 protein in each individual cell from a representative section. The curve represents a Gaussian process regression (GPR), and a 95% confidence interval is shown as shaded area.
(H) Quantification of fluorescence intensity for Cdkn1a, Trp63, and Id1 transcripts and K14 protein in each individual cell from a representative section.
(I) OncoPrint representation of Cdkn1a, Trp63, and Id1 expression in individual cells where a rectangle represents an individual cell. A color-coded rectangle indicates high expression of the corresponding marker gene. The cells are sorted based on the onor off state of the markers to show mutually exclusive expression pattern.
(J) Bar graph showing percentage of cells that exclusively express Cdkn1a, Trp63, or Id1 per total number of cells that express the particular gene (n = 3 replicates). Error bars represent mean ± SEM.
The three NP basal cell states also showed distinct molecular signatures. The GA state showed the highest expression of TNF-α signaling, hypoxia, and EMT genes and enrichment for a gene signature derived from a label-retaining basal cell population (Sada et al., 2016; Figures 4D, S4E, and S4F). In contrast, the Col17a1Hi state scored the lowest for inflammation and EMT genes but highest for a “quiescence and sternness” signature derived from tissue quiescent stem cells (Cheung and Rando, 2013; Figure 4D). There was also a stepwise increase in epidermal differentiation gene expression from Col17a1Hi to ER and to GA states (Figure 4D). These data suggest that these three basal cell states are defined by their differences in inflammation, migration, quiescence and stemness, cell cycle exit, and differentiation status.
We then used RNAScope to validate the existence of three NP basal cell states in the intact skin tissue. Co-analysis of GA marker Cdkn1a with Krt14 transcripts and K14 protein revealed several interesting points: (1) the levels of Krt14 transcript and, to a lesser extent, K14 protein fluctuated along the basal layer of the epidermis, and the location of such variation did not always coincide; and (2) Cdkn1a transcripts were present in some but not all K14-positive basal cells, and the highest expression was detected in a subset of suprabasal cells (Figures 4E and 4G). Co-analysis of Col17a1Hi markers Col17a1 and Trp63 detected basal cells that express both Col17a1 and Trp63, only Col17a1, or neither (Figure S5A). Quantitative analysis revealed that most basal cells expressing high levels of Trp63 also expressed high levels of Col17a1, but some cells only expressed high levels of Col17a1 (Figure S5B). We also validated and detected the heterogeneous expression of Col17a1 and p63 proteins in the epidermal basal layer (Figures S5C and S5D). Co-analysis of Cdkn1a with Trp63 and ER state marker Id1 revealed the presence of cells in the basal layer that uniquely expressed each of the markers (Figures 4F and 4H-4J). Few cells expressed two markers simultaneously, Trp63 and Cdkn1a expression was mutually exclusive, and none expressed all three markers (Figures 4I and 4J). Thus, epidermal basal cells exist in multiple NP transcriptional states in vivo.
Three Distinct NP Basal Cell States Exist in WO Skin and Are Spatially Partitioned
We next turned to the WO samples to assess basal cell heterogeneity during wound healing. We observed three distinct NP subclusters, which we also termed Col17a1Hi, ER, and GA subclusters (Figures 5A-5C). The corresponding UW and WO subclusters shared the highest levels of gene expression with each other compared with other subclusters (Figures S5E and S5F; Tables S5 and S6). Moreover, random forest classification provided further support for assigning the WO subclusters with similar identities as those in the UW basal cells (Figure S5G).
Figure 5. Basal Cell Heterogeneity in WO Skin.

(A) tSNE plot for NP basal cells from two WO replicates. WO-2 was not included in this analysis given its low basal cell number. The percentage of each subpopulation per total number (1,555) of basal cells was indicated.
(B) Heatmap of top 10 markers for each subcluster. A complete list of marker genes is provided in Table S6.
(C) Violin plots showing expression of key marker genes. p values are from two-sided Wilcoxon rank-sum tests.
(D) Gene scoring analysis using the indicated molecular signatures. p values are from two-sided Wilcoxon rank-sum tests.
(E) Quantitative analysis of immunofluorescence data for Snai2 protein. Percent Snai2+ cells in the basal layer of different regions in WO skin is shown here, and representative images are shown in Figure S6F.
(F) Quantitative analysis of immunofluorescence data for Fos protein. Percent Fos+ cells in the basal layer of different regions in WO skin is shown here and representative images are shown in Figure S6G.
(G) RNAScope data showing spatial distribution of Col17a1 and Trp63 transcripts and K14 protein in WO skin. DAPI stains the nuclei. Scale bar: 50 μm.
(H) Enlarged images of the boxed areas in (G). Zones 1–3 correspond to regions that are distal from the wound (zone 1), hyperproliferative (zone 2), and in migrating front (zone 3). Scale bar: 50 μm.
(I) RNAScope data showing spatial distribution of Cdkn1a and Krt14 transcripts and K14 protein in WO skin. DAPI stains the nuclei. Scale bar: 50 μm.
(J) Enlarged images of the boxed areas in (I). See legends for (H) for zone definition. Scale bar: 50 μm.
(K) Quantification of fluorescence intensity for Cdkn1a and Krt14 transcripts and K14 protein in each individual cell from a representative WO skin section. The curve represents a GPR, and a 95% confidence interval is shown as shaded area.
The WO GA cells were enriched for genes associated with α5-integrin-positive leading edge, inflammation, and hypoxia and have the lowest quiescence and stemness score but the highest epidermal differentiation score (Figures 5D and S6A; Table S6). Moreover, individual genes that are known to be enriched (Snai2, Krt16, and Hbegf) or downregulated (Cd9) in the wound migratory front (Haensel and Dai, 2018; Jiang et al., 2013; Shirakata et al., 2005) were observed in the WO GA subcluster (Figures S6B and S6C), suggesting that cells at the migrating front are predominantly in a GA state (see below). Interestingly, WO Col17a1Hi cells showed a significant enrichment for oxidative phosphorylation genes (Figure S6A), a point we will return to later.
We next examined the spatial distribution of basal cell states in WO skin. Co-analysis of Col17a1 and Trp63 transcripts revealed dramatic upregulation of single- and double-positive cells from the wound periphery to the hyperproliferative zone but a decrease of such cells in the migrating front (Figures 5G and 5H). Similarly, Col17a1 and p63 proteins were found to be downregulated in the migrating front relative to the hyperproliferative zone or distal to the wound (Figure S6D and S6E). Although Col17a1 expression was largely restricted to the basal layer, Trp63 expression was also detected in suprabasal cells. In the wound hyperproliferative zone, GA marker Cdkn1a was predominantly expressed in suprabasal cells; however, in the migrating front, its strong expression was detected in both basal and suprabasal cells (Figures 5I-5K). Quantification of Cdkn1a signals along the entire basal layer of the WO area revealed a clear increase toward the migrating front (Figure 5K). WO GA marker Snai2 (Slug) protein was enriched in basal cells at the migrating front relative to those in the hyperproliferative zone or distal to the wound (Figures 5E and S6F). ER marker Fos protein was enriched in the proliferative zone, but its expression dissipated in the migratory zone or UW area (Figures 5F and S6G). The overall trend is enrichment of Col17a1Hi and ER markers in the proliferative zone and wound periphery and enrichment of GA marker in the migrating front.
Collectively, our data show that basal cells in WO skin also exist in three major distinct states, with the Col17a1Hi/ER states dominating the proliferative zone and the GA state similar to the previously described leading-edge population (Aragona et al., 2017) dominating the migrating front.
Metabolic Heterogeneity in Basal Cells of the Normal and WO Skin
To compare the metabolic status of the different basal cell states identified above, we scored all UW and WO basal cells for their expression of oxidative phosphorylation (OxPhos) and glycolysis genes. In UW skin, the proliferative basal cell states showed the highest, whereas the GA state showed the lowest, oxidative phosphorylation score (Figure 6A). In WO skin, oxidative phosphorylation of the Col17a1Hi state was elevated to a level similar to that of proliferative WO basal cells (Figure 6B). The WO GA state still scored the lowest for oxidative phosphorylation, which is in keeping with its highest mRNA expression of the hypoxia response factor Hif1a (Figures 6B and S6H). An apparently opposite trend was seen for glycolysis genes, as GA cells displayed higher glycolysis score than the other NP subsets in both UW and WO skin (Figure S6J and S6I).
Figure 6. FLIM Data Validating scRNA-Seq-Predicted Metabolic Heterogeneity in WO Skin.

(A) Gene scoring analysis of all four UW basal subclusters using an oxidative phosphorylation signature. p values are from two-sided Wilcoxon rank-sum tests.
(B) Gene scoring analysis of all four WO basal subclusters. p values are from two-sided Wilcoxon rank-sum tests.
(C) Sketch diagram of wound epithelium showing the areas probed with FLIM.
(D) A representative image of wound epidermal cells indicated by GFP expression.
(E) Representative images of NADH signal and NADH lifetime signals.
(F) A representative phasor plot with cell phasor fingerprint, which is a representation of the fluorescence lifetime decay of all cells in the region of interest (ROI) after fast Fourier transformation.
(G) Violin plot incorporating all cells and their corresponding free/bound NADH ratios from four biological replicates (156 cells from the outside region, 127 cells from the adjacent region, and 231 cells from the neo-epidermis).
(H) Quantification of average free/bound NADH ratios from multiple cells from the four biological replicates of various regions of the wound. For statistical analysis we used an unpaired two-tailed Student’s t test. Error bars represent mean ± SEM.
Two-photon excitation (TPE) and NADH FLIM have been used for in vivo metabolic measurements during skin wound healing (Deka et al., 2016; Jones et al., 2018). Applying this method to WO skin exercised from K14-Cre;ROSAmTmG mice, where epidermal cells were visualized by GFP expression, we measured NADH autofluorescence in individual cells within the GFP+ basal layer in three different areas: outside of the wound, portions of the proliferative zone, and regions of the migratory front deep within the wound (Figures 6C and 6D). TPE NADH intensities and lifetimes were captured and displayed in phasor plots (Figures 6E and 6F). Free-to-bound NADH ratios, indicative of the relative level of oxidative phosphorylation (Stringari et al., 2012, 2015), were calculated from each basal cell. A gourd-shaped distribution of the ratios was observed for cells in the region far from and outside of the wound (Figure 6G), suggesting that basal cells in UW skin can be classified into OxPhosHigh (less abundant) and OxPhosLow states. The ratios were elevated in basal cells immediately adjacent to and within the wound, with the highest values detected in a significant fraction of basal cells in the neo-epidermis (Figures 6G and 6H). Together, these data provide in vivo validation for scRNA-seq-revealed metabolic heterogeneity within the basal compartment and show basal cells in the wound neo-epidermis to be enriched for an OxPhosLow and glycolysisHigh state compared with their UW counterparts.
Pseudotemporal Trajectory and RNA Velocity Analyses Reveal Basal Cell State Transition Dynamics and Wound-Induced Cellular Plasticity
To pseudotemporally order the distinct basal cell states in the context of epidermal differentiation, we first applied Monocle 2 (Qiu et al., 2017b; Trapnell et al., 2014) to all interfollicular epidermal cells, which include proliferating and NP basal cells as well as spinous cells. In both UW and WO skin, we observed three paths that extend from the Col17a1Hi state: proliferating basal cells, GA state (transitioning through ER state), and spinous cells. Col17a1Hi cells contributed in part to each of the observed paths (Figures S7A and S7B).
Because Monocle 2 is unable to determine the origin of trajectory without prior knowledge, we next used our previously developed method scEpath (Jin et al., 2018) by performing uniform manifold approximation and projection (UMAP) of cells (McInnes et al., 2018), based on batch effect-corrected data (Figures 7A-7D; STAR Methods). This method, when applied to a 536-interfollicular epidermal cell dataset (Joost et al., 2016), revealed what appears to be a single-path trajectory just as reported (Figure S7C). However, in our UW dataset, we observed three distinct and largely separated clusters composed of basal, proliferative basal, and spinous cells (Figures 7A and 7B). In the WO dataset, we identified the same three distinct epidermal clusters, but noted bridges between the basal and spinous clusters with overall less dramatic basal-spinous separation (Figures 7C and 7D).
Figure 7. Pseudotemporal Dynamics Analysis of Interfollicular Epidermal Cells in UW and WO Skin.

(A) UMAP dimensional reduction of all UW epidermal cells. Cells are colored by the annotated identity.
(B) Feature plots of (A) for the indicated genes. Cells are colored by the normalized expression, with dark red indicating the highest expression.
(C) UMAP of all WO epidermal cells.
(D) Feature plots of (C) for the indicated genes.
(E) scEpath-predicted lineage differentiation diagram.
(F) Projection of non-linear RNA velocity fields onto the UMAP space in (A).
(G) Projection of non-linear RNA velocity fields onto the UMAP space in (C).
(H) Pseudotemporal dynamics of the 3,699 UW pseudotime-dependent genes along the Col17a1Hi-to-GA path in UW and WO samples. Each row (i.e., gene) is normalized to its peak value along the pseudotime. Distinct stages during pseudotime are represented by colored bars on the side. Cell identity is indicated on the top of each heatmap generated by the smoothed, normalized gene expression.
(I) Average expression patterns (left) and enriched biological processes (right) of the four gene clusters along pseudotime in (H). Solid and dashed lines indicate the average expression of a particular gene cluster in UW and WO samples, respectively. The number of genes in each gene cluster is indicated in parenthesis, and the enriched GO terms in each gene cluster are listed.
Next, we used scEpath to infer and quantify cell lineages with single-cell energies (scEnergy). We found that lower energies, although typically associated with committed and differentiated cell states (Jin et al., 2018; Teschendorff and Enver, 2017), were also associated with a quiescent cell state because the quiescent Bu-HFSC (Cd34+) population showed the lowest scEnergy of all the skin epithelial cell types (Figure S7D). Independent of the numbers of UW epidermal cells from each cell state that were used to infer lineage progression, scEpath predicted a near-linear path that originates from the Col17a1Hi basal cell state, which displayed the lowest energy of all interfollicular epidermal cells (Figures 7E and S7E). The Col17a1Hi state transitions to the ER state, to the GA state, and then to the spinous cell populations (SP2 to SP1), whereas the proliferative basal cells follow a side path that originates from the ER state (Figure 7E). This prediction was further confirmed using Monocle 3 (Cao et al., 2019; Figure 7F).
To further analyze the epidermal differentiation dynamics in UW skin, we performed RNA velocity analysis, in which the direction of state transitions and the extent of change in RNA dynamics are indicated by the vectors (arrows) and their lengths, respectively (La Manno et al., 2018; Svensson and Pachter, 2018). The Col17a1Hi and SP1 states showed small RNA velocities (short or no arrows), known to associate with both quiescent and terminally differentiated cells (Svensson and Pachter, 2018; Zywitza et al., 2018), whereas the ER, GA, and SP2 states exhibited large RNA velocities (Figure 7F). Transition from the Col17a1Hi state to the ER state was associated with increasing arrow lengths, which may reflect a rapid activation in RNA dynamics (e.g., increased RNA splicing efficiency; see below). Remarkably, the proliferative basal cells followed a cyclical trajectory, which originates from the border between Col17a1Hi and ER states, and then returns to the Col17a1Hi state. This analysis suggests that epidermal basal cells normally transition through three distinct states before embarking on spinous differentiation, and this differentiation trajectory is fueled by the active proliferation of basal cells at the junction between Col17a1Hi and ER states.
The cyclical dynamics of the proliferative UW basal cells was faithfully recapitulated in the WO sample (Figure 7G). An appreciable fraction of the WO Col17a1Hi basal cells and SP1 cells exhibited apparently more rapid RNA dynamics than their UW counterparts. RNA velocity also provided evidence for enhanced cell-fate fluidity in the WO sample, with bidirectional transitions at multiple cellular states between the basal and spinous cells that were not seen in the UW sample. Overall, these data suggest that WO skin epidermal cells not only are generally more active but also exhibit increased plasticity and relaxed cell differentiation constraints compared with their UW counterparts.
HFSCs downregulate the expression of outer bulge markers, such as Cd34, as they contribute to forming a new epidermis during wound healing (Joost et al., 2018). Compared with a single HFSC cluster marked by Cd34, Lhx2, and Postn expression (Joost et al., 2016; Rhee et al., 2006) in the UW sample, two distinct HFSC clusters—both expressing Lhx2 but with different levels of Cd34 and Postn—were called in the WO sample (Figures 2A, 2B, 2D, and 2E). Inclusion of these two HFSC clusters in RNA velocity analysis of WO epidermal basal cells revealed velocity arrows pointing from Cd34Low HFSCs to ER/GA epidermal basal cells (Figure S7G), raising the possibility that the Cd34Low subpopulation might represent those HFSCs that are in the process of becoming wound epidermal cells.
We next sought to identify key molecular changes that may be important for basal cell state transitions. scEpath identified 3,699 and 3,129 pseudotime-dependent genes (including Trp63, Fos, and Cdkn1a) from the UW and WO dataset, respectively, that changed significantly as the basal cells transitioned through the different states (Figures 7H and 7I). Interestingly, genes related to specific biochemical and cellular processes defined the Col17a1Hi to ER transition (group I and II; e.g., protein translation, rRNA processing, and cell cycle), the Col17a1Hi/ER to GA transition (group III; e.g., RNA splicing, mRNA processing, and cell cycle), and the GA state (group IV; e.g., mRNA processing, cell adhesion, and translation). Although gene expression changes were sequential and gradual during basal state transitions in the UW sample, they appeared significantly earlier, were more abrupt, and were sometimes sporadic in the WO sample (Figures 7H, 7I, S7H, S7I, and S7J). This said, transcription factors (TFs), which represent less than 1% of the total 3,699 pseudotime-dependent genes, showed little difference (Figure S7K). Thus, post-transcriptional and cell cycle events underlie the wound-induced remodeling of basal cell state transitions.
The overall emerging picture is that Col17a1Hi basal cells in homeostatic skin first become activated (an early response-like state), at which point they can either enter active cell cycle and expand as progenitor cells or undergo growth arrest and subsequently differentiate into spinous cells. Although this multi-step basal-spinous differentiation trajectory is largely maintained during wound healing, cell fate plasticity and differentiation fluidity are enhanced such that bidirectional conversions between basal and spinous cells are enabled.
DISCUSSION
To date, several studies have used scRNA-seq to make general molecular and cellular categorization of the various epithelial components of the mouse (Joost et al., 2016, 2018) and human (Cheng et al., 2018) skin to provide key insights into normal regeneration and wound healing (Joost et al., 2018; Yang et al., 2017) and to unearth p63-regulated molecular and cellular events in the developing mouse epidermis (Fan et al., 2018). Our work adds to this growing list with a comprehensive study of the transcriptional and metabolic heterogeneities of interfollicular epidermal basal cells in both normal and WO skin.
Our discovery of a Col17a1Hi basal cell state is thought-provoking. This transcriptional state is associated with high quiescence and stemness and high oxidative phosphorylation but low EMT, low differentiation, low hypoxia and inflammation, low scEnergy, and low RNA dynamics. These molecular characteristics are suggestive of a relatively quiescent, primitive stem and progenitor cell state, a notion supported by its high expression of Col17a1, which encodes a marker of long-term epidermal stem cells that can outcompete other cells and a negative regulator of epidermal proliferation (Liu et al., 2019; Watanabe et al., 2017). The Col17a1Hi state also shows heightened expression of Trp63, a master regulator of various aspects of epidermal development, including the initial specification from simple epithelia, promotion of stratification, proliferation, as well as terminal differentiation (Li et al., 2019; Mills et al., 1999; Pattison et al., 2018; Koster et al., 2007; Truong et al., 2006; Yang et al., 1999). The dynamic change in Col17a1 and Trp63 expression during wound re-epithelization implicates the mobilization of Col17a1Hi-state basal cells when there is an increased demand for cellular outputs (see below).
ER genes, such as Fos and Jun, are known as stress response genes that can be upregulated by flow cytometry (van den Brink et al., 2017). A transient upregulation of such immediate early genes during embryonic and adult wound healing has been previously suggested to represent a “kick-start” mechanism to initiate the repair process (Grose et al., 2002). We were able to detect epidermal basal cells in situ that express ER-associated Fos protein or Id1 mRNA, indicating that an ERbasal cell transcriptional state likely exists even in the intact, UW tissue. In all gene expression and lineage prediction analyses, these cells occupy an intermediate position between Col17a1Hi and GA states, raising the possibility that ER is an obligatory transition state when dormant cells become activated to proliferate or migrate.
Basal cells that have committed to differentiation (expressing early differentiation marker Involucrin [Inv]) have been identified by lineage tracing (Mascré et al., 2012). Although Inv expression was not detectable in our scRNA-seq-identified GA basal cells, it is likely that the GA state in the normal epidermis is post-mitotic, most ready to commit to differentiation, and most prone to migrate upward compared with the other basal cell states. The high expression of EMT, glycolysis, hypoxia, and inflammation genes associated with the GA state implicates it as being the most ready to respond to extracellular signals from the everchanging tissue microenvironment. Indeed, our data show that during wound healing GA-state basal cells preferentially localize to the migrating front that is closest to the hypoxic wound bed and the infiltrating immune cells. As such, our results are in keeping with the intravital imaging-revealed finding that migration and proliferation are spatially separated in the healing wound, with migrating cells at the tips of the growing neo-epidermis being generally devoid of proliferative activity (Park et al., 2017). More importantly, our data suggest that the wound repair process capitalizes on the existing transcriptional heterogeneity of normal epidermal basal cells but redirects it toward a spatially coordinated program of proliferation, migration, and metabolism.
We used four computational tools to investigate basal cell dynamics, showing a sequential progression of basal cells through the Col17a1Hi, ER, and then GA states in homeostatic skin. Interestingly, scEpath and Monocle 3 predict GA as a transitional state between basal and spinous fates, a finding consistent with ample experimental data supporting the notion that basal cells undergo cell cycle exit when terminally differentiating (Fuchs, 2008). This said, we note that in RNA velocity analysis there is an apparent scarcity of velocity arrows pointing directly from GA to SP2 cells (Figure 7F). It is possible that in adult homeostatic skin, active basal-to-spinous transition is a rare event that is not readily captured by RNA velocity analysis performed on data from a single time point. Instead, most GA basal cells in adult skin may be in a stable GA state but with rapid intrinsic RNA dynamics and readiness to differentiate. Alternatively, direct GA-SP2 transition may not associate with prominent RNA splicing events. In addition, the RNA dynamics of SP2 pointing to the GA cells in UW skin (Figure 7F) raises the possibility of uncommitted nature (Guo et al., 2018) of spinous cells before fully committing to terminal differentiation.
Significantly enhanced cell fate fluidity occurs in the WO skin, which is evident by the overall less distinct gene expression differences between basal and suprabasal fates as well as multiple RNA velocity vector paths that bridge the different states of the basal and spinous cell clusters. Cellular plasticity during wound healing is well-documented, as cells of the HF and sebaceous gland lineages can be reprogramed to gain an interfollicular epidermal fate (Park et al., 2017; Rognoni and Watt, 2018). Our findings highlight yet another layer of cell fate plasticity, namely bidirectional fluidity between basal and spinous fates at multiple transcriptional states within the interfollicular epidermal compartment. Although the predicted conversion of spinous cells back into a basal cell fate during wound repair is consistent with earlier studies (Fu et al., 2001; Mannik et al., 2010), recent lineage tracing (using Inv-CreER and tail skin wounds) and live-cell imaging (using ear skin wounds) experiments suggest that the conversion of suprabasal cells into basal cells does not occur in wound healing (Aragona et al., 2017; Park et al., 2017). It is possible that the spinous-basal cell fate fluidity that we observe here occurs only at the transcriptional level, but the actual conversion of spinous cells into bona fide basal cells does not occur or occurs as a rare event that requires a very large sample size or more extended wound healing time course (e.g., larger wounds) to experimentally detect. It is also possible that spinous cells with the ability to revert back to a basal fate are not efficiently targeted by Inv-CreER. Finally, differential cellular dynamics during the healing of wounds that incur in different body locations (e.g., tail and ear versus back skin) may contribute to the apparent discrepancy.
Also of interest is our identification of a distinct pool of proliferating basal cells as a separate path that forms a loop with, and thus fueling, the rest of the basal cells. The unique location of these cells in the lineage trajectories, namely at the border between Col17a1Hi and ER basal cell states, suggests that (1) passage through this border might be critical for the active proliferation of the otherwise dormant adult epidermal basal cells; and (2) Col17a1Hi and ER cells are responsible for generating more basal cells, whereas GA cells as a bulk population have likely reached a point of no return such that they can no longer re-enter the cell cycle to serve as a major source of basal cell self-renewal.
It is important to note that alternative models of epidermal lineage differentiation cannot be fully excluded. For example, the proximity between Col17a1Hi and SP1 states as well as between GA and SP2 states may lead to speculation that Col17a1Hi and GA basal cells independently give rise to SP1 and SP2 cells, respectively, in homeostatic and WO skin. Direct conversion of Col17a1Hi basal cells into spinous cells (i.e., without having to pass through ER and GA states) is indeed suggested by Monocle 2 analysis (Figures S7A and S7B). Wound-mobilized Col17a1Hi cells in the wound hyperproliferative zone may adopt multiple possible fates: (1) differentiating directly (skipping ER/GA states) and indirectly into suprabasal cells (SP1 and SP2, respectively) at the wound periphery; (2) converting, at higher rates than during homeostasis, into GA population, which subsequently migrate into the wound; or (3) migrating into the wound and then converting into GA cells. The scarcity of Col17a1Hi cells in the migrating front is more consistent with the second rather than last possibility. Alternatively or additionally, pre-existing GA basal cells may be preferentially recruited at the WO site to form the migrating front, where they directly differentiate into suprabasal cells within the wound bed. Clearly, future experimentation is needed to test the lineage differentiation predictions and the multiple possibilities indicated in this study.
Overall, the sequential progression of basal cells through three NP states, two of which are capable of active proliferation, suggests a revised “hierarchical-lineage” model of epidermal homeostasis that encompasses more than one possible stem and progenitor cell states. Our study lays a foundation for future investigation of the significance of a multi-step, and even multi-route, basal-spinous differentiation trajectory in the performance objectives of adult epidermis, namely maintaining a functional, homeostatic epithelium that can robustly regenerate itself upon injury—a goal that is difficult to attain under pathological conditions.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xing Dai (xdai@uci.edu).
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
K14-Cre transgenic mice (C57BL/6J background) have been previously described (Andl et al., 2004). ROSAmTmG (C57BL/6J background) and wild-type C57BL/6J mice are from the Jackson Laboratory (Stock #s 007576 and 000664, respectively). Seven-week old female mice were used for the studies. All maintenance, care, and experiments have been approved and abide by regulatory guidelines of the International Animal Care and Use Committee (IACUC) of the University of California, Irvine.
METHOD DETAILS
Wounding
For single cell experiments, 7-week-old (p49, telogen) female C57BL/6J mice were anesthetized using isoflurane (Primal Healthcare; NDC-66794-017-25), backs shaved, and then a 6-mm punch (Integra; 33-36) was used to generate a full-thickness wound on each side of the mouse. Wounds were collected 4 days later for analysis.
For FLIM-related wounding experiments, 7 week-old female K14-Cre; ROSAmTmG mice (C57BL/6J background) were anesthetized using isoflurane, backs shaved, and Nair was applied to the backs of the shaved mice for complete hair removal. A 6-mm punch was used to generate a full-thickness wound on each side of the mouse. Four days after wounding, the wound and surrounding un-wounded skin regions were excised (approximately 1.5 cm in diameter with surgical scissors) for analysis.
Single cell isolation for scRNA-Seq
For UW back skin, 7-week-old (p49, telogen) female C57BL/6J mice were shaved, back skin removed, fat scrapped off, and then skin was minced into pieces less than 1 mm in diameter. For WO back skin, skin was removed, large pieces of fat attached to underside of the wound were carefully removed, a 10-mm punch (Acuderm; 0413) was then used to capture the wound and a portion of unwounded skin adjacent to the wound. The wounds were then minced into pieces less than 1 mm in diameter. The minced samples were placed in 15-mL conical tubes and digested with 10 mL of collagenase mix [0.25% collagenase (Sigma; C9891), 0.01M HEPES (Fisher; BP310), 0.001M sodium pyruvate (Fisher; BP356), and 0.1 mg/mL DNase (Sigma; DN25)]. Samples were incubated at 37°C for 2 hours with rotation, and then filtered through 70-μm and 40-μm filters, spun down, and resuspended in 2% FBS. Cells were stained with SytoxBlue (Thermo Fisher; S34857) as per manufacturer’s instructions and live cells (SytoxBlue-negative) were sorted using BD FACSAria Fusion Sorter.
Single cell library generation
FACS-sorted cells were washed in PBS containing 0.04% BSA and resuspended at a concentration of approximately 1,000 cell/μL. Library generation was performed following the Chromium Single Cell 3′ Reagents Kits v2 (following the CG00052 Rev B. user guide) where we target 10,000 cells per sample for capture. Additional reagents included: nuclease-free water (Thermo Fisher Scientific; AM9937), low TE buffer (Thermo Fisher Scientific; 12090-015), ethanol (Millipore Sigma; E7023-500ML), SPRIselect Reagent Kit (Beckman Coulter; B23318), 10% Tween 20 (Bio-Rad; 1662404), glycerin (Ricca Chemical Company; 3290-32), QIAGEN Buffer EB (QIAGEN; 19086). Each library was sequenced on the Illumina HiSeq 4000 platform to achieve an average of approximately 50,000 reads per cell.
Processing and quality control of scRNA-seq data
FASTQ files were aligned utilizing 10x Genomics Cell Ranger 2.1.0. Each library was aligned to an indexed mm10 genome using Cell Ranger Count. Cell Ranger Aggr function was used to normalize the number of mapped reads per cells across the libraries. Quality control parameters were used to filter cells with 200-5000 genes with a mitochondrial percentage under 10% for subsequent analysis.
Doublet analysis of the scRNA-seq data was performed using the DoubletDetection Python (Gayoso and Shor, 2018) package. For each individual sample, we ran DoubletDetection with default parameters using the raw count data from CellRanger output. We then visualized the predicted singlets and doublets on the tSNE space. One small group of cells was predicted as potential doublets (Figure S1B); however, since they exhibited the medium number of genes and UMI per cell, and were identified by markers of fibroblasts (Figures 1C, S1C, and S2E), we did not attempt to remove them from subsequent analysis that primarily focused on skin epithelial cells.
Clustering analysis of scRNA-seq data
Clustering of cells was performed using the Seurat R package (Satija et al., 2015). Briefly, single cell data matrices were column-normalized and log-transformed. Replicates for UW and WO samples were merged and then corrected using the MultiCCA function. To identify cell clusters, principle component analysis (PCA) was first performed and the top 10 PCs with a resolution = 0.6 were used to obtaining 15 and 14 clusters for the UW and WO samples, respectively. For the “combined” analysis of all five samples, the top 15 PCs with a resolution = 0.8 were used to obtain 25 clusters. These clusters were also merged based on the marker genes of major cell types. For subclustering of epithelial cells, we first identified epithelial clusters from UW or WO replicate using the top 10 PCs with resolution = 0.6 and then subset out the appropriate epithelial clusters. Replicates of these epithelial clusters were then merged using MultiCCA function again using 10 PCs with resolution = 0.6.
For subclustering of epidermal basal cells, we performed batch correction using the Bayesian-based method ComBat from the sva R package (Johnson et al., 2007). The corrected data were used for further clustering analysis. Briefly, for the UW sample, the top 23 PCs were used for clustering and 3 subclusters were obtained with a resolution = 0.8. For the WO sample, the top 26 PCs were used and 3 subclusters were obtained with a resolution = 0.3. Marker genes were determined with p value < 0.01 and log(fold-change) > 0.25 as cutoff by performing differential gene expression analysis between the clusters using Wilcoxon rank sum test. To present high dimensional data in two-dimensional space, we performed t-SNE analysis using the results of PCA with significant PCs as input.
Random forest classifier
Using the Seurat R Package 2.2.0, we employed the ClassifyCells function with default parameters, which relies on the Ranger package to build a random forest suited for high dimensional data. Training class was based on identities of the basal cells from the UW sample, which was subsequently applied to the basal cells from the WO sample.
Pseudotime and trajectory analysis
We performed pseudotemporal ordering of all interfollicular epidermal cells, including proliferative and non-proliferative basal cells and spinous cells, using Monocle 2 (Qiu et al., 2017b) and scEpath (Jin et al., 2018). For Monocle 2, batch effect information was passed into the residualModelFormulaStr option in the “reduceDimension” function. The scEpath method can quantify the energy landscape using scEnergy, which quantitatively measures the developmental potency of single cells (Jin et al., 2018) and was used in our analysis to predict the initial state in pseudotime. Pseudotemporal ordering was performed on Combat-batch corrected data. The corrected data was scaled using the ScaleData function with default parameters, and then used as an input for dimension reduction using PCA and UMAP, which were performed using Seurat package. The number of significant PCs was determined by the PCEl-bowPlot function. The top six PCs were used in UMAP with the parameter min_dist being 0.35. Based on this reduced UMAP space, scEpath infers lineage relationships between cell states via predicted transition probabilities and reconstructs pseudotime by separately ordering individual cells along each lineage branch via a principal curve-based approach. The calculated pseudotime is rescaled such that it is bounded in [0, 1]. scEpath also identifies pseudotime-dependent genes that are significantly changed over the pseudotime by creating a smoothed version of gene expression using a cubic regression spline (Jin et al., 2018). To determine the pseudotime dependent genes, we compared the standard deviation of the observed smoothed expressions with a set of similarly permuted expressions by randomly permuting the cell order (1000 permutations). We considered all genes with a standard deviation greater than 0.05 and a Bonferroni-corrected p value below a significance level α = 0.01 to be pseudotime dependent. To analyze pseudotime-dependent TFs, we used TFs that are annotated in the Animal TF Database (AnimalTFDB 2.0) (Zhang et al., 2015).
We also performed pseudotemporal trajectory analysis using Monocle 3 v0.1.3 (Cao et al., 2019). As a successor of Monocle 2, the major updates in Monocle 3 include use of UMAP space to initialize trajectory inference and a better structured workflow to learn developmental trajectories. The raw count data of the highly variable genes were used in pseudotemporal trajectory analysis, which were identified using FindVariableGenes function from Seurat package (parameter y.cutoff = 0.5). The UMAP space from Seurat package was used as an input of the reduced dimensional space in Monocle 3.
RNA velocity analysis
RNA velocity was calculated based on the spliced and unspliced counts as previously reported (La Manno et al., 2018), and cells that were present in the pseudotemporal ordering were used for the analysis. We used the R implementation “velocyto” with a modified dynamical model to perform RNA velocity analysis. La Manno et al. (2018) used a linear model to relate abundance of pre-mRNA U(t) with abundance of mature mRNA S(t):
In this model, mRNA abundance over time (represented as dS/dt) is the velocity of gene expression. Given that the molecular regulatory mechanisms between pre-mRNA and mature mRNA are complicated, and in many molecular networks more commonly we observe non-linear (e.g., switch-like) responses, we also proposed a nonlinear model of RNA velocity for the effects of pre-mRNA on the abundance of mature mRNA based on Michaelis-Menten kinetics. The nonlinear RNA velocity model is formulated as:
where n is the Hill coefficient (describing cooperativity) and K is a constant. We set n and K to be 1 and 0.5 in all the analyses below. The R package implementing this non-linear dynamical model, termed as nlvelo, is available at https://github.com/sqjin/nlvelo.
RNA velocity was estimated using gene-relative model with k-nearest neighbor cell pooling (k = 30). Velocity fields were then projected onto a low dimensional space (e.g. UMAP). Parameter n-sight, which defines the size of the neighborhood used for projecting the velocity, was set to 500.
For RNA velocity analysis of basal cells and HFSCs in WO samples, the UMAP space was generated using Seurat with the top 10 PCs as inputs. Velocity fields were then projected onto this UMAP space.
FLIM and data analysis
Freshly excised skin was placed in a glass bottom microwell dish (MatTek Corporation; PG-35 g-1.5-14-C) and imaging was performed using a 63X Oil 1.4NA lens (Zeiss) on a Zeiss LSM 880 microscope coupled to a Ti:Sapphire laser system (Spectra Physics, Santa Clara CA, USA, Mai Tai HP). External hybrid photomultiplier tubes (Becker&Hickl; HPM-100-40) and ISS A320 FastFLIM system (ISS, Urbana-Champaign, Illinois) were used for Phasor Fluorescence Lifetime Imaging Microscopy (Colyer et al., 2008; Digman et al., 2008; Stringari et al., 2015). A 690 nm internal dichroic filter (Zeiss) was used to separate the fluorescence emission from the laser excitation. The fluorescence emission was reflected onto a 495LP dichroicmirror and subsequently a 460/80 nm bandpass filter (Semrock; FF02-460/80-25) before the external detector to filter the NADH fluorescence emission. Images were acquired using unidirectional scan, 16.38 us pixel dwell time, 256 × 256 pixels per frame, and 58.67um field of view. All images were acquired within 1.5 hours of animal death.
The phasor plot method provides a fit-free, unbiased way of analyzing FLIM data quantitatively. FlimBox, developed by the Laboratory for Fluorescence Dynamics at UC Irvine, records the photon counts per pixel in a number of cross-correlation phase bins called the phase histogram used for the Digital Frequency Domain FLIM method. The phase histogram is processed by the fast Fourier transform to produce the phase delay ϕ and modulation ratio m of the emission relative to the excitation from which the G and S coordinates calculated at each pixel of the image are represented in the phasor plot.
Data analysis was performed with Globals for Images (SIMFCS 4.0) software developed at the Laboratory for Fluorescence Dynamics. We used coumarin 6 (Sigma-Aldrich; 546283), with known lifetime of 2.5ns, for calibration of the instrument response function.
Quantification of the average NADH phasor per region of interest was calculated using the built-in masking feature in SimFCS 4.0. This masking feature averages the lifetime (τ) of all pixels included within a designated region of interest (ROI). SimFCS converts G and S coordinates of the phasor plot into the fraction of bound by calculating the distance of the ROI average τ to the theoretical lifetime τ of bound NADH (τ = 3.4 ns), divided by the total distance between free NADH (τ = 0.4 ns) and bound NADH. An ROI within the boundary of each cell demarked by GFP expression (but excluding the cell membrane-associated GFP signal) was drawn to estimate the free/bound NADH ratio for each cell within a field of view for all images. The fraction bound values obtained from SimFCS 4.0 were then converted to free/bound ratio NADH for each ROI as a measure of metabolism based on previous work (Cinco et al., 2016; Kim et al., 2016; Mah et al., 2018; Stringari et al., 2012, 2015).
Morphology and immunostaining
For histological analysis, mouse back skin was shaved, removed, fixed in 4% paraformaldehyde (MP; 150146) in 1X PBS, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H/E). For indirect immunofluorescence, mouse back skin was freshly frozen in OCT (Fisher; 4585), sectioned at 5 μm, and staining was performed using DAPI (Thermo Fisher; D1306: 1:1000) and the following primary antibodies: Ki67 (Cell Signaling, D3B5, 1:1000), K14 (chicken, 1:1000; rabbit, 1:1000; gift of Julie Segre, National Institutes of Health, Bethesda), Slug/Snai2 (Cell Signaling, C19G7, 1:1000), Fos (Santa Cruz Biotechnology, sc271243, 1:100), F4/80 (eBioscience, 14-4801-82, 1:200), anti-SMA (Abcam, ab5694, 1:500), Col17a1 (Abcam, ab184996, 1:200), or p63 (Santa Cruz Biotechnology, sc-8343, 1:50).
RNAScope, data analysis and presentation
RNAScope was performed using the Multiplex Fluorescent v2 system (ACD; 323100). Briefly, mouse back skin or wounds were freshly frozen in OCT (Fisher; 4585) and sectioned at 10 μm. Sections were fixed at room temperature for 1 hour with 4% paraformaldehyde (Electron Microscopy Sciences; 15715-S), which was diluted from stock with 1x DPBS (Corning Cellgro; 21-031-CM). After fixation, standard RNAScope protocols were used according to manufacturer’s instructions. The following probes were used: Krt14 (ACD; 422521-C3), Trp63 (ACD; 464591-C2), Cdkn1a (ACD; 408551-C1), and Id1 (ACD; 312221-C3). Fluorescence intensity in the basal cells (stained positive for anti-K14 antibody and adjacent to the basement membrane or wound bed) in both UW and WO (from the wound margin to the tip of the migrating front) samples was quantified in a manner that preserves spatial information.
We used Gaussian Process Regression (GPR), a non-parametric method to fit observations and to visualize the major trends of data by controlling the smoothness of the model. GPR uses kernels to measure similarity between inputs based on their distances, and inputs with high similarity should have similar output from the fitted model. We used the implementation of GPR in scikit-learn package (Pedregosa et al., 2011; Rasmussen and Williams, 1996). The Matérn kernel is used for similarity measurement and a white noise kernel is included to accommodate noise in the data.
Given a collection of values, BASC method (Hopfensitz et al., 2012) first sorts the values to obtain an initial step function representation. This step function is then iteratively refined until there are only two steps. It can be roughly understood as finding the strongest discontinuity point in data. The R implementation of this package “Binarize” is used with algorithm option B to determine thresholds for binarization of the markers.
Calculation of signature score of a gene set
For gene scoring analysis, gene sets were acquired from the MSigDB database, the MGI Gene Ontology Browser (including keratinocyte differentiation scoring) and published literatures (including α5 integrin-expressing cell and quiescence/stemness scoring) (Aragona et al., 2017; Cheung and Rando, 2013). Specific genes in each gene set are listed in Table S7. The AddModuleScore function in Seurat R package was then used to calculate the signature score of each gene set in each cell. The two-sided Wilcoxon rank sum test was used to evaluate whether there are significant differences in the computed signature scores between two groups of cells.
Analysis of gene expression overlap
To computationally analyze the potential overlap in basal cell expression of Col17a1, Trp63, Id1, and Cdkn1a in our scRNA-seq data, we binarized the expression of each gene by choosing thresholds based on the quantile of all expressed cells. We quantified the percentage of cells expressing one gene, two genes, or three genes using three different quantile (0.25, 0.5, and 0.75) thresholds.
QUANTIFICAITON AND STATISTICAL ANALYSIS
Data are presented as the mean ± standard error of mean (SEM), or the median ± interquartile range (IQR), as indicated. The sample sizes in each plot have been listed in the Results section and Figure Legends where appropriate. For data represented as violin plots, two-tailed Wilcoxon rank sum test was performed using R (https://www.r-project.org/). For comparison of percentage changes, Chi-square test was performed using MATLAB (https://www.mathworks.com/). For differential gene expression analysis between cell clusters, Wilcoxon rank sum test was performed using R. A significance threshold of p < 0.01 was used for defining marker genes of each cell cluster. For data presented in bar plot, unpaired two-tailed Student’s t test was used.
DATA AND CODE AVAILABILITY
The scRNA-seq data reported in this paper have been deposited in the GEO database under accession code GEO: GSE142471. The software of nlvelo R package is available at https://github.com/sqjin/nlvelo. The codes and walkthroughs for pseudotemporal trajectory analysis are available at https://github.com/sqjin/codes_CellReports2019.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ki67 | Cell Signaling | Cat #: D3B5; RPID: AB_2687446 |
| Purified Rabbit Anti-Mouse K14 | Gift: Juile Segre, NIH | N/A |
| Purified Chicken Anti-Mouse K14 | Gift: Juile Segre, NIH | N/A |
| Purified Mouse Anti-Mouse Fos | Santa Cruz Biotechnology | Cat #: sc271243; RPID: AB_10610067 |
| Slug (Snai2) | Cell Signaling | Cat #: C19G7; RRID:AB_2239535 |
| F4/80 | eBioscience | Cat #: 14-4801-82, RRID:AB_467558 |
| aSMA | Abcam | Cat #: ab5694 |
| Col17a1 | Abcam | Cat #: ab184996 |
| P63 | Santa Cruz Biotechnology | Cat #: sc-8343 RRID:AB_653763 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase | Sigma | Cat #: C9091 |
| HEPES | Fisher | Cat #: BP310 |
| Sodium Pyruvate | Fisher | Cat #: BP356 |
| DNase | Sigma | Cat #: DN25 |
| SytoxBlue | Thermo Fisher | Cat #: S34857 |
| Nuclease-free Water | Thermo Fisher | Cat #: AM9937 |
| Low TE Buffer | Thermo Fisher | Cat #: 12090-015 |
| Ethanol | Millipore Sigma | Cat #: E7023-500ML |
| 10% Tween 20 | Bio-Rad | Cat #: 1662404 |
| Glycerin | Ricca Chemical Company | Cat #: 3290-32 |
| QIAGEN Buffer EB | QIAGEN | Cat #: 19086 |
| Coumarin 6 | Sigma | Cat #: 546283 |
| DAPI | Thermo Fisher | Cat #: D1306 |
| DPBS | Corning Cellgro | Cat #: 21-031-CM |
| Krt14-Probe | ACD | Cat #: 422521-C3 |
| Trp63-Probe | ACD | Cat #: 464591-C2 |
| Cdkn1a-Probe | ACD | Cat #: 408551-C1 |
| Id1-Probe | ACD | Cat #: 312221-C3 |
| Col17a1-Probe | ACD | Cat #: 552141-C1 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3′ Library & Gel Bead Kit v2 | 10x Genomics | Cat #: PN-120237 |
| Chromium Single Cell A Chip Kits | 10x Genomics | Cat #: PN-120236 |
| Chromium i7 Multiplex Kit | 10x Genomics | Cat #: PN-120262 |
| SPRIselect Reagent Kit | Beckman Coulter | Cat #: B23318 |
| Multiplex Fluorescent v2 system | ACD | Cat #: 323100 |
| Deposited Data | ||
| Raw scRNA-seq data | This paper | GEO: GSE142471 |
| Experimental Models: Organisms/Strains | ||
| Mouse: K14-Cre | Andl et al., 2004 | N/A |
| Mouse: ROSAmTmG, Gt(ROSA)26 Sortm4(ACTB-tdTomato,-EGFP)Luo | The Jackson Laboratory | Cat #: JAX 007576 |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat #: JAX 000664 |
| Software and Algorithms | ||
| Cell Ranger 2.1.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Seruat v2 | Satija et al., 2015 | https://satijalab.org/seurat |
| Monocle 2 | Qiu et al., 2017b | http://cole-trapnell-lab.github.io/monocle-release/ |
| Monocle 3 | Cao et al., 2019 | https://cole-trapnell-lab.github.io/monocle3/ |
| scEpath | Jin et al., 2018 | https://github.com/sqjin/scEpath |
| nlvelo | This paper | https://github.com/sqjin/nlvelo |
| Combat | Johnson et al., 2007 | https://www.bioconductor.org/packages/release/bioc/html/sva.html |
| UMAP | McInnes et al., 2018 | https://github.com/lmcinnes/umap |
| AnimalTFDB 2.0 | Zhang et al., 2015 | http://bioinfo.life.hust.edu.cn/AnimalTFDB/#!/ |
| Velocyto | La Manno et al., 2018 | https://github.com/velocyto-team/velocyto.R |
| DoubletDetection | Gayoso and Shor, 2018 | https://github.com/JonathanShor/DoubletDetection |
| SimFCS 4.0 | omicX | https://omictools.com/simfcs-tool |
| scikit-learn | A free Python library | https://scikit-learn.org/stable/ |
| BASC | Hopfensitz et al., 2012 | https://cran.r-project.org/web/packages/Binarize/index.html |
Highlights.
scRNA-seq identifies four epidermal basal cell states in homeostatic adult skin
Computational analysis supports a “hierarchical” model of epidermal homeostasis
Basal cell states are metabolically distinct and spatially partitioned in wounded skin
Epidermal basal cells show enhanced cell fate and state plasticity during wound healing
ACKNOWLEDGMENTS
We thank the Genomics High Throughput Facility and the Institute for Immunology FACS Core Facility at University of California, Irvine (UCI) for expert service. Stock images in the graphical abstract were created through an academic subscription with BioRender. FLIM experiments were performed at the Laboratory for Fluorescence Dynamics, which is supported jointly by the NIGMS/NIH (P41GM103540) and UCI. This work was supported by NIH grants R01-AR068074 (X.D.), R01GM123731 (X.D.), and U01-AR073159 (Q.N. and X.D.); NSF grants DMS1763272 and DMS1562176; and Simons Foundation grant 594598 (Q.N.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.091.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Andl T, Ahn K, Kairo A, Chu EY, Wine-Lee L, Reddy ST, Croft NJ, Cebra-Thomas JA, Metzger D, Chambon P, et al. (2004). Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development 131, 2257–2268. [DOI] [PubMed] [Google Scholar]
- Andrianne M, Assabban A, La C, Mogilenko D, Salle DS, Fleury S, Doumont G, Van Simaeys G, Nedospasov SA, Blackshear PJ, et al. (2017).Tristetraprolin expression by keratinocytes controls local and systemic inflammation. JCI Insight 2, e92979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M, Dekoninck S, Rulands S, Lenglez S, Mascré G, Simons BD, and Blanpain C (2017). Defining stem cell dynamicsand migration during wound healing in mouse skin epidermis. Nat. Commun 8, 14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briso EM, Guinea-Viniegra J, Bakiri L, Rogon Z, Petzelbauer P, Eils R, Wolf R, Rincón M, Angel P, and Wagner EF (2013). Inflammation-mediated skin tumorigenesis induced by epidermal c-Fos. Genes Dev. 27, 1959–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JB, Sedgewick AJ, Finnegan AI, Harirchian P, Lee J, Kwon S, Fassett MS, Golovato J, Gray M, Ghadially R, et al. (2018). Transcriptional Programming of Normal and Inflamed Human Epidermis at Single-Cell Resolution. Cell Rep. 25, 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, and Rando TA (2013). Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol 14, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinco R, Digman MA, Gratton E, and Luderer U (2016). Spatial Characterization of Bioenergetics and Metabolism of Primordial to Preovulatory Follicles in Whole Ex Vivo Murine Ovary. Biol. Reprod 95, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colyer RA, Lee C, and Gratton E (2008). A novel fluorescence lifetime imaging system that optimizes photon efficiency. Microsc. Res. Tech 71, 201–213. [DOI] [PubMed] [Google Scholar]
- Deka G, Chu S-W, and Kao F-J (2016). Skin Wound Healing Revealed by Multimodal Optical Microscopies. In Microscopy and Analysis (Polytechnic University of Bucharest), pp. 151–179. [Google Scholar]
- Digman MA, Caiolfa VR, Zamai M, and Gratton E (2008). The phasor approach to fluorescence lifetime imaging analysis. Biophys. J 94, L14–L16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Wang D, Burgmaier JE, Teng Y, Romano RA, Sinha S, and Yi R (2018). Single Cell and Open Chromatin Analysis Reveals Molecular Origin of Epidermal Cells of the Skin. Dev. Cell 47, 21–37.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L, Knebel J, Zigrino P, Vonderstrass B, Mauch C, Schorpp-Kistner M, Szabowski A, and Angel P (2006). Delayed wound healing and epidermal hyperproliferation in mice lacking JunB in the skin. J. Invest. Dermatol 126, 902–911. [DOI] [PubMed] [Google Scholar]
- Fu X, Sun X, Li X, and Sheng Z (2001). Dedifferentiation of epidermal cells to stem cells in vivo. Lancet 358, 1067–1068. [DOI] [PubMed] [Google Scholar]
- Fuchs E (2008). Skin stem cells: rising to the surface. J. Cell Biol 180, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayoso Adam, and Shor Jonathan. (2018). DoubletDetection (Version v2.4). Zenodo; 10.5281/zenodo.2678042. [DOI] [Google Scholar]
- Gonzales KAU, and Fuchs E (2017). Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 43, 387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose R, Harris BS, Cooper L, Topilko P, and Martin P (2002). Immediate early genes krox-24 and krox-20 are rapidly up-regulated after wounding in the embryonic and adult mouse. Dev. Dyn 223, 371–378. [DOI] [PubMed] [Google Scholar]
- Guo J, Grow EJ, Mlcochova H, Maher GJ, Lindskog C, Nie X, Guo Y, Takei Y, Yun J, Cai L, et al. (2018). The adult human testis transcriptional cell atlas. Cell Res. 28, 1141–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, and Longaker MT (2008). Wound repair and regeneration. Nature 453, 314–321. [DOI] [PubMed] [Google Scholar]
- Haensel D, and Dai X (2018). Epithelial-to-mesenchymal transition in cutaneous wound healing: Where we are and where we are heading. Dev. Dyn 247, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond NL, Headon DJ, and Dixon MJ (2012). The cell cycle regulator protein 14-3-3σ is essential for hair follicle integrity and epidermal homeostasis. J. Invest. Dermatol 132, 1543–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- Herschman HR (1991). Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem 60, 281–319. [DOI] [PubMed] [Google Scholar]
- Hopfensitz M, Müssel C, Wawra C, Maucher M, Kühl M, Neumann H, and Kestler HA (2012). Multiscale binarization of gene expression data for reconstructing Boolean networks. IEEE/ACM Trans. Comput. Biol. Bioinformatics 9, 487–498. [DOI] [PubMed] [Google Scholar]
- Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, et al. (2006). Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet 38, 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, and Amit I (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XP, Zhang DX, Teng M, Zhang Q, Zhang JP, and Huang YS (2013). Downregulation of CD9 in keratinocyte contributes to cell migration via upregulation of matrix metalloproteinase-9. PLoS One 8, e77806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, MacLean AL, Peng T, and Nie Q (2018). scEpath: energy landscape-based inference of transition probabilities and cellular trajectories from single-cell transcriptomic data. Bioinformatics 34, 2077–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Jones JD, Ramser HE, Woessner AE, and Quinn KP (2018). In vivo multiphoton microscopy detects longitudinal metabolic changes associated with delayed skin wound healing. Commun. Biol 1, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S, Zeisel A, Jacob T, Sun X, La Manno G, Lönnerberg P, Linnarsson S, and Kasper M (2016). Single-Cell Transcriptomics Reveals that Differentiation and Spatial Signatures Shape Epidermal and Hair Follicle Heterogeneity. Cell Syst. 3, 221–237.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S, Jacob T, Sun X, Annusver K, La Manno G, Sur I, and Kasper M (2018). Single-Cell Transcriptomics of Traced Epidermal and Hair Follicle Stem Cells Reveals Rapid Adaptations during Wound Healing. Cell Rep. 25, 585–597.e7. [DOI] [PubMed] [Google Scholar]
- Kim SM, Roy SG, Chen B, Nguyen TM, McMonigle RJ, McCracken AN, Zhang Y, Kofuji S, Hou J, Selwan E, et al. (2016). Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways. J. Clin. Invest 126, 4088–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MI, Dai D, Marinari B, Sano Y, Costanzo A, Karin M, and Roop DR (2007). p63 induces key target genes required for epidermal morphogenesis. Proc. Natl. Acad. Sci. USA 104, 3255–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang Y, Torkelson JL, Shankar G, Pattison JM, Zhen HH, Fang F, Duren Z, Xin J, Gaddam S, et al. (2019). TFAP2C- and p63-Dependent Networks Sequentially Rearrange Chromatin Landscapes to Drive Human Epidermal Lineage Commitment. Cell Stem Cell 24, 271–284.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Guo X, Polak L, Lawton LN, Young RA, Zheng D, and Fuchs E (2011). Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell 9, 219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Matsumura H, Kato T, Ichinose S, Takada A, Namiki T, Asakawa K, Morinaga H, Mohri Y, De Arcangelis A, et al. (2019). Stem cell competition orchestrates skin homeostasis and ageing. Nature 568, 344–350. [DOI] [PubMed] [Google Scholar]
- Mah EJ, Lefebvre AEYT, McGahey GE, Yee AF, and Digman MA (2018). Collagen density modulates triple-negative breast cancer cell metabolism through adhesion-mediated contractility. Sci. Rep 8, 17094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannik J, Alzayady K, and Ghazizadeh S (2010). Regeneration of multilineage skin epithelia by differentiated keratinocytes. J. Invest. Dermatol 130, 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascré G, Dekoninck S, Drogat B, Youssef KK, Broheé S, Sotiropoulou PA, Simons BD, and Blanpain C (2012). Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 489, 257–262. [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, Saul N, and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw 3, 861. [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, and Bradley A (1999). p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713. [DOI] [PubMed] [Google Scholar]
- Nair M, Teng A, Bilanchone V, Agrawal A, Li B, and Dai X (2006). Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J. Cell Biol 173, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Gonzalez DG, Guirao B, Boucher JD, Cockburn K, Marsh ED, Mesa KR, Brown S, Rompolas P, Haberman AM, et al. (2017). Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice. Nat. Cell Biol 19, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison JM, Melo SP, Piekos SN, Torkelson JL, Bashkirova E, Mumbach MR, Rajasingh C, Zhen HH, Li L, Liaw E, et al. (2018). Retinoic acid and BMP4 cooperate with p63 to alter chromatin dynamics during surface epithelial commitment. Nat. Genet 50, 1658–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikitlearn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017b). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen CE, and Williams CKI (1996). Gaussian Processes for Regression. In Advances in Neural Information Processing Systems (The MIT Press), pp. 515–520. [Google Scholar]
- Rhee H, Polak L, and Fuchs E (2006). Lhx2 maintains stem cell character in hair follicles. Science 312, 1946–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittié L (2016). Cellular mechanisms of skin repair in humans and other mammals. J. Cell Commun. Signal 10, 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognoni E, and Watt FM (2018). Skin Cell Heterogeneity in Development, Wound Healing, and Cancer. Trends Cell Biol. 28, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Kawaguchi K, Park S, Gonzalez D, Brown S, Boucher J, Klein AM, and Greco V (2016). Spatiotemporal coordination of stem cell commitment during epidermal homeostasis. Science 352, 1471–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotzer D, Krampert M, Sulyok S, Braun S, Stark HJ, Boukamp P, and Werner S (2006). Id proteins: novel targets of activin action, which regulate epidermal homeostasis. Oncogene 25, 2070–2081. [DOI] [PubMed] [Google Scholar]
- Sada A, Jacob F, Leung E, Wang S, White BS, Shalloway D, and Tumbar T (2016). Defining the cellular lineage hierarchy in the interfollicular epidermis of adult skin. Nat. Cell Biol 18, 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TJ, and Martin P (2009). Wound repair at a glance. J. Cell Sci 122, 3209–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakata Y, Kimura R, Nanba D, Iwamoto R, Tokumaru S, Morimoto C, Yokota K, Nakamura M, Sayama K, Mekada E, et al. (2005). Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. J. Cell Sci. 118, 2363–2370. [DOI] [PubMed] [Google Scholar]
- Stringari C, Edwards RA, Pate KT, Waterman ML, Donovan PJ, and Gratton E (2012). Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep 2, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringari C, Wang H, Geyfman M, Crosignani V, Kumar V, Takahashi JS, Andersen B, and Gratton E (2015). In vivo single-cell detection of metabolic oscillations in stem cells. Cell Rep. 10, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson V, and Pachter L (2018). RNA Velocity: Molecular Kinetics from Single-Cell RNA-Seq. Mol. Cell 72, 7–9. [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, and Enver T (2017). Single-cell entropy for accurate estimation of differentiation potency from a cell’s transcriptome. Nat. Commun 8, 15599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topley GI, Okuyama R, Gonzales JG, Conti C, and Dotto GP (1999). p21(WAF1/Cip1) functions as a suppressor of malignant skin tumor formation and a determinant of keratinocyte stem-cell potential. Proc. Natl. Acad. Sci. USA 96, 9089–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong AB, Kretz M, Ridky TW, Kimmel R, and Khavari PA (2006). p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 20, 3185–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Natsuga K, Nishie W, Kobayashi Y, Donati G, Suzuki S, Fujimura Y, Tsukiyama T, Ujiie H, Shinkuma S, et al. (2017). Type XVII collagen coordinates proliferation in the interfollicular epidermis. eLife 6, e26635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawersik MJ, Mazzalupo S, Nguyen D, and Coulombe PA (2001). Increased levels of keratin 16 alter epithelialization potential of mouse skin keratinocytes in vivo and ex vivo. Mol. Biol. Cell 12, 3439–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, and McKeon F (1999). p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718. [DOI] [PubMed] [Google Scholar]
- Yang H, Adam RC, Ge Y, Hua ZL, and Fuchs E (2017). Epithelial-Mesenchymal Micro-niches Govern Stem Cell Lineage Choices. Cell 169, 483–496.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenz R, and Wagner EF (2006). Jun signalling in the epidermis: From developmental defects to psoriasis and skin tumors. Int. J. Biochem. Cell Biol 38, 1043–1049. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Liu T, Liu CJ, Song S, Zhang X, Liu W, Jia H, Xue Y, and Guo AY (2015). AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 43, D76–D81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu BM, Ishida Y, Robinson GW, Pacher-Zavisin M, Yoshimura A, Murphy PM, and Hennighausen L (2008). SOCS3 negatively regulates the gp130-STAT3 pathway in mouse skin wound healing. J. Invest. Dermatol. 128, 1821–1829. [DOI] [PubMed] [Google Scholar]
- Zywitza V, Misios A, Bunatyan L, Willnow TE, and Rajewsky N (2018). Single-Cell Transcriptomics Characterizes Cell Types in the Subventricular Zone and Uncovers Molecular Defects Impairing Adult Neurogenesis. Cell Rep. 25, 2457–2469.e8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data reported in this paper have been deposited in the GEO database under accession code GEO: GSE142471. The software of nlvelo R package is available at https://github.com/sqjin/nlvelo. The codes and walkthroughs for pseudotemporal trajectory analysis are available at https://github.com/sqjin/codes_CellReports2019.