

Abstract
Coumarins represent well-established structures to introduce fluorescence into tool compounds for biochemical investigations. They are valued for their small size, chemical stability and accessibility as well as their tunable photochemical properties. As components of fluorophore/quencher pairs or FRET donor/acceptor pairs, coumarins have frequently been applied in substrate mapping approaches for serine and cysteine proteases. This review also focuses on the incorporation of coumarins into the side chain of amino acids and the exploitation of the resulting fluorescent amino acids for the positional profiling of protease substrates. The protease-inhibiting properties of certain coumarin derivatives and the utilization of coumarin moieties to assemble activity-based probes for serine and cysteine proteases are discussed as well.
Keywords: Activity-based probes, Coumarins, Cysteine proteases, Serine proteases, Substrate mapping
Highlights
-
•
Coumarins represent well-established structures to introduce fluorescence into tool compounds for biochemical investigations.
-
•
They are valued for their small size, chemical stability and accessibility as well as their tunable photochemical properties.
-
•
Coumarins are components of fluorophore/quencher pairs or FRET donor/acceptor pairs in substrate mapping of proteases.
-
•
Coumarins have been incorporated into amino acids side chains to be used for the positional profiling of protease substrates.
-
•
Coumarins have protease-inhibiting properties and are used for activity-based probes for serine and cysteine proteases.
1. Introduction
Coumarin derivatives have manifold pharmacological properties showing, for example, antidiabetic [1], antiviral [2], anti-inflammatory [3], as well as anticancer and antileukemia activities [4,5] and acting as adenosine receptor antagonists [6] and monoamine oxidase inhibitors [7,8]. Beside the identification of coumarin derivatives as lead structures against different targets, coumarins are frequently used because of their fluorescent properties to label substrates or inhibitors of several enzymes [9]. Through some minor modifications at the coumarin scaffold, it is possible to obtain small molecules with excellent stability, good fluorescence, high quantum yield and sufficient water solubility [10,11].
This review will focus on coumarins as fluorescence dyes to generate substrates and probes for serine and cysteine proteases. These two classes of proteolytic enzymes share an acyl transfer mechanism of the peptide bond cleavage involving the nucleophilic attack of the active site serine hydroxyl and cysteine thiolate nucleophile, respectively, at the carbonyl carbon of the scissile bond (Fig. 1 ). Specificity of proteases is generally achieved through defined interactions of amino acid residues and certain subsites at the active site. The small size of fluorescent coumarins suggests their incorporation into the side chain of amino acids and the generation of fluorescent peptides. Internally quenched fluorescent (IQF) peptide substrates make use of this option. They consist of a fluorophore, e.g. a coumarin, and a quencher moiety, separated by several amino acids. Proteolytic cleavage of any peptide bond within this portion leads to a loss of quenching and the concomitant generation of fluorescence. Such IQF peptides have frequently been employed for the positional profiling of protease substrates to perform an active-site mapping and to elucidate substrate specificity [9].
Fig. 1.
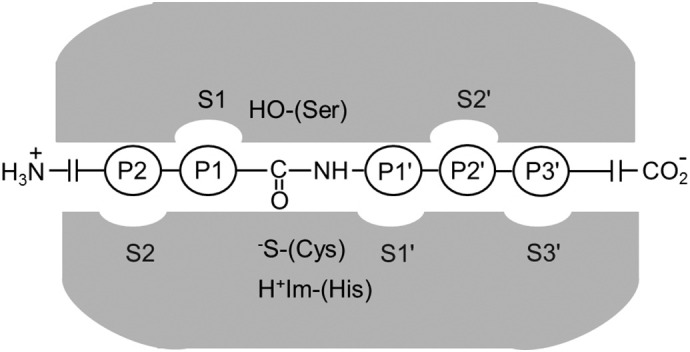
Active site of a substrate-bound serine or cysteine protease. The peptide sequence covering P2 to P3´ residues and the corresponding subsites comprising S2 to S3´ in the nomenclature of Schechter and Berger [12].
In addition to the utilization of coumarins for the reconstruction of specificity profiles for serine and cysteine proteases, this review will also highlight the role of coumarins as structural components of activity-based probes (ABPs). Serine and cysteine proteases, both of which are characterized by a covalent mode of catalysis, are particularly suitable to be targeted by ABPs. These probes elicit an irreversible inhibition of the protease because the nucleophilic attack is directed to an electrophilic moiety of the probe, the so called warhead. The sulfur of a cysteine protease represents a soft nucleophile and thus prefers soft electrophiles such as acyloxymethyl ketones, epoxysuccinates and Michael acceptors. In contrast, the serine proteases favor harder electrophiles like phosphonates or isocoumarins [13]. Next to the warhead, ABPs contain two further parts, a spacer and a tag (Fig. 2 ). The highest impact on the specificity of an ABP is caused by the spacer, which can consist of a well-accepted peptidic recognition unit. The tag permits the detection of the inhibited protease, either through biotin or fluorescent reporters, among which cyanines, fluoresceins, boron-dipyrromethene (BODIPY) derivatives and coumarins have been frequently employed [14]. The introduction of the latter fluorophore into ABPs for serine and cysteine proteases will be considered in this review. We will not discuss the detection and quantification of inorganic ions or low-molecular weight thiols by means of coumarin-containing probes, which has comprehensively reviewed elsewhere [11].
Fig. 2.
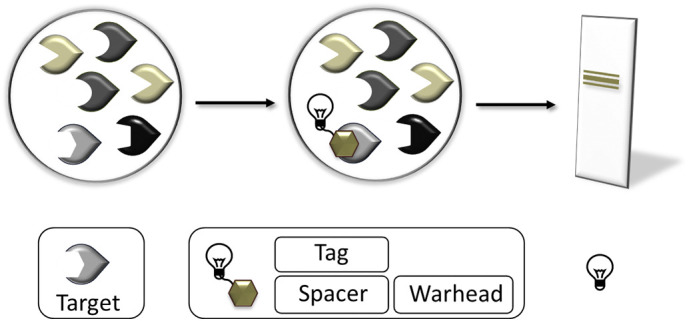
Application of fluorescent activity-based probes for the detection of target proteins on SDS-PAGE gels.
2. Some chemical and biochemical properties of coumarins
Coumarins are aromatic lactones. The aromatic character of coumarin was confirmed by topological resonance energy and nucleus independent chemical shift values and calorimetric measurement [15,16]. Thiocoumarin, in which the ring oxygen is replaced by sulfur, is somewhat more stable [15], similar to the comparison of 4H-3,1-benzoxazin-4-ones and 4H-3,1-benzothiazin-4-ones [17]. Hydrolysis of coumarins at high pH produced cis-coumarinic acids, and lactonization of this species occurred at low pH. Coumarin (1, Fig. 3 ) had a half-life of 160 min at pH 10 and 25 °C [18]. A 7-methoxy and, more pronounced, 7-diethylamino substitution of 4-methylcoumarin led to a reduced reactivity in alkaline hydrolysis [19], whereas introduction of an electron-withdrawing phenyl group at position 3 accelerated this reaction [20].
Fig. 3.

Selected fluorescent coumarin derivatives.
Besides their bioactivities, the importance of coumarins rests upon their fluorescent properties as short-wavelength reactive dyes. They have been widely applied for the detection of proteins or other biological targets. The parent compound 1 (Fig. 3) is a weak fluorophore with low quantum yield. However, a combination of an electron-withdrawing substituent at 3-position and an electron-donating group at 7-position produces high fluorescence quantum yields. The 7-methoxy derivative 2 and the 7-diethylamino analogue 3 have absorption and emission maxima of 366 nm and 478 nm (in acetonitrile) [21], and 418 nm and 461 nm (in ethanol) [22], respectively. 7-Diethylamino derivatives are established fluorophores, but their fluorescence is quenched in aqueous medium. The primary amines 5–7 are frequently used moieties for fluorogenic peptide substrates. Fluorescence is generated only when the 7-aminocoumarin is released from the 7-peptidylcoumarin through the protease-catalyzed hydrolysis of the amide bond. Compounds 4 and 5 (AMC) exhibit maxima for absorption and emission of 404 nm and 445 nm (in ethanol) and of 354 nm and 435 nm (in ethanol), respectively [23].
As coumarin derivatives exhibit a broad spectrum of biological effects, their pharmacokinetics is well known. For example, coumarin itself (1), a plant ingredient e.g. of tonka beans and sweet woodruff, is rapidly metabolized in the liver via the cytochrome CYP2A6 through hydroxylation at the free positions C3-C8, with 7-hydroxycoumarin representing the main metabolite. The hydroxylated derivatives are coupled with glucuronic acid and the conjugates are eliminated via the kidney. Other coumarin-containing xenobiotics are metabolized by other CYP enzymes, for example the vitamin K epoxide reductase (VKOR) inhibitors warfarin and phenprocoumon via CYP2C9 and CYP3A4, respectively [24,25].
Regarding the photophysical behavior of 7-aminocoumarins, a planar, highly emissive intramolecular charge-transfer (ICT) excited state and a non-fluorescent twisted intramolecular charge-transfer (TICT) state have been rationalized (Fig. 4 ). Preventing this twisting process increases the quantum yield and restores fluorescence in aqueous media, as can be achieved through rigidization of the amino group in one or two rings [11,26]. In coumarin 343 (8), longer wavelengths of excitation (440 nm) and emission (480 nm, in aqueous solution) appear [27]. The extension of the π-electron system due to coupling in position 3 with a heteroaryl substituent and sulfonation at position 6 resulted in a bathochromic shift, high quantum yield and good water solubility of the resulting dye 9 with an absorption maximum of 426 nm and an emission maximum of 481 nm (in aqueous solution) [10].
Fig. 4.

4-Substituted-7-aminocoumarins as fluorescent compounds (R = alkyl).
The incorporation of coumarin moieties into the side chain of amino acids has frequently been carried out and utilized for the assembly of fluorescent probes. Two prototypical structures are exemplary depicted in Fig. 3. In compounds 10 (X = N, O, OR; n = 1–4), the linkage is realized through an amide bond [[28], [29], [30], [31]], while in 11, an alkylidene chain connects the coumarin C-4 with the α‑carbon of the amino acid [[32], [33], [34], [35], [36], [37]].
3. Coumarin derivatives as serine protease inhibitors
The pharmacology of coumarins is closely related to hemostatic processes, mainly conducted by serine proteases, which are, however not directly affected by most of the coumarins. Until the early 1930s, cows and sheep consistently died for unexplained reasons through internal bleeding. In 1921, Schofield and Roderick independently discovered, that the disease was caused by hay contaminated with the sweet clovers Melilotus alba and Melilotus officinalis and thus was named sweet clover disease [38]. Putrefactive bacteria in the hay, such as Aspergillus fumigatus led to the formation of dicumarol (12, Fig. 5 ) [39]. This coumarin dimer turned out to be a VKOR inhibitor, hence decreasing antithrombotic proteins. In 1941, dicumarol was established as anticoagulant and was the first approved coumarin-containing drug [38]. The coumarin derivatives phenprocoumon and warfarin are still on the market as VKOR inhibitors.
Fig. 5.

Anticoagulant coumarin derivatives.
There are reports on coumarin-based compounds acting as serine protease inhibitors [40], which will be discussed in the following. The trypsin-like serine protease factor XIIa operates as the starting point for the intrinsic pathway of the secondary hemostasis. FXIIa activates plasma kallikrein which produces an amplification loop of FXII activation. In the coagulation cascade, factor XIIa ultimately leads to the activation of factor X to Xa, which itself activates, together with factor Va, prothrombin. Coumarin derivatives of the anilide type 13 and 14 (Fig. 5) have been described as active site-directed factor XIIa inhibitors. Both compounds show activity with IC50 values of 4.3 μM [41] and 5 μM [42]. The halogens from the fused benzene ring of 14 are assumed to point to a tyrosine residue of a hydrophobic region within the active site of factor XIIa and the para-chlorophenol ring to extend towards the entry of the S1 pocket. The lactone function may contribute to binding by forming hydrogen bonds [42]. Both representative inhibitors were selective for factor XIIa over the related enzymes thrombin, factor Xa, factor VIIa, and plasma kallikrein [41,42].
The 3-carboxamide-coumarins of type 13 and 14 are devoid of a latent alkylating function. In contrast, coumarins of type 15–17 are thought to display a different interaction with target serine proteases. Studies of Frédérick et al. have been focused on the inhibition of the trypsin-like enzymes factor Xa and thrombin by such coumarins [[43], [44], [45]]. Factor Xa acts in the clotting cascade by converting prothrombin into thrombin in the presence of phospholipids and calcium. Factor Xa is a connecting link between internal and external ways of coagulation. Factor Xa inhibition has become a particular successful strategy in the development of new anticoagulants. Thrombin represents the terminal serine protease of the blood clotting cascade. It acts as a stimulator of platelet activation and as the direct converter of the glycoprotein fibrinogen to fibrin. Thrombin amplifies its own generation by feedback activation of factors V, VII and IX. Moreover, thrombin also activates factor XIII by cleaving an activation peptide segment. The resultant transglutaminase introduces the covalent crosslinks into the fibrin clot [46]. Compound 15 show a pronounced inhibition of chymotrypsin [47], rather than of thrombin and factor Xa. Coumarin 16 exhibited a second-order rate constant of thrombin inactivation of 37,007 M−1 s−1. Activity against chymotrypsin was reduced in the order 15 to 17, and the latter compound was more potent against thrombin (k i/K i = 3457 M−1 s−1) than against chymotrypsin [45]. The introduction of a para-guanidino group into the phenoxy part of such coumarins led to water soluble derivatives [48].
The postulated interaction of mechanism-based inhibitors of type 15–17 is initiated by the attack of the active-site serine hydroxyl nucleophile at the lactone carbon, which leads to a ring-opened structure with a covalently bound serine (Fig. 6 ). The formation of such an acyl enzyme is not irreversible. Slow hydrolysis can occur to relieve the enzyme and produce the “destroyed”, ring-opened compound. Indispensable for the suicide mechanism is a good leaving group in position 6 linked via a methylene group. Its elimination can create a para-quinone methide derivative, able to subsequently react in an irreversible way with an active-site nucleophile. A covalent mechanism (step 1 in Fig. 6) already confirmed by the crystal structure of the complex of 7-hydroxycoumarin bound to γ-chymotrypsin where 2,4-dihydroxycinnamic acid is acylating the active site serine [49], PDB entry 1K2I.
Fig. 6.

Enzyme-catalyzed hydrolysis of coumarins and suicide mechanism of inactivation [43,50], NuH, nucleophile; LG, leaving group.
Plasma kallikrein and the fifteen closely related tissue kallikreins are part of the kallikrein family of serine proteases. The tissue kallikreins are potential drug targets for several disorders, including respiratory, cardiovascular, and dermatological diseases, as well as cancer. The expression of various tissue kallikreins was identified in the skin, among which kallikrein 7 (stratum corneum chymotryptic enzyme) is involved in epidermal desquamation by degrading corneodesmosome proteins [51]. Tan et al. have investigated coumarins of type 15–17 towards kallikrein 7 and highlighted the same suicide mechanism [50].
4. Coumarin-labeled substrates
Proteases represent an important class of enzymes found in all kingdoms of life. Thereof, approximately 37% are serine and 17% cysteine proteases [52]. In the human genome, on its own, over 600 genes for proteases have been identified [53]. On the one hand, knowledge about endogenous substrate and the respective cleavage sites allows determining the specificity of a certain protease. On the other hand, based on a given substrate specificity, so far unknown substrates of a protease can be assumed. Moreover, the preferred amino acid sequence around the scissile peptide bond can be employed to create specific and highly active peptidomimetic inhibitors for further drug development. Besides the application as drugs, pharmacological and biochemical tool compounds, in particular activity-based probes can be designed and employed to study the protease of interest in detail.
A commonly used principle for the identification of favored substrate sequences and the generation of such substrates is the implementation of a positional scanning synthetic combinatorial library (PS-SCL) [54,55]. In a peptide sequence, for example with four amino acids P1-P4, one position is fixed with a known amino acid, while the others contain an equimolar or isokinetic mixture of natural amino acids. The expansion by unnatural amino acids led to hybrid combinatorial substrate libraries (HyCoSuLs) [56,57]. In a fluorogenic PS-SCL, the C-terminal P1 amino acid is coupled to a fluorophore, which is buried in the S1´ pocket. Protease-catalyzed cleavage of the amide bond between the P1 amino acid and the dye results in time-dependent increase in fluorescence, amenable to kinetic measurements. By means of this technique, it is possible to study the impact of the known amino acid at the desired position independent of the other ones. Thus, PS-SCLs provide information on the particularly preferred amino acid residues at the corresponding positions and allow for the deconvolution to obtain an optimal peptide sequence. The resulting substrates can be characterized by the specificity constant k cat/K m with high values corresponding to suitable substrates. As a matter of course, such an active site mapping is limited to the non-primed positions [56,58].
Coumarin derivatives are prevailingly applied fluorophores for such attempts. Owing to small size, they are well acceptable in the S1´ pocket. 7-Amino-4-methylcoumarin (AMC; structure 5, see Fig. 3) was introduced in a variety of peptidic substrates for a huge number of proteases. However, the use for AMC in PS-SCLs for substrate mapping provides a clear constraint. Those libraries are produced through solid phase synthesis mainly by employing Fmoc-protected amino acids and the peptides are elongated in the C-to-N direction. AMC coupled to the C-terminus of the substrate does not allow evaluation of substrate specificity for amino acids in position P1, because the side chain of a specifically functionalized amino acid P1 has to be fixed at the solid phase.
This limitation was circumvented by the implementation of an altered solid-phase synthesis technique, employing an alkanesulfonamide “safety-catch” linker as follows. The carboxylic group of amino acid P2 is attached to a sulfonamide-modified aminomethyl resin. Subsequently, amino acids P3 and P4 are successively added. Alkylation of the acidic SO2NHCO nitrogen with a haloacetonitrile forms a highly reactive N,N-cyanomethylacylsulfonamide which undergoes regioselective nucleophilic attack of the amino group of the P1 amino acid amide possessing AMC releasing the final coumarinyl substrate. This practice allows the incorporation of the unmodified P1 amino acids and thus the positional profiling of this position [59,60]. The involvement of the P1 amino acid to reconstruct the specificity profile is also possible by applying 7-amino-4-carbamoylmethylcoumarin (ACC; structure 7, see Fig. 3). ACC is bifunctional and can be coupled to the C-terminus of the peptide via the scissile amide bond. For the connection to a Rink amide resin, for example, the 7-amino-Fmoc protected free acid of ACC can be used. Cleavage from the resin can be achieved under acidic conditions with triisopropyl silane as reducing scavenger of carbenium ions to generate the final coumarinyl peptide. ACC exhibits a 3-times higher quantum yield compared to AMC, making ACC-based assays more sensitive [54,55]. AMC and ACC-based PS-SCLs have been utilized for the substrate mapping of several proteases, also including the type II metalloprotease aminopeptidase N [61]. In the following, coumarin substrates for serine and cysteine proteases are reviewed [9,56,58], and some recent and significant examples are discussed, including human, bacterial and viral proteases.
4.1. Coumarin substrates for serine proteases
Factor VII activating protease (FSAP) is a human serine protease implicated in thrombosis, atherosclerosis, stroke and cancer. FSAP plays a dual role in hemostasis, being involved in both procoagulant and fibrinolytic pathways. The liver enzyme is secreted as inactive 70 kDa zymogen (pro-FSAP) into the blood plasma. The circulating enzyme seems to conduct an auto-activation, which is triggered through ingredients of damaged cells and on positively and negatively charged macromolecules [62,63]. Once activated, FSAP can be inhibited by several endogenous inhibitors such as α1-proteinase inhibitor, α2-plasmin inhibitor, antithrombin, C1 inhibitor, plasminogen activator inhibitor-1 [64]. The activated FSAP was reported to cleave single-chain urokinase-type plasminogen activator (scuPA) and several other substrates, such as platelet-derived growth factor, basic fibroblast growth factor/epidermal growth factor, histones, high-molecular-weight kininogen and protease activated receptor-2 [62,63,65]. Since the physiological role of FSAP is not fully understood and the significance of certain substrate cleavages has been questioned, knowledge of its substrate specificity and defined peptide substrates to specifically measure FSAP activity in the plasma are required.
Kara et al. performed an FSAP substrate mapping using a diverse PS-SCL of AMC-tetrapeptides. The assays were monitored fluorimetrically with excitation at 380 nm and emission at 460 nm. The preferred P4-P1 sequence was Ala-Lys-Nle-Arg and the corresponding fluorogenic substrate Ac-Ala-Lys-Nle-Arg-AMC exhibited a k cat/K m value of 26,300 M−1 s−1 and a high selectivity for FSAP compared to thrombin, plasmin, FXa, uPA, tPA, FXIIa. After addition of histones to plasma, turnover of this substrate was accelerated, reflecting the histone-mediated activation of pro-FSAP [64]. Rut et al. obtained a synthetic FSAP substrate by means of a HyCoSuL with arginine fixed in P1 position and 19 natural and 108 unnatural amino acids in P2-P4 position. ACC was used as fluorophore [66]. Excitation was conducted at 320 nm and the emission was measured at 460 nm. The best identified substrate, Ac-Pro-d-Tyr-Lys-Arg-ACC, showed k cat/K m values of 55,045 M−1 s−1 and 17,833 M−1 s−1 for the protease domain (amino acids 292–560) of wild-type FSAP and plasma FSAP, respectively (Fig. 7 ). Based on these outcomes, a biotin and a cyanine-5 (Cy5) containing ABPs were designed. For this purpose, the coumarin moiety was exchanged for a diphenyl phosphonate whereby the electrophilic phosphorous atom, in place of the carbonyl carbon, forms a covalent bond to the α‑carbon of the P1 arginine. The probe was assembled through the introduction of the linker-connected biotin or Cy5 substructure at the N-terminus of the peptide. A nucleophilic attack of the active-site serine at the warhead's phosphorous leads to irreversible inhibition and labeling of the target protease [66]. This prototypical stepwise approach towards an ABP starting from the identification of a preferred amino acid sequence has frequently been demonstrated. Examples include ABPs for cathepsin G [[67], [68], [69]], human neutrophil elastase (HNE) and proteinase-3 [70], the major secreted serine proteases of neutrophils and mast cells.
Fig. 7.

Exemplary illustration of a protease-catalyzed cleavage of an ACC-based peptidic substrate. Ac-Pro-d-Tyr-Lys-Arg-ACC was identified by employing a PS-SCL [66].
Matriptase-2 (MT-2), a member of the type II transmembrane serine proteases (TTSPs), is mainly expressed in the liver. It shares the primary substrate specificity for basic amino acids in P1 position with FSAP and other trypsin-like serine proteases. MT-2 cleaves membrane-bound hemojuvelin, the co-receptor of the bone morphogenic protein (BMP) receptor, resulting in a decreased activation of the SMAD signaling cascade and in a reduced expression of the hepcidin gene. Hepcidin, through interaction with ferroportin, acts as a regulator of the intestinal iron absorption. Accordingly, high activity of MT-2 results in increased iron uptake and MT-2 was considered as a target for the treatment of iron overload diseases and β-thalassemia [71,72]. ACC-containing peptides were used to elucidate the substrate specificity of MT-2 with respect to the P4-P1 positions. Deconvolution of the applied library provided Ile-Arg-Ala-Arg to comprise the sequence best accepted by MT-2 [73]. By dint of this outcome, the substrate specificity for the positions P1´-P3´ was evaluated. By using PS-SCL, IQF heptapeptides were designed, in which positions P1-P4 were occupied by the before determined sequence. N-terminal 2-aminobenzoic acid (Abz) and C-terminal 3-nitrotyrosine were introduced as fluorescence donor and acceptor, respectively. After excitation at 320 nm, cleavage of the substrate after the P1 arginine (in bold) resulted in a rise of fluorescence at 450 nm, because of the henceforward absence of quenching. The best specificity was observed for Abz-Ile-Arg-Ala-Arg-Ser-Ala-Gly-Tyr(3-NO2)-NH2 with a k cat/K m value of 454,200 M−1 s−1 [73]. Béliveau et al. incorporated the filaggrin-derived octapeptide Arg-Lys-Arg-Arg-Gly-Ser-Arg-Gly into an IQF substrate which possessed a k cat/K m value of 230,000 M−1 s−1 for MT-2 [74].
The human serine protease dipeptidyl peptidase-4 (DPP4) is expressed in several tissues and organs and occurs as membrane-associated or secreted enzyme. It cleaves and inactivates the incretins glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP). Incretins constitute metabolic hormones that stimulate, in the presence of high glucose levels, the secretion of insulin from pancreatic beta cells and inhibit the release of glucagon, leading to a downregulation of blood glucose concentration. Accordingly, DPP4 plays a crucial role in glucose homeostasis, and DPP4 inhibitors, such as sitagliptin, are used in the treatment of type-II diabetes mellitus [75]. DPP4 cleaves N-terminal dipeptides from polypeptides with a proline residue at the penultimate, i.e. P1, position. In order to identify new DPP-4 inhibitors, a suitable sensitive assay for a high-throughput screening is required. A commonly applied principle makes use of fluorescent coumarinyl dipeptides with a free amino group, e.g. H-Gly-Pro-AMC [76]. Kawaguchi et al. discussed disadvantages of such substrates, such as the excitation by a short wavelength and the short fluorescence life-time. Since the material of microplates scatters short wavelength light, undesirable interferences may occur. To overcome these limitations, a time-resolved fluorescence probe containing an aza-coumarin was developed (Fig. 8 ). The probe contains a lanthanide and is characterized by a large Stokes shift and a long luminescence lifetime, so the short background signals are negligible, resulting in an increased sensitivity for HTS. The substrate's strong luminescence is decreased after cleavage through acceptor-excited photo-induced electron transfer (PeT). The k cat/K m value of this probe for DPP4 (850,000 M−1 s−1) is similar to that of H-Gly-Pro-AMC (760,000 M−1 s−1) [77]. Likewise, a ratiometric two-photon fluorescent probe to monitor DPP4 was developed, based upon the cleavage of H-Gly-Pro-OH from 6-amino-2-butyl-1H-benzo[de]isoquinoline-1,3(2H)-dione [78].
Fig. 8.

Schematic representation of the DPP4 probe containing an aza-coumarin, a chelator moiety and the lanthanide terbium [77].
The cytoplasmic bacterial Lon proteases belong to the superfamily of the AAA+ (ATPases associated with various cellular activities) family and include the subfamily LonA, which occurs in bacteria and eukaryotes. LonA features an AAA domain and a proteolytic domain. The AAA domain forms oligomeric ring structures, and substrates are led through this pore into a separate chamber, where they are consumed by the proteolytic domain. Lon proteases are responsible for the degradation of more than 50% of the bacterial misfolded proteins. Even some sequences of cleavage sites are known, the modality of substrate recruitment is not well understood [79]. Because the Lon proteases play important roles in biofilm formation and stress tolerance, and inhibitors reduce colonization and virulence, Lon proteases are promising targets for new antibiotics. Lon proteases are serine proteases but possess a serine-lysine dyad, in contrast to most serine proteases with a catalytic triad [80]. The majority of Lon-protease inhibitors exhibits no selectivity and interacts with a huge number of serine proteases. Babin et al. introduced a well-tolerated substrate with high selectivity for Lon protease by using a HyCoSuL. Deconvolution led to the substrate Ac-hArg-nptGly-Phe-ACC with a k cat/K m value of 25,000 M−1 s−1. Neopentylglycine in P2 and homoarginine in P3 position resulted in selectivity over other serine proteases. The collected data were used for the assembly of an irreversible inhibitor. A boronic acid was inserted as a warhead, forming a covalent bond with active-site serine, and pyrazine-2‑carbonyl as capping group at the N-terminus to create Pyz-hArg-nptGly-Leu-B(OH)2 [81].
Several flaviviruses, including Dengue virus (DENV), Japanese encephalitis (JEV), West Nile (WNV) and Zika virus (ZIKV), possess a serine protease referred to as NS2B-NS3. The protein NS2B is integrated in the membrane of the endoplasmic reticulum. NS2B contains a hydrophilic part, essential for the non-covalent interaction with the serine protease NS3. Together they form the complex NS2B-NS3, which plays a crucial role in virus replication. The viral genome encodes a single polyprotein, which is cleaved into structural and non-structural proteins by host proteases and NS2B-NS3 itself [82,83]. NS2B-NS3 gets more and more in the focus as a promising drug target. By means of an ACC-based HyCoSuL, Rut et al. evaluated the substrate specificity of the P1-P4 positions with 19 natural and 108 unnatural amino acids for the positions P2-P4 and with 139 natural and unnatural amino acids for the P1 position. The best accepted substrates were Ac-D-Arg-Lys-Orn-Arg-ACC with a k cat/K m value of 1,104,000 M−1 s−1 for ZIKV NS2B-NS3 and Ac-D-Lys-Lys-Orn-Arg-ACC with a k cat/K m value of 65,300 M−1 s−1 for WNV NS2B-NS3. By considering these mapping approaches as well as selectivity requirements, ABPs for flaviviral proteases have been developed which share the characteristic dibasic recognition motif (Fig. 9 ). Probe 18 was selective for the DENV2 NS2B-NS3 protease, and 19 had a gradual activity for the proteases from ZIKV, WNF and DENV2 [84].
Fig. 9.

Activity-based probes for flaviviral serine proteases with biotin as detectable tag and diphenyl phosphonate as warhead [84].
4.2. Coumarin substrates for cysteine proteases
The cysteine cathepsin family consists of 11 members, most of them possess endopeptidase activity. Typically stored in the acid environment of the lysosome, their primary function is the nonspecific degradation of lysosomal proteins from endocytosis [[85], [86], [87]].
Cathepsin B is a ubiquitously expressed enzyme, synthesized as a 339-amino acid pre-proenzyme. Its signal peptide guides the protein to the rough endoplasmatic reticulum, where the short sequence is removed forming procathepsin B. The mature enzyme is generated through autocatalytic activation. The protease features endopeptidase and dipeptidyl carboxypeptidase activity, depending on the translocation of the occluding loop to form an open and closed conformation, respectively. Cathepsin B has been identified as an important tumor-promoting factor. In several cancer types, the enzyme is overexpressed and responsible for the activation of other proteases and the breakdown of the extracellular matrix (ECM), either by extracellular proteolysis catalyzed by the secreted enzyme or after internalization of ECM components, leading to tumor migration, proliferation, angiogenesis, and metastasis. Cytosolic cathepsin B plays a role in cell death by promoting apoptosis, triggered by the activation of caspases 3 and 9 [58,86,88]. Since cathepsin B expression is elevated in cancer cells, its proteolytic activity can be addressed for prodrug activation and drug delivery [89,90]. Such approaches can make use of coumarin fluorescence. For example, a prodrug system was designed with a dipeptide trigger and a coumarin linker. After proteolytic cleavage, a disassembling cascade was initiated resulting in the release of the anti-cancer drug melphalan and the emergence of the coumarin fluorescence [91].
The 333-amino acid pre-proenzyme of cathepsin L traverses a similar way to activation as cathepsin B, and its active form is obtained by autocatalytic cleavage or through the aspartyl protease cathepsin D. Cathepsin L has a strong tumor-promoting function. Besides cathepsin B, it is expressed at high levels in tumors and expression of both enzymes serves as a valuable biomarker. Cathepsin L is involved in endolysosomal and autophagic degradation pathways and plays a role in the accruement of liver fibrosis as well as polycystic and proteinuric kidney diseases, the latter through the cleavage of dynamin. Cathepsin L was reported to attenuate cardiac hypertrophy and to contribute to cardiac repair. Under uncontrolled diabetic conditions, a reduced cathepsin L activity impairs the invasion of circulating endothelial progenitor cells [85,88,92].
While the pre-proenzyme of cathepsin S exhibits 331 amino acids, the mature protein consists out of 217. Contrary to other cathepsins, it is expressed in antigen-presenting cells, localized in lymph and spleen, as well as other immune cells. Cathepsin S is the main processing enzyme of the major histocompatibility complex (MHC) class II-associated invariant chain, displaying a pivotal role in MHC II-dependent antigen presentation. Hence, the endolysosomal activity is crucial for regulating the immune response by B cells, dendritic cells and macrophages and connects cathepsin B to the pathophysiology of autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, myasthenia gravis and psoriasis. Moreover, the effects of cathepsin S in nociception and atherogenesis might be mediated due the cleavage of a precursor of fractalkine, a large, membrane-bound cytokine protein [85,86].
The 329-amino acid pre-proenzyme of cathepsin K is activated through auto- or heterocatalysis to the mature 215-amino acid protease. It is mainly expressed in osteoclasts, while its expression rate is triggered by receptor activator of NF-κB ligand (RANKL) and reduced by estrogen and calcitonin. Collagen type I represents 90% of the organic extracellular bone matrix. Cathepsin K, which is stored in cytosolic vesicles, is released in the resorption lacunae, and efficiently cleaves triple helix and telopeptide type I collagen as well as osteonectin. Also proteins, occurring outside of the bone matrix, are substrates of cathepsin K such as aggrecan, type II collagen, and elastin. Its involvement in bone resorption and disease states like osteoporosis and osteoarthritis turned cathepsin K into an outstanding therapeutic target. However, the development of the most promising drug candidate, odanacatib, has been discontinued because of the increased risk of cerebrovascular accidents [87,93].
Out of several reports on mapping of the non-primed substrate specificity, three will be discussed here. They all used combinatorial peptide libraries with a terminal ACC (7, see Fig. 3) fluorophore and determined the suitability of a substrate sequence by kinetic measurement. Ac-His-Arg-Leu-Arg-ACC (for cathepsin B), Ac-His-Arg-Tyr-Arg-ACC (for cathepsin L) and Ac-Leu-Arg-ACC (for cathepsin K) showed specificity constants (k cat/K m) of 15,000 M−1 s−1, 1,900,000 M−1 s−1 and 340,000 M−1 s−1, respectively [94]. Substrate profiling of cathepsin B by means of a HyCoSuL identified an N-terminal acetylated ACC peptide with cyclohexylalanine (P4), leucine (P3), glutamic acid benzyl ester (P2) and arginine (P1) to be highly selective over related cathepsins and to exhibit a specificity constants of 3,100,000 M−1 s−1 [95]. Accordingly, the substrate specificity of cathepsin L was dissected, leading to the most selective peptide with histidine (P4), d-threonine (P3), pentafluorophenylalanine (P2) and arginine (P1) characterized by a k cat/K m value of 729,200 M−1 s−1 [96]. Subsequently, Poreba et al. designed activity-based probes for cathepsin B and L equipped with an acyloxymethyl ketone warhead and a cyanine-5 fluorophore [95,96].
7-Amino-4-trifluoromethylcoumarin (AFC, 6, see Fig. 3), when released from the peptide substrates possesses a red-shift of excitation and emission, an advantageous feature for monitoring protease activity, compared to AMC and ACC. Dipeptidyl peptidase-1 (DPP1), also referred to as cathepsin C, is expressed in most mammalian tissues and reported to catalytically activate several leukocyte-derived serine proteases. DPP1 is thought to play a role in acute experimental arthritis. DPP1 cleaves dipeptides from the N-termini of substrates. Accordingly, AFC was placed at the C-terminus of an appropriate dipeptide, Gly-Phe, with a free amino group. Enzyme activity was followed at 400 nm (excitation) and 492 nm (emission) and additionally by time-dependent 19F NMR spectroscopy to evaluate DPP1 activities in serum samples [97].
Three-component probes have been reported for cathepsin B and L that produce in the cleavage step a two-component leaving group which undergoes spontaneous immolation to generate the fluorescent reporter AMC [98,99].
The SARS-Cov-2 virus out of the Coronaviridae family emerged first in the Chinese province Hubei in December 2019 and has since then spread around the world. The main protease SARS-CoV-2 Mpro, one of the coronaviral nonstructural proteins, constitutes a promising target for the treatment of the global pandemic COVID-19. SARS-CoV-2 Mpro cleaves the viral polyprotein to several proteins, which are indispensable for the virus replication. Rut et al. used a HyCoSuL to determine the substrate specificity of the Mpro using ACC-labeled substrates. The deconvolution identified the tetrapeptide Ac-Abu-Tle-Leu-Gln-ACC as the best substrate with a k cat/K M value of 859 M−1 s−1 (with Abu = (S)-2-aminobutyric acid and Tle = (S)-tert-leucine). The substrates were excited at 355 nm and the raising fluorescence was measured at 460 nm [100].
5. Internally quenched fluorescent peptide substrates
IQF peptides represent excellent tools to reconstruct the specificity profiles of proteases. Several donor/acceptor pairs have been established for the design of combinatorial libraries. These allow the deconvolution of the catalytic specificity of the protease of interest with respect to both, the non-primed and primed positions, and accordingly, the mapping of the non-primed and primed subsites. Suitable substrates for individual proteases can be obtained. Such approaches can circumvent restrictions of kinetically weak or inadequately selective substrates that only address the non-primed region. In the following, we will discuss examples in which coumarins serve as donors and/or quenchers of IQF peptide substrates of serine and cysteine proteases.
5.1. IQF substrates with a C-terminal coumarin moiety
The cleavage of plasminogen into plasmin is mediated by two types of activators, urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA). Besides their fibrinolytic profile, they are implicated in tissue proliferation and cellular adhesion. uPA, a 411-amino acids protein, consists of a serine protease domain, a kringle domain, and a growth factor domain. Active uPA is generated from the plasma-circulating zymogen prourokinase, which, after cleavage between Lys158 and Ile159, remains connected via a disulfide bond. The uPA-mediated plasminogen activation is focalized to the surface of cells expressing the urokinase receptor. uPA, its receptor and endogenous inhibitors, including plasminogen activator inhibitor type 1 (PAI-1), modulate the essential processes of tumor development. In particular, overexpression of uPA has been linked to breast cancer [101].
Roubinet et al. reported on a uPA substrate [10], which was assembled with coumarin 9 (see Fig. 3) as solubility-mediating fluorescence donor and a nitro-azobenzene derivative as acceptor with a quenching range between 394 and 506 nm. The enzymatic reaction was followed with excitation and emission wavelengths of 400 nm and 480 nm, respectively. The principle of such quenched substrates is shown for this uPA substrate in Fig. 10 .
Fig. 10.

Exemplary illustration of a protease-catalyzed cleavage of an IQF peptide substrate to monitor the activity of uPA [10].
Goldberg et al. utilized a non-scissile thioamide bond within the N-terminal part of the peptide as quencher of the fluorescence emitted by a C-terminal coumarin amino acid (11, n = 0, X = OMe, see Fig. 3). Excitation at 330 nm and emission at 390 nm was employed to analyze the activity of different proteases, including trypsin, chymotrypsin, papain and calpain. This noticeable quencher causes an only minimal perturbation of the native structure and efficient photoinduced electron transfer was achieved when the donor/acceptor pair was placed at least up to six amino acids apart [102].
Caspases are a family of cysteine proteases with a pronounced primary specificity for aspartic acid in P1 position and are involved in inflammation and apoptosis. All members of the family of caspases are initially synthesized as inactive zymogens. These contain an N-terminal prodomain of variable length, a large central subunit with the active site cysteine, and a small C-terminal domain. Caspases were grouped into three subfamilies, inflammatory, initiator of apoptosis, and effector/executioner caspases. Caspase-1 belongs to the first group and is engaged in the processing of cytokine pro-forms and the release of the maturated pro-inflammatory cytokines IL-1β and IL-18. The executioner caspases 3 and 7 have activity towards multiple substrates, such as Bid, XIAP and gelsolin and initiate a series of events that directly lead to the morphological changes associated with apoptosis [103,104].
To monitor caspase-1, Ranganathan et al. used an N-acetylated pentapeptide with Trp (P4), Glu (P3), His (P2) Asp (P1) and a coumarin amino acid of type 11 (n = 0, X = OMe, see Fig. 3) at P1´ position. The tryptophan moiety was excited at 270 nm and, upon cleavage, increased emission was detected at 345 nm. A specificity constant k cat/K m of 150,000 M−1 s−1 was reported. The coumarin uncommonly acted here as fluorescence acceptor, and the potential advantage of a ratiometric measurement was not taken [105].
To detect caspase-3 activity, Mizukami et al. established a novel dual function probe based on fluorescence and 19F NMR signal detection and synergizing the advantages of both sensitive methods (Fig. 11 ). Moreover, almost no intrinsic 19F NMR signal is detectable in living organisms. The probe consists of a gadolinium-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate (Gd3+-DOTA) complex, the caspase-3 substrate sequence Asp-Glu-Val-Asp and the coumarin derivative 6 (see Fig. 3). Substrate cleavage at the C-terminus results in the release of 6 and an increase in fluorescence. The Gd3+-DOTA complex in the intact probe quenches the 19F NMR signal through a paramagnetic relaxation enhancement (PRE) effect, which disappears upon cleavage. The dual probe exhibited a V max/K m value of 7.61 × 10−3 s−1 compared to 9.91 × 10−4 s−1 for the Ac-DEVD-AMC substrate [106].
Fig. 11.

Representation of 19F magnetic resonance imaging combined with coumarin fluorescence for detection of caspase 3 activity [106].
Apart from serine and cysteine proteases, the incorporation of a coumarin fluorophore at the C-terminus of IQF substrates has also been demonstrated for other proteases. One example included renin, an aspartyl protease secreted by the kidneys that participates in the renin–angiotensin–aldosterone system. Paschalidou et al. reported on a decapeptide with an N-terminal lysine equipped with a quenching 2,4-dinitrophenyl (DNP) substituent at the α-amino group and a C-terminal (S)-2-amino-3-(7-methoxycoumarin-4-yl)propanoic acid (AMP; 11, X = OMe, n = 0, see Fig. 3) at position P4’. DNP-Lys-His-Pro-Phe-His-Leu-Val-Ile-His-AMP exhibited a k cat/K m value for human renin of 352,000 M−1 s−1 [32].
The principle of Förster Resonance Energy Transfer (FRET) relies on the detection of the emission of an acceptor chromophore which is excited through energy transfer from a nearby donor chromophore. Obviously, FRET can be utilized for the positional profiling or the establishment of peptidic protease substrates. Okorochenkova et al. constructed a three-fluorophore system including a cleavage site for a trypsin-like protease (Ala-Lys-Ala) and a chymotrypsin-like protease (Ala-Phe-Ala, P1 amino acids are in bold) in a single compound. The authors introduced a coumarin amino acid of type 10 (n = 4, X = N(Et)2, see Fig. 3) C-terminal of the Ala-Lys-Ala motif, fluorescein between both motifs, and rhodamine B N-terminal of the Ala-Phe-Ala motif. This substrate employs a double-FRET mechanism. Following excitation of the coumarin at 435 nm and collecting the three emission signals of coumarin, fluorescein and rhodamine B, it was possible to detect and distinguish the activity of one or two proteases or their mixture. For example, in the presence of chymotrypsin and under excitation of the coumarin, energy transfer to fluorescein and a time-dependent increase of the fluorescein fluorescence occurred, which made ratiometric analyses possible [107].
5.2. IQF substrates with an N-terminal coumarin moiety
IQF peptide libraries with a coumarin positioned N-terminal of the scissile bond and, for example, 4-dimethylamino-azobenzene as quencher have also been launched for fingerprinting experiments towards protease specificity [108].
An important class of proteases occurring in neutrophilic granulocytes are the so called neutrophil serine proteases (NSPs), mainly cathepsin G, proteinase 3 and HNE. They are expressed during the myeloblast and promyelocyte stage of differentiation as inactive pre-proenzymes and are stored after N-terminal and C-terminal processing in azurophilic granules. Upon stimuli, the NSPs are released into the phagosome and the extracellular space or associated to the plasma membrane. After fusion of azurophilic granules with vacuoles carrying phagocytosed bacteria, NSPs function in intracellular host defense by degrading bacterial proteins. Once released into the extracellular space, NSPs are responsible for the destruction of proteins of the extracellular matrix including elastin, laminin, type IV collagen and fibronectin. Hence, they participate in neutrophil migration, and are also able to activate or inactivate cytokines. Altered activities of NSPs are connected to different diseases e.g. inflammation, infections, cardiovascular diseases, cancer and autoimmune vasculitis [109,110].
In a combinatorial approach to convolute the P1´ and P2´ specificities of cathepsin G, Lesner et al. acylated the terminal amino group of hexapeptides with 7-methoxy-coumarin-4-yl-acetic acid (MCA) and placed 4-guanidino-l-phenylalanine (Gnf) at P1 position and anthranilamide (ANB) as C-terminal quencher. The assay was performed with an excitation wavelength of 325 nm and an emission wavelength of 400 nm. A specificity constant of 251,920 M−1 s−1 for MCA-Phe-Val-Thr-Gnf-Ser-Trp-ANB was obtained [111]. Groborz et al. evaluated the P1´ to P3´ positions with the fluorophore ACC (7, see Fig. 3) at the N-terminus and the quencher 2,4-dinitrophenyl-lysine (Lys(DNP)) at P4´ position applying 355 nm and 460 nm and identified the cathepsin G substrate ACC-Gly-His(Bzl)-Tle-Pro-Phe-Ser-Asp-Met(O)-Gly-Lys(DNP)-Gly-NH2 (with His(Bzl) = N-benzylhistidine and Met(O) = methionine S-oxide) with a k cat/K m value of 206,854 M−1 s−1 [112].
The paracaspase MALT1 is a ubiquitously expressed cysteine protease containing a death domain, three immunoglobulin-like domains and the catalytic caspase-like domain. Its activity is triggered by MALT1-activating receptors. MALT1 acts due to a combination of its scaffolding and proteolytic functions. MALT1 plays a role in adaptive immune responses and, as part of the so-called CBA complex, regulates gene expression by the activation of NF-κB transcription factors and controls transcript stability [113]. For full-length MALT1, an IQF substrate was developed with structure ACC-AHX-Ala-Leu-Val-Ser-Arg-Gly-Thr-Lys(DNP)-Gly-OH (with AHX = 6-aminohexanoic acid, the P1 amino acid is in bold) exhibiting a k cat/K m value of 6300 M−1 s−1 [114].
In the following probe, a coumarin was again positioned at the N-terminus of the peptide. Yuan et al. employed 7-diethylamino-coumarin-3-carboxylic acid as energy donor and a tetraphenylethenethiophene moiety as energy quencher, connected through a caspase-3 specific peptide (Fig. 12 ). The probe is non-fluorescent by itself due to the energy transfer and the dissipation caused by the molecular motion of the quencher. Substrate cleavage renders not only the coumarin to a non-quenched fluorophore, but also the tetraphenylethene derivative to a fluorescent compound. In the absence of the energy transfer, its intramolecular rotation is restricted, leading to fluorescence after excitation at the same wavelength. In the cellular environment, caspases 3 and 7 were able to turn-on the fluorescence of this probe [115].
Fig. 12.

A probe to detect caspase activity with a dual signal output [115].
Fluorescence quenched peptide substrates have also been widely used to monitor the activity of metalloproteases. Matrix metalloproteinases (MMPs) and the related tumor necrosis factor converting enzyme (TACE) play important roles in cell migration, tissue remodeling and processing of signaling molecules, such as cytokines and adhesion factors. Knight et al. introduced MCA-Pro-Leu-Gly-Leu-Dap(DNP)-Ala-Arg-NH2 (with H-Dap-OH = 2,3-diaminopropionic acid) as a substrate for MMPs [116]. An N-terminal coumarin was also used in an improved and fully water soluble substrate for MMPs and TACE, i.e. MCA-Lys-Pro-Leu-Gly-Leu-Dap(DNP)-Ala-Arg-NH2 [117]. The following peptide sequence was employed for a MMP-2 substrate, Gly-Pro-Leu-Gly-Val-Arg-Gly-Lys-Gly-Gly, with 3-carboxy-7-diethylaminocoumarin coupled to the N-terminal glycine and (4-((4-(dimethylamino)phenyl)azo)benzoic acid) attached to the side chain of lysine [118]. To monitor MMP-12 activity, several ratiometric FRET substrates were developed based on the sequence Pro-Leu-Gly-Leu-Glu-Glu-Ala. The following FRET pairs were introduced, coumarin 343 (8, see Fig. 3) / TAMRA, 3-carboxy-7-diethylaminocoumarin / Oregon green and coumarin 343 / 3-carboxy-7-methoxycoumarin. The N-terminal fluorophores (excitation at 350–450 nm, emission at 410–490 nm) were amide-connected to proline. The C-terminal fluorophores (emission 490–575 nm) were coupled to the ε-amino group of a lysine moiety whose α-amino group was linked via 2-(2-(2-aminoethoxy)ethoxy)acetic acid to the alanine of the aforementioned sequence [119]. Two quenched substrates, donor-Gly-Pro-Leu-Gly-Leu-Lys(DNP)-Ala-Arg-NH2 with (7-amino-coumarin-4-yl)acetyl and (7-methoxy-coumarin-4-yl)acetyl as donors, showed k cat/K m values for MMP-2 of 278,800 M−1 s−1 and 179,900 M−1 s−1, respectively, and for MMP-9 of 89,200 M−1 s−1 and 82,400 M−1 s−1, respectively [120].
5.3. IQF substrates with two coumarin moieties
The fluorescence properties of coumarins can be fine-tuned through well-considered molecular design. Thus, it is possible to employ two different coumarins as a FRET pair positioned at both sides of the scissile peptide bond. Gehrig et al. demonstrated that such a substrate is suitable to monitor the activity of HNE and mouse neutrophil elastase. 7-Methoxy-coumarin-3-carboxylic acid was excited at 354 nm and coumarin 343 (8, see Fig. 3) emitted light of 490 nm (Fig. 13 ). The analysis of this true FRET substrate included a ratiometric approach, considering increasing fluorescence at 400 nm and the decreasing fluorescence at 490 nm in the course of substrate consumption [121].
Fig. 13.

A FRET probe for mouse and human neutrophil elastases (NEs) [121].
Wysocka et al. established an IQF-substrate based assay to simultaneously monitor the activity of the three NSPs, cathepsin G, proteinase-3 and HNE using only one single measurement. For this purpose, three different donor/acceptor pairs were incorporated into three different peptide sequences, leading to three individual and selective substrates. Substrate cleavage led to an increasing fluorescence of the respective donor at 395 nm (proteinase-3), 445 nm (cathepsin G) and 490 nm (HNE). In two cases, coumarins were incorporated both as donors and quenchers. The proteinase-3 substrate (k cat/K M = 524,100 M−1 s−1) was assembled with lysines acylated with coumarins 11 (n = 3, X = OMe and X = OH, see Fig. 3) as donor and quencher, respectively. The cathepsin G substrate (k cat/K M = 379,000 M−1 s−1) was designed with lysine acylated with 7-hydroxy-4-methyl-coumarin-3-yl-acetic acid as donor and with ornithine acylated with coumarin 343 (8, see Fig. 3) as quencher [122]. An overview on the protease substrates discussed in the review is given in the Supplementary material.
6. Activity-based probes
Fluorescent activity-based probes have become a powerful tool in enzyme research, e.g. for the localization and imaging of enzymatic activities in biological systems by virtue of the high sensitivity, nondestructive analysis, and real-time detection abilities of the probes. ABPs enable the analysis of only the active form of the target enzyme, which is beneficial, because the enzymatic activity does not necessarily correlate with the expression level of the protein [13]. Coumarins do not represent the most frequently applied fluorophores to generate ABPs for serine and cysteine proteases, but relevant recent examples will be discussed in the following.
6.1. Activity-based probes for serine proteases
In order to design an ABP for the serine protease matriptase-2, substrate mapping approaches and the consideration of the endogenous substrate were used as point of origin. Because the S1 and the S3/S4 subunits prefer basic amino acids, in particular arginine, two guanidinophenyl groups were introduced as arginine mimetics. A coumarin label was inserted into corresponding phosphonate inhibitors leading to probe 20 (Fig. 14 ) [123]. The nucleophilic attack conducted by the catalytic OH group at the warhead's phosphorus resulted in the elimination of one phenoxy group, the formation of a new P–O bond and irreversible inhibition. The “Eastern” guanidinophenyl moiety was assumed to interact with the S1 pocket and the “Western” one to extend towards the S3/S4 pocket. Compound 20 with the (R)-configured warhead and the (S)-configured amino acid possessed a k inac/K i value of 50.4 M−1 s−1. The applicability of 20 as ABP was demonstrated by in-gel fluorescence detection of matriptase-2 after treating the medium of stably transfected human embryonic kidney (HEK) cells with 20 and subjecting the mixture to SDS-PAGE.
Fig. 14.

A phosphono dipeptide ABP for matriptase-2 [123].
Matriptase, one of the best characterized TTSP and a close relative of matriptase-2, is mainly expressed in epidermis, salivary gland, thyroid, stomach, kidney, prostate and ovaries as an inactive zymogen and has to be converted in an autocatalytic manner into its active form. Matriptase processes several proteins, such as hepatocyte growth factor/scatter factor, uPA and protease-activated receptor 2 which play critical roles in tumorigenesis and trigger other signaling pathways related to cancer proliferation and metastasis [124]. An ABP for matriptase (21, Fig. 15 ) also included the diphenyl phosphonate warhead and two arginine mimetics expected to occupy the S1 and S2 subunits. The coumarin fluorophore was assumed to be orientated towards the S3/S4 region. The second-order rate constant for inactivation of matriptase (k inac/K i = 576 M−1 s−1) was one order of magnitude higher than that for matriptase-2. Probe 21 was successfully employed for direct fluorescence readout after SDS-PAGE and HPLC size-exclusion chromatography coupled to fluorescence detection [125].
Fig. 15.

A matriptase-directed ABP for in-gel and HPLC fluorescence detection [125].
Sulfonyloxyphtalimides are mechanism-based inhibitors of serine proteases whose interaction involves a nucleophilic attack of the active-site serine, ring opening, Lossen rearrangement of the O-sulfonyl hydroxamic acid intermediate, and trapping of the resulting isocyanate by water or a second adjacent active-site nucleophile [126]. This principle was adapted for the design of the ABP 22 targeting human neutrophil elastase (Fig. 16 ) [127]. The coumarin tag of the probe allowed for the in-gel fluorescence detection of HNE with a low-nanomolar probe concentration. The reaction of porcine pancreatic elastase with 22 was followed by analyzing the fluorescence kinetics. A FRET was applied by exciting the anthranilic acid fluorophore at 320 nm and detecting the emission of coumarin 343 at 490 nm. However, at an excitation wavelength of 258 nm for the enzyme's tryptophan, a double-FRET system was realized. By monitoring the later stages of the reaction, the half-life of the anthranoyl enzyme 23 (3.3 h) could be determined.
Fig. 16.

An ABP for elastases. Implementation of the Lossen rearrangement to induce Förster Resonance Energy Transfers [127].
6.2. Activity based probes for cysteine proteases
Cysteine cathepsins represent a promising target for activity-based probing. For the purpose of establishing an ABP with cathepsin S preference, an α,β-unsaturated Michael acceptor was used as a soft-electrophile warhead (Fig. 17 ). The formation of a covalent sulfur-β‑carbon bond led to an irreversible inhibition. In the predicted binding mode, the vinyl sulfone moiety extended towards the S1´ pocket and the hydrophobic S2 subsite was occupied by phenylalanine. Probe 24 was selective for cathepsins S and L (k inac/K i = 49,000 M−1 s−1 and 16,900 M−1 s−1) over cathepsins K and B (k inac/K i < 900 M−1 s−1). The feasibility of compound 24 for direct in-gel fluorescence detection and for labeling of native cathepsin S in the protein extract from human placenta was demonstrated [128].
Fig. 17.

A tripeptidomimetic ABP for cysteine cathepsins [128].
Dipeptide nitriles are well-established covalent inhibitors of cysteine proteases forming thioimidates with the active-site cysteine [87]. The inhibition is not irreversible, and the adduct can dissociate and release the enzyme. This is the reason that peptide nitriles are not suitable as ABPs for in-gel detection. However, the labelling with a coumarin can be performed to study the cellular uptake by fluorescence measurements. A prototypical cathepsin inhibitor structure was equipped with coumarin 343 leading to 25 (Fig. 18 ) [129]. The integration of an isobutylsulfonylcysteine at P2 provides particular affinity for cathepsin S, while the tetracyclic coumarin fits in the deep S3 pocket of cathepsin K. Accordingly, a dual-active, sub-micromolar inhibitor was obtained. HEK cells were treated with 25 for different incubation times and cell lysates were analyzed with an excitation wavelength of 450 nm and an emission wavelength of 492 nm. The protein concentration was used as a relative benchmark for the cellular uptake of 25 [129].
Fig. 18.

A fluorescence-labeled dipeptide nitrile for cellular uptake studies [129].
7. Conclusion
Coumarins are valued for their fluorescence properties with rather large Stokes shifts and the opportunity to fine-tune the photochemical features by the tailored control of bond rotations and introduction of substituents. The coumarin scaffold is easily accessible by chemical synthesis. A particular advantage is the small size of coumarins which predestinate their incorporation into the side chain of amino acids. The resulting fluorescent amino acids can easily be introduced into peptides. This allows for the design of fluorescent peptides as valuable tools for the active-site mapping of proteases. Fluorophore/quencher pairs or FRET donor/acceptor pairs can be placed at the termini or inserted inside of these peptides. Besides the utilization of coumarins as structural components of substrates, they can also be beneficially applied for the design of activity-based probes for serine and cysteine proteases.
Declaration of Competing Interest
None.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbapap.2020.140445.
Appendix A. Supplementary data
Supplementary material
References
- 1.Dwivedi A.P., Kumar S., Varshney V., Singh A.B., Srivastava A.K., Sahu D.P. Synthesis and antihyperglycemic activity of novel N-acyl-2-arylethylamines and N-acyl-3-coumarylamines. Bioorg. Med. Chem. Lett. 2008;18:2301–2305. doi: 10.1016/j.bmcl.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Yu D., Suzuki M., Xie L., Morris-Natschke S.L., Lee K.H. Recent progress in the development of coumarin derivatives as potent anti-HIV agents. Med. Res. Rev. 2003;23:322–345. doi: 10.1002/med.10034. [DOI] [PubMed] [Google Scholar]
- 3.Kontogiorgis C.A., Hadjipavlou-Litina D.J. Synthesis and biological evaluation of novel coumarin derivatives with a 7-azomethine linkage. Bioorg. Med. Chem. Lett. 2004;14:611–614. doi: 10.1016/j.bmcl.2003.11.060. [DOI] [PubMed] [Google Scholar]
- 4.Stefanachi A., Favia A.D., Nicolotti O., Leonetti F., Pisani L., Catto M., Zimmer C., Hartmann R.W., Carotti A. Design, synthesis, and biological evaluation of imidazolyl derivatives of 4,7-disubstituted coumarins as aromatase inhibitors selective over 17-α-hydroxylase/C17-20 lyase. J. Med. Chem. 2011;54:1613–1625. doi: 10.1021/jm101120u. [DOI] [PubMed] [Google Scholar]
- 5.Yang J., Liu G.Y., Dai F., Cao X.Y., Kang Y.F., Hu L.M., Tang J.J., Li X.Z., Li Y., Jin X.L., Zhou B. Synthesis and biological evaluation of hydroxylated 3-phenylcoumarins as antioxidants and antiproliferative agents. Bioorg. Med. Chem. Lett. 2011;21:6420–6425. doi: 10.1016/j.bmcl.2011.08.090. [DOI] [PubMed] [Google Scholar]
- 6.Matos M.J., Vilar S., Vazquez-Rodriguez S., Kachler S., Klotz K.N., Buccioni M., Delogu G., Santana L., Uriarte E., Borges F. Structure-based optimization of coumarin hA3 adenosine receptor antagonists. J. Med. Chem. 2020;63:2577–2587. doi: 10.1021/acs.jmedchem.9b01572. [DOI] [PubMed] [Google Scholar]
- 7.Mertens M.D., Hinz S., Müller C.E., Gütschow M. Alkynyl-coumarinyl ethers as MAO-B inhibitors. Bioorg. Med. Chem. 2014;22:1916–1928. doi: 10.1016/j.bmc.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 8.Pisani L., Farina R., Catto M., Iacobazzi R.M., Nicolotti O., Cellamare S., Mangiatordi G.F., Denora N., Soto-Otero R., Siragusa L., Altomare C.D., Carotti A. Exploring basic tail modifications of coumarin-based dual acetylcholinesterase-monoamine oxidase B inhibitors: identification of water-soluble, brain-permeant neuroprotective multitarget agents. J. Med. Chem. 2016;59:6791–6806. doi: 10.1021/acs.jmedchem.6b00562. [DOI] [PubMed] [Google Scholar]
- 9.Poreba M., Drag M. Current strategies for probing substrate specificity of proteases. Curr. Med. Chem. 2010;17:3968–3995. doi: 10.2174/092986710793205381. [DOI] [PubMed] [Google Scholar]
- 10.Roubinet B., Chevalier A., Renard P.Y., Romieu A. A synthetic route to 3-(heteroaryl)-7-hydroxycoumarins designed for biosensing applications. Eur. J. Org. Chem. 2015:166–182. doi: 10.1002/ejoc.201403215. [DOI] [Google Scholar]
- 11.Cao D., Liu Z., Verwilst P., Koo S., Jangjili P., Kim J.S., Lin W. Coumarin-based small-molecule fluorescent chemosensors. Chem. Rev. 2019;119:10403–10519. doi: 10.1021/acs.chemrev.9b00145. [DOI] [PubMed] [Google Scholar]
- 12.Schechter I., Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 13.Serim S., Haedke U., Verhelst S.H. Activity-based probes for the study of proteases: recent advances and developments. ChemMedChem. 2012;7:1146–1159. doi: 10.1002/cmdc.201200057. [DOI] [PubMed] [Google Scholar]
- 14.Liu H.W., Chen L., Xu C., Li Z., Zhang H., Zhang X.B., Tan W. Recent progresses in small-molecule enzymatic fluorescent probes for cancer imaging. Chem. Soc. Rev. 2018;47:7140–7180. doi: 10.1039/c7cs00862g. [DOI] [PubMed] [Google Scholar]
- 15.Jurić A., Trinajstić N. Topological resonance energies of thiocoumarins. Croat. Chem. Acta. 1983;56:215–219. [Google Scholar]
- 16.Matos M.A., Sousa C.C., Miranda M.S., Morais V.M., Liebman J.F. Energetics of coumarin and chromone. J. Phys. Chem. B. 2009;113:11216–11221. doi: 10.1021/jp9026942. [DOI] [PubMed] [Google Scholar]
- 17.Neumann U., Gütschow M. 3,1-Benzothiazin-4-ones and 3,1-benzoxazin-4-ones: highly different activities in chymotrypsin inactivation. Bioorg. Chem. 1995;23:72–88. doi: 10.1006/bioo.1995.1006. [DOI] [Google Scholar]
- 18.Garrett E.R., Lippold B.C., Mielck J.B. Kinetics and mechanisms of lactonization of coumarinic acids and hydrolysis of coumarins. J. Pharm. Sci. 1971;60:396–405. doi: 10.1002/jps.2600600312. [DOI] [PubMed] [Google Scholar]
- 19.O'Neal J.S., Schulman S.G., van der Giesen W.F., Roomer A.C. Alkaline hydrolytic lability of some hydroxy- and methoxycoumarins and related anticoagulants. Int. J. Pharm. 1982;12:355–359. doi: 10.1016/0378-5173(82)90107-7. [DOI] [Google Scholar]
- 20.Huitink G.M. Studies of 7-hydroxycoumarins. Talanta. 1988;35:973–976. doi: 10.1016/0039-9140(88)80231-5. [DOI] [PubMed] [Google Scholar]
- 21.Murata C., Masuda T., Kamochi Y., Todoroki K., Yoshida H., Nohta H., Yamaguchi M., Takadate A. Improvement of fluorescence characteristics of coumarins: syntheses and fluorescence properties of 6-methoxycoumarin and benzocoumarin derivatives as novel fluorophores emitting in the longer wavelength region and their application to analytical reagents. Chem. Pharm. Bull. 2005;53:750–758. doi: 10.1248/cpb.53.750. [DOI] [PubMed] [Google Scholar]
- 22.Corrie J.E., Munasinghe V.R., Rettig W. Synthesis and fluorescence properties of substituted 7-aminocoumarin-3-carboxylate derivatives. J. Heterocyclic Chem. 2000;37:1447–1455. doi: 10.1002/jhet.5570370608. [DOI] [Google Scholar]
- 23.Besson T., Coudert G., Guillaumet G. Synthesis and fluorescent properties of some heterobifunctional and rigidized 7-aminocoumarins. J. Heterocyclic Chem. 1991;28:1517–1523. doi: 10.1002/jhet.5570280608. [DOI] [Google Scholar]
- 24.Barot K.P., Jain S.V., Kremer L., Singh S., Ghate M.D. Recent advances and therapeutic journey of coumarins: current status and perspectives. Med. Chem. Res. 2015;24:2771–2798. doi: 10.1007/s00044-015-1350-8. [DOI] [Google Scholar]
- 25.Ufer M. Comparative pharmacokinetics of vitamin K antagonists: warfarin, phenprocoumon and acenocoumarol. Clin. Pharmacokinet. 2005;44:1227–1246. doi: 10.2165/00003088-200544120-00003. [DOI] [PubMed] [Google Scholar]
- 26.Jones G., II, Jackson W.R., Choi C.Y., Bergmark W.R. Solvent effects on emission yield and lifetime for coumarin laser dyes. Requirements for a rotatory decay mechanism. J. Phys. Chem. 1985;89:284–300. doi: 10.1021/j100248a024. [DOI] [Google Scholar]
- 27.Wang Q., Hu W., Feng Q., Cao X.H., Chai W., Yi C., Li M.J. Coumarin-modified gold nanoprobes for the sensitive detection of caspase-3. RSC Adv. 2015;5:43824–43830. doi: 10.1039/C5RA05350A. [DOI] [Google Scholar]
- 28.Kuru E., Hughes H.V., Brown P.J., Hall E., Tekkam S., Cava F., de Pedro M.A., Brun Y.V., VanNieuwenhze M.S. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew. Chem. Int. Ed. Eng. 2012;51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murase T., Yoshihara T., Yamada K., Tobita S. Fluorescent peptides labeled with environment-sensitive 7-aminocoumarins and their interactions with lipid bilayer membranes and living cells. Bull. Chem. Soc. Jpn. 2013;86:510–519. doi: 10.1246/bcsj.20120314. [DOI] [Google Scholar]
- 30.Häußler D., Gütschow M. Fluorescently labeled amino acids as building blocks for bioactive molecules. Synthesis. 2016;48:245–255. doi: 10.1055/s-0035-1560361. [DOI] [Google Scholar]
- 31.Mertens M.D., Gütschow M. Clickable coumarins as fluorescent labels for amino acids. Synthesis. 2014;46:2191–2200. doi: 10.1055/s-0033-1338636. [DOI] [Google Scholar]
- 32.Paschalidou K., Neumann U., Gerhartz B., Tzougraki C. Highly sensitive intramolecularly quenched fluorogenic substrates for renin based on the combination of L-2-amino-3-(7-methoxy-4-coumaryl)propionic acid with 2,4-dinitrophenyl groups at various positions. Biochem. J. 2004;382:1031–1038. doi: 10.1042/BJ20040729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brun M.P., Bischoff L., Garbay C. A very short route to enantiomerically pure coumarin-bearing fluorescent amino acids. Angew. Chem. Int. Ed. Eng. 2004;43:3432–3436. doi: 10.1002/anie.200454116. [DOI] [PubMed] [Google Scholar]
- 34.Kuhn S.M., Rubini M., Müller M.A., Skerra A. Biosynthesis of a fluorescent protein with extreme pseudo-stokes shift by introducing a genetically encoded non-natural amino acid outside the fluorophore. J. Am. Chem. Soc. 2011;133:3708–3711. doi: 10.1021/ja1099787. [DOI] [PubMed] [Google Scholar]
- 35.Goldberg J.M., Speight L.C., Fegley M.W., Petersson E.J. Minimalist probes for studying protein dynamics: thioamide quenching of selectively excitable fluorescent amino acids. J. Am. Chem. Soc. 2012;134:6088–6091. doi: 10.1021/ja3005094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koopmans T., van Haren M., van Ufford L.Q., Beekman J.M., Martin N.I. A concise preparation of the fluorescent amino acid L-(7-hydroxycoumarin-4-yl) ethylglycine and extension of its utility in solid phase peptide synthesis. Bioorg. Med. Chem. 2013;21:553–559. doi: 10.1016/j.bmc.2012.10.055. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt L., Doroshenko T., Barbie P., Grüter A., Jung G., Kazmaier U. Synthesis of fluorescent amino acids via palladium-catalyzed allylic alkylations. Synthesis. 2016;48:3077–3086. doi: 10.1055/s-0035-1561628. [DOI] [Google Scholar]
- 38.Gómez-Outes A., Suárez-Gea M.L., Calvo-Rojas G., Lecumberri R., Rocha E., Pozo-Hernández C., Terleira-Fernández A.I., Vargas-Castrillón E. Discovery of anticoagulant drugs: a historical perspective. Curr. Drug Discov. Technol. 2012;9:83–104. doi: 10.2174/1570163811209020083. [DOI] [PubMed] [Google Scholar]
- 39.Bye A., King H.K. The biosynthesis of 4-hydroxycoumarin and dicoumarol by Aspergillus fumigatus Fresenius. Biochem. J. 1970;117:237–245. doi: 10.1042/bj1170237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pochet L., Frédérick R., Masereel B. Coumarin and isocoumarin as serine protease inhibitors. Curr. Pharm. Des. 2004;10:3781–3796. doi: 10.2174/1381612043382684. [DOI] [PubMed] [Google Scholar]
- 41.Robert S., Bertolla C., Masereel B., Dogné J.M., Pochet L. Novel 3-carboxamide-coumarins as potent and selective FXIIa inhibitors. J. Med. Chem. 2008;51:3077–3080. doi: 10.1021/jm8002697. [DOI] [PubMed] [Google Scholar]
- 42.Bouckaert C., Serra S., Rondelet G., Dolušić E., Wouters J., Dogné J.M., Frédérick R., Pochet L. Synthesis, evaluation and structure-activity relationship of new 3-carboxamide coumarins as FXIIa inhibitors. Eur. J. Med. Chem. 2016;110:181–194. doi: 10.1016/j.ejmech.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 43.Frédérick R., Robert S., Charlier C., de Ruyck J., Wouters J., Pirotte B., Masereel B., Pochet L. 3,6-disubstituted coumarins as mechanism-based inhibitors of thrombin and factor Xa. J. Med. Chem. 2005;48:7592–7603. doi: 10.1021/jm050448g. [DOI] [PubMed] [Google Scholar]
- 44.Frédérick R., Charlier C., Robert S., Wouters J., Masereel B., Pochet L. Investigation of mechanism-based thrombin inhibitors: implications of a highly conserved water molecule for the binding of coumarins within the S pocket. Bioorg. Med. Chem. Lett. 2006;16:2017–2021. doi: 10.1016/j.bmcl.2005.12.070. [DOI] [PubMed] [Google Scholar]
- 45.Frédérick R., Robert S., Charlier C., Wouters J., Masereel B., Pochet L. Mechanism-based thrombin inhibitors: design, synthesis, and molecular docking of a new selective 2-oxo-2H-1-benzopyran derivative. J. Med. Chem. 2007;50:3645–3650. doi: 10.1021/jm061368v. [DOI] [PubMed] [Google Scholar]
- 46.Fischer P.M. Design of small-molecule active-site inhibitors of the S1A family proteases as procoagulant and anticoagulant drugs. J. Med. Chem. 2018;61:3799–3822. doi: 10.1021/acs.jmedchem.7b00772. [DOI] [PubMed] [Google Scholar]
- 47.Pochet L., Doucet C., Schynts M., Thierry N., Boggetto N., Pirotte B., Jiang K.Y., Masereel B., de Tullio P., Delarge J., Reboud-Ravaux M. Esters and amides of 6-(chloromethyl)-2-oxo-2H-1-benzopyran-3-carboxylic acid as inhibitors of alpha-chymotrypsin: significance of the "aromatic" nature of the novel ester-type coumarin for strong inhibitory activity. J. Med. Chem. 1996;39:2579–2585. doi: 10.1021/jm960090b. [DOI] [PubMed] [Google Scholar]
- 48.Bouckaert C., Zhu S., Govers-Riemslag J.W., Depoorter M., Diamond S.L., Pochet L. Discovery and assessment of water soluble coumarins as inhibitors of the coagulation contact pathway. Thromb. Res. 2017;157:126–133. doi: 10.1016/j.thromres.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 49.Ghani U., Ng K.K., Atta-ur-Rahman M.I. Choudhary, Ullah N., James M.N. Crystal structure of gamma-chymotrypsin in complex with 7-hydroxycoumarin. J. Mol. Biol. 2001;314:519–525. doi: 10.1006/jmbi.2001.5148. [DOI] [PubMed] [Google Scholar]
- 50.Tan X., Soualmia F., Furio L., Renard J.F., Kempen I., Qin L., Pagano M., Pirotte B., El Amri C., Hovnanian A., Reboud-Ravaux M. Toward the first class of suicide inhibitors of kallikreins involved in skin diseases. J. Med. Chem. 2015;58:598–612. doi: 10.1021/jm500988d. [DOI] [PubMed] [Google Scholar]
- 51.Borgoño C.A., Michael I.P., Komatsu N., Jayakumar A., Kapadia R., Clayman G.L., Sotiropoulou G., Diamandis E.P. A potential role for multiple tissue kallikrein serine proteases in epidermal desquamation. J. Biol. Chem. 2007;282:3640–3652. doi: 10.1074/jbc.M607567200. [DOI] [PubMed] [Google Scholar]
- 52.MEROPS The Peptidase Database. Released 12.1. 2020. http://www.ebi.ac.uk/merops/ (accessed 13 February 2020)
- 53.Bond J.S. Proteases: history, discovery, and roles in health and disease. J. Biol. Chem. 2019;294:1643–1651. doi: 10.1074/jbc.TM118.004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneider E.L., Craik C.S. Positional scanning synthetic combinatorial libraries for substrate profiling. Methods Mol. Biol. 2009;539:59–78. doi: 10.1007/978-1-60327-003-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris J.L., Backes B.J., Leonetti F., Mahrus S., Ellman J.A., Craik C.S. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7754–7759. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kasperkiewicz P., Poreba M., Groborz K., Drag M. Emerging challenges in the design of selective substrates, inhibitors and activity-based probes for indistinguishable proteases. FEBS J. 2017;284:1518–1539. doi: 10.1111/febs.14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poreba M., Salvesen G.S., Drag M. Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nat. Protoc. 2017;12:2189–2214. doi: 10.1038/nprot.2017.091. [DOI] [PubMed] [Google Scholar]
- 58.Schmitz J., Gilberg E., Löser R., Bajorath J., Bartz U., Gütschow M. Cathepsin B: active site mapping with peptidic substrates and inhibitors. Bioorg. Med. Chem. 2019;27:1–15. doi: 10.1016/j.bmc.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 59.Backes B.J., Ellman J.A. An alkanesulfonamide “safety-catch” linker for solid-phase synthesis. J. Organomet. Chem. 1999;64:2322–2330. doi: 10.1021/jo981990y. [DOI] [Google Scholar]
- 60.Backes B.J., Harris J.L., Leonetti F., Craik C.S., Ellman J.A. Synthesis of positional-scanning libraries of fluorogenic peptide substrates to define the extended substrate specificity of plasmin and thrombin. Nat. Biotechnol. 2000;18:187–193. doi: 10.1038/72642. [DOI] [PubMed] [Google Scholar]
- 61.Byzia A., Szeffler A., Kalinowski L., Drag M. Activity profiling of aminopeptidases in cell lysates using a fluorogenic substrate library. Biochimie. 2016;122:31–37. doi: 10.1016/j.biochi.2015.09.035. [DOI] [PubMed] [Google Scholar]
- 62.Zeerleder S. Factor VII-activating protease: Hemostatic protein or immune regulator? Semin. Thromb. Hemost. 2018;44:151–158. doi: 10.1055/s-0037-1607431. [DOI] [PubMed] [Google Scholar]
- 63.Byskov K., Le Gall S.M., Thiede B., Camerer E., Kanse S.M. Protease activated receptors (PAR)-1 and −2 mediate cellular effects of factor VII activating protease (FSAP) FASEB J. 2020;34:1079–1090. doi: 10.1096/fj.201801986RR. [DOI] [PubMed] [Google Scholar]
- 64.Kara E., Manna D., Løset G.Å., Schneider E.L., Craik C.S., Kanse S. Analysis of the substrate specificity of Factor VII activating protease (FSAP) and design of specific and sensitive peptide substrates. Thromb. Haemost. 2017;117:1750–1760. doi: 10.1160/TH17-02-0081. [DOI] [PubMed] [Google Scholar]
- 65.Baeten K.M., Richard M.C., Kanse S.M., Mutch N.J., Degen J.L., Booth N.A. Activation of single-chain urokinase-type plasminogen activator by platelet-associated plasminogen: a mechanism for stimulation of fibrinolysis by platelets. J. Thromb. Haemost. 2010;8:1313–1322. doi: 10.1111/j.1538-7836.2010.03813.x. [DOI] [PubMed] [Google Scholar]
- 66.Rut W., Nielsen N.V., Czarna J., Poreba M., Kanse S.M., Drag M. Fluorescent activity-based probe for the selective detection of factor VII activating protease (FSAP) in human plasma. Thromb. Res. 2019;182:124–132. doi: 10.1016/j.thromres.2019.08.016. [DOI] [PubMed] [Google Scholar]
- 67.Wysocka M., Legowska A., Bulak E., Jaśkiewicz A., Miecznikowska H., Lesner A., Rolka K. New chromogenic substrates of human neutrophil cathepsin G containing non-natural aromatic amino acid residues in position P(1) selected by combinatorial chemistry methods. Mol. Divers. 2007;11:93–99. doi: 10.1007/s11030-007-9063-7. [DOI] [PubMed] [Google Scholar]
- 68.Zou F., Schmon M., Sienczyk M., Grzywa R., Palesch D., Boehm B.O., Sun Z.L., Watts C., Schirmbeck R., Burster T. Application of a novel highly sensitive activity-based probe for detection of cathepsin G. Anal. Biochem. 2012;421:667–672. doi: 10.1016/j.ab.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 69.Schroeder R., Grzywa R., Wirtz C.R., Sienczyk M., Burster T. Application of a novel FAM-conjugated activity-based probe to determine cathepsin G activity intracellularly. Anal. Biochem. 2020;588:113488. doi: 10.1016/j.ab.2019.113488. [DOI] [PubMed] [Google Scholar]
- 70.Kasperkiewicz P., Altman Y., D'Angelo M., Salvesen G.S., Drag M. Toolbox of fluorescent probes for parallel imaging reveals uneven location of serine proteases in neutrophils. J. Am. Chem. Soc. 2017;139:10115–10125. doi: 10.1021/jacs.7b04394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gitlin-Domagalska A., Mangold M., Dębowski D., Ptaszyńska N., Łęgowska A., Gütschow M., Rolka K. Matriptase-2: monitoring and inhibiting its proteolytic activity. Future Med. Chem. 2018;10:2745–2761. doi: 10.4155/fmc-2018-0346. [DOI] [PubMed] [Google Scholar]
- 72.Camaschella C., Nai A., Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020;105:260–272. doi: 10.3324/haematol.2019.232124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wysocka M., Gruba N., Miecznikowska A., Popow-Stellmaszyk J., Gütschow M., Stirnberg M., Furtmann N., Bajorath J., Lesner A.K. Substrate specificity of human matriptase-2. Biochimie. 2014;97:121–127. doi: 10.1016/j.biochi.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 74.Béliveau F., Désilets A., Leduc R. Probing the substrate specificities of matriptase, matriptase-2, hepsin and DESC1 with internally quenched fluorescent peptides. FEBS. 2009;276:2213–2226. doi: 10.1111/j.1742-4658.2009.06950.x. [DOI] [PubMed] [Google Scholar]
- 75.De S., Banerjee S., Kumar S.K., Paira P. Critical role of dipeptidyl peptidase IV: A therapeutic target for diabetes and cancer. Mini-Rev. Med. Chem. 2019;19:88–97. doi: 10.2174/1389557518666180423112154. [DOI] [PubMed] [Google Scholar]
- 76.Kim D., Wang L., Beconi M., Eiermann G.J., Fisher M.H., He H., Hickey G.J., Kowalchick J.E., Leiting B., Lyons K., Marsilio F., McCann M.E., Patel R.A., Petrov A., Scapin G., Patel S.B., Roy R.S., Wu J.K., Wyvratt M.J., Zhang B.B., Zhu L., Thornberry N.A., Weber A.E. (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: A potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005;48:141–151. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- 77.Kawaguchi M., Okabe T., Terai T., Hanaoka K., Kojima H., Minegishi I., Nagano T. A time-resolved fluorescence probe for dipeptidyl peptidase 4 and its application in inhibitor screening. Chem. Eur. J. 2010;16:13479–13486. doi: 10.1002/chem.201001077. [DOI] [PubMed] [Google Scholar]
- 78.Zou L.W., Wang P., Qian X.K., Feng L., Yu Y., Wang D.D., Jin Q., Hou J., Liu Z.H., Ge G.B., Yang L. A highly specific ratiometric two-photon fluorescent probe to detect dipeptidyl peptidase IV in plasma and living systems. Biosens. Bioelectron. 2017;90:283–289. doi: 10.1016/j.bios.2016.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Voos W., Pollecker K. The mitochondrial Lon protease: novel functions off the beaten track? Biomolecules. 2020;10 doi: 10.3390/biom10020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raju R.M., Goldberg A.L., Rubin E.J. Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discov. 2012;11:777–789. doi: 10.1038/nrd3846. [DOI] [PubMed] [Google Scholar]
- 81.Babin B.M., Kasperkiewicz P., Janiszewski T., Yoo E., Drąg M., Bogyo M. Leveraging peptide substrate libraries to design inhibitors of bacterial Lon protease. ACS Chem. Biol. 2019;14:2453–2462. doi: 10.1021/acschembio.9b00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lei J., Hansen G., Nitsche C., Klein C.D., Zhang L., Hilgenfeld R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science. 2016;252:503–505. doi: 10.1126/science.aag2419. [DOI] [PubMed] [Google Scholar]
- 83.Voss S., Nitsche C. Inhibitors of the Zika virus protease NS2B-NS3. Bioorg. Med. Chem. Lett. 2020;30:126965. doi: 10.1016/j.bmcl.2020.126965. [DOI] [PubMed] [Google Scholar]
- 84.Rut W., Groborz K., Zhang L., Modrzycka S., Poreba M., Hilgenfeld R., Drag M. Profiling of flaviviral NS2B-NS3 protease specificity provides a structural basis for the development of selective chemical tools that differentiate dengue from Zika and West Nile viruses. Antivir. Res. 2020;175:104731. doi: 10.1016/j.antiviral.2020.104731. [DOI] [PubMed] [Google Scholar]
- 85.Olson O.C., Joyce J.A. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer. 2015;15:712–729. doi: 10.1038/nrc4027. [DOI] [PubMed] [Google Scholar]
- 86.Löser R., Pietzsch J. Cysteine cathepsins: their role in tumor progression and recent trends in the development of imaging probes. Front. Chem. 2015;23:37. doi: 10.3389/fchem.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cianni L., Feldmann C.W., Gilberg E., Gütschow M., Juliano L., Leitão A., Bajorath J., Montanari C.A. Can cysteine protease cross-class inhibitors achieve selectivity? J. Med. Chem. 2019;62:10497–10525. doi: 10.1021/acs.jmedchem.9b00683. [DOI] [PubMed] [Google Scholar]
- 88.Liu C.L., Guo J., Zhang X., Sukhova G.K., Libby P., Shi G.P. Cysteine protease cathepsins in cardiovascular disease: from basic research to clinical trials. Nat. Rev. Cardiol. 2018;15:351–370. doi: 10.1038/s41569-018-0002-3. [DOI] [PubMed] [Google Scholar]
- 89.Weidle U.H., Tiefenthaler G., Georges G. Proteases as activators for cytotoxic prodrugs in antitumor therapy. Cancer Genomics Proteomics. 2014;11:67–79. [PubMed] [Google Scholar]
- 90.Dheer D., Nicolas J., Shankar R. Cathepsin-sensitive nanoscale drug delivery systems for cancer therapy and other diseases. Adv. Drug Deliv. Rev. 2019;151–152:130–151. doi: 10.1016/j.addr.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 91.Weinstain R., Segal E., Satchi-Fainaro R., Shabat D. Real-time monitoring of drug release. Chem. Commun. 2010;46:553–555. doi: 10.1039/b919329d. [DOI] [PubMed] [Google Scholar]
- 92.Dana D., Pathak S.K. A review of small molecule inhibitors and functional probes of human cathepsin L. Molecules. 2020;25 doi: 10.3390/molecules25030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Drake M.T., Clarke B.L., Oursler M.J., Khosla S. Cathepsin K inhibitors for osteoporosis: biology, potential clinical utility, and lessons learned. Endocr. Rev. 2017;38:325–350. doi: 10.1210/er.2015-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choe Y., Leonetti F., Greenbaum D.C., Lecaille F., Bogyo M., Brömme D., Ellman J.A., Craik C.S. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 2006;281:12824–12832. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- 95.Poreba M., Groborz K., Vizovisek M., Maruggi M., Turk D., Turk B., Powis G., Drag M., Salvesen G.S. Fluorescent probes towards selective cathepsin B detection and visualization in cancer cells and patient samples. Chem. Sci. 2019;10:8461–8477. doi: 10.1039/c9sc00997c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Poreba M., Rut W., Vizovisek M., Groborz K., Kasperkiewicz P., Finlay D., Vuori K., Turk D., Turk B., Salvesen G.S., Drag M. Selective imaging of cathepsin L in breast cancer by fluorescent activity-based probes. Chem. Sci. 2018;9:2113–2129. doi: 10.1039/c7sc04303a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu X., Wang J., Chu Y., Zhou X. Serum based fluorescent assay for evaluating dipeptidyl peptidase I activity in collagen induced arthritis rat model. Mol. Cell. Probes. 2017;32:5–12. doi: 10.1016/j.mcp.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 98.Chowdhury M.A., Moya I.A., Bhilocha S., McMillan C.C., Vigliarolo B.G., Zehbe I., Phenix C.P. Prodrug-inspired probes selective to cathepsin B over other cysteine cathepsins. J. Med. Chem. 2014;57:6092–6104. doi: 10.1021/jm500544p. [DOI] [PubMed] [Google Scholar]
- 99.Dana D., Garcia J., Bhuiyan A.I., Rathod P., Joo L., Novoa D.A., Paroly S., Fath K.R., Chang E.J., Pathak S.K. Cell penetrable, clickable and tagless activity-based probe of human cathepsin L. Bioorg. Chem. 2019;85:505–514. doi: 10.1016/j.bioorg.2019.02.032. [DOI] [PubMed] [Google Scholar]
- 100.Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Hilgenfeld R., Drag M. Substrate specificity profiling of SARS-CoV-2 Mpro protease provides basis for anti-COVID-19 drug design. bioRxiv. 2020 doi: 10.1101/2020.03.07.981928. [DOI] [Google Scholar]
- 101.McMahon B.J., Kwaan H.C. Components of the plasminogen-plasmin system as biologic markers for cancer. Adv. Exp. Med. Biol. 2015;867:145–156. doi: 10.1007/978-94-017-7215-0_10. [DOI] [PubMed] [Google Scholar]
- 102.Goldberg J.M., Chen X., Meinhardt N., Greenbaum D.C., Petersson E.J. Thioamide-based fluorescent protease sensors. J. Am. Chem. Soc. 2014;136:2086–2093. doi: 10.1021/ja412297x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Häcker H.G., Sisay M.T., Gütschow M. Allosteric modulation of caspases. Pharmacol. Ther. 2011;132:180–195. doi: 10.1016/j.pharmthera.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 104.Brentnall M., Rodriguez-Menocal L., De Guevara R.L., Cepero E., Boise L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013;14:32. doi: 10.1186/1471-2121-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ranganathan R., Lenti G., Tassone N.M., Scannell B.J., Southern C.A., Karver C.E. Design and application of a fluorogenic assay for monitoring inflammatory caspase activity. Anal. Biochem. 2018;543:1–7. doi: 10.1016/j.ab.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 106.Mizukami S., Takikawa R., Sugihara F., Shirakawa M., Kikuchi K. Dual-function probe to detect protease activity for fluorescence measurement and 19F MRI. Angew. Chem. Int. Ed. 2009;48:3641–3643. doi: 10.1002/anie.200806328. [DOI] [PubMed] [Google Scholar]
- 107.Okorochenkova Y., Porubský M., Benická S., Hlaváč J. A novel three-fluorophore system as a ratiometric sensor for multiple protease detection. Chem. Commun. 2018;54:7589–7592. doi: 10.1039/c8cc01731j. [DOI] [PubMed] [Google Scholar]
- 108.Sun H., Panicker R.C., Yao S.Q. Activity based fingerprinting of proteases using FRET peptides. Biopolymers. 2007;88:141–149. doi: 10.1002/bip.20664. [DOI] [PubMed] [Google Scholar]
- 109.Kettritz R. Neutral serine proteases of neutrophils. Immunol. Rev. 2016;273:232–248. doi: 10.1111/imr.12441. [DOI] [PubMed] [Google Scholar]
- 110.Burster T. Processing and regulation mechanisms within antigen presenting cells: a possibility for therapeutic modulation. Curr. Pharm. Des. 2013;19:1029–1042. doi: 10.2174/1381612811319060005. [DOI] [PubMed] [Google Scholar]
- 111.Lesner A., Wysocka M., Guzow K., Wiczk W., Legowska A., Rolka K. Development of sensitive cathepsin G fluorogenic substrate using combinatorial chemistry methods. Anal. Biochem. 2008;375:306–312. doi: 10.1016/j.ab.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 112.Groborz K., Kołt S., Kasperkiewicz P., Drag M. Internally quenched fluorogenic substrates with unnatural amino acids for cathepsin G investigation. Biochimie. 2019;166:103–111. doi: 10.1016/j.biochi.2019.05.013. [DOI] [PubMed] [Google Scholar]
- 113.Juilland M., Thome M. Holding all the CARDs: how MALT1 controls CARMA/CARD-dependent signaling. Front. Immunol. 2018;9:1927. doi: 10.3389/fimmu.2018.01927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kasperkiewicz P., Kołt S., Janiszewski T., Groborz K., Poręba M., Snipas S.J., Salvesen G.S., Drąg M. Determination of extended substrate specificity of the MALT1 as a strategy for the design of potent substrates and activity-based probes. Sci. Rep. 2018;8:15998. doi: 10.1038/s41598-018-34476-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yuan Y., Zhang R., Cheng X., Xu S., Liu B. A FRET probe with AIEgen as the energy quencher: dual signal turn-on for self-validated caspase detection. Chem. Sci. 2016;7:4245–4250. doi: 10.1039/c6sc00055j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Knight C.G., Willenbrock F., Murphy G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 117.Neumann U., Kubota H., Frei K., Ganu V., Leppert D. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal. Biochem. 2004;328:166–173. doi: 10.1016/j.ab.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 118.He G., Yang L., Qian X., Li J., Yuan Z., Li C. A coumarin-based fluorescence resonance energy transfer probe targeting matrix metalloproteinase-2 for the detection of cervical cancer. Int. J. Mol. Med. 2017;39:1571–1579. doi: 10.3892/ijmm.2017.2974. [DOI] [PubMed] [Google Scholar]
- 119.Cobos-Correa A., Trojanek J.B., Diemer S., Mall M.A., Schultz C. Membrane-bound FRET probe visualizes MMP12 activity in pulmonary inflammation. Nat. Chem. Biol. 2009;5:628–630. doi: 10.1038/nchembio.196. [DOI] [PubMed] [Google Scholar]
- 120.Poreba M., Szalek A., Rut W., Kasperkiewicz P., Rutkowska-Wlodarczyk I., Snipas S.J., Itoh Y., Turk D., Turk B., Overall C.M., Kaczmarek L., Salvesen G.S., Drag M. Highly sensitive and adaptable fluorescence-quenched pair discloses the substrate specificity profiles in diverse protease families. Sci. Rep. 2017;7:43135. doi: 10.1038/srep43135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gehrig S., Mall M.A., Schultz C. Spatially resolved monitoring of neutrophil elastase activity with ratiometric fluorescent reporters. Angew. Chem. Int. Ed. Eng. 2012;51:6258–6261. doi: 10.1002/anie.201109226. [DOI] [PubMed] [Google Scholar]
- 122.Wysocka M., Lesner A., Gruba N., Korkmaz B., Gauthier F., Kitamatsu M., Łęgowska A., Rolka K. Three wavelength substrate system of neutrophil serine proteinases. Anal. Chem. 2012;84:7241–7248. doi: 10.1021/ac301684w. [DOI] [PubMed] [Google Scholar]
- 123.Häußler D., Mangold M., Furtmann N., Braune A., Blaut M., Bajorath J., Stirnberg M. M Gütschow, Phosphono bisbenzguanidines as irreversible dipeptidomimetic inhibitors and activity-based probes of matriptase-2. Chem. Eur. J. 2016;22:8525–8535. doi: 10.1002/chem.201600206. [DOI] [PubMed] [Google Scholar]
- 124.Zuo K., Qi Y., Yuan C., Jiang L., Xu P., Hu J., Huang M., Li J. Specifically targeting cancer proliferation and metastasis processes: the development of matriptase inhibitors. Cancer Metastasis Rev. 2019;38:507–524. doi: 10.1007/s10555-019-09802-8. [DOI] [PubMed] [Google Scholar]
- 125.Häußler D., Schulz-Fincke A.C., Beckmann A.M., Keils A., Gilberg E., Mangold M., Bajorath J., Stirnberg M., Steinmetzer T., Gütschow M. A fluorescent-labeled phosphono bisbenzguanidine as an activity-based probe for matriptase. Chem. Eur. J. 2017;23:5205–5209. doi: 10.1002/chem.201700319. [DOI] [PubMed] [Google Scholar]
- 126.Neumann U., Gütschow M. N-(Sulfonyloxy)phthalimides and analogues are potent inactivators of serine proteases. J. Biol. Chem. 1994;269:21561–21567. [PubMed] [Google Scholar]
- 127.Schulz-Fincke A.C., Tikhomirov A.S., Braune A., Girbl T., Gilberg E., Bajorath J., Blaut M., Nourshargh S., Gütschow M. Design of an activity-based probe for human neutrophil elastase: implementation of the Lossen rearrangement to induce Förster resonance energy transfers. Biochemistry. 2018;57:742–752. doi: 10.1021/acs.biochem.7b00906. [DOI] [PubMed] [Google Scholar]
- 128.Mertens M.D., Schmitz J., Horn M., Furtmann N., Bajorath J., Mareš M., Gütschow M. A coumarin-labeled vinyl sulfone as tripeptidomimetic activity-based probe for cysteine cathepsins. ChemBioChem. 2014;15:955–959. doi: 10.1002/cbic.201300806. [DOI] [PubMed] [Google Scholar]
- 129.Kohl F., Schmitz J., Furtmann N., Schulz-Fincke A.C., Mertens M.D., Küppers J., Benkhoff T., Tobiasch E., Bartz U., Bajorath J., Stirnberg M., Gütschow M. Design, characterization and cellular uptake studies of fluorescence-labeled prototypic cathepsin inhibitors. Org. Biomol. Chem. 2015;13:10310–10323. doi: 10.1039/c5ob01613d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material