

SUMMARY
The tumor microenvironment (TME) plays a pivotal role in cancer progression, and, in ovarian cancer (OvCa), the primary TME is the omentum. Here, we show that the diabetes drug metformin alters mesothelial cells in the omental microenvironment. Metformin interrupts bidirectional signaling between tumor and mesothelial cells by blocking OvCa cell TGF-β signaling and mesothelial cell production of CCL2 and IL-8. Inhibition of tumor-stromal crosstalk by metformin is caused by the reduced expression of the tricarboxylic acid (TCA) enzyme succinyl CoA ligase (SUCLG2). Through repressing this TCA enzyme and its metabolite, succinate, metformin activated prolyl hydroxylases (PHDs), resulting in the degradation of hypoxia-inducible factor 1α (HIF1α) in mesothelial cells. Disruption of HIF1α-driven IL-8 signaling in mesothelial cells by metformin results in reduced OvCa invasion in an organotypic 3D model. These findings indicate that tumor-promoting signaling between mesothelial and OvCa cells in the TME can be targeted using metformin.
Graphical Abstract
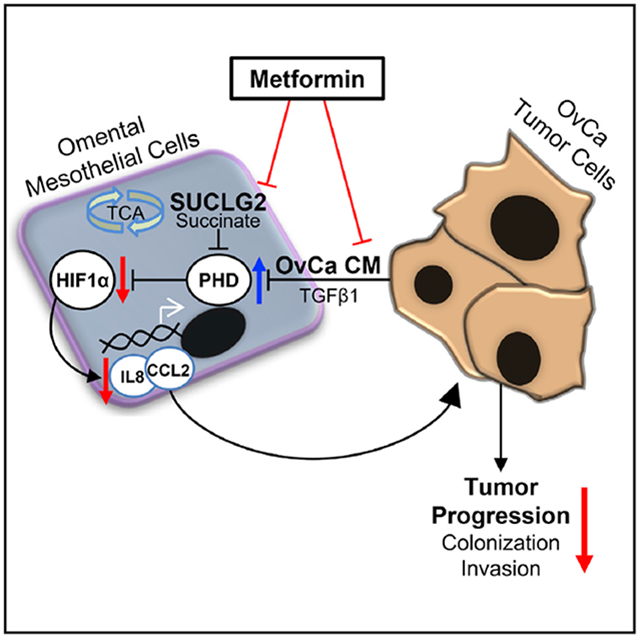
In Brief
Hart et al. identify that the type 2 diabetes drug metformin inhibits ovarian cancer invasion by targeting crosstalk between cancer cells and adjacent normal stromal mesothelial cells, making the microenvironment less hospitable to cancer growth.
INTRODUCTION
Ovarian cancer (OvCa) is a devastating disease marked by poor prognosis, as patients typically present after the disease has spread from the site of origin throughout the peritoneal cavity (Lengyel, 2010). Metastasis to the peritoneum and omentum marks a pivotal step for progression of the disease, as it provides a nutrient-rich tumor microenvironment (TME) composed of multiple cell types, with superficial mesothelial cells providing a barrier over other stromal cells, including fibroblasts and adipocytes (Kenny et al., 2009; Lengyel, 2010). Increasing evidence suggests that bidirectional signaling and nutrient exchange between cancer cells and stromal cells in the TME is critically important in supporting cancer growth (Romero et al., 2015).
Initial studies regarding the TME focused on cancer-associated fibroblasts (Gascard and Tlsty, 2016) and adipocytes (Nieman et al., 2013); however, recently, mesothelial cells have been observed to promote tumor progression. In OvCa, mesothelial cells are reprogrammed and activated toward a mesenchymal phenotype by interacting with cancer cells (Fujikake et al., 2018; Kenny et al., 2014; Rynne-Vidal et al., 2017). These activated mesothelial cells promote multiple tumorigenic processes, including adhesion (Ksiazek et al., 2009; Pakuła et al., 2018), migration (Pakuła et al., 2018; Rieppi et al., 1999), and invasion (Kenny et al., 2014), through the altered expression of surface adhesion molecules and their ligands (Cannistra et al., 1993; Lessan et al., 1999; Mikuła-Pietrasik et al., 2014; Watanabe et al., 2012), as well as extracellular matrix (ECM) production and remodeling (Heyman et al., 2010; Kenny et al., 2008; Ksiazek et al., 2009; Sandoval et al., 2013). The mechanisms by which mesothelial cell reprogramming occurs are just beginning to be elucidated, but multiple groups have reported that secreted transforming growth factor β (TGF-β) induces pro-tumorigenic changes in mesothelial cells (Falk et al., 2013; Fujikake et al., 2018; Rynne-Vidal et al., 2017). In OvCa, we have shown in mesothelial cells that blocking fibronectin signaling downstream of TGF-β-dependent activation prevented OvCa adhesion and invasion in vitro and omental colonization in vivo (Kenny et al., 2014).
Our understanding of the biology of the TME has evolved quickly; however, translating these discoveries into cancer treatments directed at vulnerabilities in the TME has been challenging. While compounds and antibodies have been identified that target mesothelial cell ECM to prevent OvCa adhesion in vitro Kenny et al., 2014), moving even the most promising TME-directed compound from pre-clinical evidence to clinical use will require many years. To accelerate the discovery of cancer therapeutics, a drug-repurposing paradigm has been proposed whereby compounds already in use for non-cancer indications are tested for efficacy in cancer (Corsello et al., 2017; Sleire et al., 2017). Metformin, a biguanide commonly used for type 2 diabetes, is a leading drug in this paradigm and is undergoing clinical testing in several types of cancer (Chae et al., 2016; Litchfield et al., 2015). A recent trial in epidermal growth factor receptor (EGFR) mutant lung adenocarcinoma reported increased progression-free survival when metformin was added to treatment with EGFR-tyrosine kinase inhibitors (Arrieta et al., 2019), further supporting the potential of metformin as a cancer treatment and warranting systematic assessment of the mechanisms of action of the drug in cancer. In OvCa, we and others have shown in retrospective analyses that 12%–17% of OvCa patients have diabetes and that patients with diabetes who use metformin have increased progression-free survival compared to patients who do not use metformin (Kumar et al., 2013; Romero et al., 2012). Complementing the retrospective clinical data, in cell culture, metformin has a direct effect on OvCa cells by altering central carbon metabolism (Liu et al., 2016) and inhibiting viability, clonogenicity, and invasion (Hart et al., 2019; Litchfield et al., 2015). Here, we ask whether, beyond the cancer compartment alone, metformin can be used to target the TME to make it inhospitable to OvCa seeding and growth. We also sought to discover any molecular or metabolic pathways in the TME that contribute to the potential efficacy of metformin as a cancer therapeutic.
RESULTS
Exposure of the TME to Metformin Reduces Adhesion, Colonization, and Invasion of OvCa Cells
The microenvironment of the omentum is uniquely conducive to OvCa metastasis (Lengyel, 2010). To assess the global effect of metformin on this metastatic site, fresh samples of omentum were obtained from patients without cancer, and ex vivo adhesion experiments were performed (Figure 1A). Patients taking metformin for diabetes had remarkably fewer OvCa cells adhere to the fresh omentum compared with control patients not using metformin (Figure 1B). Consistent with these results, when omental explants from patients without diabetes were treated with metformin before seeding OvCa cells, fewer OvCa cells were found to adhere to (1.5 h, Figure 1C) or colonize (72 h, Figure 1D) the omental explants. Similarly, the pretreatment of omental tissue decreased the number of OvCa cells that migrate toward the omentum in an invasion assay (Figure 1E). The superficial anatomic layers of the omentum are made up of mesothelial cells covering fibroblasts in a collagen-rich ECM (Kenny et al., 2014). To identify which cell type in the omental TME is responsible for the effect of metformin, we used a 3D organotypic model constructed with normal omental fibroblasts (NOFs) and human primary mesothelial cells (HPMCs) Kenny et al., 2007) and pretreated OvCa cells and each stromal cell type separately with metformin before constructing the model. All of the cell types were responsive to metformin as evidenced by decreased mitochondrial respiration (Figure S1A), and this dose of metformin was not cytotoxic in any of the cell types tested (Figure S1B). The results in the 3D organotypic model indicate that the effect of metformin in the ovarian TME is mediated by HPMCs, as only pretreatment of this cell type reduced the invasion of OvCa cells (Figures 1F and S1C). The activation of AMP kinase (AMPK) is one proposed mechanism of the anti-cancer effects of metformin (Rena et al., 2017); however, here, we found the effects of metformin to be independent of AMPK as the ability of metformin to inhibit OvCa invasion was not attenuated by co-treatment with an AMPK inhibitor, compound C (Zhou et al., 2001), and the activation of AMPK using 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) Corton et al., 1995) did not recapitulate the inhibition of OvCa cell invasion observed with metformin (Figure 1G).
Figure 1. Metformin Inhibits Colonization of the Omentum Ex Vivo and Invasion of the 3D TME In Vitro.
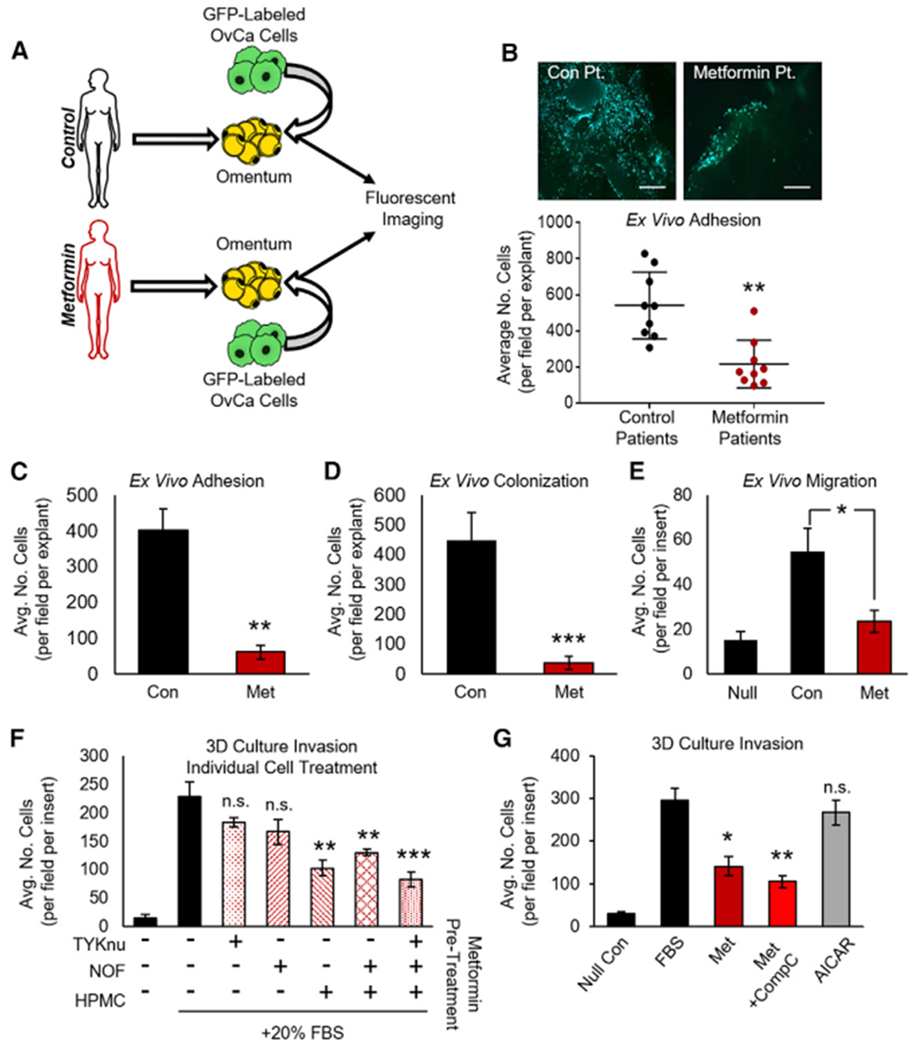
(A) Schematic: ex vivo adhesion of GFP-tagged HeyA8 OvCa cells to fresh human omental biopsies from patients without cancer taking metformin for type 2 diabetes or control patients not taking metformin.
(B) Omental explants from control or metformin patients were seeded with GFP-tagged HeyA8 OvCa cells. Tumor cells were allowed to adhere to omental explants for 1.5 h before imaging and quantification (n = 3 patients per group in triplicate). Images are representative and were taken at 10× magnification; scale bar represents 200 μm.
(C and D) Ex vivo adhesion and colonization: human omental explants were pretreated with metformin (250 μM) for 72 h before seeding of GFP-tagged HeyA8 OvCa cells, and the number of tumor cells that adhered to (1.5 h, C) or colonized (72 h, D) per omentum were quantified (n = 2 patients in triplicate for both experiments).
(E) Migration of HeyA8 OvCa cells through transwell chamber (15 h) toward omental explants that had been pretreated with metformin (250 μM, 72 h). During the assay, metformin was removed, and all media were replaced with serum-free media containing 0.1% BSA (n = 2 patients in triplicate).
(F) Invasion through 3D organotypic model (36 h) after seeding of TYKnu OvCa cells. Each cell type was individually treated with metformin (250 μM, 72 h) before the 3D model was constructed. HPMC, human primary mesothelial cells; NOF, normal omental fibroblast; n = 3 patients in triplicate.
(G) Invasion of TYKnu cells through 3D model (36 h) treated with the indicated compounds: metformin (1 mM), AMPK inhibitor compound C (CompC, 1 μM), and AMPK activator AICAR (1 mM) (n = 3 patients in triplicate).
Data represent mean values ± SDs. *p < 0.05, **p < 0.01, and ***p < 0.005; n.s., not significant.
Metformin Blocks OvCa Cell Invasion by Decreasing CCL2 and IL-8 Production
To understand how the effect of metformin on HPMCs inhibits the invasion of OvCa cells, we first sought to identify HPMC-derived cytokines that are altered by metformin. Cytokine arrays were performed on omenta from C57BL/6 mice pretreated with metformin for 3 weeks before the injection of ID8 murine OvCa cell conditioned media (CM) into the peritoneal cavity (experimental design, Figure S2A). Analysis of cytokine arrays representing 40 cytokines showed that ID8 CM-induced expression of the CXCL1 and C-C motif chemokine ligand 2 (CCL2) cytokines in the mouse omentum and that induction of these cytokines was prevented in mice that had received metformin (Figure 2A, quantified in Figure S2B). Both cytokines were validated in an independent cohort of mice by ELISA analysis of omental samples (Figure 2B). When p53 was stably knocked down in ID8 cells, CXCL1 and CCL2 were still induced by OvCa cell CM, but not significantly repressed by metformin (Figure S2C).
Figure 2. Suppression of Mesothelial Cell-Derived CCL2 and IL-8 Is Required for the Inhibition of Invasion In Vitro and the Colonization Ex Vivo by Metformin.
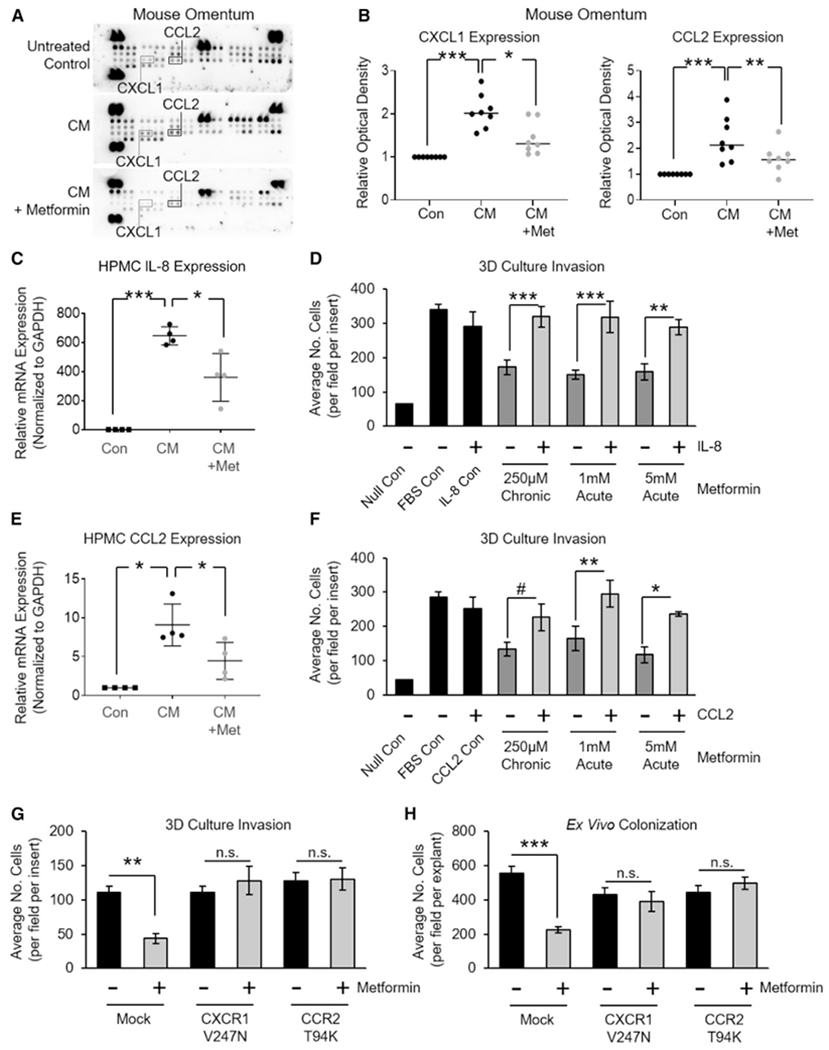
(A) A representative array membrane measuring cytokines in mouse omentum. Mice were treated with or without metformin for 3 weeks before the intraperitoneal (i.p.) injection of ID8 tumor cell CM, and the omentum was harvested 72 h later (see Figure S2A for details) (n = 2 mice per group).
(B) Validation of candidate cytokines CXCL1 and CCL2 from (A) using ELISA (n = 8 mice per group).
(C) qRT-PCR of IL-8 mRNA expression in HPMCs exposed to TYKnu OvCa CM (36 h). HPMCs were pretreated with metformin (250 μM) for 72 h before CM exposure (n = 4 patients in duplicate).
(D) Invasion of TYKnu OvCa cells (36 h) through the 3D model with or without metformin pretreatment of HPMCs (250 μM for 72 h), or simultaneous treatment ± recombinant human IL-8 protein (150 ng/mL) (n = 2 patients in triplicate).
(E) qRT-PCR of CCL2 mRNA expression in HPMCs subjected to TYKnu OvCa CM (36 h). HPMCs were pretreated with metformin (250 μM) for 72 h before CM exposure (n = 4 patients in duplicate).
(F) Invasion of TYKnu OvCa cells (36 h) through the 3D model with or without metformin pretreatment of HPMCs (250 μM for 72 h), or simultaneous treatment ± recombinant human CCL2 protein (150 ng/mL) (n = 2 patients in triplicate).
(G and H) 3D invasion (G) or omental explant colonization (H): TYKnu OvCa cells expressing constitutively active IL-8 receptor (CXCR1-V247N) or CCL2 receptor (CCR2-T94K) were added to the 3D model constructed with HPMCs pretreated with metformin (G) or to a human omental explant pretreated with metformin (H).
Data represent mean values ± SDs. #p < 0.09, *p < 0.05, **p < 0.01, and ***p < 0.005; n.s., not significant.
Next, we asked whether the effect of metformin on the cytokines that were noted in mice were also present in human cells. Murine CXCL1 is functionally homologous to human interleukin 8 (IL-8) (mice do not harbor an IL-8 gene) Lee et al., 1995), and we previously showed that IL-8 plays a pivotal role in OvCa cell colonization of the omentum (Nieman et al., 2011). Consistent with the observation in mice, CM from human OvCa cells increased IL-8 mRNA expression in HPMCs, and induction of this cytokine was prevented by metformin (Figure 2C). To understand the functional significance of IL-8 in the effect of metformin on OvCa we used the 3D organotypic model to test whether the candidate cytokines were necessary targets for the inhibition of OvCa invasion by metformin. In the 3D organotypic model, in the presence of recombinant IL-8, metformin no longer inhibited the invasion of OvCa cells, indicating that the inhibition of OvCa cell invasion by metformin was IL-8 dependent (Figure 2D). We also assessed the effect of metformin on CCL2 and found that metformin prevented the induction of CCL2 mRNA in HPMCs by OvCa CM (Figure 2E) and that recombinant CCL2 protein abrogated the ability of metformin to inhibit OvCa cell invasion in 3D culture (Figure 2F). Lastly, we tested whether IL-8 and CCL2 signaling specifically in the tumor cells is required for the effect of metformin. Here, OvCa cells were stably transfected with receptor mutants for IL-8 (CXCR1; Han, 2014) or CCL2 (CCR2; Alvarez Arias et al., 2003) that maintain constitutive activity independent of ligand binding (Figure S2D), and the effect of metformin on invasion through the 3D organotypic model and colonization of ex vivo omental explants was measured. The findings show that with constitutive activation of either the IL-8 or the CCL2 receptor in the OvCa cells, metformin no longer decreased OvCa invasion in the 3D model or the ex vivo colonization of omental explants (Figures 2G and 2H, respectively). These data indicate that the inhibition of IL-8 or CCL2 cytokine production by HPMCs and the subsequent signaling of these cytokines in the tumor cells is a requirement for metformin to impede OvCa invasion in the TME.
Metformin Prevents OvCa-Induced Stabilization of HIF1α in Mesothelial Cells
To obtain a global understanding of the impact of metformin on omental mesothelial cells exposed to OvCa, we performed unbiased proteomic profiling. The principal-component analysis of HPMCs co-cultured with OvCa cells clearly distinguished between metformin-treated and control untreated cells as numerous proteins were altered by the drug (Figures 3A and S3A). Enrichment analysis of these proteins revealed that metformin repressed pathways involved in pro-metastatic signaling, including hypoxia signaling, ECM remodeling, and epithelial-to-mesenchymal transition (Table S1). In addition, in HPMCs co-cultured with OvCa, unsupervised hierarchical clustering confirmed that a majority of hypoxia-related proteins were downregulated by metformin (Figure 3B). Since hypoxic signaling pathways lead to stromal cell reprogramming (Ammirante et al., 2014), and we have identified hypoxia-inducible factor 1α (HIF1α) as a metformin target in OvCa (Hart et al., 2019), we focused our evaluation on the effect of metformin on HIF1α in the ovarian TME. Western blotting demonstrated that OvCa CM increased HIF1α but not HIF2α expression in HPMCs, and the increase in HIF1α was prevented by metformin (Figure 3C). However, when HPMCs were transfected with a constitutively stable form of HIF1α (P402A/P564A) that escapes hydrolysis-mediated degradation by prolyl hydroxylases (PHDs) Jaakkola et al., 2001), metformin’s reduction in HIF1α expression was attenuated (Figure 3D). Next, we sought to understand whether HIF1α signaling played a role in metformin’s inhibition of IL-8 or CCL2 production by HPMCs. It was noted that when HPMCs overexpress HIF1α, CCL2 and IL-8 mRNA expression is increased, and metformin no longer represses these cytokines (Figure 3E).
Figure 3. Metformin Prevents Tumor-Cell-Induced Stabilization of HIF1α in Mesothelial Cells.
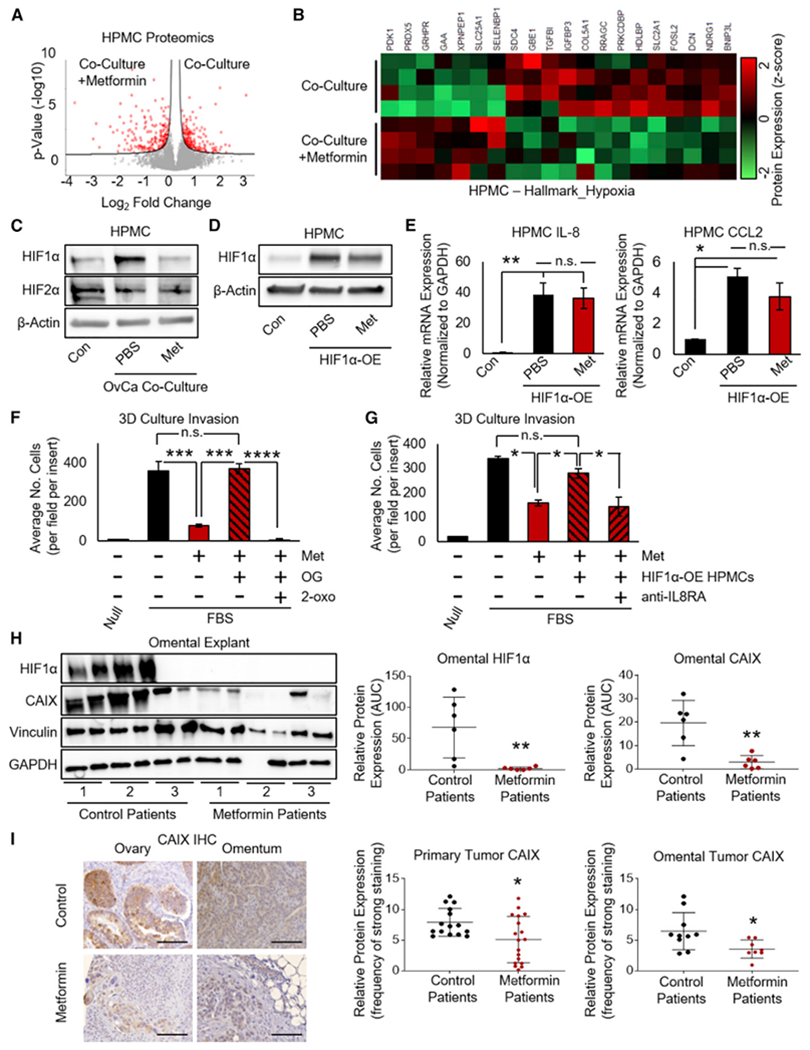
(A) Mass spectrometry (MS) analysis of mesothelial cells (HPMCs) co-cultured with OvCa cells and treated with metformin (1 mM, 48 h). Cell lysates were collected and measured on a Q Exactive HF mass spectrometer, and the quantification of proteins significantly altered by metformin is shown by volcano plot (Perseus), with the significantly altered proteins highlighted in red. Significance was defined by a false discovery rate (FDR) of 0.05 and an S0 value of 0.05 (n = 4 patients in triplicate).
(B) Unsupervised hierarchical clustering (Perseus) was performed on the dataset from (A), assessing hypoxia-related proteins in HPMCs following exposure to TYKnu OvCa co-culture in the presence or absence of metformin (1 mM, 48 h) (n = 4 patients per group in triplicate).
(C) Western blot of HIF1α and HIF2α protein expression in HPMCs following exposure to TYKnu OvCa co-culture in the presence of metformin (1 mM, 48 h) (representative blot from n = 2 patients in duplicate).
(D) Western blot of HIF1α protein expression in HPMCs expressing constitutively stable HIF1α (P402A/P564A substitutions) ± metformin (1 mM, 48 h) (representative blot from n = 2 patients in duplicate).
(E) qRT-PCR of IL-8 (left) and CCL2 (right) mRNA expression in HPMCs transfected with constitutively stable HIF1α (P402A/P564A) with or without metformin (1 mM) (n = 2 patients in duplicate).
(F) Invasion of TYKnu OvCa cells (36 h) in 3D culture with metformin (pretreatment of HPMCs with 250 μM for 72 h) in the presence of N-oxalylglycine (OG, 1 mM) and 2-oxoglutarate (2-oxo, 5 mM) (n = 2 patients per group in triplicate).
(G) Invasion of TYKnu OvCa cells (36 h) in 3D culture containing control or HPMCs expressing constitutively stable HIF1α (P402A/P564A) treated with metformin (pretreatment of HPMCs with 250 μM for 72 h), with concurrent exposure to IL-8RA/CXCR1 neutralizing antibody (150 ng/mL) during the invasion assay (n = 2 patients in duplicate).
(H) Western blot of HIF1α and CAIX expression in normal/benign omental explants from patients using metformin for diabetes compared to control patients without diabetes not using metformin. Rightside, densitometry (area under the curve [AUC]) of western immunoblot bands of HIF1α and CAIX was quantified and normalized to vinculin (n = 3 patients per group in duplicate).
(I) CAIX immunohistochemical staining in primary ovarian and omental tumors from patients with OvCa who were using metformin for diabetes compared to non-diabetic patients with OvCa not taking metformin. Quantification of primary tumor (n = 15 and 18 for control and metformin, respectively) or omental metastasis (n = 15 and 12 for control and metformin, respectively) was performed using ImageScope software. Images are representative and were taken at 20× magnification; scale bar represents 100 μm.
Data represent mean values ± SDs. *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001; n.s., not significant.
Our next goal was to understand whether HIF1α is necessary for the metformin’s inhibition of OvCa invasion in the TME. When HIF1α was stabilized through competitive inhibition of PHD activity with N-oxalylglycine, metformin did not reduce the invasion of OvCa cells, and the effect of metformin was restored when PHD activity was rescued using the PHD substrate 2-oxoglutarate (Figures 3F and S3B). Likewise, the overexpression of HIF1α in HPMCs prevented metformin from reducing OvCa cell invasion in the 3D culture; however, if the IL-8 receptor was inhibited using a neutralizing antibody while HIF1α was overexpressed in HPMCs, then metformin once again markedly reduced OvCa invasion (Figure 3G). This finding suggests that metformin’s inhibition of OvCa invasion in the TME was a result of HIF1α inhibition in HPMCs and mediated, at least in part, by downstream IL-8 signaling. To understand the clinical relevance of the in vitro findings indicating the metformin-targeted hypoxia in the TME, we analyzed tissue samples from patients. Here, we obtained fresh omental samples from patients without cancer who were taking metformin for diabetes and control patients without diabetes not taking metformin. Omentum from patients taking metformin had a lower expression of HIF1α and its downstream target, carbonic anhydrase IX (CAIX; Giatromanolaki et al., 2001), compared to omentum from controls (Figure 3H). We also established a cohort of patients with OvCa (clinical data described in Table S2). In this cohort, patients with OvCa taking metformin for diabetes had lower CAIX protein expression in both the primary tumor and the omental metastases compared to patients without diabetes not taking metformin (Figure 3I).
Mesothelial Cell SUCLG2 Is Repressed by Metformin to Prevent Metabolically Driven HIF1α Stabilization
Given that HIF1α protein expression is stabilized by excess tricarboxylic acid (TCA) intermediates (Selak et al., 2005), and we have shown that metformin alters TCA metabolism in OvCa (Liu et al., 2016), we hypothesized that metformin regulates TCA intermediates in HPMCs to prevent the induction of HIF1α expression. As a first step in testing this hypothesis, we sought to understand whether metformin changed the baseline protein expression and metabolic state of HPMCs independent of the presence of cancer cells. An integrated metabolomic and proteomic analysis of OvCa-naive HPMCs treated with metformin was performed. In the metabolomics experiment, several metabolites in HPMCs were altered by metformin, and principal-component analysis (PCA) showed a clear separation of the metabolic profiles in metformin-treated and -untreated HPMCs (Figure 4A; Table S3). In a parallel proteomic experiment, PCA showed a clear separation of the proteomic profile of metformin-treated and -untreated HPMCs (Figure 4B; Table S4). Integrating the metabolomic and proteomic datasets using joint pathway analysis revealed that metformin altered metabolites and proteins involved in cell metabolism–most prominently, the TCA cycle, but also amino acid and nucleotide metabolism (Figure S4A). Further analysis of specific metabolites showed that in metformin-treated HPMCs, there were lower levels of succinate and fumarate and malate downstream of succinate; conversely, there was an increase in one of the primary carbon sources of succinate, glutamine (Figure 4C; Table S3). Analysis of the proteomic dataset identified inhibition of the β subunit of succinate-coenzyme A (CoA) ligase (SUCLG2) in metformin-treated HPMCs (Figure 4D); this is the enzyme subunit responsible for the guanosine diphosphate (GDP)-dependent conversion of succinyl-CoA to succinate (Johnson et al., 1998; Ostergaard, 2008). A reduction in both SUCLG2 protein and mRNA expression with metformin treatment was validated by western blot and qRT-PCR, respectively (Figure S4B). The majority of the other TCA enzymes were not markedly inhibited by metformin (Figure S4C).
Figure 4. Metformin Represses SUCLG2 and TCA Intermediates to Inhibit HIF1α in Mesothelial Cells.
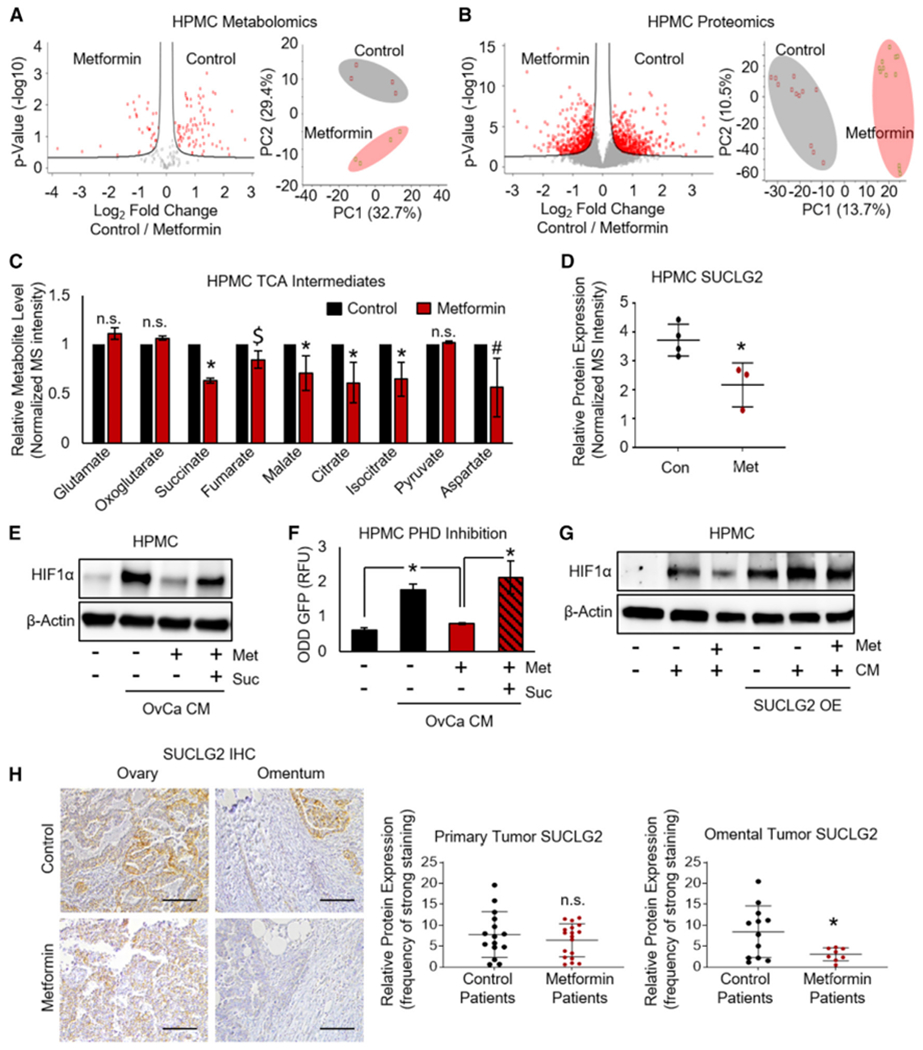
(A) Metabolomics. MS of metabolites extracted from mesothelial cells (HPMCs) treated with metformin (1 mM, 48 h)were analyzed using a two-sided t test with a FDR of 0.25 and an S0 value of 0.125. Left: volcano plot showing significant metabolites by red dots (n = 2 patients in duplicate). Right: principal-component analysis (PCA) assessing metabolite profiles of HPMCs treated with metformin compared to control. Open circles represent each group (n = 2 patients in duplicate).
(B) Proteomics. MS of proteins extracted from HPMCs treated with metformin were analyzed using a two-sided t test with an FDR of 0.05 and an S0 value of 0.1. Left: volcano plot showing significant proteins by red dots (n = 4 patients in triplicate). Right: PCA assessing protein profiles of HPMCs treated with metformin compared to control. Open circles represent each group (n = 4 patients in triplicate).
(C) Relative MS intensity of TCA metabolites in HPMCs treated with metformin from (A) was analyzed using a paired t test.
(D) Relative protein expression of the TCA enzyme SUCLG2 in HPMCs treated with metformin from (B) was analyzed using a paired t test.
(E) Western blot of HIF1α expression. HPMCs treated with metformin (250 μM, 72 h) followed by the addition of TYKnu OvCa CM ± succinate (suc, 5 mM) (representative blot from n = 2 patients in duplicate).
(F) Prolyl hydroxylase (PHD) activity using a GFP reporter construct. A GFP-tagged HIF1α oxygen-dependent degradation domain construct (ODD-GFP) was expressed in HPMCs; cells were then treated with TYKnu OvCa CM ± metformin (1 mM) ± succinate (suc, 5 mM) for 16 h. An increased GFP signal indicates depleted PHD activity (n = 2 patients in duplicate).
(G) Western blot of HIF1α expression. HPMCs were transfected with SUCLG2 and then treated with TYKnu OvCa CM ± metformin (1 mM, 48 h) (representative blot from n = 3 patients in duplicate).
(H) SUCLG2 immunohistochemical staining in primary ovarian and omental metastasis from OvCa patients with diabetes using metformin compared to non-diabetic patients with OvCa not taking metformin. Quantification of primary tumor (n = 15 and 17 for control and metformin, respectively) or omental metastasis (n = 12 and 8 for control and metformin, respectively) was performed using ImageScope software. Images are representative and were taken at 20× magnification; scale bar represents 100 μm.
Data represent mean values ± SDs. #p < 0.09, *p < 0.05; $p < 0.05 in one but not both patients tested; n.s., not significant.
Since succinate can inhibit PHD activity and thus stabilize HIF expression (Koivunen et al., 2007; Selak et al., 2005), we asked whether succinate could reverse the effect of metformin on HIF1α in HPMCs exposed to OvCa CM. Western blotting demonstrated that in the presence of succinate, metformin no longer inhibited OvCa CM-induced HIF1α protein expression in HPMCs (Figure 4E). To understand this finding further, we used a GFP reporter of the HIF1α oxygen-dependent degradation (ODD) domain. Hydroxylation of the reporter by active PHD results in lysosomal degradation of the reporter and thus suppressed the GFP signal; therefore, the ODD-GFP signal is inversely correlated with PHD activity (Yan et al., 2007). Mirroring the findings in the HIF1α western blot, the results using this reporter show that metformin reduces OvCa CM-induced PHD activity in HPMCs, but not in the presence of succinate (Figure 4F). Furthermore, the overexpression of SUCLG2 in HPMCs (Figure S4D) resulted in a striking induction of HIF1α, and in this setting, metformin no longer reduced HIF1α expression (Figure 4G). Finally, in the OvCa cohort, patients taking metformin for diabetes had lower SUCLG2 expression in omental metastases, but not in primary tumors, compared to OvCa patients without diabetes not taking metformin (Figure 4H). These findings suggest that metformin inhibits OvCa-driven stabilization of HIF1α in HPMCs, in part by preventing the metabolic inactivation of PHD by succinate downstream of the TCA enzyme, SUCLG2.
Metformin Inhibits TGF-β-Driven Metabolic Upregulation of HIF1α via PHD Inhibition
The previous results focused on the effect of metformin on mesothelial cells, but it was still unclear what, if any, signals originating from OvCa cells are targeted by the drug. To address this question in an unbiased manner, we performed a proteomics analysis of OvCa cells treated with metformin. The results established several signaling pathways affected by metformin, including TGF-β, its cognate receptor (TGF-bR2), and tumor necrosis factor α (TNF-α) (Figure 5A; Table S5). Based on previous studies showing that TGF-β1 activates HPMCs (Kenny et al., 2014; Shin et al., 2017), we tested whether TGF-β signaling was involved in the effect of metformin on HIF-dependent reprogramming of HPMCs by OvCa cells. In three different OvCa cell lines, metformin reduced the expression of TGF-β2R and the activation of the downstream target of TGF-β SMAD2/3 (Figures 5B and S5A), as well as TGF-β1 mRNA expression (Figures 5C and S5B). The induction of SMAD2/3 phosphorylation in HPMCs by OvCa co-culture was blocked by metformin or a TGF-βR inhibitor, while neither agent affected TGF-βR2 expression (Figure 5D). In addition, in all three OvCa cell lines, the inhibition of OvCa invasion by metformin pretreatment of HPMCs was reversed by recombinant human TGF-β1 (Figures 5E and S5C).
Figure 5. Metformin Targets OvCa TGF-β1 Signaling to Inhibit HIF1α Stabilization in HPMCs.
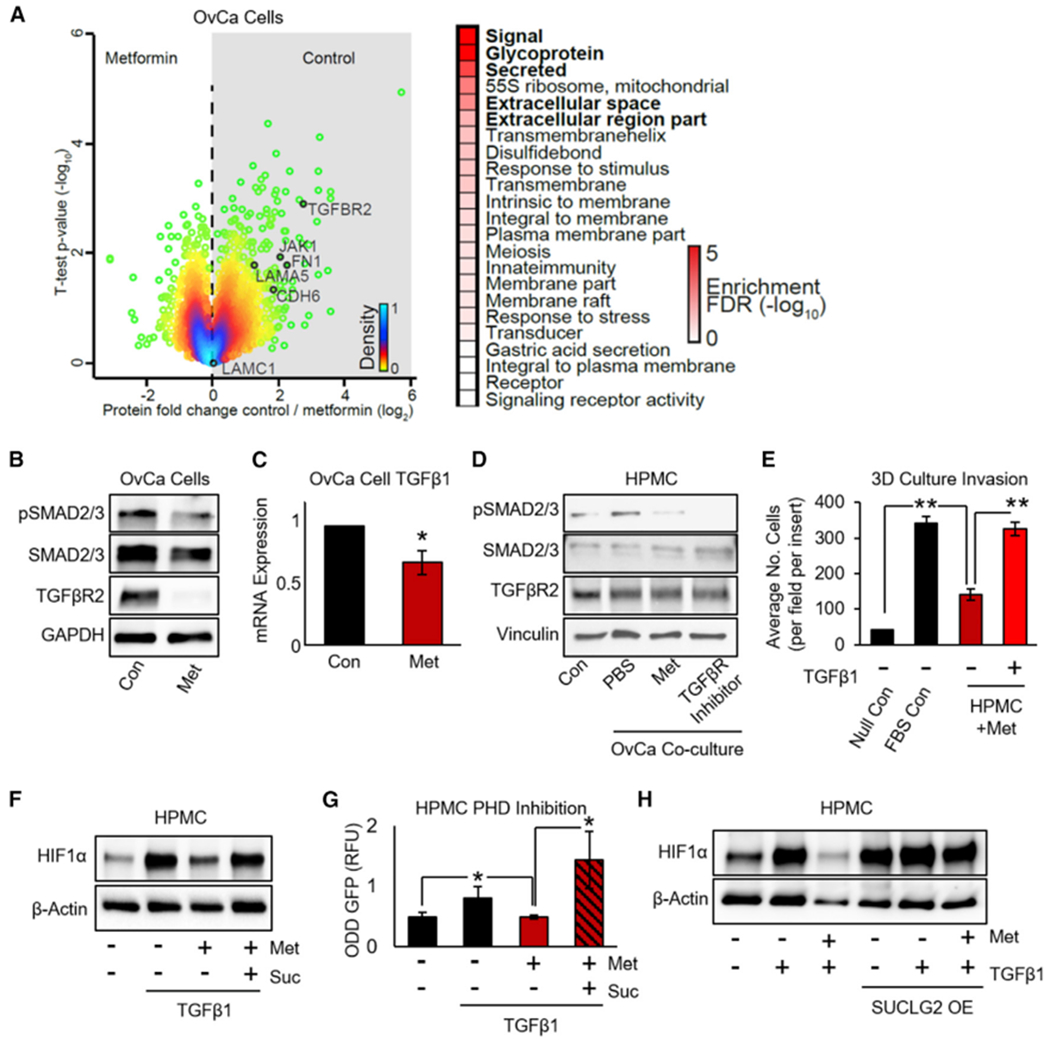
(A) Proteomics. MS of proteins extracted from DOV13 OvCa cells treated with metformin (1 mM, 72 h) were analyzed using a two-sided t test. Color gradient indicates the density of the distribution, with green dots as outer 5%. Proteins related to the TGF-β pathway are annotated for comparison. Top pathways regulated by metformin were determined by one-dimensional pathway enrichment analysis using FDR <0.02.
(B) Western blot of TGF-β-related proteins in DOV13 cells ± metformin (1 mM, 72 h) (representative blot from n = 2 experiments with 2 biological replicates each).
(C) qRT-PCR of TGF-β1 mRNA expression in DOV13 OvCa cells ± metformin treatment (1 mM, 72 h) (n = 2 samples per group in duplicate).
(D) Western blot of TGF-β-related proteins in HPMCs co-cultured with TYKnu OvCa cells ± metformin (1 mM, 48 h) (representative blot from n = 2 patients in duplicate).
(E) Invasion of TYKnu OvCa cells (36 h) through the 3D model ± metformin pretreatment of HPMCs (250 mM for 72 h) ± recombinant human TGF-β1 protein (n = 2 patients in triplicate).
(F) Western blot of HIF1α expression in HPMCs treated with metformin (250 μM, 72 h) followed by the addition of recombinant human TGF-β1 protein (TGF-β1; 10 ng/mL) ± succinate (suc, 5 mM) (representative blot from n = 3 patients in duplicate).
(G) PHD activity using the ODD-GFP reporter construct. HPMCs expressing the ODD-GFP construct were treated with TYKnu OvCa CM ± metformin (1 mM) ± succinate (suc, 5 mM) for 16 h. An increased GFP signal indicates depleted PHD activity (n = 2 patients in duplicate).
(H) Western blot of HIF1α expression. HPMCs were transfected with SUCLG2 and then treated with recombinant human TGF-β1 (10 ng/mL) ± metformin (1 mM, 48 h) (representative blot from n = 3 patients in duplicate).
Data represent mean values ± SDs. *p < 0.05 and **p < 0.01.
Building on the findings in Figure 4, we sought to understand how the inhibition of TGF-β signaling in OvCa cells by metformin was involved in the stabilization of HIF1α by succinate in HPMCs. TGF-β1 induced HIF1α protein expression in HPMCs and metformin reversed this effect, but not in the presence of succinate (Figure 5F). To understand this further, we once again used a GFP reporter of the HIF1α ODD domain. The results show that TGF-β1 decreased PHD activity in HPMCs, as indicated by the accumulation of its target ODD-GFP and that metformin reversed this effect. Again, in the presence of succinate, metformin was unable to overcome the effect of TGF-β1 on PHD activity (Figure 5G). Complementing this finding, when SUCLG2 was overexpressed in HPMCs, metformin no longer inhibited HIF1α expression induced by TGF-β1 (Figure 5H). These findings indicate that in addition to targeting HPMCs, metformin targets OvCa cells by inhibiting TGF-β1.
DISCUSSION
Emerging evidence indicating that the TME is critically important for tumor growth (Joyce and Pollard, 2009) presents the opportunity for therapeutic strategies directed against tumor-promoting cross-talk between the stroma and cancer cells. In the present study, we report that metformin makes the TME inhospitable to OvCa by interrupting bidirectional signaling between tumor and mesothelial cells. We had previously shown that metformin prevents OvCa tumor growth in a xenograft mouse model (Lengyel et al., 2015) and reduces the number of peritoneal and omental OvCa implants in a syngeneic mouse model (Litchfield et al., 2015). Although no mouse model of OvCa accurately recapitulates human disease and each model has unique limitations (Garson et al., 2012), the findings in our early studies suggested that metformin could alter both the omental stroma and the tumor cells. The findings reported here add to these studies by defining a mechanism whereby metformin targets the OvCa TME by inhibiting OvCa-induced metabolic reprogramming of mesothelial cells, resulting in decreased stromal HIF1α signaling, thereby reducing OvCa invasion (Figure 6).
Figure 6. Metformin Prevents Pro-tumorigenic Bidirectional Signaling between Tumor and Mesothelial Cells.
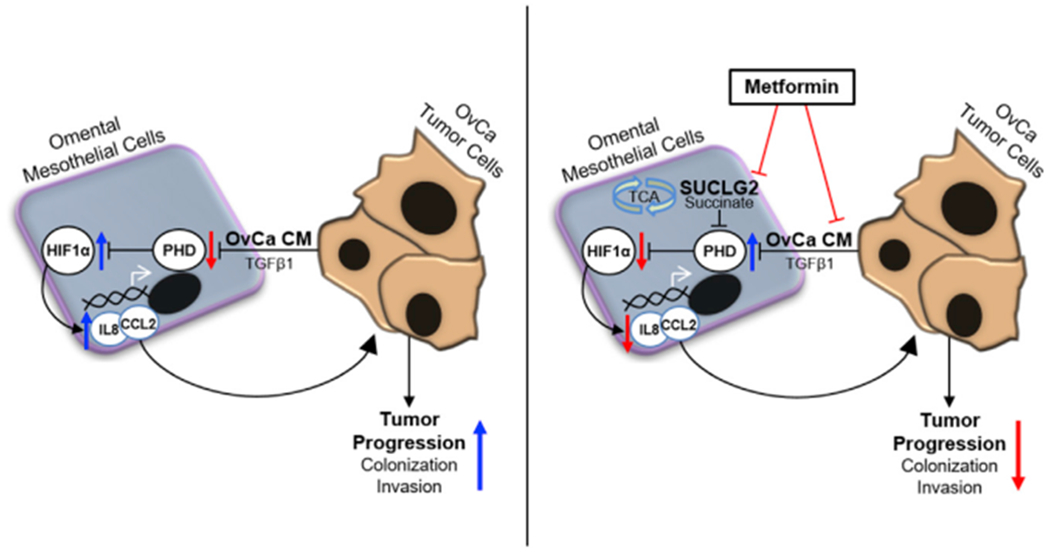
Modulation of tumor-stromal crosstalk by metformin inhibits OvCa tumor progression. Left: OvCa-derived CM or TGF-β1 inhibits PHD activation in mesothelial cells, resulting in HIF1α stabilization and the subsequent induction of IL-8 and CCL2, promoting tumor progression. Right: metformin inhibits TGF-β1 from OvCa cells to impede tumor-mesothelial cell crosstalk, thereby maintaining PHD activity and the concomitant degradation of HIF1α in mesothelial cells. This effect of metformin is mechanistically linked to a reduction in SUCLG2 and succinate and ultimately prevents the stroma-derived signaling required for OvCa colonization and invasion of the omentum.
With the goal of defining how metformin targets pro-tumorigenic reprogramming in OvCa TME, we used several integrated unbiased approaches focused on both tumor and mesothelial cells. The findings show that exposure of mesothelial cells to OvCa represses PHD enzymatic activity in mesothelial cells, resulting in increased HIF1α stabilization. Metformin prevented this OvCa-induced HIF1α signaling in mesothelial cells. The effect of metformin on PHD activity and HIF1α expression in mesothelial cells was directly related to a decreased production of TCA intermediates. This finding in OvCa-naive HPMCs complements our finding in 2016 that TCA intermediates are decreased in tumors from OvCa patients taking metformin for diabetes (Liu et al., 2016) . The integrated metabolomic and proteomic pathway analysis reported here indicates that the inhibition of the TCA cycle in mesothelial cells by metformin was primarily due to the suppression of SUCLG2, a TCA enzyme with an undefined role in cancer. SUCLG2 in mesothelial cells was also critically important in the inhibition of HIF1α by metformin. These findings demonstrate that metformin targets crosstalk in the ovarian TME by suppressing cancer-cell-induced HIF1α stabilization in mesothelial cells through the decreased production of TCA intermediates. Hulea et al. (2018) found that in breast cancer in vitro, the biguanide phenformin repressed succinate but induced lactate, fumarate, and malate, suggesting that metabolic adaptations in response to biguanides result in increased glycolysis and potentially reductive carboxylation. In the present study, our finding that metformin caused the global repression of succinate and downstream TCA intermediates while glutamine accumulated indicates an inhibition of oxidative metabolism. These contrasting data demonstrate why, moving forward, further metabolic assessment using biguanides at physiologic doses in normoglycemic conditions is critically important.
By focusing on the TME, our approach overcomes the limitations of prior studies that have focused on cancer cells in isolation using high glucose cell culture conditions. Here, using fresh human omental tissue ex vivo from patients taking metformin for diabetes, we begin to understand more clearly the global impact of metformin in the TME. The results show that in patients using metformin for diabetes, there is a profound reduction in the adhesion of OvCa cells to the omentum, the primary TME of OvCa. Furthermore, using our 3D organotypic model in normoglycemic culture conditions (5 mM glucose) and near-physiologic doses (250 μM) of metformin, we identified the molecular effects of the drug on the ovarian TME. We have previously shown that mesothelial cell-ECM interactions through fibronectin play an essential role in adhesion and invasion of OvCa cells to the omentum (Kenny et al., 2014); here, we identify that CCL2 and IL-8 are cytokines that serve as important targets of metformin in the TME. Consistent with a recent report by Gao et al., showing that metformin represses nuclear factor κB (NF-κB)-dependent IL-6 secretion in fibroblasts (Xu et al., 2018), metformin reduced the induction of CCL2 and IL-8 in the mouse omentum exposed to CM from ID8 OvCa cells with wild-type p53, but not from p53 knockout cells. While a comprehensive dissection of the role of p53 in the effect of metformin in the TME is beyond the scope of the present study, it is an important consideration in future work, as others have reported that the inhibition of colon cancer cell growth by metformin is p53 dependent (Buzzai et al., 2007). As metformin progresses to clinical use as a cancer therapeutic, we need to contend with the fact that not all patients will respond to this treatment and we will need to be able to identify a priori which patients should be offered metformin. Previous reports have suggested that CCL2 and IL-8 receptor expression is upregulated in several cancers (David et al., 2016; Yao et al., 2019). The findings reported here merit further evaluation of these cytokines as predictive biomarkers in ongoing metformin clinical trials.
Protection against stromal reprogramming may be an important mechanism of action for metformin in cancer and other conditions. In peritoneal fibrosis, metformin blocks the reprogramming of peritoneal mesothelial cells by suppressing TGF-β signaling (Shin et al., 2017). In the present study, we show that the drug also targets TGF-β-dependent tumor-mesothelial cell interaction in OvCa. The findings indicate that in the context of metformin, TCA metabolism plays an important role in the effect of TGF-β1 on PHD activity and HIF1α stability, adding to previous studies that suggest that TGF-β stabilizes HIF1α through altered PHD expression (Marushima et al., 2011; McMahon et al., 2006). The inhibition of HIF1α and CAIX by metformin in the TME was also observed in patient samples. This suggests that reduced HIF signaling in the omentum may be a mechanism by which metformin could prevent cancer progression, as previous studies have shown that increased hypoxia in high-grade serous OvCa (HGSOC) tumors was associated with worse survival (Hynninen et al., 2006). The results presented here add to the continuously growing understanding of the potential roles of HIF signaling in the TME (Casazza et al., 2014), as our data identify the importance of HIF signaling in mesothelial cells as a critical process for OvCa progression that can be effectively targeted using metformin.
In summary, while the story of metformin and cancer began with the seed (the cancer cells), the findings reported here show that the true path forward may actually lie in the soil (the adjacent stromal cells). Further dissection of the effects of metformin on tumor-stromal interactions in vivo will require the use of improved preclinical models that recapitulate the physiologic events representing the progression of the disease. Ultimately, the most rigorous test of the efficacy of metformin as a cancer treatment and the effect of the drug on the TME lies ahead in the results of prospective randomized clinical trials that are testing metformin as a cancer treatment in patients without diabetes and of the coupled tumor analysis, such as our ongoing prospective randomized neoadjuvant clinical trial in OvCa (ClinicalTrials.gov: NCT02122185).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Iris L. Romero (iromero@bsd.uchicago.edu). All unique reagents generated during this study (CXCR1 V247N and CCR2 T94K plasmids) will be made available upon reasonable request from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient Samples and Cultivation of Primary Fibroblasts and Mesothelial Cells
Whole omentum, normal omental fibroblasts (NOFs) and peritoneal mesothelial cells (HPMCs) were obtained from female patients undergoing surgery for benign indications (e.g., abdominal cerclage, hysterectomy). Patient samples used for primary cell isolation, as well as those designated as controls (e.g., for ex vivo experiments and in tissue microarray), were not diagnosed with diabetes or on any drug used for diabetes (e.g., biguanides, sulfonylureas) at the time of surgery. Patient samples used for primary cell isolation or ex vivo experiments also did not have a history of gynecologic cancer and did not present with abdominal inflammation at the time of surgery. For patients with diabetes taking metformin, metformin (500-1000mg twice/day) was administered within 24h of omental sample collection. Primary omental fibroblasts and mesothelial cells were extracted from omentum as previously described (Kenny et al., 2014) and maintained in standard RPMI supplemented with 20% FB-Essence (VWR) with 1% penicillin/streptomycin (Corning), 1% MEM non-essential amino acids (Corning) and 1% MEM vitamins (Corning). Primary cells were passaged no more than once prior to performing experiments. Written informed consent was obtained as part of an approved IRB protocol at the University of Chicago, Department of Obstetrics and Gynecology.
Human Subjects
Tumor samples from female patients with ovarian cancer were provided by the Human Tissue Resource Center at the University of Chicago. Tumors were collected from patients undergoing debulking surgery performed by a gynecologic oncologist at the University of Chicago, Section of Gynecologic Oncology. Informed consent was obtained prior to surgery in accordance with IRB approval by the University of Chicago.
Cell Lines
TYKnu, HeyA8 and DOV13 human OvCa cell lines and ID8 murine OvCa cell lines were maintained in standard DMEM supplemented with 10% FB-Essence (VWR) with 1% penicillin/streptomycin (GE Healthcare), 1% MEM non-essential amino acids (Corning) and 1% MEM vitamins (Corning). All OvCa cell lines were tested for mycoplasma and authenticated by short tandem repeat marker profiling every 3-6 months (CellCheck, IDEXX Bioresearch, Columbia, MO).
Mouse Models
Female C57BL/6 mice (Charles River Laboratories, Wilmington, MA) were 8 weeks of age and weighed approximately 25 g at the initiation of the study, and were housed in groups of 5 throughout the duration of the study. Mice were intraperitoneal (i.p.) injected with OvCa conditioned media and sacrificed for omental harvesting as described below.
METHOD DETAILS
Reagents, Plasmids, and Cell Culture
All experiments were performed in low glucose (5mM glucose) DMEM (Corning, Corning, NY), with 10% FB-Essence (VWR Life Science, Radnor, PA), 1% penicillin/streptomycin (GE Healthcare, Helsinki, Finland), 1% MEM non-essential amino acids and 1% MEM vitamins (Corning). GFP was stably expressed in OvCa cells using EF1a-GFP-IRES-puro (Zou et al., 2009), a gift from Linzhao Cheng obtained through Addgene (#26777), with selection using puromycin for several passages prior to further use. The constitutively stable HIF1α (HA-HIF1alpha P402A/P564A-pcDNA3) plasmid, which cannot be hydroxylated by PHD to target VHL-linked ubiquitination and degradation of HIF1α, was a gift from William Kaelin (Yan et al., 2007) obtained through Addgene (#338299). The pHIV-oxygen-dependent degradation (ODD) -EGFP-IRES-dTomato used to measure PHD activity was a gift from Violaine See (Bagnall et al., 2014) obtained through Addgene (#395015). SUCLG2 plasmid DNA was obtained through DNASU Plasmid Repository (Arizona State University, Tempe, AZ). CXCR1 and CCR2 plasmid DNA was obtained from Sino Biological (Beijing, China), and amino acid substitutions were performed using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs (NEB), Beverly, MA) to generate constitutively active mutants CXCR1-V247N (Han et al., 2012) and CCR2-T94K (Alvarez Arias et al., 2003), as detailed below. Transfections were performed using 1μg plasmid with Lipofectamine 2000 purchased from ThermoFisher Scientific (Waltham, MA). Metformin, AICAR, dorsomorphin (compound C), N-oxalylglycine, 2-oxoglutarate and dimethyl succinate were purchased from Sigma Aldrich (St. Louis, MO) and prepared fresh prior to each experiment. Recombinant human IL-8 and CCL2 were obtained from Peprotech (Rocky Hill, NJ), and recombinant human TGF-β1 was obtained from ThermoFisher (Waltham, MA). Neutralizing antibody for CXCR1/IL8RA was obtained from R&D Systems (Minneapolis, MN). TGFBR1 inhibitor LY364947 was obtained from Tocris Bioscience (Bristol, United Kingdom).
Conditioned Media and Co-culture Assays
To generate OvCa conditioned media (CM) for treatment of HPMCs, TYKnu cells were plated on 10cm dishes at 1,000,000 cells per plate and allowed to grow for 24h in standard growth media. Media was then replaced with low glucose DMEM (supplemented with 10% FB-Essence, 1% pen/strep, 1% MEM non-essential amino acids and 1% MEM vitamins; Corning) and cells were allowed to grow for 72h prior to collection. Media was centrifuged and moved to a new tube to avoid cell debris, and stored at −20°C until use. For analysis of mesothelial cells co-cultured with OvCa cells, HPMCs were plated at 300,000 cells per well on a 6-well plate and TYKnu cells were plated at 100,000 cells per transwell polyester membrane insert (0.4μm pore size, for use with 6-well plate; Corning) on a separate plate. Cells were cultured separately for 24h priorto moving inserts to co-culture TYKnu OvCa cells with HPMCs for 48h. Treatments for CM and co-culture experiments were performed as described in the figure legends. Conditioned media or transwell insert containing OvCa cells was then discarded and HPMCs were lysed and prepared for western blot or proteomics, as described below.
Invasion Assay
OvCa cell invasion through 3D organotypic culture was performed as described previously (Kenny et al., 2009). Briefly, 15μg of collagen type 1 (Corning) in 200μL was added to 8μm pore transwell inserts (for use with 24-well plates) and allowed to set overnight. Inserts were decanted and washed once briefly with 1X PBS, and then 4,000 NOFs were plated and allowed to adhere for 2h prior to seeding 20,000 HPMCs. Stromal cells were then allowed to grow for an additional 24h. Following stromal cell culture, media was removed and GFP-tagged HeyA8 cells (60,000), GFP-tagged TYKnu (100,000) or GFP-tagged DOV13 (100,000) cells were seeded in serum-free low-glucose DMEM (using 300μL per insert). OvCa cells were allowed to invade through the 3D culture for 15h (HeyA8) or for 36h (TYKnu and DOV13). Low-glucose DMEM containing 10% FB-Essence was used as a chemoattractant in the lower compartment (700μL per well), and serum free low-glucose DMEM was used for negative controls. Transwell inserts were then inverted and scraped using Q-tip to remove stromal cells and OvCa cells that had not migrated, fixed in 4% formaldehyde for 5min and then washed with 1X PBS for 5min. GFP of cancer cells was imaged using a Nikon Eclipse Ti2 (Nikon Inc., Melville, NY) with 3 images per insert. OvCa cell GFP was then manually counted using FIJI/ImageJ software (NIH, Bethesda, MD), the mean number of OvCa cells per insert was quantified, and then the average number of cells invaded per group was statistically analyzed. For pretreatment with metformin, each cell type was treated individually for 72h with 250μM metformin prior to formation of the 3D model; metformin was not present during the invasion assay. All other treatments were added at the time of OvCa seeding at the doses indicated.
Cytokine Measurement of Murine Omentum
To generate conditioned media (CM), wild-type or ID8 p53−/− murine tumor cells (Walton et al., 2016) were seeded at 1,000,000 per 15cm dish and allowed to grow for 24h prior to replacement of media with 15mL serum-free DMEM. CM was then collected at 48h, concentrated using centrifugal filter units with 3K membrane (Millipore, Burlington MA) by centrifugation at 4000rpm for 1h at 4°C, and then the top portion was collected and used fresh per experiment at each time point indicated. Female C57BL/6 mice (Charles River Laboratories) were treated with 300mg/kg metformin in drinking water (p.o., ad libitum) for 3 weeks, with the treatment water replaced every other day, prior to intraperitoneal (i.p.) injection of 500μL ID8 CM. To capture both early/intermediate and delayed/late cytokine production of the murine omentum, mice were injected i.p. with ID8 CM at both 4h and 72h (Bisiaux et al., 2017) prior to sacrifice. Injection of CM, rather than tumor cells, was used specifically in order to isolate cytokines originating from the murine omentum. Omental tissue was harvested, immediately frozen at −80°C and stored until preparation for cytokine array or ELISA. Expression of murine omental cytokines were assessed using the Proteome Profiler Mouse Cytokine Panel A (R&D Systems), using two mice per group on separate membranes with each cytokine in duplicate per membrane. Samples were prepared and assayed according to manufacturer’s instructions. Protein expression was analyzed using FIJI/ImageJ (NIH) by quantifying the area under the curve (AUC). Potential candidates of metformin’s action were identified as cytokines that had both consistent induction from ID8 CM as well as repression back toward baseline expression level with metformin treatment. For validation of omental CXCL1 and CCL2 expression, using eight mice per group, mouse CXCL1/KC and CCL2/MCP-1 Quantikine ELISA kits were used according to manufacturer’s instructions (R&D Systems) with each sample run in duplicate. Round-bottom Eppendorf tubes each containing a 5mm stainless steel bead (QIAGEN, Germantown MD) were pre-chilled on dry ice for 15min prior to addition of mouse omentum; samples were chilled on dry ice for an additional 30min. Samples were then moved to TissueLyser LT (QIAGEN) and allowed to thaw for 2min prior to addition of 250μL lysis buffer (100mM Tris/HCl [pH 7.4], 150mM NaCl, 1mM EDTA, 0.5% sodium deoxycholate, 1% Triton X-100; diluted 1:2 in 1X PBS and supplemented with phosphatase/protease inhibitors immediately prior to use) and homogenization at 50 oscillations/second for 5min (QIAGEN). All animal procedures were approved by the University of Chicago IACUC and were in accordance with the University of Chicago’s policies on the care, welfare, and treatment of laboratory animals.
Omental Explant Ex Vivo Adhesion, Colonization, Invasion, and Protein Expression
Omental tissue for Western immunoblotting was immediately frozen at −80°C prior to homogenization, which was carried out as described above for murine omenta. Whole omental explants for ex vivo experiments were incubated in low glucose DMEM immediately following removal from the patient. Omental ex vivo adhesion/colonization experiments were completed by seeding HeyA8-GFP/ Luciferase or TYKnu-GFP (500,000) OvCa cells directly onto omental explant in a 96-well low adherence plate (ThermoFisher, Waltham, MA). For examining omenta directly from patients who were on metformin, omental tissue from both nondiabetic controls and diabetic patients on metformin undergoing surgery for benign indications were collected within 2h of each other. OvCa cells were seeded and allowed to adhere for 1.5h; omenta were then washed with 1X PBS for 5min to remove non-adherent cells and GFP was imaged. For treatment of omenta from benign patients using exogenous metformin, explants were incubated in low glucose DMEM with 250μM metformin for 72h prior to seeding OvCa cells. The cancer cells were allowed to adhere or colonize the omenta for 1.5h or 72h, respectively, prior to washing with 1X PBS and imaging of GFP. For migration of OvCa toward explant, omental tissue was excised (50mg/well) and treated for 72h in a 24-well plate; media was replaced with serum free DMEM (containing 0.1 % BSA) and then GFP-tagged HeyA8 cells (60,000) cells were seeded on top of 8μm pore transwell inserts and allowed to invade toward omental explant for 15h. In all experiments, OvCa cell GFP was imaged on Nikon Eclipse Ti2 (Nikon) and analysis was performed using at least 3 images per explant/insert, and then averaged across replicate inserts to determine mean value per group.
Site-Directed Mutagenesis of CXCR1 and CCR2
Constitutively active mutants CXCR1-V247N and CCR2-T94K show basal receptor activity even in the absence of ligand and demonstrate enhanced activity in response to ligand binding when compared to cells expressing wild-type receptor (Han et al., 2012 and Alvarez Arias et al., 2003, respectively). CXCR1 and CCR2 ORF plasmid DNA (Sino Biological) were obtained and amino acid substitutions were performed using the Q5 Site-Directed Mutagenesis Kit (NEB). Mutagenesis primers were designed using NEBaseChanger (NEB) and synthesized by Integrated DNATechnologies (IDT, Coralville, IA). Sequences are provided in the supplementary Key Resources Table. The Q5 Hot Start High-Fidelity driven PCR was performed according to manufacturer’s instructions, using an annealing temperature of 65 or 60°C for CXCR1 or CCR2, respectively, for 30 s each cycle and then PCR product was treated with KLD enzyme as directed. Following bacterial transformation, 10 colonies were selected for mini-prep and sequenced at the University of Chicago DNA Sequencing Facility using 3730XL 96-capillary DNA sequencer (Applied Biosystems). Sequences were selected based on alignment using NCBI BLAST (NIH) and plasmids with desired substitutions were amplified by PureLink HiPure Plasmid Maxiprep Kit (ThermoFisher Scientific). CXCR1-V247N and CCR2-T94K were then transfected into GFP-tagged TYKnu cells using Lipofectamine 2000 (ThermoFisher Scientific) and at 36h post-transfection stably selected using 50mg hygromycin for 14d prior to use in functional assays.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CAIX | Abcam | 2D3, RRID:AB_2066533 |
| SMAD2/3 | BD Biosciences | RUO; RRID:AB_398161 |
| SMAD2/3 phospho-Ser465/467 | CST | D6G10; RRID:AB_2631089 |
| HIF1α | CST | D2U3T; RRID:AB_2622225 |
| HIF2α | Novus Biologicals | NB100; RRID:AB_10002593 |
| TGFBR2 | Abcam | EPR14673; RRID:AB_2818975 |
| Akt | CST | 9272; RRID:AB_329827 |
| Akt phosphor-Ser473 | CST | D9E; RRID:AB_2315049 |
| SUCLG2 | ThermoFisher Scientific | PA5-61289; RRID:AB_2648068 |
| Vinculin | Sigma | VIN115; RRID:AB_477617 |
| GAPDH | CST | 14C10; RRID:AB_561053 |
| β-Actin | Sigma | AC15; RRID:AB_476744 |
| Anti-Mouse IgG HRP-linked secondary | CST | 7076; RRID:AB_330924 |
| Anti-Rabbit IgG HRP-linked secondary | CST | 7074; RRID:AB_2099233 |
| Biological Samples | ||
| Primary omental tissue (for ex vivo studies, and isolation of NOF/HPMC) | University of Chicago Department of Obstetrics and Gynecology | https://www.uchicagomedicine.org/conditions-services/obgyn |
| Ovarian cancer tissue micro-array | University of Chicago Ovarian Cancer Research Laboratory | https://obgyn.uchicago.edu/research/lengyel-ovarian-cancer-laboratory |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Low glucose (5mM) DMEM | Life Technologies | 11885-084 |
| DMEM | Corning | 10-013-CV |
| RPMI1640 | Corning | 10-040-CV |
| FB-Essence | VWR | 10803-034 |
| Penicillin/Streptomycin (100X) | Corning | 30-002-CI |
| MEM non-essential amino acids | Corning | 25-025-CI |
| MEM vitamins | Corning | 25-020-CI |
| Metformin | Sigma | D150959 |
| AICAR | Sigma | A9978 |
| Dorsomorphin (Compound C) | Sigma | P5499 |
| N-oxalylglycine | Sigma | O9390 |
| 2-oxoglutarate | Sigma | 349631 |
| Dimethyl succinate | Sigma | W239607 |
| Q5 Site-Directed Mutagenesis Kit | NEB | E0554S |
| Lipofectamine 2000 | ThermoFisher Scientific | 11-668-500 |
| Recombinant human IL-8 | Peprotech | 200-08M |
| Recombinant human CCL2 | Peprotech | 300-04 |
| Recombinant human TGFβ1 | Life Technologies | PHG9204 |
| CXCR1/IL8RA neutralizing antibody | R&D Systems | MAB330-SP |
| LY364947 | Tocris Bioscience | 2718 |
| Critical Commercial Assays | ||
| Quantikine ELISA – murine CXCL1 | R&D Systems | MKC00B |
| Quantikine ELISA – murine CCL2 | R&D Systems | MJE00B |
| Proteome Profiler Mouse Cytokine Array Kit – Panel A | R&D Systems | ARY006 |
| Seahorse XF96 Cell Mito Stress Test | Seahorse Bioscience | 103015-100 |
| Experimental Models: Cell Lines | ||
| TYKnu | University of California Los Angeles | Koneczny Laboratory |
| HeyA8 | MD Anderson Cancer Center | Mills Laboratory |
| DOV13 | University of Illinois at Chicago | Burdette Laboratory |
| Experimental Models: Organisms/Strains | ||
| C57BL/6NCrl Inbred Mice | Charles River | 027 |
| Oligonucleotides | ||
| Please see Table S6 for oligonucleotide sequences. | ||
| Recombinant DNA | ||
| HA-HIF1α P402A/P564A-pcDNA3 | Yan et al., 2007 | Addgene: 338299 |
| pH IV-ODD-EGFP-IRES-dTomato | Bagnall et al., 2014 | Addgene: 395015 |
| SUCLG2 | DNASU | HsCD00445557 |
| CXCR1 | Sino Biological | HG10856-NF |
| CCR2 | Sino Biological | HG16084-NF |
| EF1a-GFP-IRES-Puro (pEGIP) | Zou et al., 2009 | Addgene: 26777 |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | RRID: SCR_002798 |
| FIJI [ImageJ] | National Institutes of Health | RRID:SCR_002285 |
| Aperio ScanScope / Pixel Count v9 | Leica Biosystems | RRID: SCR_014311 |
| Thermo Xcalibur | ThermoFisher Scientific | RRID: SCR_014593 |
| MaxQuant | Max Planck Institute | RRID: SCR_014485 |
| Perseus | Max Planck Institute | RRID: SCR_015753 |
| Molecular Signatures Database | Broad Institute, MIT | RRID: SCR_016863 |
| MetaboAnalyst | McGill University | RRID: SCR_015539 |
| Other | ||
| Invasion transwell insert (8μm pore) | ThermoFisher Scientific | 353097 |
| Co-culture transwell insert (0.4μm pore) | Thomas Scientific | 3450 |
| Collagen type 1 [rat tail] | Corning | 354236 |
| PureLink HiPure Plasmid Maxiprep Kit | ThermoFisher Scientific | K2100-06 |
| IDEXX CellCheck | IDEXX Bioresearch | https://www.idexxbioanalytics.com/cellcheck |
Western Immunoblotting
Cell lysates were prepared in 125μL RIPA buffer (100mM Tris/HCl [pH 7.4], 150mM NaCl, 1mM EDTA, 0.1% (w/v) sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1% Triton X-100 and supplemented with phosphatase/protease inhibitors immediately prior to use). Immunoblotting was performed as previously described (Lengyel et al., 2015). CAIX [2D3] antibody was purchased from Abcam (Cambridge, MA), HIF2α [NB100] from Novus Biologicals (Littleton, CO), and SMAD2/3 [RUO] from BD Biosciences (San Jose, CA). Vinculin [VIN115] and β-Actin [AC15] were from Sigma-Aldrich. HIF1α [D2U3T], TGFBR2 [EPR14673], SMAD2/3 pi-Ser465/467 [D6G10], Akt [9272], Akt pi-Ser473 [D9E], GAPDH [14C10] and HRP-linked secondary antibodies were from Cell Signaling Technology (Beverly, MA). Gel electrophoresis was performed using 10-well 4%–20% Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad, Hercules, CA) at 120V for 1h. Following transfer of proteins to Amersham Protran Premium Nitrocellulose Paper (GE Healthcare), primary antibodies diluted at 1:1000 in 1XTBS-T were used overnight at 4°C under agitation. Secondary antibodies diluted at 1:2000 in 1XTBS-T and supplemented with 5% non-fat milk (LabScientific, Highlands, NJ) were used at room temperature for 2h prior to developing using Clarity Western ECL Substrate (Bio-Rad). Membranes were then imaged using G:BOX Chemi XT4 for up to 10min. FIJI/ImageJ (NIH) was used to perform densitometry to quantify the area under the curve (AUC).
Quantitative Reverse Transcription-Polymerase Chain Reaction
RNA was isolated from cells with the use of 1mL TRIzol (Invitrogen, Carlsbad, CA) and transcribed into cDNA using high-capacity complementary DNA kit (Applied Biosystems, Waltham, MS). Quantitative real-time reverse transcriptase–polymerase chain reaction (qRT-PCR) was performed as described (Sawada et al., 2008) using the Applied Biosystems 7500 Real Time PCR system. IL-8, CCL2, TGFβ1 and GAPDH Taqman probes were obtained from Applied Biosystems; CXCL1, CXCR1, CCR2, SUCLG2 and GAPDH probes for SYBR Green were obtained from IDT (sequences are provided in Key Resources Table). For each individual experiment, at least 2 biological replicates were run in triplicate for each primer set. Relative levels of messenger RNA (mRNA) expression were calculated by the 2-ΔΔCT method with mRNA expression of experimental groups expressed relative to control, using GAPDH as the housekeeping gene for normalization. Expression per data point or bar represents the mean of biological replicates, with the error bars indicating standard deviation between biological replicates.
Confocal Microscopy Analysis of Oxygen-Dependent Degradation-GFP to Detect PHD Inactivation
Prolyl hydroxylase (PHD) activity was measured using a GFP-tagged reporter construct for the HIF1α oxygen-dependent degradation (ODD) domain (Bagnall etal., 2014). HPMCs were plated at 300,000 cells per well in a 6-well dish and allowed to grow for 24h prior to transduction with the pHIV-ODD-EGFP-IRES-dTomato lentiviral vector for 16h. At 48h post-infection, media was replaced with low-glucose DMEM and treatments (metformin, succinate) were added. At 16h post-treatment, cells were washed with 1x PBS, fixed with 4% paraformaldehyde for 5min, neutralized with 0.3M glycine, and then washed again with 1xPBS. ODD-GFP was imaged using LSM 510META (Zeiss, Oberkochen, Germany) and analyzed with FIJI/ImageJ (NIH), with uninfected HPMCs and GFP-tagged cells serving as negative and positive controls, respectively.
Immunohistochemistry
Tissue microarrays were assembled from paraffin-embedded tumor blocks from patients with OvCa as previously described (Sawada et al., 2007) under a protocol approved by the University of Chicago IRB. Following antigen retrieval, slides were blocked with fetal bovine serum albumin (BSA, Sigma) priorto incubation with primary antibody. Carbonic Anhydrase IX (CAIX [2D3], Abcam) was used at 1:200 in TBS-T overnight and succinate-CoA ligase beta (SUCLG2 [PA5-61289], ThermoFisher) was used at 1:2000 in TBS-T overnight. Both were then incubated with HRP-linked secondary antibody at 1:5000 in TBS-T containing 1% BSA for 1h at room temperature. Slides were scanned at 20x magnification using Aperio ScanScope XT (Leica Biosystems, Buffalo Grove, IL), and intensity and frequency of antibody staining was determined using ImageScope (Leica). Aperio Pixel Count Algorithm (version 9) was used to quantify the frequency of high and low intensity staining of pixels across each sample per slide. The relative abundance of each protein was defined by comparing the frequency of high intensity staining, to avoid background from potentially confounding the intensity data.
Liquid Chromatography-Mass Spectrometry Proteomic Analysis
For proteomic analysis of mesothelial cells co-cultured with OvCa cells, HPMCs were plated at 150,000 cells per well on a 6-well plate and OvCa cells were plated at 100,000 cells per transwell polyester membrane insert (0.4μm pore size, for use with 6-well plate; Corning) on a separate plate. Cells were cultured separately for 24h prior to moving inserts to co-culture TYKnu OvCa cells with HPMCs for 48h. For proteomic analysis of mesothelial cells alone, HPMCs from 5 separate patients not taking metformin were plated at 150,000 cells per well in triplicate in a 6-well dish and allowed to grow for 48h prior to metformin exposure (1mM, 48h). For proteomic analysis of OvCa cells alone, DOV13 cells were plated at 700,000 cells per well in triplicate per condition in 10cm dishes and allowed to grow for 24h prior to treatment with metformin (1mM, 72h). Following treatments, cells were collected with a MS-suitable lysis buffer (100mM Tris HCl, 40mM 2-chloroacetamide, 10mM Tris (2-carboxyethyl) Phosphine [TCEP], 1% [w/v] sodium deoxycholate; buffered to pH 8.5 and supplemented with phosphatase/protease inhibitors immediately prior to use) using 100μL per 100,000 cells. Q-Exactive HF mass spectrometer coupled to EASY-nLC 1000 HPLC system (ThermoFisher) was used to perform label-free shotgun proteomics as previously described in detail (Coscia et al., 2016). Briefly, peptides were separated in buffer B (80% acetonitrile, 0.5% formic acid) at 200nl/min-1 over 100min, with an isolation window of 2.2Th and resolution of 17,500 at m/z 200. For the experiments analyzing HPMCs co-cultured with TYKnu cells ± metformin, 8,945 proteins were identified. For the experiment analyzing DOV13 cells ± metformin, 10,608 proteins were identified. Data were acquired using Xcalibur software (ThermoFisher), raw data were analyzed using MaxQuant (Cox and Mann, 2008) and statistical analyses were carried out using Perseus (MaxQuant Software Suite (Tyanova et al., 2016); Max Planck Institute of Biochemistry, Martinsried, Germany). Using Perseus, LFQ Intensity data were log transformed [log2(x)], categorically annotated per group, filtered by valid values (at least 2 of 3 samples per group required a valid value per protein), missing values were imputed from normal distribution per column, and then volcano plots were generated using two-sided t test to identify significantly altered proteins. Principal component analysis (PCA) was performed using category enrichment in components (5 components) and the Benjamini-Hochberg cutoff method with FDR of 0.05. For each PCA plot, each box represents an individual patient sample/replicate. For unsupervised hierarchical clustering, data were first normalized to z-score and then analyzed using k-means preprocessing and Euclidean distances. Pathway enrichment for OvCa-exposed HPMCs was assessed by using the Molecular Signatures Database (MSigDB, GSEA, UCSD Broad Institute, San Diego, CA). For proteomic analysis of DOV13 cells, the 2D annotation enrichment algorithm was used (Cox and Mann, 2012).
Gas Chromatography-MS Metabolic Profiling
For steady-state analysis of metabolites, HPMCs from 2 separate patients were plated at 1,000,000 cells per 15cm dish in duplicate and allowed to grow for 72h prior to metformin exposure (1mM, 48h). Cells were lifted using Trypsin EDTA (1X, 0.25%; ThermoFisher Scientific) and then pelleted by centrifugation at 0.5rcf. for 3min. Cell pellets were flash frozen in liquid nitrogen and then prepared for downstream mass spectrometric analysis, as described previously (Lai et al., 2018). The Agilent 6530 Accurate-Mass Q-TOF LC/MS quadrupole analyzer and Agilent 6890 GC/MS (Agilent, Santa Clara, CA) were used to perform untargeted metabolomics. 617 metabolites were identified in total, and of those 189 known metabolites were used in subsequent analyses. Comparisons of relative abundance were performed using Perseus (Max Planck) as described above, and significantly altered metabolites were analyzed in conjunction with proteomics data using MetaboAnalyst Joint Pathway Analysis (metaboanalyst.ca)(Chong et al., 2018) to indicate pathways altered by metformin.
Extracellular Flux Analysis
TYKnu OvCa cells, NOFs and HPMCs were pretreated with metformin (250μM) for 72h prior to seeding 10,000 cells per well, with 4 replicates per group, on Seahorse XF96 custom microplates. Cells were allowed to recover for 16h overnight prior to measurement of oxygen consumption rate (OCR), an indicator of mitochondrial electron transport chain complex I activity, and analyzed by Seahorse XFe96 Extracellular Flux Analyzer using the Cell Mito Stress Test Kit (Agilent), as described previously (Hart et al., 2015). OCR was then normalized to total protein concentration as determined by bicinchoninic acid assay (ThermoFisher).
QUANTIFICATION AND STATISTICAL ANALYSIS
Specific analyses performed for each assessment are described in the figure legends. In all analyses, data were evaluated using a one-way ANOVA with post hocTukey, or two-tailed t test, as appropriate; p < 0.05 was considered statistically significant. Number of biological replicates or animals used (n) is included in the Figure Legends per experiment. Sample sizes were determined based on previous experience with each experiment. All statistical analyses were carried out using GraphPad Prism 7 (GraphPad Software, La Jolla, CA) or Perseus.
DATA AND CODE AVAILABILITY
All data supporting the findings of this study are available within the paper, in the Figures S1, S5 and in the Tables S1, S2, S3, S4, S5, and S6. Source data (e.g., representative Western immunoblots) are available from the Lead Contact upon reasonable request. All supplementary figures and data are available online. No unique software or code was used in this study.
Supplementary Material
Highlights.
Metformin targets tumor-mesothelial cell signaling to inhibit ovarian cancer invasion
Metformin abrogates tumor-cell-induced stabilization of HIF1α in mesothelial cells
Suppression of succinate and SUCLG2 are required for metformin’s reduction of HIF1α
ACKNOWLEDGMENTS
This work was supported by the Mayo Clinic Ovarian Cancer SPORE grants (P50CA136393-06A1 and UOC-208331). I.L.R. was supported by grants from the NIH, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (2K12HD000849-26), the American Board of Obstetrics and Gynecology, and the SU2C-Ovarian Cancer Research Fund Alliance-National Ovarian Cancer Coalition. E.L. is supported by grants from the National Cancer Institute (5R01CA111882-07 and 1R01CA169604-01A1). P.C.H. was supported by the Mayo Clinic Ovarian Cancer SPORE grant, Colleen’s Dream Foundation, the NIH Loan Repayment Program, and the University of Chicago Institute for Translational Medicine Core Subsidy Program (UL1 TR000430). Support was provided by a NIH-University of Chicago Cancer Center support grant (CA014599). N.G., F.C., and M.M. were supported by the Max Planck Society for the Advancement of Science and the Körber Foundation (Korber European Science Prize). N.G. was also supported by a travel grant by the Boehringer Ingelheim Fonds. The authors would like to thank Dr. I. McNeish for the contribution of the ID8 (wild-type [WT] and p53−/−) murine tumor cell model.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.079.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alvarez Arias D, Navenot JM, Zhang WB, Broach J, and Peiper SC (2003). Constitutive activation of CCR5 and CCR2 induced by conformational changes in the conserved TXP motif in transmembrane helix 2. J. Biol. Chem 278, 36513–36521. [DOI] [PubMed] [Google Scholar]
- Ammirante M, Shalapour S, Kang Y, Jamieson CA, and Karin M (2014). Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl. Acad. Sci. USA 111, 14776–14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta O, Barrón F, Padilla MS, Avilés-Salas A, Ramírez-Tirado LA, Arguelles Jiménez MJ, Vergara E, Zatarain-Barrón ZL, Hernández-Pedro N, Cardona AF, et al. (2019). Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 5, e192553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnall J, Leedale J, Taylor SE, Spiller DG, White MR, Sharkey KJ, Bearon RN, and Sée V (2014). Tight control of hypoxia-inducible factor-α transient dynamics is essential for cell survival in hypoxia. J. Biol. Chem 289, 5549–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisiaux A, Boussier J, Duffy D, Quintana-Murci L, Fontes M, Albert ML, and Milieu Intérieur C; Milieu Interieur Consortium (2017). Deconvolution of the Response to Bacillus Calmette-Guérin Reveals NF-κB-Induced Cytokines As Autocrine Mediators of Innate Immunity. Front. Immunol 8, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, and Thompson CB (2007). Systemic treatment with the antidiabetic drug metformin selectively impairs p53-defcient tumor cell growth. Cancer Res. 67, 6745–6752. [DOI] [PubMed] [Google Scholar]
- Cannistra SA, Kansas GS, Niloff J, DeFranzo B, Kim Y, and Ottensmeier C (1993). Binding of ovarian cancer cells to peritoneal mesothelium in vitro is partly mediated by CD44H. Cancer Res. 58, 3830–3838. [PubMed] [Google Scholar]
- Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, and Mazzone M (2014). Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene 88, 1743–1754. [DOI] [PubMed] [Google Scholar]
- Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, Kaplan J, Kalyan A, Altman JK, Platanias L, and Giles F (2016). Repurposing metformin for cancer treatment: current clinical studies. Oncotarget 7, 40767–40780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, and Xia J (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46 (W1), W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, Johnston SE, Vrcic A, Wong B, Khan M, et al. (2017). The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med 23, 405–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, and Hardie DG (1995). 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem 229, 558–565. [DOI] [PubMed] [Google Scholar]
- Coscia F, Watters KM, Curtis M, Eckert MA, Chiang CY, Tyanova S, Montag A, Lastra RR, Lengyel E, and Mann M (2016). Integrative proteomic profiling ofovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun 7, 12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2012). 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 13 (Suppl 16), S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David JM, Dominguez C, Hamilton DH, and Palena C (2016).The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines (Basel) 4, E22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk P, Angenete E, Bergstrom M, and Ivarsson ML (2013). TGF-β1 promotes transition of mesothelial cells into fibroblast phenotype in response to peritoneal injury in a cell culture model. Int. J. Surg 11, 977–982. [DOI] [PubMed] [Google Scholar]
- Fujikake K, Kajiyama H, Yoshihara M, Nishino K, Yoshikawa N, Utsumi F, Suzuki S, Niimi K, Sakata J, Mitsui H, et al. (2018). A novel mechanism of neovascularization in peritoneal dissemination via cancer-associated mesothelial cells affected by TGF-β derived from ovarian cancer. Oncol. Rep 39, 193–200. [DOI] [PubMed] [Google Scholar]
- Garson K, Gamwell LF, Pitre EMG, and Vanderhyden BC (2012). Technical challenges and limitations of current mouse models of ovarian cancer. J. Ovarian Res 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascard P, and Tlsty TD (2016). Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 30, 1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, and Harris AL (2001). Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res. 61, 7992–7998. [PubMed] [Google Scholar]
- Han X (2014). Constitutively active chemokine CXC receptors. Adv. Pharmacol 70, 265–301. [DOI] [PubMed] [Google Scholar]
- Han X, Tachado SD, Koziel H, and Boisvert WA (2012). Leu128(3.43) (l128) and Val247(6.40) (V247) of CXCR1 are critical amino acid residues for g protein coupling and receptor activation. PLoS One 7, e42765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PC, Mao M, deAbreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A, Diamond AM, Minshall RD, Consolaro ME, et al. (2015). MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun 6, 6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PC, Chiyoda T, Liu X, Weigert M, Curtis M, Chiang CY, Loth R, Lastra R, McGregor SM, Locasale JW, et al. (2019). SPHK1 is a novel target of metformin in ovarian cancer. Mol. Cancer Res 17, 870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman L, Leroy-Dudal J, Fernandes J, Seyer D, Dutoit S, and Carreiras F (2010). Mesothelial vitronectin stimulates migration of ovarian cancer cells. Cell Biol. Int 34, 493–502. [DOI] [PubMed] [Google Scholar]
- Hulea L, Gravel SP, Morita M, Cargnello M, Uchenunu O, Im YK, Lehuede C, Ma EH, Leibovitch M, McLaughlan S, et al. (2018).Translational and HIF-1alpha-Dependent Metabolic Reprogramming Underpin Metabolic Plasticity and Responses to Kinase Inhibitors and Biguanides. Cell Metab. 28, 817–832.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynninen P, Vaskivuo L, Saarnio J, Haapasalo H, Kivelä J, Pastoreková S, Pastorek J, Waheed A, Sly WS, Puistola U, and Parkkila S (2006). Expression of transmembrane carbonic anhydrases IX and XII in ovarian tumours. Histopathology 49, 594–602. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. (2001). Targeting of HIF-alphatothevon Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Mehus JG,Tews K, Milavetz BI, and Lambeth DO (1998). Genetic evidence for the expression of ATP- and GTP-specific succinyl-CoA synthetases in multicellular eucaryotes. J. Biol. Chem 273, 27580–27586. [DOI] [PubMed] [Google Scholar]
- Joyce JA, and Pollard JW (2009). Microenvironmental regulation of metastasis. Nat. Rev. Cancer 9, 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, Krausz T, Yamada SD, and Lengyel E (2007). Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int. J. Cancer 121, 1463–1472. [DOI] [PubMed] [Google Scholar]
- Kenny HA, Kaur S, Coussens LM, and Lengyel E (2008). The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J. Clin. Invest 118, 1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, Dogan S, Zillhardt M, K Mitra A,Yamada SD, Krausz T, and Lengyel E (2009). Organotypic models of metastasis: a three-dimensional culture mimicking the human peritoneum and omentum for the study of the early steps of ovarian cancer metastasis. Cancer Treat. Res 149, 335–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, Ladanyi A, Penicka CV, George J, Matlin K, et al. (2014). Mesothelial cells promote earlyovarian cancer metastasis through fibronectin secretion. J. Clin. Invest 124, 4614–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, and Myllyharju J (2007). Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J. Biol. Chem 282, 4524–4532. [DOI] [PubMed] [Google Scholar]
- Ksiazek K, Mikula-Pietrasik J, Korybalska K, Dworacki G, Jorres A, and Witowski J (2009). Senescent peritoneal mesothelial cells promote ovarian cancer cell adhesion: the role of oxidative stress-induced fibronectin. Am. J. Pathol 174, 1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Meuter A, Thapa P, Langstraat C, Giri S, Chien J, Rattan R, Cliby W, and Shridhar V (2013). Metformin intake is associated with better survival in ovarian cancer: a case-control study. Cancer 119, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Tsugawa H, Wohlgemuth G, Mehta S, Mueller M, Zheng Y, Ogiwara A, Meissen J, Showalter M, Takeuchi K, et al. (2018). Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 15, 53–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Cacalano G, Camerato T, Toy K, Moore MW, and Wood WI (1995). Chemokine binding and activities mediated by the mouse IL-8 receptor. J. Immunol 155, 2158–2164. [PubMed] [Google Scholar]
- Lengyel E (2010). Ovarian cancer development and metastasis. Am. J. Pathol 177, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel E, Litchfield LM, Mitra AK, Nieman KM, Mukherjee A, Zhang Y, Johnson A, Bradaric M, Lee W, and Romero IL (2015). Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am. J. Obstet. Gynecol 212, 479.e1–479.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessan K, Aguiar DJ, Oegema T, Siebenson L, and Skubitz AP (1999). CD44 and β1 integrin mediate ovarian carcinoma cell adhesion to peritoneal mesothelial cells. Am. J. Pathol 154, 1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield LM, Mukherjee A, Eckert MA, Johnson A, Mills KA, Pan S, Shridhar V, Lengyel E, and Romero IL (2015). Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget 6, 23548–23560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Romero IL, Litchfield LM, Lengyel E, and Locasale JW (2016). Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 24, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marushima H, Shibata S, Asakura T, Matsuura T, Maehashi H, Ishii Y, Eda H, Aoki K, lida Y, Morikawa T, and Ohkawa K (2011). Three dimensional culture promotes reconstitution of the tumor-specific hypoxic microenvironment under TGFβ stimulation. Int. J. Oncol 39, 1327–1336. [DOI] [PubMed] [Google Scholar]
- McMahon S, Charbonneau M, Grandmont S, Richard DE, and Dubois CM (2006). Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem 287,24171–24181. [DOI] [PubMed] [Google Scholar]
- Mikuła-Pietrasik J, Sosińska P, and Książek K (2014). Resveratrol inhibits ovarian cancer cell adhesion to peritoneal mesothelium in vitro by modulating the production of α5β1 integrins and hyaluronic acid. Gynecol. Oncol 734, 624–630. [DOI] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, et al. (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med 77, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman KM, Romero IL, Van Houten B, and Lengyel E (2013). Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 7837, 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard E (2008). Disorders caused by deficiency of succinate-CoA ligase. J. Inherit. Metab. Dis 37, 226–229. [DOI] [PubMed] [Google Scholar]
- Pakuła M, Mikuła-Pietrasik J, Stryczyński Ł, Uruski P, Szubert S, Moszyński R, Szpurek D, Sajdak S, Tykarski A, and Książek K (2018). Mitochondria-related oxidative stress contributes to ovarian cancer-promoting activity of mesothelial cells subjected to malignant ascites. Int. J. Biochem. Cell Biol 98, 82–88. [DOI] [PubMed] [Google Scholar]
- Rena G, Hardie DG, and Pearson ER (2017). The mechanisms of action of metformin. Diabetologia 60, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieppi M, Vergani V, Gatto C, Zanetta G, Allavena P, Taraboletti G, and Giavazzi R (1999). Mesothelial cells induce the motility of human ovarian carcinoma cells. Int. J. Cancer 80, 303–307. [DOI] [PubMed] [Google Scholar]
- Romero IL, McCormick A, McEwen KA, Park S, Karrison T, Yamada SD, Pannain S, and Lengyel E (2012). Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet. Gynecol 779, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IL, Mukherjee A, Kenny HA, Litchfield LM, and Lengyel E (2015). Molecular pathways: trafficking of metabolic resources in the tumor microenvironment. Clin. Cancer Res 27, 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rynne-Vidal A, Au-Yeung CL, Jiménez-Heffernan JA, Pérez-Lozano ML, Cremades-Jimeno L, Bárcena C, Cristóbal-García I, Fernández-Chacón C, Yeung TL, Mok SC, et al. (2017). Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J. Pathol 242, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval P, Jiménez-Heffernan JA, Rynne-Vidal Á, Pérez-Lozano ML, Gilsanz Á, Ruiz-Carpio V, Reyes R, García-Bordas J, Stamatakis K, Dotor J, et al. (2013). Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol 237,517–531. [DOI] [PubMed] [Google Scholar]
- Sawada K, Radjabi AR, Shinomiya N, Kistner E, Kenny H, Becker AR, Turkyilmaz MA, Salgia R, Yamada SD, Vande Woude GF, et al. (2007). c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 67, 1670–1679. [DOI] [PubMed] [Google Scholar]
- Sawada K, Mitra AK, Radjabi AR, Bhaskar V, Kistner EO, Tretiakova M, Jagadeeswaran S, Montag A, Becker A, Kenny HA, et al. (2008). Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 68, 2329–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, and Gottlieb E (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85. [DOI] [PubMed] [Google Scholar]
- Shin HS, Ko J, Kim DA, Ryu ES, Ryu HM, Park SH, Kim YL, Oh ES, and Kang DH (2017). Metformin ameliorates the Phenotype Transition of Peritoneal Mesothelial Cells and Peritoneal Fibrosis via a modulation of Oxidative Stress. Sci. Rep 7, 5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleire L, Førde HE, Netland IA, Leiss L, Skeie BS, and Enger PO (2017). Drug repurposing in cancer. Pharmacol. Res 724, 74–91. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 73, 731–740. [DOI] [PubMed] [Google Scholar]
- Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, Tookman LA, Orange C, Athineos D, Mason S, et al. (2016). CRISPR/Cas9-Mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res. 76, 6118–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Hashimoto T, Sugino T, Soeda S, Nishiyama H, Morimura Y, Yamada H, Goodison S, and Fujimori K (2012). Production of IL1-beta by ovarian cancer cells induces mesothelial cell beta1-integrin expression facilitating peritoneal dissemination. J. Ovarian Res 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Yang Z, Jin P, Yang X, Li X, Wei X, Wang Y, Long S, Zhang T, Chen G, et al. (2018). Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol. Cancer Ther 77, 1291–1302. [DOI] [PubMed] [Google Scholar]
- Yan Q, Bartz S, Mao M, Li L, and Kaelin WG Jr. (2007). The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol. Cell. Biol 27, 2092–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Fang W, Smart C, Hu Q, Huang S, Alvarez N, Fields P, and Cheng N (2019). CCR2 Chemokine Receptors Enhance Growth and Cell-Cycle Progression of Breast Cancer Cells through SRC and PKC Activation. Mol. Cancer Res 77, 604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest 708, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Maeder ML, Mali P, Pruett-Miller SM, Thibodeau-Beganny S, Chou BK, Chen G, Ye Z, Park IH, Daley GQ, et al. (2009). Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 5, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper, in the Figures S1, S5 and in the Tables S1, S2, S3, S4, S5, and S6. Source data (e.g., representative Western immunoblots) are available from the Lead Contact upon reasonable request. All supplementary figures and data are available online. No unique software or code was used in this study.