

SUMMARY:
Genomic studies of cell differentiation and function within a whole organism depend on the ability to isolate specific cell types from a tissue, but this is technically difficult. We developed a method called INTACT (for isolation of nuclei tagged in specific cell types) that allows affinity-based isolation of nuclei from individual cell types of a tissue, thereby circumventing the problems associated with mechanical purification techniques. In this method nuclei are affinity labeled through transgenic expression of a biotinylated nuclear envelope protein in the cell type of interest. Total nuclei are isolated from transgenic plants and biotin-labeled nuclei are then purified using streptavidin-coated magnetic beads without the need for specialized equipment. INTACT gives high yield and purity of nuclei from the desired cell type, which can be used for genome-wide analysis of gene expression and chromatin features. The entire procedure, from nuclei purification through cDNA preparation or chromatin immunoprecipitation (ChIP), can be completed within two days. The protocol we present assumes that transgenic lines are already available, and includes procedural details for amplification of cDNA or ChIP DNA prior to microarray or deep sequencing analysis.
Keywords: cell sorting, histone modification, epigenome, development, root
INTRODUCTION:
The tissues and organs of a multicellular organism are made up of a large number of highly specialized cell types, each of which has a specific phenotype and function. These specialized cell types are produced from undifferentiated progenitors through epigenetic reprogramming of the stem cell genome to establish the specific transcriptional program that underlies the unique characteristics of each cell type. Defining the mechanisms of cellular differentiation is critical for understanding how multicellular organisms build their bodies and for learning how to treat diseases such as cancer that can result from malfunctions in maintenance of the differentiated state. Despite the importance of this problem, our knowledge of the mechanics of cell differentiation within whole organisms is still quite limited, in large part due to the technical difficulty associated with purifying individual cell types from a tissue for transcriptional and epigenomic profiling.
Existing methods for studying specific cell types include the use of cultured cell lines1,2, ex vivo differentiation from progenitor cells3,4, laser capture microdissection (LCM) of fixed tissue sections5–7, and fluorescence-activated cell sorting (FACS) of fluorescently labeled cells or nuclei8–12. Of these techniques, LCM and FACS are the only ones amenable to use with whole tissue, but both methods are limited in that they use harsh treatment conditions, require highly complex and expensive equipment, and offer less than optimal yield and purity of the target cell type. Several recently developed methods, such as cell type-specific chemical modification of RNA13 and affinity tagging of ribosomal proteins or poly(A)-binding proteins14–16 have also been used to measure gene expression profiles in individual cell types of a tissue, but these techniques are not applicable to the study of chromatin or other nuclear components.
Given the limitations of existing methods, we developed a straightforward and generally applicable method for studying gene expression and chromatin profiles in individual cell types. Our strategy was to transgenically tag nuclei in specific cell types and then use affinity isolation to purify them from the total pool of nuclei derived from a tissue, thereby avoiding the need for dissociating and mechanically purifying whole cells. We refer to this strategy as INTACT, for isolation of nuclei tagged in specific cell types. We previously used the INTACT system to study each of the two cell types of the Arabidopsis root epidermis and found that the method gave high yield and purity of nuclei from each cell type, and these nuclei could be used for both genome-wide gene expression and chromatin profiling17. INTACT has several important advantages in that, unlike LCM and FACS, it provides nearly 100% purity of nuclei from the desired cell type in quantities sufficient for epigenomic profiling, it is a rapid procedure that avoids harsh tissue treatment, and it does not require specialized skills or instrumentation. The INTACT method is limited only by the need for a promoter or enhancer trap that is active in the cell type of interest, and the time required to make transgenic organisms. This method should be adaptable to any organism that is amenable to transformation.
Constructs and transgenic lines for INTACT
The strategy for enabling purification of nuclei from individual cell types is to affinity label the nuclear surface through transgenic expression of a biotinylated nuclear envelope protein in the cell type of interest. This nuclear targeting fusion protein (NTF) consists of three parts: the WPP domain of Arabidopsis RAN GTPASE ACTIVATING PROTEIN 1 (RanGAP1, At3g63130; amino acids 1–111, inclusive), which is necessary and sufficient for nuclear envelope association in plants18, green fluorescent protein (GFP) for visualization, and the biotin ligase recognition peptide (BLRP), which serves as a substrate for the E. coli biotin ligase BirA19 (Fig. 1a). Thus, expression of BirA and the NTF in the same cell type produces biotin-labeled nuclei exclusively in those cells. These nuclei can then be purified from the total nuclear pool by virtue of the interaction between biotin and streptavidin. We and others have previously shown that the total and nuclear RNA pools are essentially comparable in terms of their mRNA makeup17,20,21, therefore the nuclear RNA obtained can be used for accurate global analysis of gene expression in the cell type of interest. In addition, the high yield of purified nuclei provides enough material for genome-wide profiling of chromatin features.
Figure 1. Nuclear targeting fusion (NTF) protein and transgenic lines for the INTACT system.
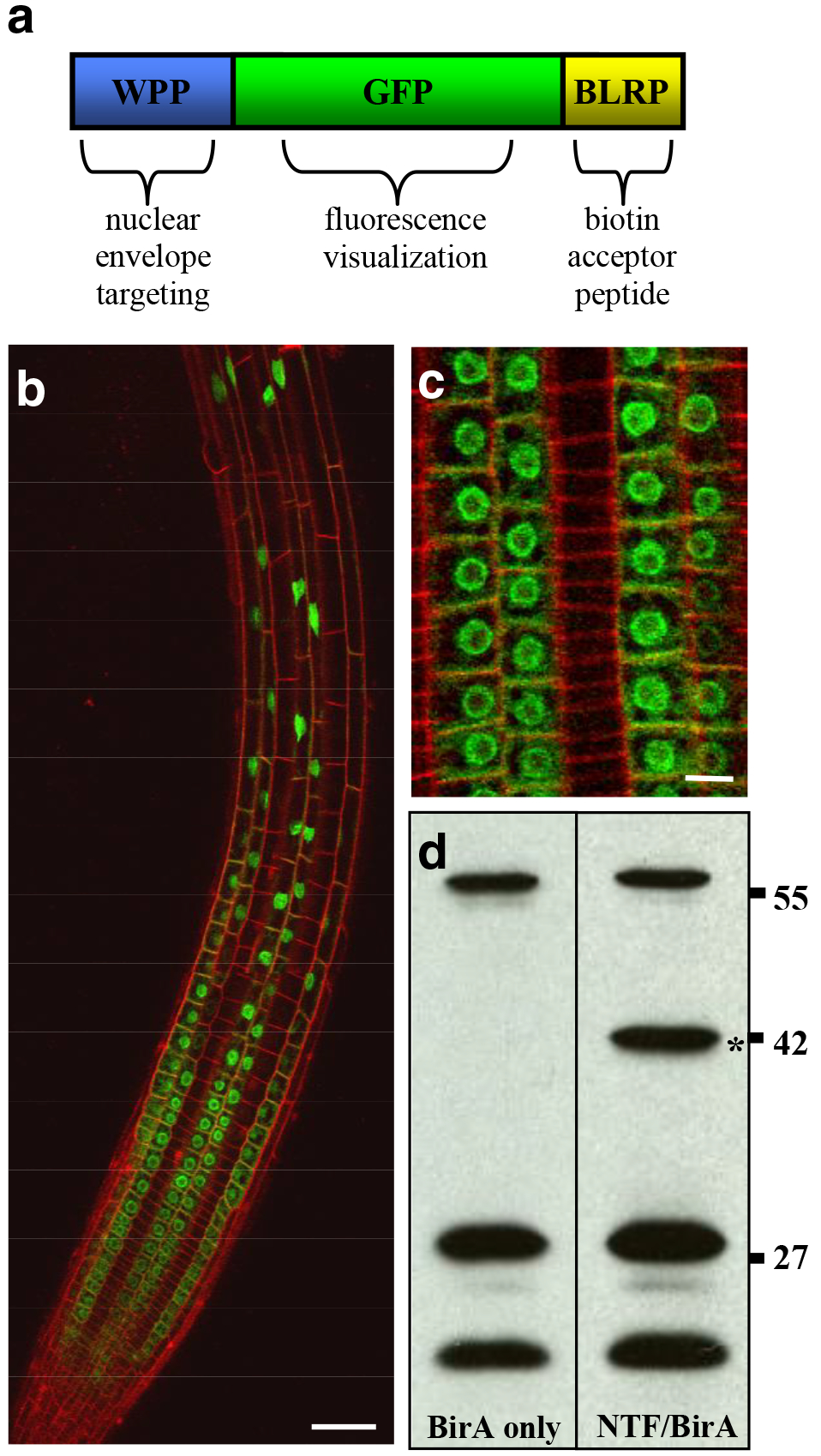
(a) The three-part structure of the NTF is shown. The chimeric protein consists of the WPP domain of RanGAP1 for nuclear envelope targeting, green fluorescent protein (GFP) to allow visualization, and the biotin ligase recognition peptide (BLRP), which is biotinylated by BirA. (b) Confocal projection image of an Arabidopsis root expressing the NTF in the epidermal non-hair cells. GFP is shown in green and cell walls are shown in red. Scale bar is approximately 20 μm. (c) Confocal section of a root expressing the NTF in non-hair cells, showing localization to the nuclear envelope. Scale bar is approximately 5 μm. (d) Streptavidin western blots of protein extracts from plants expressing BirA only or both the NTF and BirA. Asterisk indicates the position of the 42 kDa NTF. Bands found in both BirA only and NTF/BirA protein extracts are endogenous biotinylated proteins. Molecular weights (in kDa) are indicated on the right.
Use of the INTACT system in animals will require a nuclear envelope targeting sequence other than the RanGAP1 WPP domain because this protein uses a plant-specific envelope targeting mechanism22. However, a variety of other nuclear envelope proteins could possibly be used, including the nucleoporin proteins or the C-terminus of animal RanGAP.
In order to label nuclei in a cell type of interest, the NTF and BirA must both be expressed in that cell type. This can be achieved by expressing the NTF gene from a cell type-specific promoter or enhancer trap while expressing BirA constitutively. Alternatively, both components could be expressed from the same cell type-specific promoter, and this should work equally well. However, maintaining a transgenic line with one component constitutively expressed is convenient in that the second component can be placed under cell type-specific regulatory sequences and transformed into this line, thereby avoiding the need to make both components specific to each cell type to be examined. We previously used the ACTIN2 (ACT2) promoter to drive constitutive expression of BirA in all vegetative tissues23,24, and the NTF gene was expressed in either root epidermal hair cells or non-hair cells using the ACTIN DEPOLYMERIZING FACTOR 8 (ADF8) promoter25 or the GLABRA2 (GL2) promoter26, respectively17.
Once transgenic plants expressing both the NTF and BirA have been produced, they need to be analyzed to confirm expected expression of the transgene, proper protein localization to the nuclear envelope, and biotinylation of the NTF. Tissue-level expression pattern and nuclear envelope localization of the NTF can both be evaluated by fluorescence microscopy since the NTF contains GFP. Fluorescence of the NTF should be detectable exclusively in the targeted cell type and should be clearly concentrated around the nuclear envelope when viewed by confocal microscopy, although a small fraction of the protein may be cytoplasmic (Fig. 1b and c). Testing for biotinylation of the NTF can be done by analyzing protein extracts from plants expressing NTF and BirA by western blotting with streptavidin. In addition to several endogenous biotinylated proteins, the 42 kDa NTF band should also be present if the protein is being biotinylated to a sufficient extent. As a negative control, it is important to also include protein extract from plants expressing only BirA in this experiment (Fig. 1d). If the transgenic lines show proper expression, localization, and biotinylation of the NTF then they are suitable for purifying nuclei from the target cell type.
Purification of labeled nuclei
The procedure for purifying labeled nuclei involves preparation of total nuclei from the tissue, binding of streptavidin-coated magnetic beads to the labeled nuclei, and collection of the bead-bound nuclei by allowing the mixture of beads and nuclei to flow past a magnet. The apparatus used for this flow purification is shown in Fig. 2a. The binding of streptavidin beads to the biotinylated nuclei is done in a small volume to maintain a close proximity of beads and nuclei, thereby enhancing binding efficiency (see Fig. 2b and Box 1). However, once binding of beads to the nuclei is complete, the mixture is diluted 10-fold prior to collection of bound nuclei on the magnet. This dilution provides greater spatial separation of bead-bound nuclei from other nuclei and debris, which is essential for obtaining a high level of purity. To ensure that nuclei are of the highest possible purity, two rounds of flow purification are employed. We have found that this liquid flow-based scheme gives much higher purity of nuclei than a stationary setup in which the tube containing bead-bound nuclei is simply placed against a magnet to collect the beads and then decanted.
Figure 2. Purification of tagged nuclei with the INTACT system.
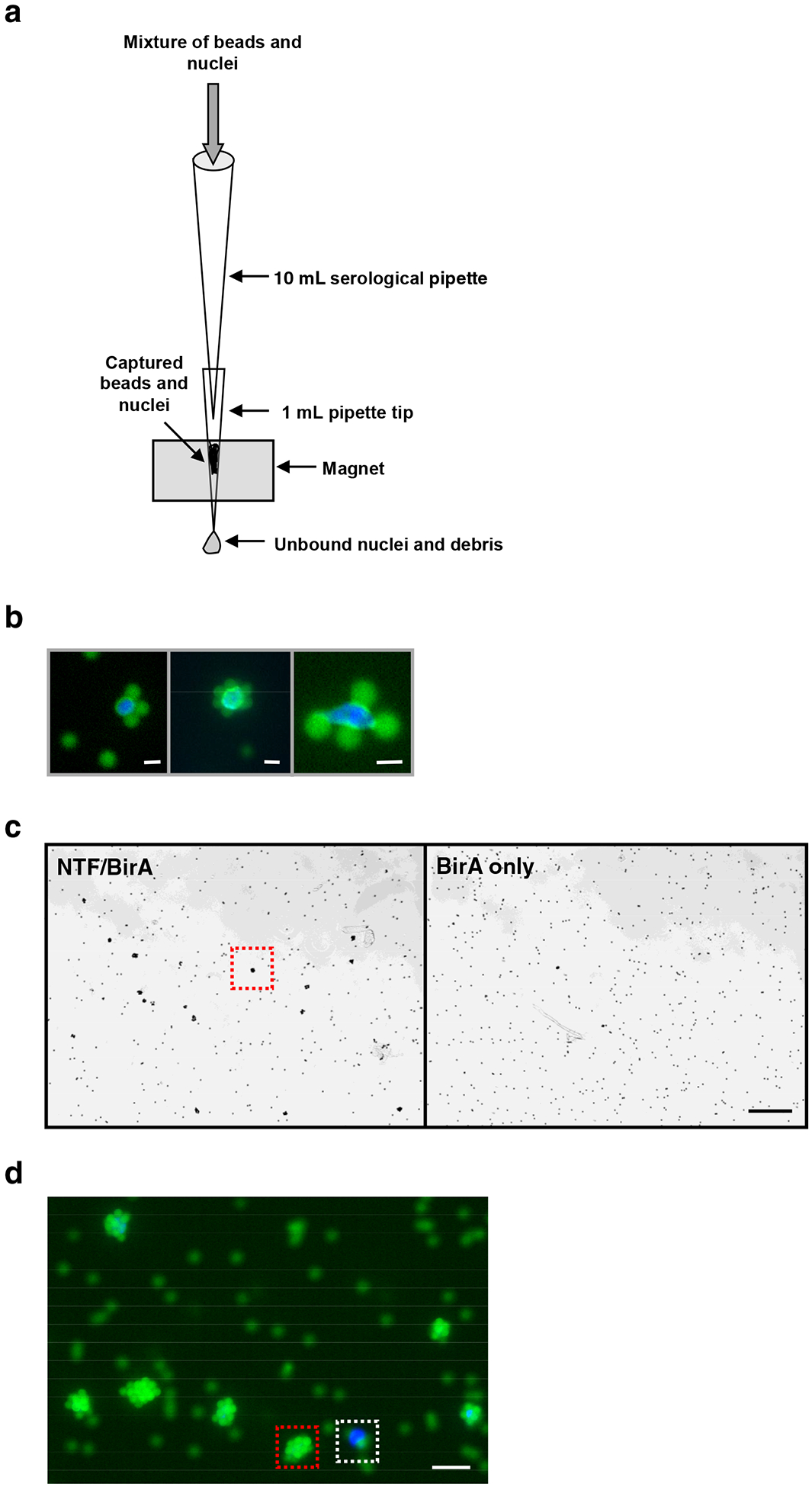
(a) Diagram of the apparatus used for purification of bead-bound nuclei. The core of the device consists of a 1 ml micropipette tip inserted into a groove in the surface of a miniMACS magnet. After binding of magnetic streptavidin-coated beads to biotinylated nuclei in solution, this mixture of beads and nuclei is diluted and drawn into a 10 ml serological pipette, and the pipette is then attached to the 1 ml tip in the magnet. The mixture of beads and nuclei in the 10 ml pipette is allowed to flow slowly past the magnet, allowing capture of beads and nuclei on the wall of the 1 ml tip. Once the liquid is drained, the 1 ml tip is attached to a pipette and the purified nuclei and beads are released into solution by drawing liquid into and out of the pipette tip. (b) Fluorescence microscopy images of NTF-labeled nuclei that have been bound by streptavidin-coated magnetic beads prior to capture on a magnet. DNA is shown in blue and GFP fluorescence of the NTF, as well as fluorescence from the beads themselves, are shown in green. Scale bar is approximately 3 μm in each panel. The right panel is at a higher magnification than the left and center panels. (c) Low magnification brightfield images of bead-bound nuclei and free beads after magnetic purification. The left panel shows the purified material from a transgenic line expressing NTF and BirA in root epidermal non-hair cells, while the right panel shows that from a line expressing only BirA. Labeled nuclei become fully coated with beads during capture on the magnet and therefore appear as conspicuous clusters of beads after purification. One such bead cluster is highlighted by a red dotted box in the NTF/BirA panel. No beads clusters are seen in the BirA only panel, indicating that the purification specifically recovers biotinylated nuclei from the cell type of interest. Scale bar is approximately 100 μm in each panel. (d) Higher magnification fluorescence image of purified material from the NTF/BirA line, as shown in the left panel of (c). The fluorescence of DNA is shown in blue and the fluorescence of beads is shown in green. Purity of the nuclei can be assessed by comparing the number of bead-bound nuclei that show green fluorescence from the beads (red box) to the number of free nuclei that show only DAPI fluorescence and are not bound by beads (white box). Scale bar is approximately 10 μm.
BOX 1: Analysis of starting nuclei preparation and bead binding.
The nuclei preparation is stained with DAPI in step 7 so that nuclei yield and quality as well as streptavidin bead binding to NTF-tagged nuclei can be assessed. This procedure requires a fluorescence microscope with visible light and both DAPI and GFP fluorescence channels.
Assess initial nuclei preparation and binding of beads to nuclei as follows:
Place 10 μl of the nuclei/bead mixture (of the 20 μl sample taken at step 10) into the chamber of the hemacytometer.
Using the hemacytometer grid, count the number of DAPI-stained nuclei by observation on a fluorescence microscope using DAPI excitation along with just enough visible light to allow the grid lines to be seen. Determine the yield of total nuclei and compare to the expected yield considering the type and amount of starting tissue. The yield of nuclei should be in the expected range and the nuclei should appear mostly undamaged.
-
Using DAPI excitation and visible light together, the nuclei and beads will both be visible. Inspect the field of DAPI-stained nuclei and identify those that appear to be in direct contact with multiple beads. Switch to the GFP excitation channel and confirm that each of these nuclei also has GFP fluorescence around the nuclear envelope (beads will also fluoresce green at the GFP excitation wavelength). These arrangements represent bead binding to NTF-labeled nuclei (see Fig. 2b). The total number of bead-bound nuclei can be determined with the hemacytometer grid, and this number should be in the range expected based on the abundance of the targeted cell type.
? TROUBLESHOOTING
The purity of the isolated nuclei can be assessed at the end of the experiment by calculating the percentage of contaminating nuclei present in the final preparation (Box 2). The NTF-labeled nuclei become fully encrusted with beads during capture on the magnet, therefore it is not possible to simply measure the number of GFP-positive versus GFP-negative nuclei in order to calculate purity, as the GFP signal from the NTF is obscured by beads. However, by staining the total nuclei preparation with the fluorescent DNA-binding dye DAPI at the beginning of the procedure, purity can be determined by measuring the ratio of bead-bound nuclei, which appear as conspicuous clusters of beads, to unbound nuclei which are bead-free and have DAPI fluorescence (Fig. 2c and d).
BOX 2: Determining final purity and yield of tagged nuclei.
The bead-bound nuclei become fully encrusted with beads during capture on the magnet, therefore GFP fluorescence from the NTF is obscured by beads in the purified nuclei preparation. In addition, the beads themselves are green fluorescent under GFP excitation. For these reasons it is not possible to calculate nuclear purity at the end of the procedure by simply comparing the number of GFP-positive versus GFP-negative nuclei. However, by staining the total nuclei preparation with DAPI at the beginning of the procedure (step 7), purity can be determined by measuring the ratio of bead-bound nuclei, which appear as conspicuous clusters of beads, to unbound nuclei which are bead-free and show strong DAPI fluorescence (seeFig. 2c and d).
Determine the final purity and yield of tagged nuclei as follows:
Place 10 μl of the mixture of beads and nuclei (of the 20 μl sample taken at step 20) into the chamber of the hemacytometer.
Observe the slide using DAPI excitation along with just enough visible light to allow the hemacytometer grid and beads to be seen. Count the number of large bead clusters (purified target nuclei) and the number of DAPI-stained nuclei that are not associated with beads (contaminating nuclei). DAPI fluorescence can generally be seen emanating from between the beads covering the target nuclei as well.
Measure purity by calculating the ratio of target nuclei to contaminating nuclei.
-
Use the hemacytometer grid to determine the total yield of purified nuclei.
? TROUBLESHOOTING
If the goal of an experiment is chromatin profiling using standard crosslinked chromatin immunoprecipitation (X-ChIP) protocols, then the tissue must be fixed with formaldehyde prior to nuclei purification. However, this pre-treatment is omitted for gene expression and DNA methylation profiling, as well as for chromatin-based assays such as native ChIP, nucleosome salt-fractionation, or DNAseI hypersensitivity mapping. When purifying nuclei for expression analysis it is important to ensure that all reagents and equipment that come into contact with the nuclei are free of RNases.
Whether tissue is pre-treated with formaldehyde or not, the procedure for nuclei purification is the same. For each expression or chromatin profiling experiment it is desirable to obtain 1–5 × 105 purified nuclei from the cell type of interest. In our experience a typical INTACT purification gives 50–70% of the theoretical yield of labeled nuclei. Therefore, it is important to take into account the abundance of the cell type of interest and the expected yield when determining the amount of starting tissue that will be needed.
Gene expression and chromatin profiling
The methods presented here for preparation of nucleic acids for profiling of global gene expression and chromatin features are essentially scaled-down versions of standard protocols for these types of experiments. Commercially available kits are used at several points in each protocol, and step-by-step instructions are given when the use of a kit deviates from the manufacturer’s directions. Otherwise, users are referred to the standard instructions that are provided with each kit.
For gene expression profiling, RNA is isolated from nuclei and cDNA is prepared by random priming, followed by PCR-based amplification using the Sigma Whole Transcriptome Amplification kit. The ChIP protocol we present is a modified version of the procedure developed by Gendrel et al.27, in which the immunoprecipitated DNA is amplified using the Sigma Single Cell Whole Genome Amplification kit at the end of the procedure. Each of these protocols assumes that microarrays will be used as the readout platform for the experiment, and therefore adjustments to the amplification methods may need to be made if deep sequencing technologies are to be used.
MATERIALS:
REAGENTS
Transgenic plants expressing NTF and BirA in the cell type of interest (NTF and BirA DNA constructs are available from the authors, as are transgenic lines that express NTF and BirA in root hair or non-hair cells of Arabidopsis17)
Appropriate plant growth media
37% Formaldehyde (J.T. Baker cat no. 2106–01) ! CAUTION Formaldehyde is harmful if inhaled or absorbed through skin. Waste should be disposed of according to local regulations.
Glycine (Fisher cat. no. BP381)
Liquid nitrogen (LN2)
Nuclei purification buffers (see REAGENT SETUP)
3-(N-Morpholino) propanesulfonic acids (MOPS) buffer (Sigma cat. no. M9381)
NaCl (Sigma cat no. S3014)
KCl (Sigma cat. no. P9333)
Ethylenediaminetetraacetic acid (EDTA) (Sigma cat. no. 431788)
Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) (Sigma cat. no. E3899)
Spermine (Sigma cat. no. S1141)
Spermidine (Sigma cat. no. S2626)
Complete Mini Protease Inhibitor tablets, EDTA-free (Roche cat. no. 11 836 170 001)
! CAUTION Protease inhibitor solutions are toxic.
4’,6-diamidino-2-phenylindole (DAPI) (Invitrogen cat. no. D1306) ! CAUTION Handle DAPI with care, as it is both toxic and mutagenic. CRITICAL Store DAPI stock solutions in the dark to avoid photobleaching.
M-280 Streptavidin Dynabeads (Invitrogen cat. no. 112–05D)
Triton X-100 (Sigma cat. no. X100)
Bovine Serum Albumin (BSA), 30% solution (Sigma cat. no. A8577)
RNeasy micro kit (Qiagen cat. no. 74004)
RiboGreen RNA quantitation kit (Invitrogen cat. no. R11490)
Turbo DNA-free kit (Ambion cat. no. AM1907M)
Whole transcriptome amplification kit (Sigma cat. no. WTA2)
Absolute ethanol (Sigma cat. no. 459844)
Antibody to chromatin protein of interest
Protein A agarose (Millipore cat. no. 16–157)
Tris (hydroxymethyl) aminomethane (Tris) (Fisher cat. no. BP152)
LiCl (Fluka cat. no. 62476)
NP-40 substitute (Fluka cat. no. 74385)
Sodium dodecyl sulfate (SDS) (Sigma cat. no. L4390) ! CAUTION Handle solid SDS with care when making solutions as inhalation is harmful.
Sodium deoxycholate (Sigma cat. no. D6750)
NaHCO3 (Sigma cat. no. S6297)
DNAse-free RNase (Roche cat. no. 11 119 915 001)
Proteinase K (Invitrogen cat. no. 25530–049)
MinElute PCR purification kit (Qiagen cat. no. 28004)
QIAquick PCR purification kit (Qiagen cat. no. 28104)
PicoGreen DNA quantitation kit (Invitrogen cat. no. P7589)
Single cell whole genome amplification kit (Sigma cat. no. WGA4)
EQUIPMENT
70 μM cell strainer (BD Falcon cat. no. 352340)
0.6 ml tubes (Axygen cat. no. MCT-060-C)
0.6 ml low retention tubes (Fisher 02-681-311)
1.5 ml tubes (Axygen cat. no. MCT-150-C)
15 ml tubes (BD Falcon cat. no. 352097)
50 ml tubes (BD Falcon cat. no. 352098)
1 ml micropipette tips (Rainin cat. no. RT-L1000S)
10 ml serological pipettes (BD Falcon cat. no. 357551)
Mini MACS separation magnet (Miltenyi Biotec cat. no. 130-042-102)
Ring stand with clamp (Enasco cat. nos. SA04429M and SB18967M)
Tygon tubing (Fisher cat. no. 14-169-1C)
Two-way stopcock (Bio Rad cat. no. 732–8102)
Refrigerated microcentrifuge (Eppendorf model 5415R or equivalent)
Refrigerated centrifuge (Sorvall model RC5C or equivalent)
Mortar and pestle
Cold room
Sonicator (Diagenode Bioruptor)
Rotating mixer for 1.5 ml tubes (Thermo Scientific Labquake or equivalent)
Electronic serological pipetting device (Eppendorf Easypet or equivalent)
100 ml glass beaker (Fisher cat. no. FB-102–100)
Vortex mixer (VWR Vortex Genie 2 or equivalent)
Fluorescence microplate reader (BioTek Synergy HT or equivalent)
Thermal Cycler (MJ Research PTC-100 or equivalent)
Agarose gel electrophoresis equipment
Hausser Bright-Line Hemacytometer (Fisher cat. no. 02-671-10)
Fluorescence microscope with DAPI and GFP filters
REAGENT SETUP
Nuclei purification buffer (NPB)
20 mM MOPS (pH 7), 40 mM NaCl, 90 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 0.5 mM spermidine, 0.2 mM spermine, 1X Complete protease inhibitors. Spermidine, spermine, and Complete protease inhibitors are added just before use and the solution is kept on ice. Without spermidine, spermine, and Complete protease inhibitors the solution can be filter sterilized, stored at 4° C for up to three months, and used as a stock to prepare each of the other nuclei purification buffers (NPBf, NPBd, NPBt, and NPBb).
Nuclei purification buffer containing 1% formaldehyde (NPBf)
20 mM MOPS (pH 7), 40 mM NaCl, 90 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 1% (vol/vol) formaldehyde. Formaldehyde is added just prior to use and the solution is kept at room temperature (RT; 25° C). Do not store this solution.
Nuclei purification buffer containing 2 ug/ml DAPI (NPBd)
20 mM MOPS (pH 7), 40 mM NaCl, 90 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 0.5 mM spermidine, 0.2 mM spermine, 1X Complete protease inhibitors, 2 μg/ml DAPI. Spermidine, spermine, Complete protease inhibitors, and DAPI are added just before use. Protect from light after addition of DAPI and keep on ice. Do not store this solution.
Nuclei purification buffer containing 0.1% Triton X-100 (NPBt)
20 mM MOPS (pH 7), 40 mM NaCl, 90 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 0.5 mM spermidine, 0.2 mM spermine, 0.1 % (vol/vol) Triton X-100. Spermidine, spermine, and Triton X-100 are added just before use and the solution is kept at 4° C. Do not store this solution.
Nuclei purification buffer with 1% BSA (NPBb)
20 mM MOPS (pH 7), 40 mM NaCl, 90 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 0.5 mM spermidine, 0.2 mM spermine, 1% (vol/vol) BSA. BSA is added just before use and the solution is kept at room temperature. Do not store this solution.
NPBb-treated 1 ml pipette tips
Treat 1 ml pipette tips to be used for nuclei purification by first drawing 1 ml of NPBb into the tip, then remove the tip from the micropipette so that the liquid is retained, and rest the tip horizontally at RT for at least 30 min. Two tips are needed for each nuclei purification, but it is best to prepare more than the number that are needed in case one is damaged during setup of the flow purification column in Step 12. Treating the tips with NPBb is necessary to prevent the nuclei and beads from sticking too tightly to the wall of the pipette tip during flow purification.
Nuclei lysis buffer
50 mM Tris (pH 8), 10 mM EDTA, 1% (wt/vol) SDS, 1X Complete protease inhibitors. This solution should be prepared just before use and kept at room temperature. Do not store this solution.
X-ChIP dilution buffer
16.7 mM Tris (pH 8), 167 mM NaCl, 1.1% (vol/vol) Triton X-100, 1.2 mM EDTA. This solution should be prepared just before use and kept on ice. Do not store this solution.
Low salt wash buffer
20 mM Tris (pH 8), 150 mM NaCl, 0.1% (wt/vol) SDS, 1% (vol/vol) Triton X-100, 2 mM EDTA. This solution should be prepared just before use and kept on ice. Do not store this solution.
High salt wash buffer
20 mM Tris (pH 8), 500 mM NaCl, 0.1% (wt/vol) SDS, 1% (vol/vol) Triton X-100, 2 mM EDTA. This solution should be prepared just before use and kept on ice. Do not store this solution.
LiCl wash buffer
10 mM Tris (pH 8), 250 mM LiCl, 1% (wt/vol) sodium deoxycholate, 1% (vol/vol) NP-40 substitute, 1 mM EDTA. This solution should be prepared just before use and kept on ice. Do not store this solution.
TE
10 mM Tris (pH 8), 1 mM EDTA. This solution can be filter sterilized and stored at 4° C for up to three months. Place on ice before use.
X-ChIP elution buffer
100 mM NaHCO3, 1% (wt/vol) SDS. This solution should be prepared just before use and kept at room temperature. Do not store this solution.
PROCEDURE:
Purification of labeled nuclei. TIMING 2 h
-
1)
Collect 1–5 g of fresh plant tissue onto ice.
CRITICAL STEP The amount of tissue needed depends on the abundance of the targeted cell type. Assuming that the yield of nuclei at the end of the purification will be ~ 50% of the theoretical yield, harvest enough tissue to give at least 1 × 105 purified nuclei.
-
2)
If purified nuclei will be used for gene expression analysis, skip to step 4. If nuclei are to be used for X-ChIP, place harvested tissue into a 50 ml conical tube and add 35 ml of NPBf. Mix well, remove lid and place under vacuum at RT for 15 min. Treatment of the tissue with NPBf will crosslink proteins to DNA so that X-ChIP can be performed.
-
3)
Add 2 M glycine to the solution from step 2, to give a final concentration of 125 mM. Mix well and return the uncapped tube to vacuum for 5 additional minutes. Decant solution, wash tissue twice with water, and blot dry with a paper towel. Proceed immediately to step 4. The addition of glycine stops the formaldehyde crosslinking reaction.
! CAUTION Formaldehyde is harmful if inhaled or absorbed through skin. Waste should be disposed of according to local regulations.
-
4)
Freeze tissue in a mortar with LN2 and grind to a fine powder with a pestle. Move the frozen powdered tissue to a clean mortar containing 10 ml of ice-cold NPB and grind gently with a clean pestle to suspend the homogenized tissue.
-
5)
Filter the suspension through a 70 μM cell strainer and into a 15 ml tube on ice.
-
6)
Centrifuge the suspension at 1000g at 4° C for 10 min, and carefully decant the supernatant.
CRITICAL STEP Always centrifuge nuclei suspensions at low speed to avoid damage to nuclei.
-
7)
Gently resuspend the nuclei and cell debris pellet in 1 ml of ice-cold NPBd, move to a new 1.5 ml tube, and place on ice for 3 min.
CRITICAL STEP In this step nuclei are stained with the DNA-binding dye DAPI so that initial quality and final purity of nuclei can be determined at the end of the procedure, as described in Box 1 and Box 2. Once the procedure is established and known to give high purity nuclei from a given cell type it is not necessary to measure purity each time. If purity is not to be measured, use NPB rather than NPBd for this step and simply invert the tube several times to mix after resuspension of nuclei rather than placing on ice for 3 min.
-
8)
Centrifuge the suspension at 1000g at 4° C for 5 min and carefully decant the supernatant. Gently resuspend the nuclei pellet in 1 ml of ice-cold NPB and move to a new 1.5 ml tube.
-
9)
Place the needed amount of M-280 streptavidin-coated Dynabeads (25 μl or ~1.5 × 107 beads per purification) into a 1.5 ml tube, then add 1 ml of NPB and invert the tube several times to wash the beads. Pellet the beads by centrifugation at 3,500g for 2 min at 4° C. Decant the supernatant and resuspend beads in their original volume with NPB.
-
10)
Add 25 μl of the bead slurry from step 9 to the nuclei suspension from step 8. Place the tube on a rotating mixer and rotate at 4° C for 30 min. After the 30 min incubation, remove 20 μl of this solution for analysis of nuclei preparation and bead binding, as described in Box 1. During the 30 min incubation, prepare NPBb-treated pipette tips as described in Reagent Setup.
? TROUBLESHOOTING
-
11)
Transfer the 1 ml mixture of nuclei and beads from step 10 into a 15 ml tube. Gently add 9 ml of ice-cold NPBt to the 1 ml nuclei/bead suspension. Invert gently to mix and keep this suspension at 4° C while performing steps 12–14.
-
12)
Begin setting up the flow purification column in the 4° C cold room. With the miniMACS magnet oriented horizontally on the bench, slide a NPBb-filled 1 ml pipette tip through the groove in the magnet until the narrow end of the tip protrudes 2.5 cm from the other side of the magnet. The part of the pipette tip running through the groove should be in full contact with the surface of the magnet. All subsequent steps of the nuclei purification are also performed at 4° C. (See the video abstract in reference 17 for a visual guide to assembly and operation of the purification column.)
CRITICAL STEP Keeping the tip full with NPBb while sliding it into the magnet groove helps to prevent the wall of the tip from cracking during this process. If the wall of the tip does crack, start over with a new tip. Keeping the NPBb-filled tips at RT until use will help to prevent cracking.
-
13)
Place the magnet with inserted 1 ml tip into the ring stand clamp so that the tip is vertical, and allow the NPBb solution in the tip to drain into a beaker. Attach a stopcock to the narrow end of the tip via a 2 cm length of tubing.
-
14)
Close the stopcock and fill the 1 ml pipette tip with NPBt until the level of liquid is 1 cm from the top of the tip.
-
15)
Gently invert the nuclei/bead mixture from Step 11 several times to mix, and then draw the solution into a 10 ml serological pipette using an electronic serological pipetting device.
CRITICAL STEP Avoid drawing any bubbles into the pipette, even if some of the nuclei/bead mixture must be left behind. Large bubbles flowing past the magnet can cause release of beads and nuclei captured on the magnet, resulting in dramatically reduced yield.
-
16)
With the 10 ml pipette still attached to the pipetting device, gently but firmly secure the narrow end of the 10 ml pipette into the wide end of the 1 ml pipette tip in the magnet. Carefully remove the pipetting device from the top of the 10 ml pipette. The flow purification device is now completely assembled.
CRITICAL STEP If bubbles were formed at the top of the liquid column, these can be removed when the column is nearly drained. Simply remove the 10 ml pipette just after it empties, but before 1 ml tip fully is drained, and draw the bubbles off the surface of the liquid column with a pipette before they come into contact with the captured beads.
-
17)
Gradually open the stopcock on the purification device until a flow rate of ~ 0.75 ml/min is reached. Allow the column to drain completely, with the flowthrough draining into a beaker. A collection of beads should become visible on the wall of the 1 ml tip as the liquid flows past.
CRITICAL STEP The slow flow rate is essential for capturing all the bead-bound nuclei, and higher flow rates can result in reduced yield .
-
18)
Once all liquid has drained from the column, including the 1 ml pipette tip, disassemble the column by removing the 10 ml pipette and tubing from the ends of the 1 ml pipette tip, and then gently removing the 1 ml tip from the groove of the magnet.
CRITICAL STEP Perform this step immediately after the column has drained so that the captured nuclei do not begin to dry out.
-
19)
Place 1 ml of ice-cold NPBt into a 1.5 ml tube. Insert the 1 ml pipette tip containing collected beads and nuclei onto a micropipette and use it to gently and repeatedly draw the 1 ml of NPBt into and out of the tip until all beads are released into solution.
-
20)
Using the 1 ml of nuclei/bead mixture from step 19, perform a second round of flow purification by repeating steps 11–19. Remove 20 μl from the 1 ml of final purified nuclei suspension for analysis of purity as described in Box 2.
CRITICAL STEP It is necessary to repeat the flow purification a second time in order to achieve maximum purity of the desired nuclei.
? TROUBLESHOOTING
-
21)
Centrifuge the 1 ml of purified nuclei/bead mixture at 1000g for 5 min at 4° C to collect bead-bound nuclei. Carefully decant the supernatant and resuspend the pellet in 20 μl of ice-cold NPB and place on ice.
PAUSE POINT Purified nuclei can be stored at −80° C for at least several days prior to proceeding with RNA purification or ChIP. Do not freeze nuclei if native chromatin assays are to be performed (e.g. native ChIP, DNase mapping, etc.)
-
22)
OPTION A: Isolation of nuclear RNA and cDNA preparation. TIMING 5 h
Step 22 Option A is used if the goal of the experiment is to perform gene expression analysis, whereas Step 22 Option B is used if the intent is to perform X-ChIP analysis. Option B can only be performed if the tissue was treated with NPBf in step 2.- Purify the nuclear RNA using the Qiagen RNeasy micro kit. Start by adding 350 μl of lysis buffer RLT to the 20 μl of purified nuclei from step 21. Vortex vigorously for 2 minutes.
- Centrifuge the lysate at 1,000g for 2 min at RT to pellet the beads. Move the supernatant to a new 1.5 ml tube, add 350 μl of 70% (vol/vol) ethanol and vortex several times to mix.
- Pipette the lysate/ethanol mixture into a RNeasy MinElute spin column resting in a 2 ml collection tube and centrifuge at 10,000g for 1 min at RT. Discard the flowthrough.
- Add 350 μl of buffer RW1 to the column. Centrifuge at 10,000g for 1 min at RT. Discard the flowthrough and move the column to a new 2 ml collection tube.
- Add 500 μl of buffer RPE to the column. Centrifuge at 10,000g for 1 min at RT. Discard the flowthrough.
- Add 500 μl of 80% (vol/vol) ethanol to the column and centrifuge at 10,000g for 1 min at RT. Discard the flowthrough and move the column to a new 2 ml collection tube.
-
Open the column lid and centrifuge at 16,000 g for 5 min at RT. Discard the flowthrough and place the column into a new 1.5 ml tube.CRITICAL STEP This step ensures that all residual ethanol is removed from the column prior to elution of RNA. Carryover of ethanol can interfere with cDNA synthesis.
- Add 14 μl of water directly to the column membrane and allow to stand for 1 min. Centrifuge at 16,000g for 1 min at RT.
-
Discard the column and place the RNA solution on ice.PAUSE POINT The RNA can now be stored at −80° C if needed.
-
Measure the concentration of the RNA using the RiboGreen RNA quantitation kit according to the manufacturer’s instructions. Expected yield of RNA is ~100–500 ng.? TROUBLESHOOTING
- Remove contaminating genomic DNA from the RNA by treating with the Turbo DNA-free kit in a 0.6 ml tube according to the manufacturer’s instructions. Move treated RNA to a new 0.6 ml tube.
-
Preparation and amplification of cDNA will be done using reagents provided with the Whole Transcriptome Amplification kit. Set up a library synthesis reaction by combining and mixing the following reagents in a 0.6 ml tube on ice:
Component Amount RNA 50 ng Library Synthesis Solution 2.5 μl H2O To 16.6 μl CRITICAL STEP Also set up a separate negative control reaction without RNA to test for nucleic acid contamination. - Place the tube in a thermal cycler programmed to 70° C for 5 min, followed by 18° C for 5 min. When the thermal cycler block reaches 18° C, add the following reagents to the reaction and mix well by pipetting up and down several times:
Component Amount Library Synthesis Buffer 2.5 μl H2O 3.9 μl Library Synthesis Enzyme 2 μl - Incubate the reaction under the following temperature conditions:
Temperature Time 18° C 10 min; in addition to the 5 min at 18° C in step xiii 25° C 10 min 37° C 30 min 42° C 10 min 70° C 20 min 4° C Until removed from thermal cycler - Centrifuge the tube at 16,000g for 30 sec to collect condensation and place on ice.
- Set up the cDNA amplification reactions by mixing the following reagents in a 1.5 ml tube on ice:
Component Amount Library synthesis reaction (from step xiv) 25 μl H2O 240 μl Amplification Mix 30 μl WTA dNTP mix 6 μl Amplification Enzyme 3 μl - Divide the amplification reaction from step xvi evenly into four 0.6 ml tubes (75 μl in each tube). Incubate the reactions in a thermal cycler under the following conditions:
Step number Denature Anneal/Extend Hold 1 94° C, 2 min 2–18 94° C, 30 sec 70° C, 5 min 19 4° C, until removed from thermal cycler -
Combine the four 75 μl reactions together in a 1.5 ml tube, mix well, and analyze 5 μl of the mixture by electrophoresis on a 1.5% (wt/vol) agarose gel. The amplified DNA product should be visible as a smear ranging in size from ~100 to 800 bp and there should be no product in the negative control lane.? TROUBLESHOOTING
-
Purify the amplified cDNA using the QIAquick PCR purification kit according to the manufacturer’s instructions. This material can now be labeled for hybridization to the appropriate microarray or used to prepare libraries for deep sequencing.PAUSE POINT The amplified DNA can now be stored at −20° C if needed.
-
22)
OPTION B: X-ChIP and DNA amplification. TIMING 8h-24h
CRITICAL STEP Tissue must have been treated with NPBf in step 2 in order to perform X-ChIP.- Add 120 μl of nuclei lysis buffer (at RT) to the 20 μl of purified nuclei from step 21, transfer solution to a 0.6 ml low-retention tube and vortex vigorously for 2 min.
-
Sonicate the lysed nuclei at 4° C in a Bioruptor water bath sonicator for 30 min with power set to ‘high’ and sonication intervals set to 30 sec on/30 sec off. Replace the water with fresh 4° C water every 10 minutes.CRITICAL STEP Lysed nuclei can be maintained at 4° C, but do not place on ice until after dilution in step v, as SDS will precipitate from the lysis buffer.
- Centrifuge at 16,000g at RT for 2 min to pellet beads and debris. Move supernatant containing the chromatin to a new 1.5 ml tube.
-
Analyze 5 μl of the sonicated chromatin on a 1.5 % (wt/vol) agarose gel or a Agilent Bioanalyzer. The DNA should be fragmented to a size range of roughly 100–600 bp.? TROUBLESHOOTING
- Add 1.2 ml of ice-cold X-ChIP dilution buffer to the fragmented chromatin from step iii, mix gently by inverting several times and place on ice.
- Place the needed amount of protein A agarose beads (50 μl of the 50% bead suspension per chromatin sample) into a 1.5 ml tube, add 1 ml of X-ChIP dilution buffer, and invert tube several times to wash the beads. Pellet the beads by centrifugation at 3,500g for 2 min at 4° C. Decant the supernatant and resuspend the beads in their original volume with X-ChIP dilution buffer. Keep the resuspended beads on ice.
-
Add 20 μl of washed beads (i.e. 40 μl of the 50% bead suspension) to the diluted chromatin from step v. Place the tube on a rotating mixer and rotate at 4° C for 30 min.CRITICAL STEP This step clears the chromatin preparation of proteins or nucleic acids that bind to the beads non-specifically.
-
Centrifuge at 3,500g for 2 min at 4° C to pellet beads. Move the supernatant containing the chromatin to a new 1.5 ml tube. Remove 15 μl of the chromatin and save this as ‘input’ DNA to be used as a reference sample in the array hybridization.CRITICAL STEP This ‘input’ DNA should be used as the reference sample only if non-histone proteins are to be immunoprecipitated. If the X-ChIP is for a histone modification or variant it is best to use a total histone H3 X-ChIP sample as the reference in order to equalize for nucleosome occupancy.
-
Divide the diluted chromatin evenly between as many as four 0.6 ml low retention tubes, each of which will be used for a different X-ChIP.CRITICAL STEP The number of X-ChIP experiments that can be performed with a single chromatin sample depends on the yield of nuclei and the abundance of the chromatin protein to be immunoprecipitated. Assuming a yield of 2 × 105 nuclei, 4 X-ChIPs can be done for relatively abundant histone modifications, such as trimethylation of histone H3 lysine 4, whereas if a low-abundance transcription factor is the target, more chromatin sample should be used for a single X-ChIP.
- Add the appropriate amount of antibody to each tube and place the tubes on a rotating mixer at 4° C for 4 to 16 hours.
- Wash the needed amount of protein A agarose beads (i.e. 70 μl of the 50% bead suspension per X-ChIP) with X-ChIP dilution buffer as in step vi, and resupend beads in their original volume with X-ChIP dilution buffer. Add 30 μl of the washed protein A agarose beads (i.e. 60 μl of the 50% bead suspension) to each X-ChIP sample from step x. Place the tubes on the mixer and rotate at 4° C for 1.5 hours..
- Centrifuge the tubes at 3,500g for 2 min at 4° C to pellet the beads, and decant the supernatants.
- Wash the beads on a rotator at 4° C with each of the following sequence of buffers:
Buffer Volume Time 1. Low salt wash 0.5 ml 5 min 2. High salt wash 0.5 ml 5 min 3. LiCl wash 0.5 ml 5 min 4. TE 0.5 ml 5 min - After the 5 min TE wash, move the bead suspension to a new 0.6 ml low retention tube and centrifuge at 3,500g for 2 min at 4° C to pellet beads. Discard the supernatant.
- Add 200 μl of X-ChIP elution buffer to the beads and vortex vigorously for 5 min. Perform this and all subsequent steps, including amplification, on the ‘input DNA’ sample also, if one was taken.
- Centrifuge at 3,500g for 2 min at RT. Move the supernatant containing eluted chromatin to a new 0.6 ml low-retention tube.
- Add 20 μl of 5M NaCl to the 200 μl eluate, mix well and heat to 100° C for 15 min to reverse formaldehyde crosslinks. Centrifuge briefly at 16,000g to collect condensation.
- Add 1 μl RNase A and incubate for 15 min at 37°, then add 1 uL Proteinase K and incubate for 15 min at 55°.
- Purify ChIP DNA with the Qiaquick MinElute PCR purification kit. Start by adding 1 ml of buffer PBI to the 220 μl eluate and vortexing briefly to mix.
- Add 700 μl of this solution to a MinElute column resting in a 2 ml collection tube. Centrifuge at 10,000g for 1 min at RT and discard the supernatant.
- Add remaining solution from step xix to the same column. Centrifuge at 10,000g for 1 min at RT and discard supernatant.
- Add 750 μl of buffer PE to the column. Centrifuge at 10,000g for 1 min at RT and discard supernatant.
-
Centrifuge at 16,000g for 2 min at RT to remove any remaining buffer PE from the column.CRITICAL STEP Carryover of buffer PE can interfere with subsequent DNA amplification.
- Place column in a new 1.5 ml tube and add 12 μl of elution buffer EB to the center of the column membrane. Let stand 1 min.
- Centrifuge at 16,000g for 1 min at RT. Discard column and place eluted DNA on ice.
-
Measure the DNA concentration using the PicoGreen DNA quantitation kit according to the manufacturer’s instructions.? TROUBLESHOOTING
-
Amplify ChIP DNA using the Single Cell Whole Genome Amplification kit as follows. Perform 2 reactions per X-ChIP sample if possible. Set up a library synthesis reaction by combining and mixing the following components:
Component Amount ChIP DNA 10–100 pg Single cell library preparation buffer 2 μl Library stabilization solution 1 μl H20 To 13 μl CRITICAL STEP 1) The amount of X-ChIP DNA added to the reaction depends on the DNA yield. Use as much as possible within the indicated range. 2) Set up a separate negative control reaction without DNA to test for nucleic acid contamination. - Heat the reaction in a thermal cycler to 95° C for 2 min, then immediately place on ice for 2 min. Centrifuge briefly at 16,000g at 4° C to collect condensation and place the tube on ice.
- Add 1 μl of the library preparation enzyme to the reaction, mix well and incubate under the following temperature conditions in a thermal cycler:
Temperature Time 16° C 20 min 24° C 20 min 37° C 20 min 75° C 5 min 4° C Until removed from thermal cycler - Centrifuge briefly at 16,000g for 1 min at 4° C to collect condensation, then place the reaction on ice.
- Set up the amplification reaction by adding the following the components to the 14 μl library synthesis reaction and mixing well:
Component Amount Amplification master mix 7.5 μl H20 48.5 μl WGA DNA polymerase 5 μl - Place the reaction in a thermal cycler set to the following conditions:
Step number Denature Anneal/Extend Hold 1 94° C, 3 min 2–26 94° C, 30 sec 65° C, 5 min 27 4° C, until removed from thermal cycler -
Analyze 5 μl of the reaction by electrophoresis on a 1.5% (wt/vol) agarose gel. The amplified DNA product should be visible as a smear ranging in size from ~100 to 800 bp and there should be no product in the negative control lane.? TROUBLESHOOTING
-
Purify the amplified ChIP DNA using the QIAquick PCR purification kit according to the manufacturer’s instructions. This material can now be labeled for hybridization to the appropriate microarray or used to prepare libraries for deep sequencing.PAUSE POINT The amplified DNA can now be stored at −20° C if needed.TROUBLESHOOTING:
Step Problem Possible reason Solution 10 and Box 1 Lower than expected yield of total nuclei Poor release of nuclei from tissue Make sure that frozen tissue is ground to a fine powder and homogenized thoroughly when resuspended in NPB at step 4. Loss of nuclei due to breakage Handle nuclei more gently during resuspension and washing, or use alternative tissue disruption method (e.g. chopping with a razor blade28). Poor or no binding of beads to labeled nuclei NTF degraded by protease Increase concentration of protease inhibitors, or use a different inhibitor cocktail. NTF lost from nuclear envelope surface Handle nuclei more gently during resuspension and washing, or use alternative tissue dissociation method (e.g. chopping with razor blade28). Step 20 and Box 2 Lower than expected yield of purified nuclei Flow rate too high Reduce the flow rate so that all beads are captured on the magnet. Binding of beads to nuclei was not efficient enough Add a greater amount of beads at step 9, or increase incubation time at step 10. Bubbles in liquid column Use care to avoid drawing bubbles into the 10 ml pipette. Alternatively, remove the 10 ml pipette as it empties, but before 1 ml tip fully is drained, and draw out bubbles with a pipette before they flow past the beads collected on the wall of the tip. Lower than expected purity of nuclei Too much debris in starting nuclei preparation Filter tissue extract through a 40 μM cell strainer in addition to the 70 μM strainer, further dilute bead/nuclei mix at step 12, or start with less tissue and perform multiple purifications. Step 22 Option A, step x Lower than expected yield of RNA RNA degradation Ensure that all solutions, equipment, and work areas are free of RNases. If tissue is suspected to be rich in RNases, use an RNase inhibitor in steps 4–10. Step 22 Option A, step xviii Low yield of amplified cDNA (> 1.5 μg) Insufficient amount of RNA added to library synthesis reaction, or RNA is partially degraded Start with more RNA in the library synthesis reaction or add 1 to 2 more cycles to the PCR step. Step 22 Option B, step iv Average size of sonicated DNA is larger than expected Sonication was not efficient enough Increase sonication time in 10 min increments. Step 22 Option B,
xxviLow yield of ChIP DNA (less than 10 pg) Insufficient amount of starting chromatin Use more of the diluted chromatin from Step 22 Option B step v, perhaps the entire 1.3 ml for a single ChIP. Poor performance of antibody Use more antibody, different antibody, or reduce stringency of washes Step 22 Option B,
xxxiiiLow yield of amplified ChIP DNA (> 1.5 μg) Insufficient amount of ChIP DNA added to library synthesis reaction Use more DNA in the library synthesis reaction or add 1 to 2 more cycles to the PCR step.
ANTICIPATED RESULTS:
Purification of labeled nuclei (Steps 1–21)
Purification of the NTF tagged nuclei is a very robust process, but the success of an experiment hinges primarily on starting with a high quality total nuclei preparation and efficient capture of the bead-bound nuclei on the magnet without contamination by other nuclei and debris. A high yield of undamaged nuclei can be obtained from many tissues by using the freezing and grinding method presented here, but if problems arise with obtaining good nuclei from the tissue of interest then alternative isolation methods, such as tissue chopping28, can be used. Once magnetic beads are bound to the tagged nuclei they will be efficiently captured by the magnet as long as the mixture is flowing past the magnet slowly enough. Avoiding bubbles at the top of the liquid column is also critical, because the bubbles can actually pull the beads and nuclei away from the magnet, resulting in the loss of most or all of the beads. Otherwise, there should be no beads visible in the column flowthrough.
Users should expect to obtain 50–70% of the theoretical yield of tagged nuclei with a purity of 90–98%. For very low abundance cell types it may be necessary to start with more tissue than is suggested in this protocol. If this is the case, the purity may be reduced due to the increased amount of debris and contaminating nuclei in the flow column. This problem can be overcome by reducing the column flow rate, increasing the volume of the diluted nuclei mixture prior to flow purification, or simply performing multiple smaller-scale purifications in parallel.
We previously found that in the purification of root hair cell nuclei, which represent ~10% of the cells in the root, we could routinely recover an average of 1.5 × 105 nuclei at 93% purity from 3 g of wet roots17.
Isolation of nuclear RNA and cDNA preparation (Step 22, Option A)
Starting with 1–5 × 105 purified nuclei, the expected yield of total nuclear RNA is ~100–500 ng, which is sufficient for several cDNA preparations. The cDNA synthesis and amplification is quite robust and will generally yield 3–8 μg of DNA when starting with 50 ng of RNA. If amplification problems are encountered, these can usually be overcome by increasing the amount of RNA in the library synthesis reaction. If necessary, yield can be increased by adding 1 or 2 more PCR cycles, with the caveat that increasing the number of cycles can exacerbate any amplification biases.
Chromatin immunoprecipitation and DNA amplification (Step 22, Option B)
The expected yield of DNA from an X-ChIP experiment is variable and depends on the amount of chromatin used for the ChIP, the antibody used, and the abundance of the target protein. Starting with chromatin from 5 × 104 nuclei (25% of the chromatin from 2 × 105 purified nuclei), an X-ChIP for a relatively abundant histone modification, like trimethylation of histone H3 lysine 4, will yield 20–50 pg of DNA, whereas an X-ChIP for total H3 will yield 150–250 pg of DNA. A successful amplification will yield 5–10 μg amplified DNA, and this can be achieved by starting with as little as 10 pg of the X-ChIP DNA. However, the amplification reaction will sometimes fail when such small amounts are used. Therefore, if the X-ChIP is for a low abundance target it is advisable to start with a larger amount of chromatin.
ACKNOWLEDGEMENTS:
We thank Mary Gehring, Florian Steiner, and Paul Talbert for helpful suggestions on improving the manuscript. This work was supported by funding from the Howard Hughes Medical Institute to S.H. and a Ruth L. Kirschstein Postdoctoral Fellowship from the National Institutes of Health to R.B.D.
Footnotes
COMPETING FINANCIAL INTERESTS:
The authors declare no competing financial interests.
REFERENCES:
- 1.Mito Y, Henikoff JG & Henikoff S Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet 37, 1090–1097 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Rao RR & Stice SL Gene expression profiling of embryonic stem cells leads to greater understanding of pluripotency and early developmental events. Biol Reprod 71, 1772–1778 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Irion S, Nostro MC, Kattman SJ & Keller GM Directed differentiation of pluripotent stem cells: from developmental biology to therapeutic applications. Cold Spring Harb Symp Quant Biol 73, 101–110 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Bhattacharya B, Puri S & Puri RK A review of gene expression profiling of human embryonic stem cell lines and their differentiated progeny. Curr Stem Cell Res Ther 4, 98–106 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Nakazono M, Qiu F, Borsuk LA & Schnable PS Laser-capture microdissection, a tool for the global analysis of gene expression in specific plant cell types: identification of genes expressed differentially in epidermal cells or vascular tissues of maize. Plant Cell 15, 583–596 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiao Y et al. A transcriptome atlas of rice cell types uncovers cellular, functional and developmental hierarchies. Nat Genet 41, 258–263 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Brunskill EW et al. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell 15, 781–791 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birnbaum K et al. A gene expression map of the Arabidopsis root. Science 302, 1956–1960 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Birnbaum K et al. Cell type-specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nat Methods 2, 615–619 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Gifford ML, Dean A, Gutierrez RA, Coruzzi GM & Birnbaum KD Cell-specific nitrogen responses mediate developmental plasticity. Proc Natl Acad Sci U S A 105, 803–808 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y et al. Identification of genes expressed in C. elegans touch receptor neurons. Nature 418, 331–335 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Zhang C, Barthelson RA, Lambert GM & Galbraith DW Global characterization of cell-specific gene expression through fluorescence-activated sorting of nuclei. Plant Physiol 147, 30–40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller MR, Robinson KJ, Cleary MD & Doe CQ TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat Methods 6, 439–441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mustroph A et al. Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc Natl Acad Sci USA, 106, 18843–18848 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heiman M et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy PJ, Stuart JM, Lund J & Kim SK Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418, 975–979 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Deal RB & Henikoff S A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell 18, 1030–1040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rose A & Meier I A domain unique to plant RanGAP is responsible for its targeting to the plant nuclear rim. Proc Natl Acad Sci U S A 98, 15377–15382 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beckett D, Kovaleva E & Schatz PJ A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci 8, 921–929 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barthelson RA, Lambert GM, Vanier C, Lynch RM & Galbraith DW Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells. BMC Genomics 8, 340 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacob Y, Mongkolsiriwatana C, Veley KM, Kim SY & Michaels SD The nuclear pore protein AtTPR is required for RNA homeostasis, flowering time, and auxin signaling. Plant Physiol 144, 1383–1390 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meier I, Xu XM, Brkljacic J, Zhao Q & Wang H-J Going green: plants’ alternative way to position the Ran gradient. Journal of microscopy 231, 225–233 (2008). [DOI] [PubMed] [Google Scholar]
- 23.An YQ et al. Strong, constitutive expression of the Arabidopsis ACT2/ACT8 actin subclass in vegetative tissues. Plant J 10, 107–121 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Zilberman D, Coleman-Derr D, Ballinger T & Henikoff S Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456, 125–129 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruzicka DR, Kandasamy MK, McKinney EC, Burgos-Rivera B & Meagher RB The ancient subclasses of Arabidopsis Actin Depolymerizing Factor genes exhibit novel and differential expression. Plant J 52, 460–472 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Masucci JD et al. The homeobox gene GLABRA2 is required for position-dependent cell differentiation in the root epidermis of Arabidopsis thaliana. Development 122, 1253–1260 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Gendrel AV, Lippman Z, Martienssen R & Colot V Profiling histone modification patterns in plants using genomic tiling microarrays. Nat Methods 2, 213–218 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Galbraith DW et al. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220, 1049–1051 (1983). [DOI] [PubMed] [Google Scholar]