

ABSTRACT
Induction or selection of radioresistant cancer (stem) cells following standard radiotherapy is presumably one of the major causes for recurrence of metastatic disease. One possibility to prevent tumor relapse is the application of targeted immunotherapies including, e.g., chimeric antigen receptor (CAR) T cells. In light of long-term remissions, it is highly relevant to clarify whether radioresistant cancer cells are susceptible to CAR T cell-mediated killing. To answer this question, we evaluated the anti-tumor activity of the switchable universal chimeric antigen receptor (UniCAR) system against highly radioresistant head and neck squamous cell carcinoma cells both in vitro and in vivo. Following specific UniCAR T cell engagement via EGFR or CD98 target modules, T cell effector mechanisms were induced including secretion of pro-inflammatory cytokines, up-regulation of granzyme B and perforin, as well as T cell proliferation. CD98- or EGFR-redirected UniCAR T cells further possess the capability to efficiently lyse radioresistant tumor cells. Observed anti-tumor effects were comparable to those against the radiosensitive parental cell lines. Finally, redirected UniCAR T cells significantly inhibited the growth of radioresistant cancer cells in immunodeficient mice. Taken together, our obtained data underline that the UniCAR system is able to overcome radioresistance. Thus, it represents an attractive technology for the development of combined radioimmunotherapeutic approaches that might improve the outcome of patients with metastatic radioresistant tumor diseases.
KEYWORDS: radioresistance, CD98, EGFR, adaptor CAR, T cell immunotherapy
Introduction
Radiotherapy mostly applied as external beam therapy or brachytherapy is one of the standard procedures for cancer treatment. Over the last century, significant technological progress has been made to increase anti-tumor effects, while reducing the risk for damage to healthy tissues [summarized in.1 Radiotherapy has achieved high success rates especially for the management of primary tumors, whereby in combination with chemotherapy up to 80% of tumors can now be locally controlled.1 However, the main obstacle of ionizing radiation is still the induction or selection of radioresistant cancer (stem) cell clones that are responsible for the recurrence of the disease.1–4 This clearly emphasizes the clinical need for adjuvant treatment options able to overcome tumor radioresistance. Different strategies of radiosensitization need to be pursued in preclinical and clinical studies including, e.g., hypoxic cell radiosensitizers, inhibitors of DNA repair mechanisms, stimulators of cell death pathways or suppressors of cell proliferation and survival pathways.2,3,5
Another strategy to prevent tumor relapse after acquired radioresistance entails targeted immunotherapies. Among them, chimeric antigen receptor (CAR) T cell technology represents one of the most promising approaches. Its high potential is reflected by the impressive success of two CD19-specific CAR T cell therapeutics, axicabtagene ciloleucel and tisagenlecleucel that were approved by the U.S. Food and Drug Administration in 2017 for treatment of B cell malignancies.6–8 CARs are composed of an extracellular tumor-specific antigen binding domain linked to intracellular signaling domains from activating immune receptors (e.g. CD3ζ, CD28, 4-1BB).9,10 After genetic modification, T cells are able to bind tumor cells via their CAR independently of their endogenous TCR specificity. This results in CAR-mediated T cell activation and subsequent tumor cell killing.9,10 However, aside from their high anti-tumor activity, CAR T cells can also trigger severe or even fatal side effects.11,12 As conventional CAR T cells lack self-limiting control mechanisms, management of adverse reactions is mainly restricted to immunosuppressive drugs so far.12 Hence, adapter CAR platforms were developed to improve the safety profile of CAR T cells [e.g.13-19]. In 2014, we introduced the switchable UniCAR system in which UniCAR T cell activity can be directly controlled by separated short-lived target modules (TMs) (Figure 1a).16–20 TMs are tumor-specific binding molecules, e.g., small peptides,21 nanobodies22,23 or single-chain fragment variables (scFv) [e.g.24-27] equipped with the E5B9 peptide epitope from the nuclear protein La/SS-B.28 UniCAR T cells in turn are armed with an E5B9-specific CAR (UniCAR) predicating its anti-tumor activity on the availability of TMs. Consequently, side effects caused by UniCAR T cells are likely to be manageable by TM titration.
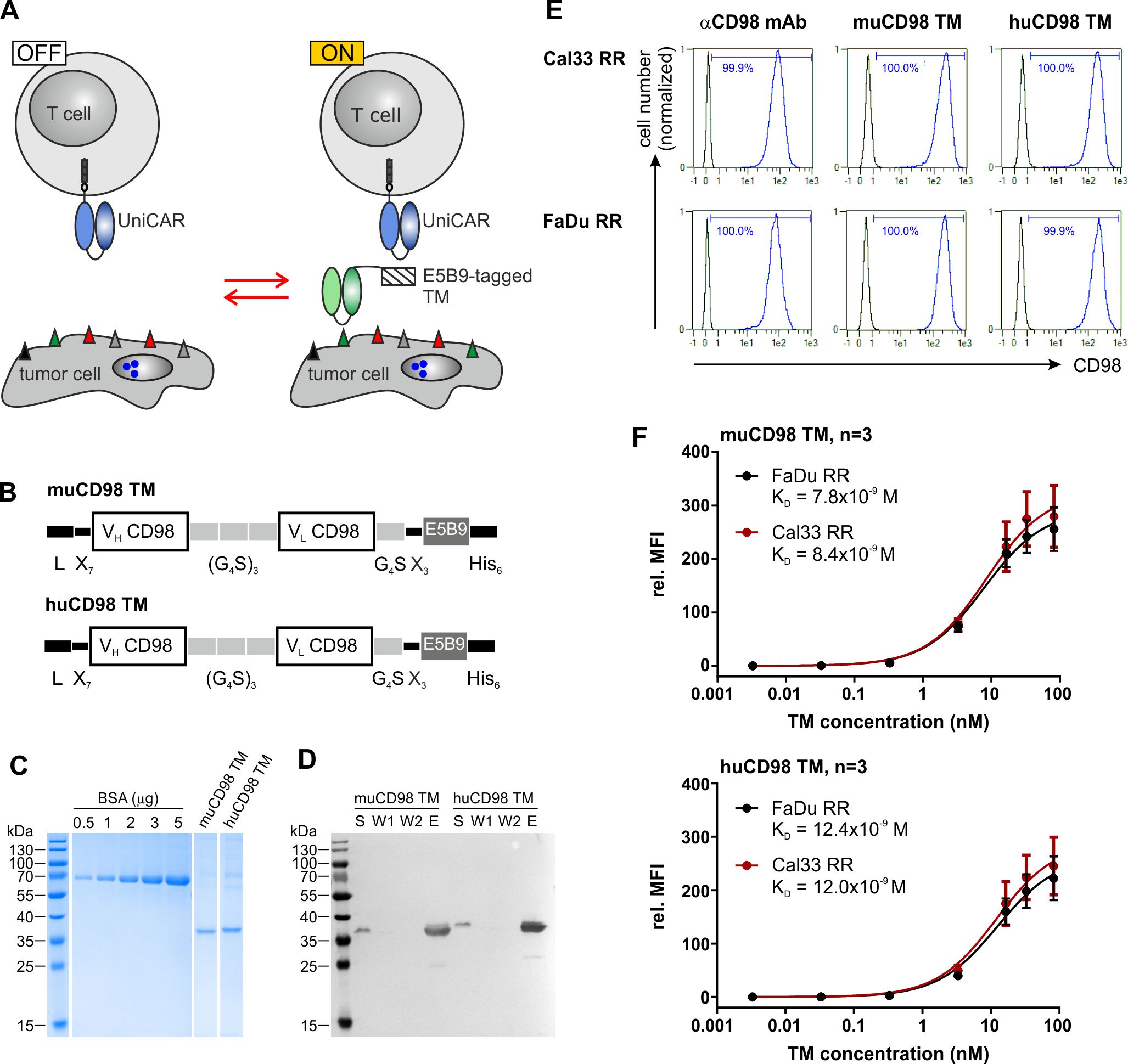
Figure 1.
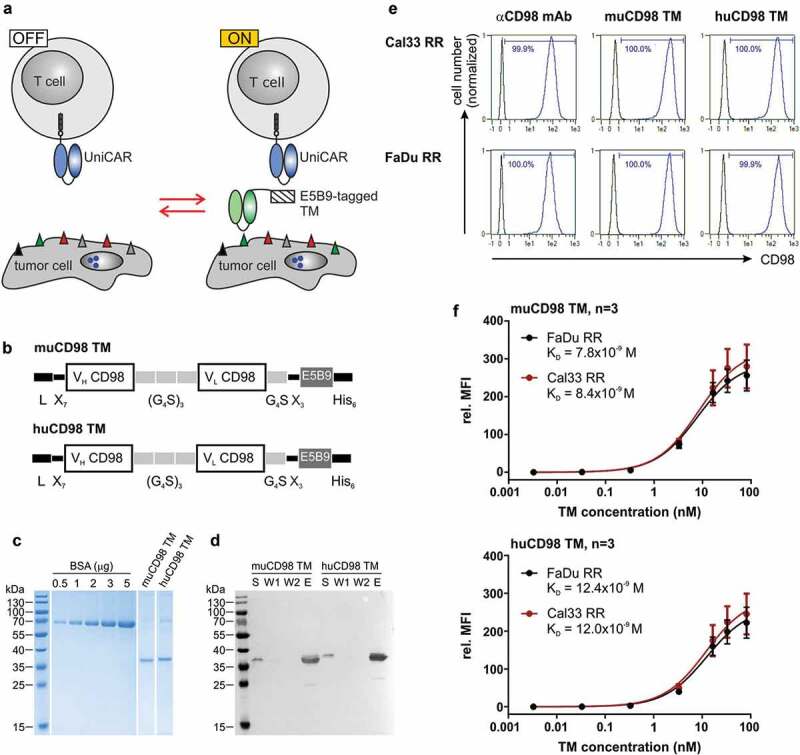
Expression and binding properties of CD98-specific TMs. (a) Antitumor activity of UniCAR T cells can be repeatedly switched ‘ON’ and ‘OFF’ in dependence of E5B9-tagged target modules (TMs). (b) The novel murine (mu) and humanized (hu) CD98 TM were generated by fusing the variable light (VL) and variable heavy (VH) domains of the αCD98 IgG1 mAb MEM-108 via flexible peptide linkers to the UniCAR epitope E5B9. The N-terminal murine Ig kappa leader sequence (L) mediates secretion, while the C-terminal hexahistidine (His6)-tag facilitates purification and detection of the recombinant proteins. (c, d) Ni-NTA purified TMs were separated by SDS-PAGE. (c) After staining with Coomassie Brilliant Blue G250, TM concentration was estimated based on a BSA standard. (d) Cell culture supernatant (S), wash fraction (W)1, W2 and eluate (E) were transferred to a nitrocellulose membrane. Recombinantly expressed TMs were subsequently detected via their C-terminal His6-Tag. (e, f) TM binding was analyzed by flow cytometry. (e) After incubation of tumor cells with 5 ng/µl of TM, TM binding was detected via the UniCAR epitope. As positive control, tumor cells were stained with an αCD98-APC-Vio770 Ab. Histograms show stained cells (blue) and respective negative controls (black). Numbers represent the percentage of CD98+ cells. Results of one out of three experiments are shown. (f) Tumor cells were incubated with increasing concentrations of muCD98 TM (upper panel) or huCD98 TM (lower panel) and subsequently stained with αHis-PE Ab. In order to determine TM affinity toward CD98, TM concentrations were plotted against the relative median fluorescence intensity (rel. MFI). Mean ± SEM of three independent experiments are shown.
For combined radioimmunotherapeutic approaches, it is of significant interest as to whether radioresistant tumor cells are still sensitive for targeted immunotherapies. Thus, the aim of the current study is to elucidate if UniCAR T cells can be redirected for efficient killing of radioresistant cancer cells. In order to provide proof of concept head and neck squamous cell carcinoma (HNSCC) was chosen as a model tumor entity. For the herein proposed T cell retargeting approach we selected the epidermal growth factor receptor (EGFR) and CD98 as suitable target antigens. The tyrosine kinase receptor EGFR is physiologically, i.e., an important regulator of cell growth, survival, differentiation and apoptosis.29 Consequently, many epithelial tumors are characterized by defects in the EGFR signaling pathway. CD98 represents a heterodimeric transmembrane protein consisting of a heavy chain (CD98hc) and one of the six permease-type amino acid transporters (CD98 light chain, e.g. LAT-1 and LAT-2).30–32 The heterodimer plays not only an important role as transporter for essential amino acids but also controls cell survival and growth via interaction with integrin-β.30 Both EGFR and CD98 were found to be overexpressed in HNSCC,29,30 while high expression levels were shown to correlate with a poor prognosis.33–37 Furthermore, CD98 was identified as a marker and regulator of cancer stem cells in HNSCC.38 Ionizing radiation can boost EGFR as well as CD98hc expression. This is in line with the fact that their downstream signaling pathways contribute to an increased level of radioresistance in tumor cells.3,33,39,40
In this study, novel murine and humanized TMs directed against CD98hc were developed. Together with established EGFR TM, they were used to test the capability of UniCAR T cells to mediate anti-tumor responses against radioresistant HNSCC cells both in vitro and in vivo.
Materials and methods
Cell lines
In this study we used established HNSCC cell lines Cal33 originated from squamous cell carcinomas of the tongue (Deutsche Sammlung von Mikroorganismen und Zellkulturen DSMZ GmbH, Braunschweig, Germany) and FaDu originated from pharyngeal squamous cell carcinoma (ATCC, Manassas, USA). The radioresistant Fadu RR and Cal33 RR derivatives were generated in the lab of Prof. Dr. Anna Dubrovska (OncoRay – National Center for Radiation Research Oncology, Faculty of Medicine, University Hospital Carl Gustav Carus, Dresden, Germany).4 The radioresistant phenotype is stable for a few months and thus was stable during the time frame in which the herein presented data were collected. The cell lines were genotyped using microsatellite polymorphism analyses and tested for radioresistant phenotype and mycoplasma prior to experimentation. For in vivo studies, Cal33 RR cells were genetically modified to express the red fluorescent protein mCherry using lentiviral transduction. The resulting cell line was termed Cal33 RRmCherry. To ensure that the cells keep their radioresistant phenotype including after their stable transduction with mCherry-encoding DNA plasmid, we have tested their radioresistance directly prior to experimentation. Tumor cell lines and 3T3 cell lines genetically modified to express TMs were maintained in DMEM complete media.41 All cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Humanization and cloning of novel muCD98 TM and huCD98 TM
The novel murine (mu) and humanized (hu) CD98 TM are based on the αCD98 IgG1 monoclonal antibody (mAb) MEM-108. Identification and humanization of VH and VL domains were performed as published previously.42,43 MuCD98 TM and huCD98 TM were developed in silico by fusion of the αCD98 mAb MEM-108 VH and VL domains to the UniCAR epitope E5B9. Whole DNA sequences were subsequently purchased from Eurofins Genomics. After digestion of TM-containing pEX-A128 vectors with NheI (ThermoFisher Scientific, #ER0971) and MssI (ThermoFisher Scientific, #ER1341) the open reading frames of muCD98 TM and huCD98 TM were cloned in the lentiviral expression vector p6NST50 which was beforehand digested with XbaI (ThermoFisher Scientific, #ER0681) and KspAI (ThermoFisher Scientific, #ER1032).
Purification and expression of TMs
All TM-producing 3T3 cell lines were generated by lentiviral gene transfer. In order to purify recombinant proteins via their C-terminal His-tag from cell culture supernatants, Ni-NTA affinity chromatography was used.41 Hereafter, purified TMs were separated via SDS-PAGE. To estimate TM concentration and purity, Coomassie Brilliant Blue G250 staining or immunoblotting of separated protein fractions were conducted as described previously.41,42,44
Generation of UniCAR T cells
Blood samples were obtained from the German Red Cross (Dresden, Germany) with informed consent of voluntary donors. After isolation of peripheral blood mononuclear cells (PBMCs) from whole blood via density gradient centrifugation, T cells were magnetically separated using the Pan T Cell Isolation Kit, human (Miltenyi Biotec GmbH, #130-096-535).45 Subsequently, T cells were genetically modified with EGFP vector control, UniCAR Stop or UniCAR 28/ζ construct by lentiviral transduction according to previously published protocols.24,46,47 Briefly, CD3/CD28 bead-stimulated T cells were transduced 5 times with the respective lentiviral particles. After 1 week of culture, genetically modified T cells were sorted using FACS technology by means of co-translated EGFP marker protein [see also 48]. During transduction and expansion, T cells were cultured in RPMI 1640 complete media41 supplemented with IL-15 (ImmunoTools, #11340155), IL-7 (ImmunoTools, #11340075) and Proleukin® (Novartis Pharmaceuticals, #2238131). One day prior to experiments, genetically modified T cells were cultured in medium without additional cytokines.
Flow cytometry
For binding analysis, 1 × 105 cells were incubated with 5 ng/μl of TM for 1 h. TM binding was detected via the UniCAR epitope E5B9. For this purpose, cells were incubated consecutively with 10 μg/ml of αLa mAb (5B9)28 and Goat anti-Mouse IgG Fc Cross-Adsorbed Secondary Antibody, PE (ThermoFisher Scientific, #31861). Surface staining with CD98-APC-Vio770, human (Miltenyi Biotec GmbH, #130-105-711) served as a positive control. To analyze CD98 expression on UniCAR T cell subpopulations, cells were additionally stained with CD3-PE-Vio770, human (Miltenyi Biotec GmbH, #130-096-749), CD4-VioBlue, human (Miltenyi Biotec GmbH, #130-114-534) and CD8-PE, human (Miltenyi Biotec GmbH, #130-091-084). For determination of TM affinity, CD98-expressing cell lines were incubated with increasing TM concentrations for 1 h followed by a 30 min-incubation with Anti-His-PE (Miltenyi Biotec GmbH, #130-120-718). GraphPad Prism 7 software (GraphPad Software Inc.) was used to calculate KD values based on the respective affinity curves. CD98 antigen density on tumor cell lines and UniCAR T cells was determined using the QifiKit® (Agilent, #K007811-8) in combination with purified anti-human CD98 Ab (BioLegend, #315602), as published elsewhere.23
Live and dead cells were distinguished by adding 1 μg/ml of propidium iodide/PBS solution (ThermoFisher Scientific, #P3566) shortly before flow cytometric measurements. Data acquisition and analysis were conducted with a MACSQuant Analyzer 10 and the MACSQuantify Software (Miltenyi Biotec GmbH).
Chromium release assay
Tumor cell killing was analyzed via standard chromium release assay as previously published.41 Briefly, 5 × 103 51Cr-labeled target cells and genetically modified T cells were incubated with or without TM at 1:1 or 5:1 effector-to-target cell (E:T) ratios. After 24 h, activity of released 51Cr was determined in co-culture supernatants using a MicroBeta2 Microplate Counter (PerkinElmer LAS GmbH).
Cytokine release, proliferation and activation status of UniCAR T cells
UniCAR T cells were cultured with or without tumor cells in the presence or absence of 50 nM TM in a 96-well plate (E:T = 5:1). Total volume was adjusted with RPMI complete media to 200 μl. After 48 h, cell culture plates were centrifuged for 5 min at 360xg and cell-free supernatant was collected. Concentration of TNF, IFN-γ, IL-2 and GM-CSF was determined by enzyme-linked immunosorbent assay (ELISA). Human TNF ELISA Set (#555212), Human IFN-Gamma ELISA Set (#555142), Human IL-2 ELISA Set (#555190), Human GM-CSF ELISA Set (#555126) as well as BD OptEIA Reagent Set B (#550534) were obtained from BD Biosciences. In addition, intracellular staining of perforin and granzyme B was performed after 48 h as described previously.49–51 In order to monitor UniCAR T cell numbers in co-cultures, cells were labeled with Cell Proliferation Dye eFluor®670 (ThermoFisher Scientific, #65-0840-85) according to previously published protocols.49,52 After 24 h and 96 h of incubation, 20 μl of samples was transferred to a 96-well plate and mixed with 80 μl of 1 μg/ml propidium iodide/PBS solution (ThermoFisher Scientific, #P3566). Cell numbers were calculated using a MACSQuant Analyzer 10 (Miltenyi Biotec GmbH).
In vivo killing assay
All animal procedures were conducted in accordance with the ARRIVE guidelines, the guidelines set by the European Communities Council Directive (86/609 EEC). The local Workplace Animal Ethical Committee (MÁB) approved the animal facilities and the protocol according to institutional guidelines at the Semmelweis University (Budapest, Hungary). Six-week old male Rj:NMRI-Foxn1nu/nu mice (herein referred to as NMRInu/nu) (Janvier Labs, Le Genest-Saint-Isle, France) served as an immunocompromised animal model. Mice were housed in a pathogen-free facility, in sterile cages with 12 h light/dark cycle. Health status of mice was monitored daily by husbandry staff. In order to assess in vivo efficacy of the UniCAR system against radioresistant tumor cells, 1 × 106 Cal33 RRmCherry cells were mixed with 1 × 106 UniCAR T cells and 10 μg of TM. Total volume was adjusted to 100 μl per mouse with PBS. Mixtures were subcutaneously injected into the right hind leg. Control group 1 received tumor cells alone, whereas control group 2 was treated with Cal33 RRmCherry cells plus UniCAR T cells. Each group consisted of five mice. Prior to optical imaging, mice were anesthetized as published previously.22,47 Fluorescent signal of living Cal33 RRmCherry cells was monitored over a period of 3 days with the In Vivo Multispectral Imaging System (Bruker, USA). Data analysis was performed using the MI 5.3 and MS 1.3 software (Bruker, USA).
Statistics
Data were statistically evaluated using GraphPad Prism 7 software (GraphPad Prism Inc.). One-way or two-way ANOVA was applied for column or group analyses, respectively. Statistical analyses of in vitro data were performed with post hoc Tukey multiple comparison test, and for in vivo data post-hoc, Sidak multiple comparison test was used. p values below 0.0332 were considered significant.
Results
Expression and purification of novel CD98 TMs
In order to retarget UniCAR T cells to radioresistant HNSCC cells, the tumor-associated antigens (TAAs) EGFR and CD98 were selected. For this study, we employed an improved EGFR TM that was developed based on findings from our previous studies.22,23 In order to establish a novel muCD98 TM, the variable domains of the heavy (VH) and light chains (VL) of the αCD98 monoclonal antibody (Ab) MEM-108 were connected to the UniCAR epitope E5B9 via flexible peptide linkers (Figure 1b). The immunogenic potential of this TM was further reduced by humanization. Therefore, the murine framework regions (FWR) of the VH and VL domain were replaced by human sequences possessing the highest degree of homology: IGHV1-46*01 and IGHJ6*01 for VH or IGKV4-1*01 and IGKJ2*02 for VL. Except for these human sequences, structural features of the resulting huCD98 TM are identical to the murine counterpart (Figure 1b).
The novel muCD98 TM and huCD98 TM were expressed by stably transduced 3T3 cell lines. Due to an N-terminal Igκ leader sequence, recombinant proteins were secreted into the cell culture media. As shown in Figure 1c, both CD98-specific TMs could be successfully isolated via Ni-NTA affinity chromatography with high purity. Eluates showed mainly one single, hexahistidine (His)-tagged band of approximately 35 kDa corresponding to the calculated size of 30.6 kDa and 30.3 kDa for the muCD98 TM and huCD98 TM, respectively.
Binding properties of muCD98 TM and huCD98 TM
Flow cytometry-based surface staining revealed that both muCD98 TM and huCD98 TM were able to specifically bind 100% of the radioresistant HNSCC sub-cell lines FaDu RR and Cal33 RR (Figure 1e). Binding of the TMs was verified via the E5B9 tag. This method demonstrated that the UniCAR epitope of cell-bound TMs was accessible for Ab binding and thus most probably also for UniCAR T cell interactions. Despite humanization, the huCD98 TM maintained its high affinity toward CD98 (Figure 1f). By performing surface staining of FaDu RR and Cal33 RR cells with titrated amounts of TM, KD values of approximately 8 × 10−9 M and 12 × 10−9 M were calculated for the muCD98 TM (Figure 1f, upper panel) and huCD98 TM (Figure 1f, lower panel), respectively.
CD98 expression profile
CD98 is a common amino acid transporter and also known as a T cell activation marker.30 Therefore, we investigated CD98 expression on both tumor cell lines and UniCAR T cells. As shown in Figure 2a, all examined cells were CD98+. Median fluorescence intensity (MFI) values of stained Cal33 RR and FaDu RR were higher in comparison to the parental radiosensitive cell lines Cal33 and FaDu. This is in line with recently published data of Digomann and colleagues33 also showing increased CD98 protein expression levels for these radioresistant cells. CD98 antigen density of all investigated tumor cell lines is above 106 molecules/cell (Figure 2b). Although both CD4+ and CD8+ UniCAR T cells showed CD98 surface expression (Figure 2a), antigen density was reduced by a factor of 50 to 125 in comparison to HNSCC cell lines (Figure 2b).
Figure 2.
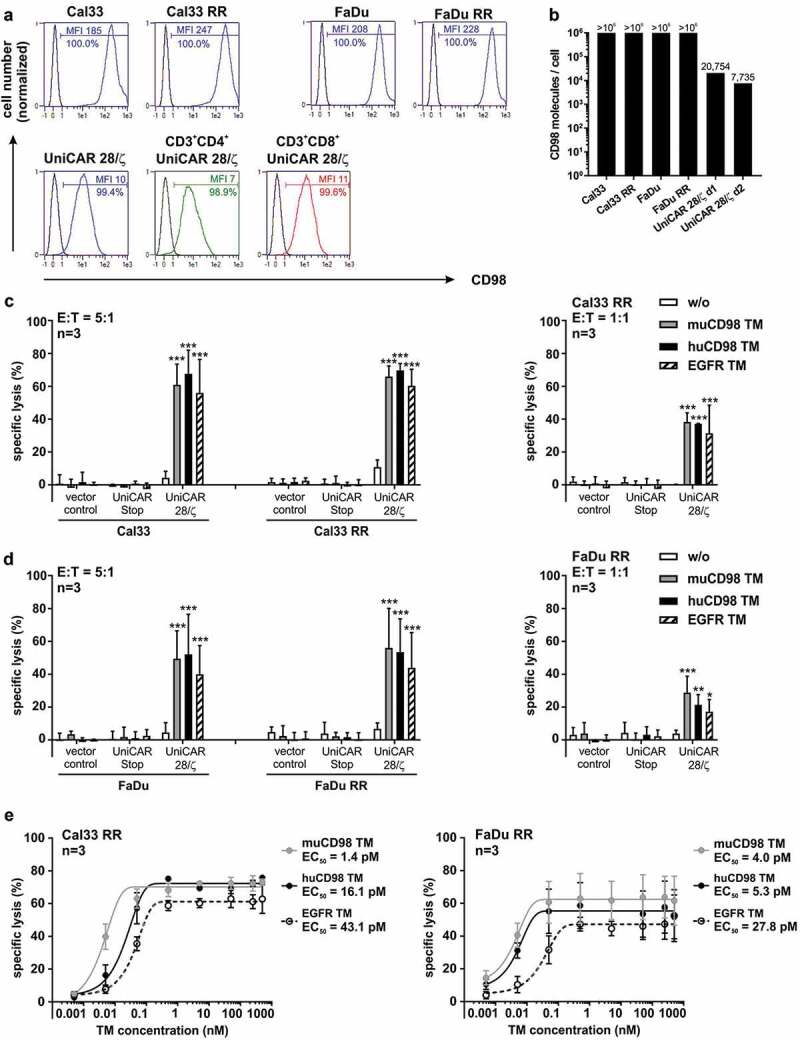
CD98 expression level and UniCAR T cell-mediated lysis of parental and radioresistant HNSCC cell lines. (a) Surface expression of CD98 on UniCAR T cells as well as on different tumor cells and their radioresistant (RR) derivatives was analyzed by flow cytometry using an αCD98-APC-Vio770 Ab. Histograms show the percentage of CD98+ cells and the median fluorescence intensity (MFI) of stained cells (blue) in comparison to unstained controls. (b) CD98 density on tumor and UniCAR T cells was determined by fluorescent-based QIFIKIT® (n = 1). (c-e) In order to analyze UniCAR T cell-mediated tumor cell killing, 24 h-chromium release assays were performed. (c, d) Genetically modified T cells were incubated with 51Cr-labeled tumor cells in the absence or presence of 50 nM TM for 24 h at indicated E:T ratios. Experiments were performed in triplicates. Diagrams show mean specific lysis + SD for three independent experiments using different T cell donors. (*p < .0332, **p < .0021, ***p < .0002 with respect to samples w/o TM, Two-way ANOVA with posthoc Tukey Multiple Comparison Test) (e) UniCAR T cells were co-cultured with tumor cells in the presence of different TM concentrations (E:T = 5:1). Based on the resulting dose–response curves half-maximal effective concentration (EC50) values were calculated. Experiments were performed in triplicates. Summarized data of three independent experiments with different T cell donors are shown (mean ± SEM).
Engagement of UniCAR T cells via EGFR TM and CD98 TM for killing of radioresistant HNSCC cell lines
The capacity of TM-redirected UniCAR T cells to kill radioresistant tumor cells was investigated by 24 h-standard chromium release assays. The HNSCC cell lines Cal33 RR and FaDu RR, which possess substantially increased or long-term moderate radioresistance in comparison to their respective parental cell lines, were selected.4 As shown in Figure 2c-d, in the presence of either the EGFR TM, the muCD98 TM or the huCD98 TM, UniCAR T cells were able to induce significant tumor cell lysis (40-65%). Radioresistant cell lines were killed with similar efficiency as the parental Cal33 and FaDu cells irrespective of the selected TM. Importantly, humanization of the muCD98 TM did not negatively influence its ability to trigger the anti-tumor activity of UniCAR T cells. In the absence of TMs, tumor cells were not significantly eliminated, emphasizing that the functionality of the UniCAR system was strictly TM-dependent. Chromium release assays under limiting conditions further revealed that the EGFR TM as well as both CD98 TMs were able to engage UniCAR T cells for significant lysis at an E:T ratio of 1:1 (Figure 2c-d) and at low TM concentrations (Figure 2e). All tested TMs exhibited EC50 values in the low picomolar range (1–43 pM).
Activating UniCAR T cell effector mechanisms upon TM-mediated cross-linkage with radioresistant HNSCC cells
Having proven the ability of the UniCAR system to effectively eliminate different radioresistant cancer cells, we elucidated UniCAR T cell-mediated effector mechanisms in more detail. For this purpose, 48 h-co-cultivation assays of UniCAR T cells with or without tumor cells were performed. TMs were added at a concentration of 50 nM. Experiments were conducted with Cal33 RR cells showing the highest degree of radioresistance.4
Upon cross-linkage with Cal33 RR via the muCD98 TM, the huCD98 TM or the EGFR TM, UniCAR T cells secreted significant amounts of the pro-inflammatory cytokines TNF, IFN-γ and GM-CSF, whereas substantial concentrations of growth-promoting IL-2 could be detected only in the presence of the CD98-specific TMs (Figure 3). However, in the absence of TMs, no cytokines were measured in co-culture supernatants. As UniCAR T cells express CD98 on their surface (see Figure 2a-b), we also investigated whether CD98-specific TMs per se can induce cytokine secretion of UniCAR T cells. As shown in Figure 3, both the muCD98 TM and huCD98 TM but not the EGFR TM stimulated UniCAR T cells to release TNF, IFN-γ, IL-2 and GM-CSF. However, cytokine concentrations were considerably lower in comparison to co-cultures with Cal33 RR target cells.
Figure 3.
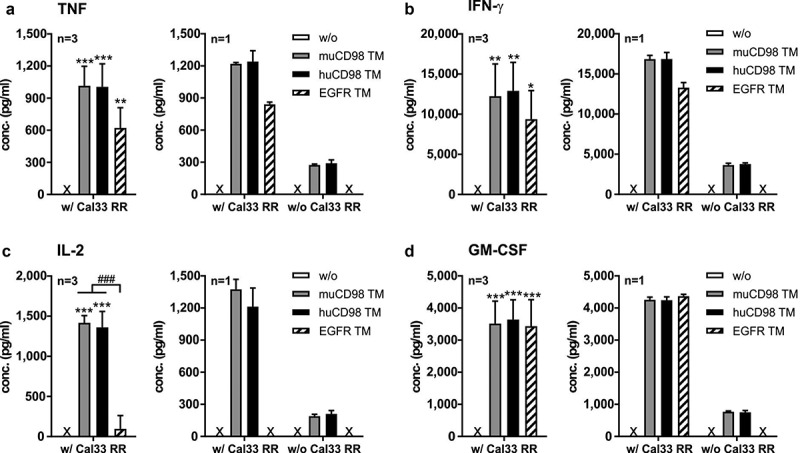
Cytokine profile of TM-redirected UniCAR T cells. UniCAR T cells were incubated with (w/) or without (w/o) Cal33 RR cells in the presence or absence of 50 nM TM (E:T = 5:1). After 48 h, cell-free supernatants were analyzed by ELISA for (a) TNF, (b) IFN-γ, (c) IL-2 and (d) GM-CSF. Experiments were performed in triplicates. (a-d) Left panels show summarized data (mean + SD) of three different T cell donors, right panels show mean + SD from triplicates of one individual experiment (*p < .0332, **p < .0021, **p < .0002 compared to samples w/o TM; One-way ANOVA with post hoc Tukey multiple comparison test). x, not detectable
Aside from cytokine secretion, we further assessed upregulation of perforin and granzyme B by co-cultured UniCAR T cells via intracellular staining. As shown in Figure 4a, already a minor population of UniCAR T cells (20-36%) expressed intracellular perforin in a resting state, whereas all cells were positive for granzyme B. In the presence of either the CD98- or EGFR-specific TMs, not only the percentage of perforin+ UniCAR T cells increased but also the intracellular granzyme B expression was upregulated as shown by higher MFI values (Figure 4a). Overall, CD4+ and CD8+ UniCAR T cell populations behaved in a similar way (Figure S1). In line with the cytokine expression profile, perforin and granzyme B levels increased in CD4+ and CD8+ UniCAR T cells upon incubation with muCD98 TM or huCD98 TM alone (Figure 4a and Figure S1). Similar results were not observed with the EGFR TM. This raised the question, whether CD98 TMs could not only mediate specific elimination of tumor cells but would also lead to fratricide of UniCAR T cells. Quantitative assays revealed that in the presence of tumor cells, only the muCD98 TM mediated a 20% decrease in UniCAR T cell numbers after 24 h (Figure 4b). In the absence of target cells, this effect is more pronounced and triggered by the huCD98 TM as well. Long-term cultivation (96 h) with muCD98 TM and huCD98 TM resulted in a considerable decrease of UniCAR T cell numbers independent of the presence or absence of tumor cells. In contrast, upon cross-linkage with Cal33 RR cells via the EGFR TM, UniCAR T cells showed a threefold expansion within 96 h while numbers of UniCAR T cells were not altered in the absence of target cells (Figure 4b).
Figure 4.
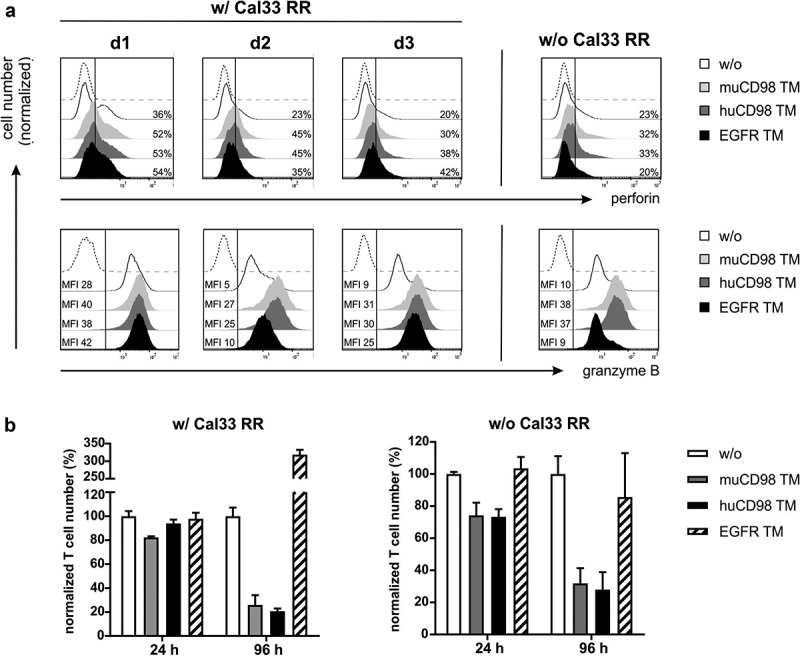
Perforin/granzyme B production and expansion of UniCAR T cells after TM-mediated cross-linkage with radioresistant tumor cells. UniCAR T cells were co-cultured with (w/) or without (w/o) Cal33 RR cells in the presence or absence of 50 nM TM at an E:T ratio of 5:1. (a) After 48 h, UniCAR T cells were stained for intracellular perforin and granzyme B expression and analyzed by flow cytometry. Histograms show the percentage of perforin+ UniCAR T cells (upper panel) or median fluorescence intensity (MFI) of granzyme B stained UniCAR T cells (lower panel). Marker was set according to the respective Fluorescence Minus One (FMO) control (dashed line). Each histogram (w/ Cal33 RR) shows the results of one individual donor (d1, d2 or d3). For samples w/o Cal33 RR cells, data of one representative donor are shown. (b) After 24 h and 96 h, eFluor670+ UniCAR T cell numbers were determined by flow cytometry. Control samples w/o TM were set to 100%. Normalized UniCAR T cell numbers (mean + SD of triplicates) are shown for one donor (n = 1).
Killing of radioresistant tumor cells in vivo via the UniCAR system
Finally, the ability of the UniCAR system to efficiently eliminate radioresistant tumor cells was studied in vivo. In order to allow visualization of tumor growth in mice, Cal33 RRmCherry cells were used.
Prior to subcutaneous injection into immunodeficient NMRInu/nu mice, 1 × 106 Cal33 RRmCherry cells and 1 × 106 UniCAR T cells were mixed with 10 μg of TM. The study was conducted only with the EGFR TM and muCD98 TM as both CD98-specific TMs showed similar efficacy in vitro. Control mice received tumor cells alone (control group 1) or in combination with UniCAR T cells (control group 2). As summarized in Figure 5, treatment of mice with the EGFR- or CD98-specific UniCAR system led to significant inhibition of tumor growth in comparison to control groups. Already after 3 days, nearly all tumor cells were eradicated. Even though control group 2 (Cal33 RRmCherry + UniCAR T) showed an initial reduction in tumor growth compared to control group 1 (Cal33 RRmCherry), the tumor size stabilized over time. Most likely this effect can be attributed to donor-dependent rejection reactions against alloantigens present on the tumor cells.
Figure 5.
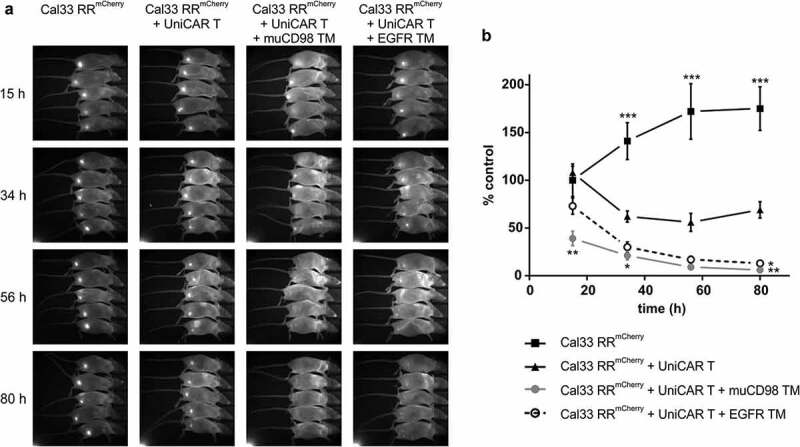
In vivo killing of radioresistant cancer cells using the UniCAR system. Cal33 RRmCherry and UniCAR T cells were mixed at an E:T ratio of 1:1 together with muCD98 TM or EGFR TM and subsequently injected s.c. into the right hind leg of NMRInu/nu mice. As a control, Cal33 RRmCherry cells alone or together with UniCAR T cells were administered. Over a period of 3 days, optical imaging was performed to follow tumor growth based on the mCherry signal. Each group consisted of five mice. Results from one experiment are shown. (a) Each image was scaled to maximum intensity. (b) Quantitative analysis of the mCherry signal. Values were normalized to control group (Cal33 RRmCherry only) at day 0. (*p < .0332, **p < .0021, ***p < .0002 compared to the control group receiving tumor and UniCAR T cells; One-way ANOVA with post hoc Sidak multiple comparison test).
Discussion
Combination of standard treatment modalities, including radiotherapy with novel adjuvant targeted immunotherapies, is a recent and rapidly growing field in cancer management. Advances are especially expected for the treatment of solid tumors. For example, in preclinical glioblastoma and pancreatic tumor mouse models, the combination of radiation with CAR T cell therapy has proven to act synergistically.53–55 In terms of successful implementation of such combined radioimmunotherapeutic approaches, the rise and induction of radioresistant tumor cells are a significant aspect that should be taken into consideration. Therefore, the major focus of this work was to analyze the effect of (Uni)CAR T cells on radioresistant HNSCC cells. HNSCC is still difficult to treat, especially in the case of recurrent or metastatic diseases.56 The prognosis of HNSCC patients with relapsed radioresistant tumors is poor with overall survival rates less than 6 months.57,58
In this study, highly chemo- and radioresistant FaDu RR and Cal33 RR tumor cells were selected.33 Compared to their parental counterparts they show a higher expression level of CD98hc.33 As recently published, higher expression of CD98hc is associated with a poor prognosis after radiochemotherapy in patients with HNSCC.33 Thus, these findings suggest that high level of CD98hc expression supports cell survival and tumor growth after radiotherapy. Here, we demonstrate for the first time that these chemo- and radioresistant tumor cells4 can be killed by (Uni)CAR T cells both in vitro and in vivo despite their more aggressive phenotype. This was demonstrated using different TMs directed either against EGFR or CD98 reported to be overexpressed on HNSCC.29,30 UniCAR T cells eradicate radioresistant tumor cells as efficiently as radiosensitive parental cells in an antigen-specific and TM-dependent manner. Both EGFR TM and CD98 TMs activated UniCAR T cells for tumor cell killing at low E:T ratios and picomolar concentrations which is in the same concentration range or even better than previously described TMs.21,22,24
For clinical applications, the immunogenic potential of novel therapeutic drugs should be reduced to a minimum. This is especially important for continuously and repeatedly infused components including TMs. Consequently, the CD98 TM described in this study was humanized to minimize the risk for induction of human anti-mouse antibodies (HAMA) that could potentially lead to neutralization/inactivation of a TM, or might even trigger severe, life-threatening responses, e.g., renal failure.59 As shown by comparative functional analyses, humanization does not impair the capability of the CD98 TM to engage UniCAR T cells for tumor cell killing. Hence, the novel huCD98 TM provides an improved immunogenic profile while maintaining its functional properties.
In contrast to EGFR, CD98 is found to be also expressed on UniCAR T cells. This finding is in line with the fact that CD98 is a well-known activation marker of immune cells including T cells.30,60 However, compared to HNSCC radioresistant cancer cell lines, CD98 antigen density on UniCAR T cells is considerably lower. Our in vitro co-cultivation experiments suggest that CD98-expressing UniCAR T cells can be cross-linked with each other via CD98 TMs which finally results in fratricide (Figure 4b). Nevertheless, substantial loss of UniCAR T cells in the presence of target cells could be observed only after 5 days. Thus, we assume that first killing of CD98high tumor cells and thereafter lysis of CD98low (UniCAR T) cells will take place. Prevalence for killing of high over low antigen-expressing cells was previously demonstrated for other T cell immunotherapies61 and is further supported by our data. In the presence of CD98high target cells, the huCD98 TM does not mediate fratricide of UniCAR T cells after 24 h, although 30% of UniCAR T cells were eliminated in the absence of target cells (Figure 4b). This underlines that targeting of ubiquitously expressed biomarkers including CD98 (but also EGFR) requires special safety management as provided by the UniCAR system to avoid unwanted “on-target, off-tumor” effects. After the elimination of tumor cells via redirected UniCAR T cells, long-term destruction of healthy tissues/cells can be prevented by stopping the TM supply. In order to further improve therapeutic safety, CD98 and EGFR are also promising candidates for “AND” gate targeting strategies in combination with other tumor-specific antigens.
Since both EGFR and CD98 were identified to positively correlate with a radioresistant phenotype and are promising biomarkers for radiosensitization,3,33,39,40 immunotargeting of these molecules might be a mutual profit for future combination with radiation. In HNSCC, complementary advances have been shown for EGFR-targeted immunotherapy with Cetuximab plus radiotherapy resulting in significantly prolonged progression-free survival of patients with locoregionally advanced HNSCC.62,63 Besides the induction of tumor cell death by DNA damage, ionizing radiation possesses immune-modulating properties that might exert synergistic effects on immunotherapeutic approaches like (Uni)CAR T cell therapy. Radiation of tumors results not only in the upregulation of certain TAAs as discussed for EGFR and CD98,3,33,39,40 but also remodels the vascular system, induces the secretion of T cell attracting chemokines and leads to upregulation of adhesion molecules.64,65 This in turn increases the antigenicity of tumors, improves tumor infiltration with therapeutic effector cells including (Uni)CAR T cells and facilitates an increased diffusion of antibody derivatives (e.g. TMs). In particular, combination of radiotherapy with EGFR- or CD98-redirected UniCAR T cells may further reduce the risk for tumor evasion. Potential immunotherapy-induced downregulation of EGFR and/or CD98 will presumably sensitize tumor cells to ionizing radiation. Reversion of radioresistance depending on the EGFR and CD98 expression levels was previously proven by other groups [e.g. 33,39]. Conversely, radiotherapy-induced upregulation of CD98 and EGFR may augment anti-tumor effects of the UniCAR system. Moreover, simultaneous or consecutive redirection of UniCAR T cells to two antigens (CD98 and EGFR) is assumed to reduce the risk for therapy-induced target antigen loss and subsequent tumor relapse as clinically observed for conventional, monospecific CAR T cell therapies.66,67
Overall, we successfully showed that radioresistant tumor cells can be killed via the UniCAR system by targeting both EGFR and CD98 in a highly efficient and antigen-specific manner. Thus, immunotherapy with UniCAR T cells might be able to overcome radioresistance.
Funding Statement
This work was supported by the National Center for Tumor Diseases (NCT), Dresden, Germany: German Cancer Research Center (DKFZ), Heidelberg, Germany; Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany; Helmholtz-Zentrum Dresden-Rossendorf (HZDR), Dresden, Germany under Grant number 1030000128.
Acknowledgments
We thank Julia Lagler for excellent technical assistance with in vitro assays as well as David Szöllösi and Ildikó Horvát for their support with studies in immunocompromised mice.
Disclosure of potential conflict of interest
MB has filed patents related to the UniCAR system. MB is a shareholder of the company GEMoaB which owns the IP related to the UniCAR system. The other authors have declared that no competing interest exists.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
{kind=link}
References
- 1.Baumann M, Krause M, Overgaard J, Debus J, Bentzen SM, Daartz J, Richter C, Zips D, Bortfeld T.. Radiation oncology in the era of precision medicine. Nat Rev Cancer. 2016. April;16(4):234–12. Epub 2016 Mar 18. doi: 10.1038/nrc.2016.18. [DOI] [PubMed] [Google Scholar]
- 2.Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers (Basel). 2019. June 21;11(6):E862. doi: 10.3390/cancers11060862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. 2016. January 14;17(1):E102. doi: 10.3390/ijms17010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurth I, Hein L, Mäbert K, Peitzsch C, Koi L, Cojoc M, Kunz-Schughart L, Baumann M, Dubrovska A. Cancer stem cell related markers of radioresistance in head and neck squamous cell carcinoma. Oncotarget. 2015. October 27;6(33):34494–34509. doi: 10.18632/oncotarget.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeman EM. 2012. Biologic basis of radiation oncology, in clinical radiation oncology. 3ed.UK: Saunders Elsevier. [Google Scholar]
- 6.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018. July 5;379(1):64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, Blumenthal GM, Bryan W, McKee AE, Pazdur R. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019. March 15;25(6):1702–1708. Epub 2018 Nov 9. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 8.Freyer CW. Tisagenlecleucel: the first CAR on the highway to remission for acute lymphoblastic leukemia. J Adv Pract Oncol. 2018. Jul-Aug;9(5):537–544. Epub 2018 Jul 1. [PMC free article] [PubMed] [Google Scholar]
- 9.Gill S, Maus MV, Porter DL. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016. May;30(3):157–167. Epub 2015 Nov 6. doi: 10.1016/j.blre.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, Temme A, Schmitz M. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol. 2010;2010:956304. Epub 2010 May 5. doi: 10.1155/2010/956304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng PP, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today. 2018. June;23(6):1175–1182. Epub 2018 Mar 1. doi: 10.1016/j.drudis.2018.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018. January;15(1):47–62. Epub 2017 Sep 19. doi: 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ Jr.. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012. April 1;72(7):1844–1852. Epub 2012 Feb 7. doi: 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015. March 4;137(8):2832–2835. Epub 2015 Feb 24. doi: 10.1021/jacs.5b00106. [DOI] [PubMed] [Google Scholar]
- 15.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016. January 26;113(4):E459–68. Epub 2016 Jan 12. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachmann M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunology Letters. 2019. May;211:13–22. doi: 10.1016/j.imlet.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Arndt C, Bachmann M, Bergmann R, Berndt N, Feldmann A, Koristka S. Theranostic CAR T cell targeting: A brief review. J Labelled Comp Radiopharm. 2019. June 30;62(8):533–540. Epub 2019 Jun 6. doi: 10.1002/jlcr.3727. [DOI] [PubMed] [Google Scholar]
- 18.Feldmann A, Arndt C, Koristka S, Berndt N, Bergmann R, Bachmann M. Conventional CARs versus modular CARs. Cancer Immunol Immunother. 2019. October;68(10):1713–1719. Epub 2019 Sep 21. Review. PMID: 31542798. doi: 10.1007/s00262-019-02399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koristka S, Cartellieri M, Feldmann A, Arndt C, Loff S, Michalk I, Aliperta R, von Bonin M, Bornhäuser M, Ehninger A, et al. Flexible antigen-specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood. 2014;124(21):3494. doi: 10.1182/blood.V124.21.3494.3494. [DOI] [Google Scholar]
- 20.Cartellieri M, Loff S, von Bonin M, Bejestani EP, Ehninger A, Feldmann A, Koristka S, Arndt C, Ehninger G, Bachmann M. Unicar: A novel modular retargeting platform technology for CAR T cells. Blood. 2015;126(23):5549. doi: 10.1182/blood.V126.23.5549.5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arndt C, Feldmann A, Koristka S, Schäfer M, Bergmann R, Mitwasi N, Berndt N, Bachmann D, Kegler A, Schmitz M, et al. A theranostic PSMA ligand for PET imaging and retargeting of T cells expressing the universal chimeric antigen receptor UniCAR. Oncoimmunology. 2019. September 7;8(11):1659095. eCollection 2019. PMID: 31646084. doi: 10.1080/2162402X.2019.1659095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, Ludwig F, Ziller-Walter P, Kegler A, Gärtner S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017. February 6;6(4):e1287246. eCollection 2017. doi: 10.1080/2162402X.2017.1287246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, Schmitz M, Pietzsch J, Steinbach J, Bachmann M. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget. 2018. May 22;9(39):25597–25616. eCollection 2018 May 22. doi: 10.18632/oncotarget.25390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016. August 12;6(8):e458. doi: 10.1038/bcj.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachmann D, Aliperta R, Bergmann R, Feldmann A, Koristka S, Arndt C, Loff S, Welzel P, Albert S, Kegler A, et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget. 2017. December 21;9(7):7487–7500. eCollection 2018 Jan 26. doi: 10.18632/oncotarget.23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loureiro LR, Feldmann A, Bergmann R, Koristka S, Berndt N, Arndt C, Pietzsch J, Novo C, Videira P, Bachmann M. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018. August 22;8(9):81. doi: 10.1038/s41408-018-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jureczek J, Bergmann R, Berndt N, Koristka S, Kegler A, Puentes-Cala E, Soto JA, Arndt C, Bachmann M, Feldmann A. An oligo-His-tag of a targeting module does not influence its biodistribution and the retargeting capabilities of UniCAR T cells. Sci Rep. 2019. July 22;9(1):10547. doi: 10.1038/s41598-019-47044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koristka S, Cartellieri M, Arndt C, Bippes CC, Feldmann A, Michalk I, Wiefel K, Stamova S, Schmitz M, Ehninger G, et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J Autoimmun. 2013. May;42:105–116. Epub 2013 Jan 22. doi: 10.1016/j.jaut.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014. January;79:34–74. Epub 2013 Nov 20. doi: 10.1016/j.phrs.2013.11.002.. [DOI] [PubMed] [Google Scholar]
- 30.Cantor JM, Ginsberg MH. CD98 at the crossroads of adaptive immunity and cancer. J Cell Sci. 2012. March 15;125(Pt 6):1373–1382. Epub 2012 Apr 12. doi: 10.1242/jcs.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Napolitano L, Scalise M, Galluccio M, Pochini L, Albanese LM, Indiveri C. LAT1 is the transport competent unit of the LAT1/CD98 heterodimeric amino acid transporter. Int J Biochem Cell Biol. 2015. October;67:25–33. Epub 2015 Aug 6. doi: 10.1016/j.biocel.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Rosell A, Meury M, Álvarez-Marimon E, Costa M, Pérez-Cano L, Zorzano A, Fernández-Recio J, Palacín M, Fotiadis D. Structural bases for the interaction and stabilization of the human amino acid transporter LAT2 with its ancillary protein 4F2hc. Proc Natl Acad Sci U S A. 2014. February 25;111(8):2966–2971. Epub 2014 Feb 10. doi: 10.1073/pnas.1323779111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Digomann D, Kurth I, Tyutyunnykova A, Chen O, Löck S, Gorodetska I, Peitzsch C, Skvortsova II, Negro G, Aschenbrenner B, et al. The CD98 heavy chain is a marker and regulator of head and neck squamous cell carcinoma radiosensitivity. Clin Cancer Res. 2019. May 15;25(10):3152–3163. Epub 2019 Jan 22. doi: 10.1158/1078-0432.CCR-18-2951. [DOI] [PubMed] [Google Scholar]
- 34.Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90(11):824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 35.Ang KK, Andratschke NH, Milas L. Epidermal growth factor receptor and response of head-and-neck carcinoma to therapy. Int J Radiat Oncol Biol Phys. 2004;58(3):959–965. doi: 10.1016/j.ijrobp.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 36.Eriksen JG, Steiniche T, Askaa J, Alsner J, Overgaard J. The prognostic value of epidermal growth factor receptor is related to tumor differentiation and the overall treatment time of radiotherapy in squamous cell carcinomas of the head and neck. Int J Radiat Oncol Biol Phys. 2004;58(2):561–566. doi: 10.1016/j.ijrobp.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 37.Ang KK, Berkey BA, Tu X, Zhang H-Z, Katz R, Hammond EH, Fu KK, Milas L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–7356. [PubMed] [Google Scholar]
- 38.Martens-de Kemp SR, Brink A, Stigter-van Walsum M, Damen JM, Rustenburg F, Wu T, van Wieringen WN, Schuurhuis GJ, Braakhuis BJ, Slijper M, et al. CD98 marks a subpopulation of head and neck squamous cell carcinoma cells with stem cell properties. Stem Cell Res. 2013. May;10(3):477–488. Epub 2013 Feb 18. doi: 10.1016/j.scr.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Liang K, Ang KK, Milas L, Hunter N, Fan Z. The epidermal growth factor receptor mediates radioresistance. Int J Radiat Oncol Biol Phys. 2003;57(1):246–254. doi: 10.1016/S0360-3016(03)00511-X. [DOI] [PubMed] [Google Scholar]
- 40.Bonner JA, Maihle NJ, Folven BR, Christianson TJ, Spain K. The interaction of epidermal growth factor and radiation in human head and neck squamous cell carcinoma cell lines with vastly different radiosensitivities. Int J Radiat Oncol Biol Phys. 1994;29(2):243–247. doi: 10.1016/0360-3016(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 41.Feldmann A, Stamova S, Bippes CC, Bartsch H, Wehner R, Schmitz M, Temme A, Cartellieri M, Bachmann M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: comparison of different antibody formats. Prostate. 2011. June 15;71(9):998–1011. Epub 2010 Dec 28. doi: 10.1002/pros.21315. [DOI] [PubMed] [Google Scholar]
- 42.Feldmann A, Arndt C, Töpfer K, Stamova S, Krone F, Cartellieri M, Koristka S, Michalk I, Lindemann D, Schmitz M, et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J Immunol. 2012. September 15;189(6):3249–3259. Epub 2012 Aug 8. doi: 10.4049/jimmunol.1200341. [DOI] [PubMed] [Google Scholar]
- 43.Stamova S, Cartellieri M, Feldmann A, Arndt C, Koristka S, Bartsch H, Bippes CC, Wehner R, Schmitz M, von Bonin M, et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol Immunol. 2011. December;49(3):474–482. Epub 2011 Oct 19. doi: 10.1016/j.molimm.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 44.Arndt C, Koristka S, Feldmann A, Bergmann R, Bachmann M. Coomassie brilliant blue staining of polyacrylamide gels. Methods Mol Biol. 2018;1853:27–30. doi: 10.1007/978-1-4939-8745-0_4. [DOI] [PubMed] [Google Scholar]
- 45.Arndt C, Feldmann A, Töpfer K, Koristka S, Cartellieri M, Temme A, Ehninger A, Ehninger G, Bachmann M. Redirection of CD4+ and CD8+ T lymphocytes via a novel antibody-based modular targeting system triggers efficient killing of PSCA+ prostate tumor cells. Prostate. 2014. September;74(13):1347–1358. Epub 2014 Jul 22. doi: 10.1002/pros.22851. [DOI] [PubMed] [Google Scholar]
- 46.Cartellieri M, Koristka S, Arndt C, Feldmann A, Stamova S, von Bonin M, Töpfer K, Krüger T, Geib M, Michalk I, et al. A novel ex vivo isolation and expansion procedure for chimeric antigen receptor engrafted human T cells. PLoS One. 2014. April 3;9(4):e93745. eCollection 2014. doi: 10.1371/journal.pone.0093745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017. May 9;8(19):31368–31385. doi: 10.18632/oncotarget.15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S, Kegler A, Jureczek J, Hoffmann A, Ehninger A, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. 2017. September 18;8(65):108584–108603. eCollection 2017 Dec 12. doi: 10.18632/oncotarget.21017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koristka S, Kegler A, Bergmann R, Arndt C, Feldmann A, Albert S, Cartellieri M, Ehninger A, Ehninger G, Middeke JM, et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J Autoimmun. 2018. June;90:116–131. Epub 2018 Mar 2. doi: 10.1016/j.jaut.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 50.Koristka S, Ziller-Walter P, Bergmann R, Arndt C, Feldmann A, Kegler A, Cartellieri M, Ehninger A, Ehninger G, Bornhäuser M, et al. Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol Immunother. 2019. August 14;68(9):1401–1415. Epub ahead of print. doi: 10.1007/s00262-019-02376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fasslrinner F, Arndt C, Koristka S, Feldmann A, Altmann H, von Bonin M, Schmitz M, Bornhäuser M, Bachmann M. Midostaurin abrogates CD33-directed UniCAR and CD33-CD3 bispecific antibody therapy in acute myeloid leukaemia. Br J Haematol. 2019. September;186(5):735–740. Epub 2019 May 23. doi: 10.1111/bjh.15975. [DOI] [PubMed] [Google Scholar]
- 52.Arndt C, Feldmann A, von Bonin M, Cartellieri M, Ewen EM, Koristka S, Michalk I, Stamova S, Berndt N, Gocht A, et al. Costimulation improves the killing capability of T cells redirected to tumor cells expressing low levels of CD33: description of a novel modular targeting system. Leukemia. 2014. January;28(1):59–69. Epub 2013 Aug 20. doi: 10.1038/leu.2013.243. [DOI] [PubMed] [Google Scholar]
- 53.Minn I, Rowe SP, Pomper MG. Enhancing CAR T-cell therapy through cellular imaging and radiotherapy. Lancet Oncol. 2019. August;20(8):e443–e451. Epub 2019 Jul 29. doi: 10.1016/S1470-2045(19)30461-9. [DOI] [PubMed] [Google Scholar]
- 54.DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, Sadelain M. Low-dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol Ther. 2018. November 7;26(11):2542–2552. Epub 2018 Sep 13. doi: 10.1016/j.ymthe.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018. February 15;78(4):1031–1043. Epub 2017 Dec 8. doi: 10.1158/0008-5472.CAN-17-1788. [DOI] [PubMed] [Google Scholar]
- 56.Lo Nigro C, Denaro N, Merlotti A, Merlano M. Head and neck cancer: improving outcomes with a multidisciplinary approach. Cancer Manag Res. 2017. August 18;9:363–371. eCollection 2017. doi: 10.2147/CMAR.S115761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saloura V, Cohen EE, Licitra L, Billan S, Dinis J, Lisby S, Gauler TC. An open-label single-arm, phase II trial of zalutumumab, a human monoclonal anti-EGFR antibody, in patients with platinum-refractory squamous cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 2014. June;73(6):1227–1239. Epub 2014 Apr 9. doi: 10.1007/s00280-014-2459-z. [DOI] [PubMed] [Google Scholar]
- 58.Ling DC, Bakkenist CJ, Ferris RL, Clump DA. Role of immunotherapy in head and neck cancer. Semin Radiat Oncol. 2018. January;28(1):12–16. doi: 10.1016/j.semradonc.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 59.Hwang WY1, Foote J. Immunogenicity of engineered antibodies. Methods. 2005. May;36(1):3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 60.Haynes BF, Hemler ME, Mann DL, Eisenbarth GS, Shelhamer J, Mostowski HS, Thomas CA, Strominger JL, Fauci AS. Characterization of a monoclonal antibody (4F2) that binds to human monocytes and to a subset of activated lymphocytes. J Immunol. 1981. April;126(4):1409–1414. [PubMed] [Google Scholar]
- 61.Krupka C, Kufer P, Kischel R, Zugmaier G, Bögeholz J, Köhnke T, Lichtenegger FS, Schneider S, Metzeler KH, Fiegl M, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood. 2014. January 16;123(3):356–365. Epub 2013 Dec 3. doi: 10.1182/blood-2013-08-523548. [DOI] [PubMed] [Google Scholar]
- 62.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006. February 9;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 63.Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010. January;11(1):21–28. Epub 2009 Nov 10. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 64.Flynn JP, O’Hara MH, Gandhi SJ. Preclinical rationale for combining radiation therapy and immunotherapy beyond checkpoint inhibitors (i.e., CART). Transl Lung Cancer Res. 2017. April;6(2):159–168. doi: 10.21037/tlcr.2017.03.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eckert F, Gaipl US, Niedermann G, Hettich M, Schilbach K, Huber SM, Zips D. Beyond checkpoint inhibition - Immunotherapeutical strategies in combination with radiation. Clin Transl Radiat Oncol. 2017. February 4;2:29–35. eCollection 2017 Feb. doi: 10.1016/j.ctro.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiaoqing L, Weihong C. Mechanisms of failure of chimeric antigen receptor T-cell therapy. Curr Opin Hematol. 2019. November;26(6):427–433. doi: 10.1097/MOH.0000000000000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Timmers M, Roex G, Wang Y, Campillo-Davo D, Van Tendeloo VFI, Chu Y, Berneman ZN, Luo F, Van Acker HH, Anguille S. Chimeric antigen receptor-modified T cell therapy in multiple myeloma: beyond B cell maturation antigen. Front Immunol. 2019. July 16;10:1613. eCollection 2019. doi: 10.3389/fimmu.2019.01613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.