

Abstract
Titanium(IV) complexes of amino-tris(phenolate) ligands (LTiX, X = chloride, isopropoxide) together with bis(triphenylphosphine)iminium chloride (PPNCl) are active catalyst systems for the ring-opening copolymerization of carbon dioxide and cyclohexene oxide. They show moderate activity, with turnover frequency values of ∼60 h–1 (0.02 mol % of catalyst, 80 °C, 40 bar of CO2) and high selectivity (carbonate linkages >90%), but their absolute performances are lower than those of the most active Ti(IV) catalyst systems. The reactions proceed with linear evolution of polycarbonate (PCHC) molar mass with epoxide conversion, consistent with controlled polymerizations, and evolve bimodal molar mass distributions of PCHC (up to Mn = 42 kg mol–1). The stoichiometric reaction between [LTiOiPr] and tetraphenylphosphonium chloride, PPh4Cl, allows isolation of the putative catalytic intermediate [LTi(OiPr)Cl]−, which is characterized using single-crystal X-ray diffraction techniques. The anionic titanium complex [LTi(OR)Cl]− is proposed as a model for the propagating alkoxide intermediates in the catalytic cycle.
Introduction
The copolymerization of carbon dioxide (CO2) and epoxides is an attractive means to valorize an abundant, renewable feedstock and to produce useful polymers with reduced greenhouse gas emissions.1−3 Coupling reactions between CO2 and epoxides yield cyclic carbonates as the thermodynamic products,4,5 but with a suitable catalyst the kinetic polycarbonate product may also be accessible.6−8 Catalyst selection, therefore, is critical to governing reaction selectivity and rates, as well as providing a means to control polymer properties such as molar mass and dispersity.9,10 In this field of homogeneous catalysis, the majority of the research has focused on metal complexes of cobalt(III), chromium(III), and zinc(II), with the best catalysts showing high activity and excellent polycarbonate selectivity.11 In many cases, the metal complexes are applied together with an ionic cocatalyst with the combination being referred to as a binary catalyst system.6,10 The cocatalyst significantly improves the catalyst activity and selectivity, although its role is somewhat complex. Different researchers propose that the cocatalyst is important to labilize the propagating alkoxide and carbonate intermediates, to provide counterions for coordination with metal-free polymer chains, to “hold” the propagating ionic (metal-free) chains close to the metal center, and to limit backbiting reactions to form cyclic carbonates.6,10
Here, Ti(IV) catalyst systems are selected for investigation due to their earth abundance, lack of color, and precedence in related catalytic processes.12−14 In particular, tripodal M(IV) complexes have an excellent track record in lactide and lactone ring-opening polymerization (M = Ti(IV), Zr(IV), Hf(IV)).13,15−20 The complexes combine outstanding rates, high tolerance, operation at low loadings, and stability under industrially relevant high-temperature polymerization conditions.13,16−20 Although the mechanism for epoxide/CO2 ROCOP is different from lactone polymerization, there are some similarities in catalyst requirements. For example, both cycles apply Lewis acidic metal centers to bind the monomers (epoxide or lactone), and both invoke labile metal alkoxide intermediates as propagating species. To deliver catalysts relevant for larger-scale deployment, the metal applied should be colorless, thermally stable, and earth abundant, show low toxicity, and lack redox chemistry under the conditions of catalysis; Ti(IV) complexes fit these criteria, and furthermore, there is already some precedent for their application in cyclohexene oxide (CHO)/CO2 ROCOP.21−28 For example, Nozaki and co-workers pioneered group IV catalyst systems, applying a tetradentate Ti(IV) complex, with bis(triphenylphosphine)iminium chloride (PPNCl), to achieve a turnover frequency (TOF) of 76 h–1 (0.05 mol %, 50 °C, 20 bar of CO2) with quantitative polycarbonate selectivity (>99%) (Figure 1).27 Ti(IV) complexes coordinated by phenolate-N-heterocyclic carbene26 or salalen24,29 ligands also showed good catalytic activities of 55 h–1 (0.04 mol %, 80 °C, 10 bar of CO2) and 41 h–1 (0.2 mol %, 90 °C, 40 bar of CO2), respectively (Figure 1). Encouraged by these prior successes for Ti(IV) complexes, we reasoned that Ti(IV) amino-tris(phenolate) complexes could be promising catalyst systems for CHO/CO2 ROCOP. The target Ti(IV) complexes are also related to the successful Al(III) and Fe(III) catalyst systems for epoxide/CO2 ROCOP developed by Kleij and co-workers (Figure 1).30−34 Kleij’s catalysts show outstanding activity for CO2/epoxide coupling to cyclic carbonates and a CHO/CO2 ROCOP TOF of ∼6 h–1 (0.5 mol %, 85 °C, 2 bar of CO2).31,34 The Al(III)–amino tris(phenolate) catalyst system is also notable, as it is one of only two catalysts active for CO2/limonene oxide ROCOP.33,35
Figure 1.

Structures of reported Ti(IV) catalysts along with an active Al(III)−amino tris(phenolate) complex for ROCOP of CO2/CHO.24,26,27,32
Results and Discussion
Two ligands were targeted, H3LtBu and H3LMe, and their syntheses and subsequent complexation reactions to Ti(IV) to yield complexes 1–3 were carried out according to literature procedures (see the Supporting Information for full experimental protocols and Figures S1–S12).14,19,36,37 The successful ligand complexation to Ti(IV) was confirmed using 1H NMR spectroscopy; one diagnostic feature is the formation of diastereotopic benzylic resonances at 3.96 and 2.49 ppm. Complexes 1–3 show 1H and 13C NMR spectra that are fully consistent with previous reports,37 and elemental analysis results indicate acceptable analytical purity.
The catalysts were tested under a standard set of conditions using 40 bar of CO2 pressure, 80 °C, a catalyst loading of 0.05 mol % (1:2000 catalyst:CHO) in neat cyclohexene oxide (CHO) (9.9 M), and catalyst:cocatalyst = 1:1 (Table 1). All of the complexes displayed high CO2 and polymer selectivity (>99%) and showed moderate polymerization activities from 10 to 35 h–1 (Table 1, entries 1, 3, and 5). Complexes 2 and 3, featuring methyl ligand substituents, have higher activity than 1, featuring tert-butyl substituents, and this higher activity may arise from a reduced steric hindrance at Ti(IV) accelerating rates of epoxide coordination or insertion (vide infra). The similar activities of 2 and 3 suggest that the coligand does not substantially accelerate the rate, and this is consistent with its proposed function as a polymer chain initiating group. When the polymerization temperature was increased to 100 °C, the catalytic activity increased significantly, reaching a TOF of 81 h–1 (catalyst 2), but these conditions also resulted in a significant reduction in polymer selectivity (<65%) and in the formation of cis and trans cyclic carbonate products (Table 1, entries 2 and 4). The performance of complexes 1–3 showed significantly enhanced activity and selectivity in comparison to the homoleptic titanium alkoxide precursor complex Ti(OiPr)4 and better performance than a catalyst system comprising Ti(OiPr)4/PPNCl (Table S1). This finding demonstrates the importance of the ancillary ligand in mediating Lewis acidity, oxophilicity, and lability of the Ti(IV) active site.
Table 1. ROCOP of CO2/CHO Using Catalysts 1–4/PPNCla.
| entry | catalyst | T (°C) | time (h) | conversn (%)b | CO2 (%)c | polym (%)d | TONe | TOF (h–1)f | Mn [Đ] (kg mol–1)g |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 80 | 24 | 13 | >99 | >99 | 263 | 11 | 17.1 [1.04] |
| 7.6 [1.16] | |||||||||
| 2 | 1 | 100 | 16 | 17 | >99 | 80 | 342 | 21 | 7.5 [1.21] |
| 3 | 2 | 80 | 24 | 42 | >99 | >99 | 840 | 35 | 36.8 [1.05] |
| 10.7 [1.33] | |||||||||
| 4 | 2 | 100 | 16 | 65 | >99 | 65 | 845 | 81 | 37.6 [1.06] |
| 8.5 [1.56] | |||||||||
| 5 | 3 | 80 | 24 | 40 | >99 | >99 | 800 | 33 | 37.9 [1.11] |
| 16.1 [1.07] | |||||||||
| 6 | 4 | 80 | 24 | 32 | >99 | >99 | 640 | 27 | 21.6 [1.02] |
| 9.7 [1.04] | |||||||||
| 7 | [(Boxdipy)TiIVCl]h | 60 | 12 | 45 | >99 | >99 | 900 | 76 | 13.0 [1.27] |
| 8 | [(NHC)TiIVCl2]i | 80 | 24 | 53 | >99 | >99 | 1325 | 55 | 8.3 [1.53] |
| 9 | [(Salalen)TiIVCl]j | 70 | 10 | 44 | 98 | 98 | 220 | 22 | 4.2 [1.11] |
Reaction conditions unless specified otherwise: catalyst (0.05 mol %), cocatalyst (0.05 mol %), CHO (8 mL, 9.9 M), 40 bar of CO2.
Expressed as a percentage of CHO conversion vs the theoretical maximum (100%), determined from the 1H NMR spectrum by comparison of the relative integrals of the resonances assigned to the carbonate (4.65 ppm for PCHC and 4.00 ppm for trans cyclic carbonate) and ether (3.45 ppm) linkages against CHO (3.00 ppm).
Expressed as a percentage of CO2 uptake vs the theoretical maximum (100%), determined by comparison of the relative integrals of the 1H NMR resonances due to carbonate (4.65 ppm for the polymer and 4.00 ppm for the trans cyclic carbonate) and ether (3.45 ppm) linkages.
Expressed as a percentage of polymer formation vs the theoretical maximum (100%), determined by comparison of the relative integrals of the 1H NMR resonances due to the polymer (4.65 ppm) and trans cyclic carbonate (4.00 ppm).
Turnover number (TON) = number of moles of cyclohexene oxide consumed/number of moles of catalyst.
Turnover frequency (TOF) = TON/time (h).
Determined by GPC, in THF, calibrated using narrow-Mn polystyrene standards.
Catalyst (0.01 mmol), cocatalyst (0.01 mmol), CHO (20 mmol).27
Catalyst (0.008 mmol), cocatalyst (0.008 mmol), CHO (20 mmol), 10 bar of CO2.26
Catalyst (0.2 mol %), cocatalyst (0.2 mol %), CHO (9.9 M), 40 bar of CO2.29
Catalyst systems 1–3 all produce poly(cyclohexene carbonate) (PCHC) showing bimodal molar mass distributions, as evidenced by GPC analysis of the crude polymer samples. The bimodal distributions arise due to polymer chains being initiated from both 1,2-cyclohexanediol, formed by reaction between the epoxide and residual water, and from chloride, present in the cocatalyst. The presence of both catalyst-initiated chains and telechelic chain transfer agent-initiated chains gives rise to two molar mass distributions, one of approximately double the molar mass of the other.38 To understand the influence, if any, of water on the catalysis and on the putative catalytic intermediates, complex 2 was reacted with 10 equiv of water, in THF at 25 °C, and stirred for 2 h. This reaction evolved the known oxo-bridged Ti(IV) dimer complex [(LMeTi)2O] (4) in 32% yield (Scheme 1).39 The formation of 4 was confirmed using 1H and 13C NMR spectroscopy, where the loss of the isopropoxide coligand peaks, at 5.34 ppm (1H NMR) and 78.1 ppm (13C NMR), in comparison to precursor complex 2 was clearly observed (Figures S11 and S12). To establish whether the dimer showed any catalytic activity, it was subjected to identical polymerization conditions, with the addition of a cocatalyst, where it showed performance nearly identical with that of catalyst system 2/PPNCl (Table 1, entry 6). This result was somewhat surprising, as the dimeric complex does not feature any obvious initiating groups.
Scheme 1. Structures of the Ti(IV) Complexes 1–4 Used in This Work.

The catalytic performance of 2/PPNCl was compared to three of the most active Ti(IV) catalysts reported in the literature. Although detailed comparisons are premature, as it is not clear that all of the catalysts function with identical rate laws and mechanisms, it can be seen that qualitatively these new catalysts show activity values which are consistent with previous investigations. The most active Ti(IV) catalyst system remains that reported by Nozaki and co-workers, [(Boxdipy)TiCl] (1:2000 catalyst:CHO), which shows approximately double the activity of 2 and functions at a lower temperature. The phenolate-carbene Ti(IV) complex [(NHC)TiIVCl2] is also slightly more active than 2 and shows a TOF of 55 h–1. In contrast, 2 is more active than [(Salalen)TiIVCl] but considerably less active than a related Ti(III) salen catalyst reported by the same group (TOF = 161 h–1, 0.1 mol % catalyst, 40 bar of CO2, 80 °C).24
With a successful catalyst system in hand, it was of interest to understand the limits of the catalyst concentration on copolymerization. Holding the catalyst:cocatalyst ratio constant at 1:1 and using progressively greater quantities of epoxide resulted in effective catalysis even at 0.025 mol % catalyst loading (Table 2, entry 4).
Table 2. Effect of Catalyst Loading and Cocatalyst on the ROCOP of CO2/CHO Using Catalyst 2a.
| entry | cocat | cat:cocat:CHO | time (h) | conversn (%)b | CO2 (%)c | polym (%)d | TONe | TOF (h–1)f | Mn [Đ] (kg mol–1)g |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PPNCl | 1:1:500 | 4 | 48 | >99 | >99 | 235 | 59 | 15.7 [1.02] |
| 6.8 [1.05] | |||||||||
| 2 | PPNCl | 1:1:1000 | 9 | 42 | >99 | >99 | 420 | 47 | 19.1 [1.03] |
| 8.1 [1.06] | |||||||||
| 3 | PPNCl | 1:1:2000 | 24 | 42 | >99 | >99 | 840 | 35 | 36.8 [1.05] |
| 10.7 [1.33] | |||||||||
| 4 | PPNCl | 1:1:4000 | 48 | 31 | >99 | >99 | 1240 | 26 | 13.1 [1.05] |
| 5.4 [1.09] | |||||||||
| 5 | N/A | 1:0:2000 | 24 | 0 | - | - | - | - | - |
| 6 | DMAP | 1:1:2000 | 24 | 21 | >99 | >99 | 420 | 18 | 8.4 [1.10] |
| 3.1 [1.14] | |||||||||
| 7 | PPh4Cl | 1:1:2000 | 24 | 59 | >99 | >99 | 1180 | 49 | 24.5 [1.04] |
| 10.9 [1.05] |
Reaction conditions: 80 °C, neat CHO (8 mL, 9.9 M), 40 bar of CO2.
Expressed as a percentage of CHO conversion vs the theoretical maximum (100%), determined from the 1H NMR spectrum by comparison of the relative integrals of the resonances assigned to the carbonate (4.65 ppm for PCHC and 4.00 ppm for trans cyclic carbonate) and ether (3.45 ppm) linkages against CHO (3.00 ppm).
Expressed as a percentage of CO2 uptake vs the theoretical maximum (100%), determined from the 1H NMR spectrum by comparison of the relative integrals of the resonances assigned to the carbonate (4.65 ppm for PCHC and 4.00 ppm for trans cyclic carbonate) and ether (3.45 ppm) linkages.
Expressed as a percentage of polymer formation vs the theoretical maximum (100%), determined from the 1H NMR spectrum by comparison of the relative integrals due to the polymer (4.65 ppm) and trans cyclic carbonate (4.00 ppm).
Turnover number (TON) = number of moles of CHO consumed/number of moles of catalyst.
Turnover frequency (TOF) = TON/time (h).
Determined by GPC, in THF, calibrated using narrow-Mn polystyrene standards.
Also, all the copolymerizations proceed with quantitative CO2 and polymer selectivity (>99%) and there is no detectable formation of ether linkages or cyclic carbonate byproducts by 1H NMR spectroscopy. The catalytic activity decreases as progressively greater amounts of CHO are used because the overall catalyst concentration falls. It is clearly essential to apply a cocatalyst, since using only complex 2 resulted in no polymerization at all. The cocatalyst selection was also important in controlling activity, and the most effective systems comprised complex 2 in combination with ionic cocatalysts such as PPNCl and Ph4PCl. These ionic cocatalyst systems were about twice as active as when Lewis base cocatalysts such as DMAP were applied. The finding that ionic cocatalysts show superior activity is also consistent with previous studies of Ti(IV) or other metal complexes for CO2/epoxide ROCOP, but an understanding of the catalyst/cocatalyst speciation remains somewhat ill-defined.40,41
Next the influence of copolymerization temperature on the activity of catalyst 2 was assessed (Table 3). The lowest temperature at which effective catalysis occurred was 60 °C, and as expected, the activity increases upon increasing the temperature to 80 °C (20 to 35 h–1). Increasing the temperature from 100 to 120 °C continued to increase the activity but compromised the reaction selectivity with the production of significant quantities of cyclic carbonate byproduct (35–66%, respectively). The cyclic carbonate comprised both cis and trans isomers, which indicates that polymer chain backbiting occurs from both metal alkoxide and carbonate intermediates. The temperature selectivity behavior is consistent with backbiting from the metal carbonate intermediate occurring more quickly than epoxide insertion and forming the cis-cyclohexene carbonate and, concomitantly, with backbiting from the alkoxide intermediate to form the trans-cyclohexene carbonate. To rule out a change in reaction selectivity being caused by catalyst degradation at elevated temperatures, complex 2 was heated for 24 h over the temperature range 80–120 °C and the speciation monitored by 1H NMR spectroscopy. No observable change in the 1H NMR spectrum of complex 2 was measured over the temperature range explored, indicating good catalyst stability over the typical conditions of the polymerizations (Figure S13).
Table 3. Effect of Temperature on the ROCOP of CO2/CHO Using Catalyst 2/PPNCla.
| entry | temp (°C) | time (h) | conversn (%)b | CO2 (%)c | polym (%)d | TONe | TOF (h–1)f | Mn [Đ] (kg mol–1)g |
|---|---|---|---|---|---|---|---|---|
| 1 | 60 | 48 | 47 | >99 | >99 | 940 | 20 | 36.8 [1.05] |
| 15.4 [1.55] | ||||||||
| 2 | 80 | 24 | 42 | >99 | >99 | 820 | 35 | 36.8 [1.05] |
| 10.7 [1.33] | ||||||||
| 3 | 100 | 16 | 65 | >99 | 65 | 320 | 81 | 37.6 [1.06] |
| 8.5 [1.56] | ||||||||
| 4 | 120 | 8 | 54 | >99 | 34 | 160 | 135 | 13.8 [1.07] |
| 5.1 [1.15] |
Reaction conditions: 2 (0.05 mol %), cocatalyst (0.05 mol %), CHO (8 mL, 9.9 M), 40 bar of CO2.
Expressed as a percentage of CHO conversion vs the theoretical maximum (100%), determined from the 1H NMR spectrum by comparison of the relative integrals of the resonances assigned to the carbonate (4.65 ppm for PCHC and 4.00 ppm for trans cyclic carbonate) and ether (3.45 ppm) linkages against CHO (3.00 ppm).
Expressed as a percentage of CO2 uptake vs the theoretical maximum (100%), determined from the 1H NMR spectrum by comparison of the relative integrals due to carbonate (4.65 ppm for PCHC and 4.00 ppm for trans cyclic carbonate) and ether (3.45 ppm) linkages.
Expressed as a percentage of polymer formation vs the theoretical maximum (100%), determined from the 1H NMR spectrum by comparison of the relative integrals due to polymer (4.65 ppm) and trans cyclic carbonate (4.00 ppm).
Turnover number (TON) = number of moles of cyclohexene oxide consumed/number of moles of catalyst.
Turnover frequency (TOF) = TON/time (h).
Determined by GPC, in THF, calibrated using narrow-Mn polystyrene standards.
Controlled polymerization catalysis is useful because it allows for prediction of the polycarbonate molar mass from the starting concentrations of catalyst and monomer conversion. Catalysts showing high polymerization control can also be useful for the preparation of block polymers and, more recently, are important in switch catalysis, where multiple polymerization cycles are accessed by a single catalyst to enchain monomer mixtures into multiblock polymers.42,43 To assess the polymerization control, a set of identical copolymerizations, using 2/PPNCl, was stopped at progressively increasing time periods. The crude polymer, PCHC, was analyzed by GPC and NMR spectroscopy. A plot of PCHC molar mass vs CHO conversion shows a linear fit and the resulting polymers show molar masses with narrow dispersity (Đ < 1.15). Overall the molar mass distributions are all bimodal, consistent with the growth of both α-chloro-PCHC and hydroxyl telechelic PCHC (Figure 2).
Figure 2.

(left) Plot of CHO conversion (%) vs PCHC molar mass (kg mol−1) (black squares) for the upper and lower molar mass distributions vs average polymer dispersity, Đ (blue triangles). (right) GPC data showing progressively increasing molar mass with conversion and the bimodal mass distributions.
To understand the speciation of the key propagating species, a single crystal, suitable for X-ray diffraction, was obtained by vapor diffusion of pentane into a saturated solution of complex 2 and PPh4Cl, in THF at −40 °C under a nitrogen atmosphere. Structural elucidation revealed a hexacoordinate titanium anionic complex whereby the titanium center is coordinated by the ligand, the isopropoxide coligand, and an additional chloride, from the added cocatalyst Ph4PCl (Figure 3 and Figure S15). The titanium atom adopts an octahedral geometry and is coordinated to the four donor atoms of the ligand (O, O, O, N), the isopropoxide coligand and the chloride. It is important to note that the formation of such an anionic metal “ate” catalytic intermediate has been proposed in several ROCOP mechanisms but there are only a few examples of the isolation of metal complexes showing this coordination mode.26,44−47 One recent example, reported by Le Roux and co-workers, features a hafnium “ate” complex, formed by reaction between a hafnium diphenoxy-carbene chloride complex and PPNCl, and the anionic species was proposed as the active site for epoxide ring opening in the ROCOP mechanism.48
Figure 3.
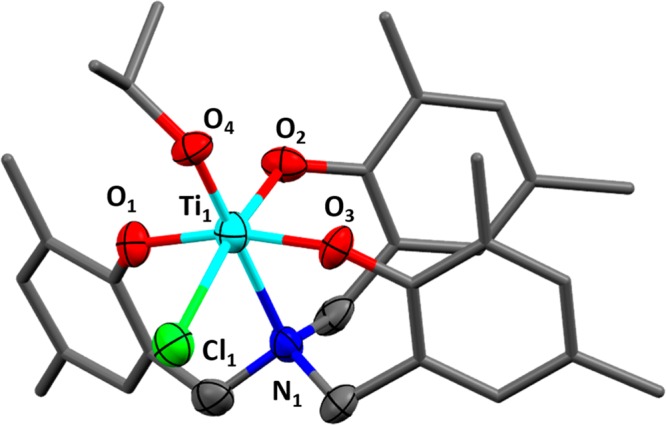
Molecular structure of the product of the reaction between complex 2 and PPh4Cl. X-ray diffraction gave insufficient data to fully resolve the structure, though its connectivity could still be obtained. The hydrogen atoms and PPh4 cation (along with some disorder) are not illustrated for clarity (for detailed information on the X-ray crystallography see Figure S14 and Table S2 in the Supporting Information).
To rationalize the performance of catalyst systems 1–3 and the molecular structure of the putative intermediate, a polymerization catalytic pathway is proposed (Figure 4). The mechanism relates closely to that proposed by Kleij and co-workers to rationalize the performance of Al(III) catalyst systems and which was substantiated by DFT calculations.31 Accordingly, the pathway requires two Ti(IV) complexes for catalysis and has parallels with bicomponent mechanisms proposed for other metal complex/cocatalyst systems.31,33,48 The initiation reaction involves a neutral Ti(IV) complex coordinating the epoxide, while a second anionic Ti(IV) “ate” complex ([LTi(OR)Cl]−) attacks the coordinated epoxide using the chloride nucleophile. This reaction generates a titanium alkoxide “ate” intermediate ([LTi(OR)2]−) which can undergo carbon dioxide insertion to form a titanium carbonate “ate” intermediate ([LTi(OR)(O2COR)]−). The carbonate intermediate propagates the chain by attacking and ring-opening an activated epoxide (coordinated at a second Ti(IV) center) and by re-forming the titanium alkoxide intermediate. Propagation reactions involve repeating the cycles between anionic Ti alkoxide and Ti carbonate intermediates. At higher reaction temperatures, the catalytic selectivity is reduced as barriers to chain backbiting reactions are overcome both from the Ti alkoxide (to form cis-cyclohexene carbonate) and Ti carbonate (to form trans-cyclohexene carbonate) intermediates. Under optimized conditions (i.e., temperature ∼80 °C), the backbiting pathways to cyclic carbonate are not accessible and the propagation cycles continue until the monomer is consumed or until the reaction is quenched. Reactions are quenched by exposure to air and the addition of solvents (not predried) to precipitate the polymer. The model studies into the reaction between 2 and excess water showed that a dimeric titanium μ-oxide species, 4, formed. This species would be expected to be catalytically inactive, but surprisingly 4/PPNCl showed performance nearly equivalent to that of the system using 2. It is clear that there is water present during the copolymerizations, since GPC data show bimodal mass distributions consistent with initiation from 1,2-cyclohexanediol (formed by a reaction between epoxide and water), which is a chain-transfer agent. Thus, a key question that remains unanswered is whether that residual water reacts more quickly with epoxide to form 1,2-cyclohexanediol (CHD) or with catalyst to form the dititanium μ-oxide species 4. Both possibilities would result in the formation of bimodal PCHC distributions, and on the basis of the catalysis using 4, both processes allow onward propagation. The mechanism by which 4 propagates could be similar to that proposed for 2/PPNCl: i.e., 4 reacts with PPNCl to generate the anionic intermediate [L2Ti2(O)Cl-alkoxide]−, which coordinates an epoxide and ring-opens it by nucleophilic chloride coligand attack. The anionic dititanium alkoxide intermediate [L2Ti2(O)(OR)]− inserts carbon dioxide to form a carbonate intermediate, [L2Ti2(O)(O2COR)]−. Another possibility is that the “ate” complexes are in equilibrium with neutral Ti(IV) complexes and anionic polymer chains which propagate “off-metal” (Figure S16). Future investigations should focus on a detailed analysis of the polymerization kinetics and use of operando analytical techniques to fine-tune the mechanistic hypotheses.
Figure 4.

Illustration of a possible catalytic cycle, involving two Ti(IV) complexes, to rationalize the formation of PCHC and cyclic carbonates using a catalyst system comprising 2 and PPNCl.
Conclusions
The utilization and transformation of CO2 into value-added products remains a significant challenge in order to reduce greenhouse gas emissions and fossil fuel consumption involved in polymer production. The sequestering of CO2 into polycarbonate polyols, for subsequent use in resin or polyurethane production, is a credible utilization method and delivers products suitable for a range of applications. Titanium(IV) catalysts confer advantages including low toxicity, high elemental abundance, good temperature stability, high stability to oxidation/reduction side reactions, and a lack of color. The titanium complexes reported herein, with the addition of cocatalyst, are active catalyst systems for the ROCOP of carbon dioxide and cyclohexene oxide. They show activity values which are similar to those of other reported titanium(IV) complexes. The isolation of a rare anionic titanium complex, formed by a stoichiometric reaction with the cocatalyst, supports the proposed role of the cocatalyst as an activated nucleophile. The complexes are the bedrock upon which future ligand and catalyst optimization studies should be conducted to improve activity and allow for yet further insight into the polymerization mechanism. For example, Le Roux and co-workers recently demonstrated higher rates of CO2/CHO ROCOP by changing the active metal in the catalysts from Ti(IV) to Hf(IV).48 They also observed much better activity at low CO2 pressure (1 bar). The use of larger ionic radii metals may also allow for mononuclear mechanisms, free from the need for cocatalyst,49 and investigations into heavier group IV congeners are warranted.
Experimental Section
General Experimental Details
All manipulations involving air- or moisture-sensitive compounds were carried out using standard Schlenk line or glovebox techniques. 2,4-Dimethylphenol, 2,4-di-tert-butylphenol, hexamethylenetetramine, and bis(triphenylphosphine)iminium chloride (PPNCl) were purchased from Sigma-Aldrich. Cyclohexene oxide (CHO) was purchased from Acros Organics and dried over CaH2 before being fractionally distilled and degassed, through several freeze–pump–thaw cycles, and stored under an inert atmosphere. Bis(triphenylphosphine)iminium chloride (PPNCl) and tetraphenylphosphonium chloride (Ph4PCl) were purified by crystallization from chloroform and hexane. Dimethylaminopyridine (DMAP) was crystallized from toluene. The ligands H3LMe and H3LtBu and the catalysts 1–4 were synthesized by following previously reported methods.14,19,36,37,39
NMR spectra were recorded on Bruker AV-400 and Bruker AV-500 instruments. All spectra were processed using MestreNova or Topspin software. GPC analysis was carried out on a Shimadzu LC-20AD instrument, equipped with a refractive index (RI) detector and two PSS SDV 5 μm linear M columns. HPLC-grade THF was used as the eluent, at 1.0 mL/min, at 30 °C. Samples were filtered through 0.2 μm PTFE filters prior to analysis. Narrow-Mn polystyrene standards were used to calibrate the instrument. Elemental analysis was carried out by Mr. Steven Boyer and Mr. Eric Coleman at the London Metropolitan University.
Representative CHO/CO2 Copolymerization (ROCOP)
All polymerizations were carried out in a 25 mL high pressure Parr reactor, fitted with a mechanical stirrer and aluminum block heating control. Catalyst (0.040 mmol), cocatalyst (0.040 mmol), and cyclohexene oxide (8 mL, 80 mmol) were measured inside a glovebox and transferred into the reactor. The reactor was filled with 20 bar of CO2 and heated to 80 °C with continuous stirring. The CO2 pressure was then increased to 40 bar. The polymerization was stopped after the desired time by cooling the reactor to room temperature using an ice bath before the excess carbon dioxide pressure was released. The crude product was purified by dissolving it in dichloromethane (10 mL) and by precipitation with the addition of methanol (3 × 100 mL) to form a white solid. The isolated polymer was dried under vacuum at 50 °C for 48 h.
General Synthesis of Tris-phenolates
A mixture of hexamethylenetetramine (2.58 g, 18.4 mmol, 1 equiv), 2,4-dialkylphenol (82.8 mmol, 4.5 equiv), and p-toluenesulfonic acid hydrate (0.07 g, 0.36 mmol, 0.02 equiv) was heated at 110 °C for 20 h. Then, an additional quantity of 2,4-dialkylphenol (27.6 mmol, 1.5 equiv) was added and the solution heated for a further 20 h. The reaction mixture was cooled to 25 °C, and MeOH (20 mL) was added to the yellow slurry. The solution was sonicated until a pale yellow solid evolved from the solution. The solid was filtered, washed with cold MeOH (−78 °C), and subsequently dried under vacuum. The pure ligand was crystallized from acetone. All spectroscopic data were consistent with the data in the literature (Figures S1–S4).
General Synthesis of LTiIVX
Titanium isopropoxide (1.20 mmol, for complexes 1 and 2) or chloro-tris(iso-propoxide)titanium(IV) (1.20 mmol, for complex 3) was added to a solution of the proligand (1.20 mmol) in THF (10 mL) and a stirrer for 16 h. All volatiles were removed in vacuo, and the product was washed with hexane (3 × 10 mL), affording a white solid.
Complex 1, (L)tBuTiOiPr
Yield: 0.54 g, 0.70 mmol, 58%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 7.45 (s, 3H), 6.83 (s, 3H), 5.47 (hept, J = 6.05 Hz, 1H), 4.03 (d, J = 13.71 Hz, 3H), 2.62 (d, J = 13.71 Hz, 3H), 1.71 (d, J = 6.05 Hz, 6H), 1.66 (s, 27H), 1.35 (s, 27H). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ (ppm) 161.19, 142.62, 135.63, 124.93, 124.53, 123.22, 79.84, 59.34, 35.43, 34.57, 31.99, 30.02, 27.04. Anal. Calcd for C48H73NO4Ti (775.50 g mol–1): C, 74.30; H, 9.48; N, 1.81. Found: C, 74.04; H, 8.94; N, 1.88.
Complex 2, (L)MeTiOiPr
Yield: 0.38 g, 0.73 mmol, 73%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 6.79 (s, 3H), 6.49 (s, 3H), 5.34 (hept, J = 6.12 Hz, 1H), 3.96 (d, J = 10.60 Hz, 3H), 2.49 (d, J = 10.60 Hz, 3H), 2.34 (s, 9H), 2.17 (s, 9H), 1.63 (d, J = 6.12 Hz, 6H). 13C{1H} NMR (126 MHz, C6D6, 298 K): δ (ppm) 160.39, 131.81, 129.19, 128.36, 124.54, 124.00, 78.09, 59.44, 25.98, 21.29, 16.60. Anal. Calcd for C30H37NO4Ti (523.22 g mol–1): C, 68.83; H, 7.12; N, 2.68. Found: C, 68.47; H, 7.23; N, 2.80.
Complex 3, (L)MeTiCl
Yield: 0.42 g, 0.84 mmol, 70%. 1H NMR (500 MHz, CDCl3, 298 K): δ (ppm) 6.89 (s, 3H), 6.74 (s, 3H), 3.99 (br s, 3H), 2.84 (br s, 3H), 2.26 (s, 9H), 2.23 (s, 9H). 13C {1H} NMR (126 MHz, CDCl3, 298 K): δ (ppm) 160.36, 131.42, 131.27, 127.36, 124.51, 122.95, 58.62, 20.82, 15.99. Anal. Calcd for C27H30ClNO3Ti (499.14 g mol–1): C, 64.88; H, 6.05; N, 2.80. Found: C, 64.76; H, 6.18; N 2.83.
Complex 4, [(LMeTi)2O]
Water (69 μL, 3.8 mmol) was added to a solution of complex 2 (0.2 g, 0.38 mmol) in THF (10 mL) and the mixture stirred for 16 h. All volatiles were removed in vacuo, and the product was washed with hexane (3 × 5 mL), affording a yellow solid. Yield: 0.11 mg, 0.12 mmol, 32%. 1H NMR (400 MHz, C6D6, 298 K): δ (ppm) 6.72 (s, 3H), 6.54 (s, 3H), 4.07 (br s, 3H), 2.51 (br s, 3H), 2.38 (s, 9H), 2.15 (s, 9H). 13C{1H} NMR (126 MHz, C6D6, 298 K): δ (ppm) 160.89, 131.04, 129.30, 128.35, 125.04, 124.36, 58.86, 20.80, 16.20. Anal. Calcd for C54H60N2O7Ti2 (944.34 g mol–1): C, 68.65; H, 6.40; N, 2.97. Found: C, 68.70; H, 6.34; N, 3.00.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.9b00845.
Complete experimental and characterization data for all compounds and polymerizations (PDF)
Accession Codes
CCDC 1970304 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The EPSRC (EP/K014668/1; EP/L017393/1), EIT Climate KIC (EnCO2re), and Econic Technologies (A.D.) are acknowledged for research funding.
The authors declare the following competing financial interest(s): C.K.W. is a director of Econic Technologies.
Supplementary Material
References
- Hepburn C.; Adlen E.; Beddington J.; Carter E. A.; Fuss S.; Mac Dowell N.; Minx J. C.; Smith P.; Williams C. K. The technological and economic prospects for CO2 utilization and removal. Nature 2019, 575 (7781), 87–97. 10.1038/s41586-019-1681-6. [DOI] [PubMed] [Google Scholar]
- Artz J.; Müller T. E.; Thenert K.; Kleinekorte J.; Meys R.; Sternberg A.; Bardow A.; Leitner W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118 (2), 434–504. 10.1021/acs.chemrev.7b00435. [DOI] [PubMed] [Google Scholar]
- Grignard B.; Gennen S.; Jerome C.; Kleij A. W.; Detrembleur C. Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers. Chem. Soc. Rev. 2019, 48 (16), 4466–4514. 10.1039/C9CS00047J. [DOI] [PubMed] [Google Scholar]
- Kamphuis A. J.; Picchioni F.; Pescarmona P. P. CO2-fixation into cyclic and polymeric carbonates: principles and applications. Green Chem. 2019, 21 (3), 406–448. 10.1039/C8GC03086C. [DOI] [Google Scholar]
- Martin C.; Fiorani G.; Kleij A. W. Recent Advances in the Catalytic Preparation of Cyclic Organic Carbonates. ACS Catal. 2015, 5 (2), 1353–1370. 10.1021/cs5018997. [DOI] [Google Scholar]
- Wang Y. Y.; Darensbourg D. J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. 10.1016/j.ccr.2018.06.004. [DOI] [Google Scholar]
- Longo J. M.; Sanford M. J.; Coates G. W. Ring-Opening Copolymerization of Epoxides and Cyclic Anhydrides with Discrete Metal Complexes: Structure–Property Relationships. Chem. Rev. 2016, 116 (24), 15167–15197. 10.1021/acs.chemrev.6b00553. [DOI] [PubMed] [Google Scholar]
- Klaus S.; Lehenmeier M. W.; Anderson C. E.; Rieger B. Recent advances in CO2/epoxide copolymerization—New strategies and cooperative mechanisms. Coord. Chem. Rev. 2011, 255 (13), 1460–1479. 10.1016/j.ccr.2010.12.002. [DOI] [Google Scholar]
- Coates G. W.; Moore D. R. Discrete Metal-Based Catalysts for the Copolymerization of CO2 and Epoxides: Discovery, Reactivity, Optimization, and Mechanism. Angew. Chem., Int. Ed. 2004, 43 (48), 6618–6639. 10.1002/anie.200460442. [DOI] [PubMed] [Google Scholar]
- Darensbourg D. J. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem. Rev. 2007, 107 (6), 2388–2410. 10.1021/cr068363q. [DOI] [PubMed] [Google Scholar]
- Paul S.; Zhu Y. Q.; Romain C.; Brooks R.; Saini P. K.; Williams C. K. Ring-opening copolymerization (ROCOP): synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51 (30), 6459–6479. 10.1039/C4CC10113H. [DOI] [PubMed] [Google Scholar]
- Chmura A. J.; Davidson M. G.; Jones M. D.; Lunn M. D.; Mahon M. F.; Johnson A. F.; Khunkamchoo P.; Roberts S. L.; Wong S. S. F. Group 4 complexes with aminebisphenolate ligands and their application for the ring opening polymerization of cyclic esters. Macromolecules 2006, 39 (21), 7250–7257. 10.1021/ma061028j. [DOI] [Google Scholar]
- Chmura A. J.; Davidson M. G.; Jones M. D.; Lunn M. D.; Mahon M. F. Group 4 complexes of amine bis(phenolate)s and their application for the ring opening polymerisation of cyclic esters. Dalton Trans. 2006, (7), 887–889. 10.1039/B513345A. [DOI] [PubMed] [Google Scholar]
- Kol M.; Shamis M.; Goldberg I.; Goldschmidt Z.; Alfi S.; Hayut-Salant E. Titanium(IV) complexes of trianionic amine triphenolate ligands. Inorg. Chem. Commun. 2001, 4 (4), 177–179. 10.1016/S1387-7003(01)00157-5. [DOI] [Google Scholar]
- Sauer A.; Kapelski A.; Fliedel C.; Dagorne S.; Kol M.; Okuda J. Structurally well-defined group 4 metal complexes as initiators for the ring-opening polymerization of lactide monomers. Dalton Trans. 2013, 42 (25), 9007–9023. 10.1039/c3dt00010a. [DOI] [PubMed] [Google Scholar]
- Chmura A. J.; Davidson M. G.; Frankis C. J.; Jones M. D.; Lunn M. D. Highly active and stereoselective zirconium and hafnium alkoxide initiators for solvent-free ring-opening polymerization of rac-lactide. Chem. Commun. 2008, (11), 1293–1295. 10.1039/b718678a. [DOI] [PubMed] [Google Scholar]
- Jeffery B. J.; Whitelaw E. L.; Garcia-Vivo D.; Stewart J. A.; Mahon M. F.; Davidson M. G.; Jones M. D. Group 4 initiators for the stereoselective ROP of rac-β-butyrolactone and its copolymerization with rac-lactide. Chem. Commun. 2011, 47 (45), 12328–12330. 10.1039/c1cc15265c. [DOI] [PubMed] [Google Scholar]
- Forder T. R.; Mahon M. F.; Davidson M. G.; Woodman T.; Jones M. D. Synthesis and characterisation of unsymmetrical Zr(IV) amine tris(phenolate) complexes and their application in ROP of rac-LA. Dalton Trans. 2014, 43 (31), 12095–12099. 10.1039/C4DT01260G. [DOI] [PubMed] [Google Scholar]
- Johnson A. L.; Davidson M. G.; Pérez Y.; Jones M. D.; Merle N.; Raithby P. R.; Richards S. P. Synthesis and structure of aluminium amine-phenolate complexes. Dalton Trans. 2009, (28), 5551–5558. 10.1039/b904534a. [DOI] [PubMed] [Google Scholar]
- Chmura A. J.; Cousins D. M.; Davidson M. G.; Jones M. D.; Lunn M. D.; Mahon M. F. Robust chiral zirconium alkoxide initiators for the room-temperature stereoselective ring-opening polymerisation of rac-lactide. Dalton Trans. 2008, (11), 1437–1443. 10.1039/b716304e. [DOI] [PubMed] [Google Scholar]
- Quadri C. C.; Lalrempuia R.; Hessevik J.; Tornroos K. W.; Le Roux E. Structural Characterization of Tridentate N-Heterocyclic Carbene Titanium(IV) Benzyloxide, Silyloxide, Acetate, and Azide Complexes and Assessment of Their Efficacies for Catalyzing the Copolymerization of Cyclohexene Oxide with CO2. Organometallics 2017, 36 (22), 4477–4489. 10.1021/acs.organomet.7b00705. [DOI] [Google Scholar]
- Lalrempuia R.; Breivik F.; Tornroos K. W.; Le Roux E. Coordination behavior of bis-phenolate saturated and unsaturated N-heterocyclic carbene ligands to zirconium: reactivity and activity in the copolymerization of cyclohexene oxide with CO2. Dalton Trans. 2017, 46 (25), 8065–8076. 10.1039/C7DT01117B. [DOI] [PubMed] [Google Scholar]
- Garden J. A.; White A. J. P.; Williams C. K. Heterodinuclear titanium/zinc catalysis: synthesis, characterization and activity for CO2/epoxide copolymerization and cyclic ester polymerization. Dalton Trans. 2017, 46 (8), 2532–2541. 10.1039/C6DT04193K. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Qin Y.; Wang X.; Wang F. Trivalent Titanium Salen Complex: Thermally Robust and Highly Active Catalyst for Copolymerization of CO2 and Cyclohexene Oxide. ACS Catal. 2015, 5 (1), 393–396. 10.1021/cs501719v. [DOI] [Google Scholar]
- Su C. K.; Chuang H. J.; Li C. Y.; Yu C. Y.; Ko B. T.; Chen J. D.; Chen M. J. Oxo-Bridged Bimetallic Group 4 Complexes Bearing Amine-Bis(benzotriazole phenolate) Derivatives as Bifunctional Catalysts for Ring-Opening Polymerization of Lactide and Copolymerization of Carbon Dioxide with Cyclohexene Oxide. Organometallics 2014, 33 (24), 7091–7100. 10.1021/om500784a. [DOI] [Google Scholar]
- Quadri C. C.; Le Roux E. Copolymerization of cyclohexene oxide with CO2 catalyzed by tridentate N-heterocyclic carbene titanium(iv) complexes. Dalton Trans. 2014, 43 (11), 4242–4246. 10.1039/C3DT52804A. [DOI] [PubMed] [Google Scholar]
- Nakano K.; Kobayashi K.; Nozaki K. Tetravalent Metal Complexes as a New Family of Catalysts for Copolymerization of Epoxides with Carbon Dioxide. J. Am. Chem. Soc. 2011, 133 (28), 10720–10723. 10.1021/ja203382q. [DOI] [PubMed] [Google Scholar]
- Mandal M.; Chakraborty D. Group 4 complexes bearing bis(salphen) ligands: Synthesis, characterization, and polymerization studies. J. Polym. Sci., Part A: Polym. Chem. 2016, 54 (6), 809–824. 10.1002/pola.27918. [DOI] [Google Scholar]
- Wang Y.; Qin Y.; Wang X.; Wang F. Coupling reaction between CO2 and cyclohexene oxide: selective control from cyclic carbonate to polycarbonate by ligand design of salen/salalen titanium complexes. Catal. Sci. Technol. 2014, 4 (11), 3964–3972. 10.1039/C4CY00752B. [DOI] [Google Scholar]
- Kindermann N.; Cristofol A.; Kleij A. W. Access to Biorenewable Polycarbonates with Unusual Glass Transition Temperature (T-g) Modulation. ACS Catal. 2017, 7 (6), 3860–3863. 10.1021/acscatal.7b00770. [DOI] [Google Scholar]
- González-Fabra J.; Castro-Gómez F.; Kleij A. W.; Bo C. Mechanistic Insights into the Carbon Dioxide/Cyclohexene Oxide Copolymerization Reaction: Is One Metal Center Enough?. ChemSusChem 2017, 10 (6), 1233–1240. 10.1002/cssc.201601520. [DOI] [PubMed] [Google Scholar]
- Martín C.; Kleij A. W. Terpolymers Derived from Limonene Oxide and Carbon Dioxide: Access to Cross-Linked Polycarbonates with Improved Thermal Properties. Macromolecules 2016, 49 (17), 6285–6295. 10.1021/acs.macromol.6b01449. [DOI] [Google Scholar]
- Peña Carrodeguas L.; González-Fabra J.; Castro-Gómez F.; Bo C.; Kleij A. W. AlIII-Catalysed Formation of Poly(limonene)carbonate: DFT Analysis of the Origin of Stereoregularity. Chem. - Eur. J. 2015, 21 (16), 6115–6122. 10.1002/chem.201406334. [DOI] [PubMed] [Google Scholar]
- Whiteoak C. J.; Martin E.; Belmonte M. M.; Benet-Buchholz J.; Kleij A. W. An Efficient Iron Catalyst for the Synthesis of Five- and Six-Membered Organic Carbonates under Mild Conditions. Adv. Synth. Catal. 2012, 354 (2–3), 469–476. 10.1002/adsc.201100752. [DOI] [Google Scholar]
- Byrne C. M.; Allen S. D.; Lobkovsky E. B.; Coates G. W. Alternating Copolymerization of Limonene Oxide and Carbon Dioxide. J. Am. Chem. Soc. 2004, 126 (37), 11404–11405. 10.1021/ja0472580. [DOI] [PubMed] [Google Scholar]
- Gendler S.; Segal S.; Goldberg I.; Goldschmidt Z.; Kol M. Titanium and Zirconium Complexes of Dianionic and Trianionic Amine–Phenolate-Type Ligands in Catalysis of Lactide Polymerization. Inorg. Chem. 2006, 45 (12), 4783–4790. 10.1021/ic052120j. [DOI] [PubMed] [Google Scholar]
- Bull S. D.; Davidson M. G.; Johnson A. L.; Robinson D. E. J. E.; Mahon M. F. Synthesis, structure and catalytic activity of an air-stable titanium triflate, supported by an amine tris(phenolate) ligand. Chem. Commun. 2003, (14), 1750–1751. 10.1039/B304704K. [DOI] [Google Scholar]
- Darensbourg D. J. Chain transfer agents utilized in epoxide and CO2 copolymerization processes. Green Chem. 2019, 21 (9), 2214–2223. 10.1039/C9GC00620F. [DOI] [Google Scholar]
- Nielson A. J.; Shen C.; Waters J. M. Molecular engineering of coordination pockets in chloro-tris-phenoxo complexes of titanium(IV). Polyhedron 2006, 25 (10), 2039–2054. 10.1016/j.poly.2005.12.027. [DOI] [Google Scholar]
- Cohen C. T.; Chu T.; Coates G. W. Cobalt Catalysts for the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide: Combining High Activity and Selectivity. J. Am. Chem. Soc. 2005, 127 (31), 10869–10878. 10.1021/ja051744l. [DOI] [PubMed] [Google Scholar]
- Darensbourg D. J.; Mackiewicz R. M.; Rodgers J. L.; Phelps A. L. (Salen)CrIIIX Catalysts for the Copolymerization of Carbon Dioxide and Epoxides: Role of the Initiator and Cocatalyst. Inorg. Chem. 2004, 43 (6), 1831–1833. 10.1021/ic0352856. [DOI] [PubMed] [Google Scholar]
- Stößer T.; Williams C. K. Selective Polymerization Catalysis from Monomer Mixtures: Using a Commercial Cr-Salen Catalyst To Access ABA Block Polyesters. Angew. Chem., Int. Ed. 2018, 57 (21), 6337–6341. 10.1002/anie.201801400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T. T. D.; Zhu Y.; Williams C. K. Pentablock Copolymer from Tetracomponent Monomer Mixture Using a Switchable Dizinc Catalyst. Macromolecules 2018, 51 (14), 5346–5351. 10.1021/acs.macromol.8b01224. [DOI] [Google Scholar]
- Fieser M. E.; Sanford M. J.; Mitchell L. A.; Dunbar C. R.; Mandal M.; Van Zee N. J.; Urness D. M.; Cramer C. J.; Coates G. W.; Tolman W. B. Mechanistic Insights into the Alternating Copolymerization of Epoxides and Cyclic Anhydrides Using a (Salph)AlCl and Iminium Salt Catalytic System. J. Am. Chem. Soc. 2017, 139 (42), 15222–15231. 10.1021/jacs.7b09079. [DOI] [PubMed] [Google Scholar]
- Abel B. A.; Lidston C. A. L.; Coates G. W. Mechanism-Inspired Design of Bifunctional Catalysts for the Alternating Ring-Opening Copolymerization of Epoxides and Cyclic Anhydrides. J. Am. Chem. Soc. 2019, 141 (32), 12760–12769. 10.1021/jacs.9b05570. [DOI] [PubMed] [Google Scholar]
- Cohen C. T.; Chu T.; Coates G. W. Cobalt Catalysts for the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide: Combining High Activity and Selectivity. J. Am. Chem. Soc. 2005, 127 (31), 10869–10878. 10.1021/ja051744l. [DOI] [PubMed] [Google Scholar]
- Darensbourg D. J.; Mackiewicz R. M. Role of the Cocatalyst in the Copolymerization of CO2 and Cyclohexene Oxide Utilizing Chromium Salen Complexes. J. Am. Chem. Soc. 2005, 127 (40), 14026–14038. 10.1021/ja053544f. [DOI] [PubMed] [Google Scholar]
- Lalrempuia R.; Underhaug J.; Törnroos K. W.; Le Roux E. Anionic hafnium species: an active catalytic intermediate for the coupling of epoxides with CO2?. Chem. Commun. 2019, 55 (50), 7227–7230. 10.1039/C9CC02695A. [DOI] [PubMed] [Google Scholar]
- Thevenon A.; Cyriac A.; Myers D.; White A. J. P.; Durr C. B.; Williams C. K. Indium Catalysts for Low-Pressure CO2/Epoxide Ring-Opening Copolymerization: Evidence for a Mononuclear Mechanism?. J. Am. Chem. Soc. 2018, 140 (22), 6893–6903. 10.1021/jacs.8b01920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.