

Abstract
Background
The Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) trial targeted young sarcomeric gene mutation carriers with early stage hypertrophic cardiomyopathy (HCM) to test whether valsartan can modify disease progression. We describe the baseline characteristics of the VANISH cohort, and compare to previous trials evaluating angiotensin receptor blockers (ARB).
Methods
Applying a randomized, double-blinded, placebo-controlled design, 178 participants with non-obstructive HCM (age 23.3±10.1 years, 61% male) were randomized in the primary cohort, and 34 (age 16.5±4.9 years, 50% male) in the exploratory cohort of sarcomeric mutation carriers without left ventricular (LV) hypertrophy.
Results:
In the primary cohort, maximal LV wall thickness was 17±4 mm for adults and z-score 7.0±4.5 for children. Nineteen percent had late gadolinium enhancement on CMR. Mean peak oxygen consumption was 33 ml/kg/min and 92% of participants were New York Heart Association (NYHA) functional class I. NYHA class II was associated with older age, myosin heavy chain (MYH7) variants, and more prominent imaging abnormalities. Six previous trials of ARBs in HCM enrolled a median of 24 patients (range 19–133) with mean age 51.2 years; 42% of patients were in NYHA class ≥II, and sarcomeric mutations were not required.
Conclusions
The VANISH cohort is much larger, younger, less heterogeneous and has less advanced disease than prior ARB trials in HCM. Participants had relatively normal functional capacity and mild HCM features. NYHA functional class II symptoms were associated with older age, more prominent imaging abnormalities, and MYH7 variants, suggesting both phenotype and genotype contribute to disease manifestations.
Clinical trial registration
Subject terms: ACE/Angiotensin Receptors/Renin Angiotensin System, Clinical Studies, Genetics, Cardiomyopathy, Hypertrophy
Introduction
Hypertrophic cardiomyopathy (HCM) is one of the most common inherited cardiac diseases with a reported prevalence of 1 in 500 adults1–3. Increased left ventricular (LV) wall thickness that cannot be explained by extrinsic factors (such as increased afterload), myocardial fibrosis and myocyte disarray are hallmarks of the disease4, 5. Disease-causing (pathogenic) variants in genes encoding the cardiac sarcomere are the predominant cause of HCM and can be identified in approximately 60% of patients with familial disease6, 7. Patients with HCM are at an increased risk of sudden cardiac death, stroke and heart failure. Moreover, HCM is a progressive condition with increasing disease burden throughout life8. Thus, there is a clear need to identify treatments that can halt disease progression and, ultimately, prevent disease emergence by counteracting the pathobiology of the underlying sarcomeric gene variant. However, there is a paucity of randomized clinical trials in HCM and no pharmacological treatment has yet been reliably demonstrated to alter disease progression or improve outcomes9.
Activation of the renin-angiotensin system is known to play a pivotal role in the development of myocardial hypertrophy and fibrosis caused by pressure overload10, 11. Preclinical studies in animal models of HCM caused by sarcomeric variants have suggested that transforming growth factor-beta (TGF-β) activation is critically involved in the development of hypertrophy and fibrosis early in the pathogenesis of HCM12. Although treatment with TGF-β neutralizing antibodies and angiotensin II receptor blockers (ARB) at doses adequate to inhibit TGF-β activation appeared to dramatically attenuate development of hypertrophy and fibrosis in animal models of HCM if given early in disease evolution, these agents were unable to reverse established changes once myocardial abnormalities were present. Moreover, the role of the related pathways and the efficacy of these treatments in human disease are unclear12–21. While prior clinical trials of angiotensin converting enzyme inhibitors (ACEi) and ARBs in HCM have not shown convincing benefit16–21, these trials have generally been quite small in scale, included middle-aged or older adult patients with well-established disease, and have not specifically targeted patients with proven sarcomeric HCM.
The Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) Trial is testing the hypothesis that valsartan attenuates disease progression in sarcomeric variant carriers with early stage disease, based on young age and absence of pronounced left ventricular hypertrophy (LVH) or substantial symptoms22. The underlying rationale for VANISH is that disease may be modifiable early in its evolution while reversal of established pathology may not be feasible. Here we report and compare the baseline characteristics of the participants randomized into VANISH with those in previous clinical trials of ARBs in HCM.
Methods
Participants and trial design
A detailed description of the trial design and rationale has been published previously22. In brief, VANISH is a randomized, double-blinded, placebo-controlled trial, targeting individuals with pathogenic or likely pathogenic HCM-associated sarcomeric gene variants23 (Supplemental Table 1). The study consists of both a primary analysis cohort of patients with early disease (8–45 years of age with non-obstructive HCM) and an exploratory cohort of young sarcomeric mutation carriers with preclinical disease (10–25 years, normal LV wall thickness [no diagnosis of HCM] but evidence of early phenotypic manifestations such as impaired relaxation [reduced tissue Doppler e’ velocity] or electrocardiographic (ECG) abnormalities [Q waves or ST changes]).
Participants in both groups had to be asymptomatic or mildly symptomatic at entry (New York Heart Association (NYHA) class I-II). NYHA class was designated by the site principal investigator, based on subject self-report and clinical evaluation. Sarcomeric variants were classified as pathogenic, likely pathogenic, of uncertain significance, or likely benign/benign according to standard criteria23, 24accounting for segregation, conservation, literature review, review of publicly available databases (ClinVar, [URL: https://www.ncbi.nlm.nih.gov/clinvar]), and very low frequency in appropriate ethnically-matched control populations (Exome Aggregation Consortium [ExAC], Cambridge, MA [URL: http://exac.broadinstitute.org]). If a variant was determined to be pathogenic or likely pathogenic by one of three experienced commercial laboratories (Laboratory for Molecular Medicine, GeneDx, Invitae), the assertion was reviewed and generally accepted. For variants not previously reviewed by the three acknowledged laboratories, not present in ClinVar, and for all variants of uncertain significance, a panel with expertise in genotyping, led by the principal investigator, assessed each questionable variant and approved or denied eligibility by consensus based on the above criteria and private patient/family-specific segregation data when available.
Participants in the primary analysis cohort and exploratory cohort were randomized and analyzed independently. The primary efficacy endpoint is a composite of nine variables in three domains of surrogate endpoints reflecting myocardial structure, function and injury/stress (Supplemental table 2). The trial is registered at https://clinicaltrials.gov/ct2/show/NCT01912534. All participants or legal guardians provided written informed consent and youth assent prior to inclusion. The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Enrollment
Study participants were recruited from HCMNet sites (17 United States centers, one Canadian, one Brazilian and one Danish center) or self-identified after learning about the trial from relatives or outreach efforts with local physicians, social media, and patient advocacy groups25. A total of 588 individuals were identified as potentially eligible by the sites and were approached for consent. Baseline evaluation for eligibility, consisting of review of the sarcomeric gene variant, echocardiography, cardiopulmonary exercise testing (CPET), ECG, and cardiac magnetic resonance (CMR) imaging was performed in the 259 participants who consented to the trial (Figure 1). Participants who fulfilled eligibility criteria for either the primary or exploratory cohort entered into an active run-in phase during which increasing doses of valsartan were administered as tolerated until reaching weight-based target dose (80 to 320 mg daily). Participants were randomly assigned to receive valsartan or matching placebo for two years. Compliance was assessed by pill count at all visits and at quarterly telephone calls and also by bottle logs where study coordinators reconciled the number of pills distributed and returned22. Participants who could not tolerate titration to the target dose of valsartan were not randomized.
Figure 1.
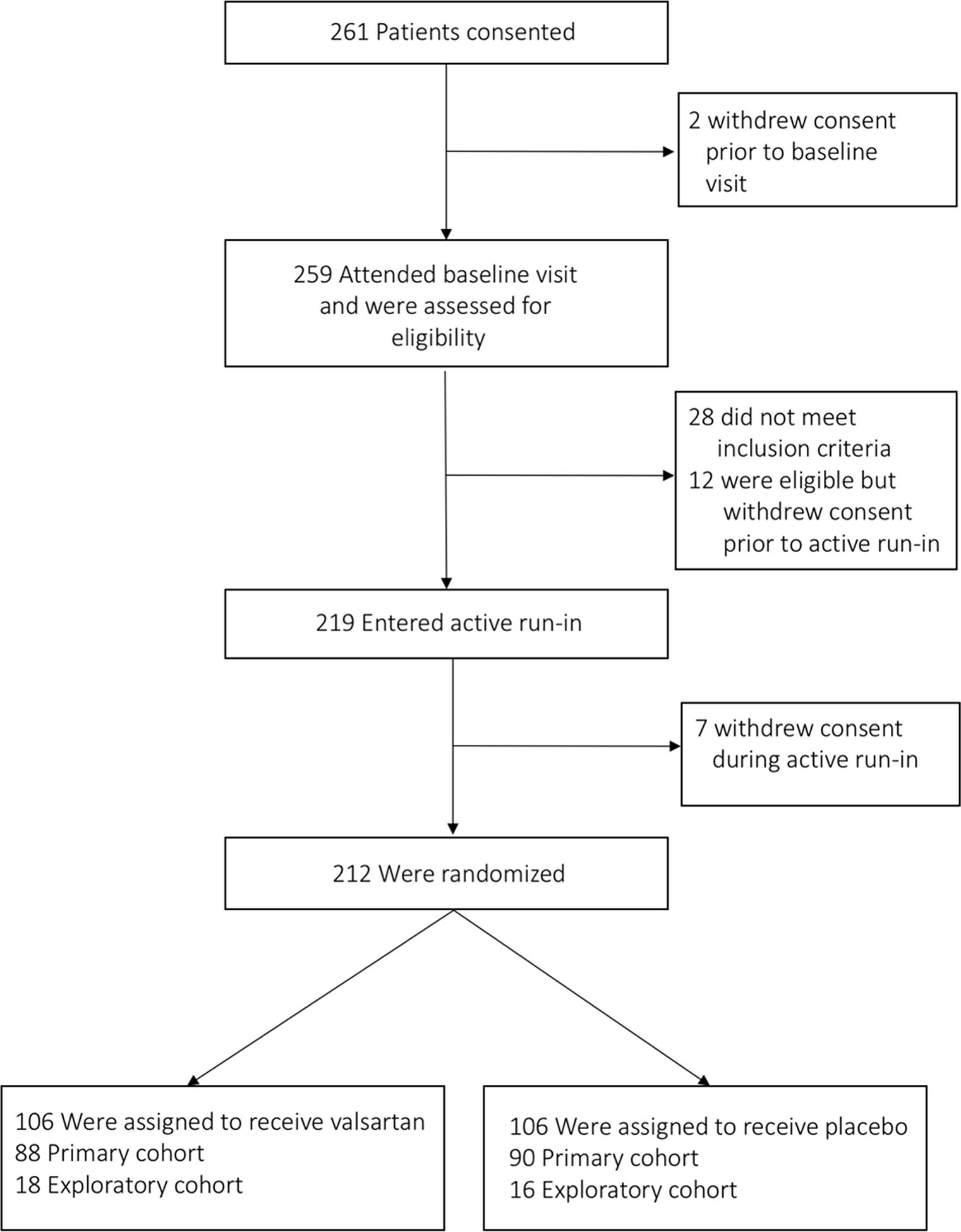
Screening, run-in and randomization in the VANISH trial.
Study procedures for the assessment of baseline characteristics
Baseline echocardiography was performed to verify eligibility and to assess values of LV morphologic and functional components of the primary outcome. Study sonographers were trained and certified by the Echo Core Laboratory prior to the start of the study to ensure data quality and standardization. Standard two- and three- dimensional images, spectral and color Doppler interrogation were obtained to assess cardiac dimensions, morphology, function, valvular function, and to characterize hemodynamics (mitral inflow patterns, estimated pulmonary artery systolic pressure, and obstructive physiology related to HCM). All analyses were performed at the Echo Core Laboratory by personnel blinded to treatment arm, including rapid analysis (<1 week from baseline visit) to determine eligibility and phenotypic status for stratified randomization for the primary or exploratory cohorts, based on the defined LV wall thickness, e’ criteria and thickness:dimension ratio. All measurement values were obtained by averaging three cardiac cycles and in accordance with established criteria of the American Society of Echocardiography26, 27.
A CMR study was performed for the assessment of baseline values of LV morphologic and functional components of the primary outcome and for the assessment of myocardial fibrosis. Imaging was performed on 1.5T to 3T systems available at the participating centers using a cardiac phased-array receiver coil gated to the ECG. CMR technicians were trained and certified by the CMR Core Laboratory prior to the start of the study to ensure data quality and standardization. CMR was not performed on children if sedation was required, or in individuals with contraindications to CMR. Standard short- and long- axis cine images were acquired using Steady-State-Free-Precession for quantification of cardiac dimensions, mass and systolic function. In addition, tagging sequences were obtained to assess regional function. For the assessment of late gadolinium enhancement T1-weighted gradient echo images were obtained as a short-axis stack and as standard long-axis images 15–20 minutes after the injection of gadolinium contrast. For late gadolinium enhancement the presence/absence, location, geographic extent, and threshold values (6 standard deviations) above mean tissue intensity were assessed. All CMR studies were stored digitally for blinded offline CMR Core Laboratory analysis. CMR image analysis was performed using CVI42 (Circle Cardiovascular Imaging; Calgary, Canada).
A standard 12-lead ECG was obtained to verify eligibility at enrollment and measurements determined by the ECG Core Laboratory.
A cardiopulmonary exercise test (CPET) was performed for the assessment of peak VO2, ventilatory efficiency and other indicators of fitness. Technicians were trained and certified by the CPET Core Laboratory prior to the start of the study to ensure data quality and standardization. Additionally, standardized calibration of equipment was required. Cycle ergometry was preferentially used with a 3-minute unloaded warm-up followed by a ramped workload of 20 watts/min. The protocol was modified to a 10 watt/min ramp for subjects younger than 14 or if site staff considered that a subject would not be able to perform a 20 watt/min ramp protocol. The goal respiratory exchange ratio was ≥1.10. All studies were transferred for blinded analysis at the CPET Core Laboratory. For analysis, the highest 30 second average VO2 during the final 90 seconds of incremental exercise was identified. Primary breath-by-breath data were used to calculate ventilatory anaerobic threshold, by the V slope method.
Structured literature review
We searched PubMed in January 2012 in preparation for the trial and again in and April 2018 and April 2019 during the preparation of this report. We included articles published in English until 22 April 2019 using the search terms ‘hypertrophic cardiomyopathy’ AND ‘angiotensin receptor blocker’. The search yielded four reports of randomized trials. In addition, two further reports were found in the reference lists of these reports.
Statistical analysis
Baseline continuous variables are presented as mean and standard deviation (SD) or median and interquartile range (IQR), depending on distribution. Categorical variables are presented as number and percent. Continuous variables considered dependent on body growth are presented as Z-score (SD from population mean) in pediatric participants28. Between-group differences were compared using t-test, ANOVA or Chi-Square depending on type of variable and number of groups. For all analyses, we regard a two-sided p-value of less than 0.05 as significant. No adjustment was made to account for comparing multiple outcome variables. When many outcomes are analyzed, it is not unexpected that one or more might have a statistically significant difference just by chance.
Results
Baseline Characteristics of the VANISH Cohort
The baseline characteristics of participants by cohort are summarized in Table 1. In total, 212 participants from 176 different families were found to be eligible, successfully completed active run-in, and were randomized into the study. Mean age was 22.2 ± 9.7 years, 40.6% were female, and 93.4% were NYHA class I. The most common reasons for not progressing to randomization were: (1) failure to meet eligibility criteria during baseline screening, including not meeting specified echo LV wall thickness criteria (N=23), not meeting age criteria (N=3), having medical conditions that contraindicated ARB administration (N=1) and not meeting specific ECG or echocardiographic requirements for the exploratory cohort (N=1); (2) withdrawal of consent prior to active run-in (N=12); and (3) withdrawal during active run-in (N=7). The majority of participants (82%) were from sites in North America, 13% were from Brazil and 6% from Denmark.
Table 1.
Baseline characteristics of participants in VANISH by cohort
| Total (n=212) | Overt HCM – Primary Analysis Cohort (n=178) | Preclinical HCM- Exploratory Cohort (n=34) | |
|---|---|---|---|
| Age, years | 22.2 (9.7) | 23.3 (10.1) | 16.5 (4.9) |
| Female gender (%) | 86 (40.6) | 69 (38.8) | 17 (50.0) |
| Race (%) | |||
| White | 203 (95.8) | 169 (94.9) | 34 (100.0) |
| Black | 3 (1.4) | 3 (1.7) | 0 (0.0) |
| Other | 6 (2.8) | 6 (3.4) | 0 (0.0) |
| NYHA class (%) | |||
| I | 198 (93.4) | 164 (92.1) | 34 (100.0) |
| II | 14 (6.6) | 14 (7.9) | 0 (0.0) |
| Cardiac medications (%) | |||
| Beta-blocker | 36 (17.0) | 34 (19.1) | 2 (5.9) |
| Calcium channel blocker | 5 (2.4) | 5 (2.8) | 0 (0.0) |
| Family history (%) | 180 (85) | 146 (82) | 34 (100) |
| No. of affected relatives (median, q1, q3) | 2 (1, 4) | 2 (1, 3) | 3 (2, 4) |
| Heart rate, bpm | 70 (14) | 70 (14) | 74 (13) |
| Blood pressure, mmHg | |||
| systolic | 117 (12) | 118 (11) | 114 (12) |
| diastolic | 68 (10) | 69 (10) | 66 (8) |
| Echocardiographic findings | |||
| Max. LV Wall Thickness, mm | 15 (4) | 16 (4) | 10 (1) |
| Max LV Wall Thickness Z-Score | 6.4 (4.2) | 7.3 (4.0) | 2.1 (0.6) |
| e’ lateral Z-Score | −1.7 (1.5) | −2.0 (1.4) | −0.4 (1.0) |
| e’ septum Z-Score | −1.6 (1.4) | −1.9 (1.3) | −0.4 (1.0) |
| Average e’, cm/s | 12.0 (3.6) | 11.3 (3.5) | 15.4 (2.0) |
| E/e’ average | 7.3 (2.4) | 7.6 (2.5) | 6.0 (1.2) |
| LVEF, % | 66 (8) | 66 (8) | 64 (4) |
| LA Diameter, cm | 3.4 (0.6) | 3.4 (0.7) | 3.1 (0.4) |
| Peak left ventricular outflow tract gradient at rest, mmHg | 7 (3) | 7 (4) | 6 (2) |
| Peak left ventricular outflow tract gradient during Valsalva maneuver, mmHg | 7 (4) | 7 (4) | 6 (2) |
| Cardiopulmonary exercise test | |||
| Peak VO2, ml/kg/min | 33.0 (9.2) | 31.7 (8.9) | 39.0 (8.1) |
| % predicted maximal VO2 | 82 (20) | 81 (21) | 87(17) |
| CMR findings | |||
| LGE present (%) | 29 (15.3) | 29 (18.6) | 0 (0.0) |
| LGE mass, g | 19 (18) | 19 (18) | - |
| LGE mass, % of LV | 13 (13) | 13 (13) | - |
| LVEDV, ml | 108 (31) | 108 (31) | 108 (27) |
| LVESV, ml | 37 (14) | 37 (14) | 40 (12) |
| LV mass, g | 116 (42) | 122 (43) | 89 (27) |
| LV mass indexed to BSA, g/m2 | 63 (17) | 65 (18) | 53 (9) |
| LA volume diastolic, ml | 29 (19) | 31 (20) | 19 (6) |
| LA volume systolic, ml | 66 (30) | 70 (31) | 51 (17) |
| Max LV wall thickness, mm* | 12 (4) | 12 (4) | 8 (1) |
Results are presented as % or as mean (SD) or median (IQR). CMR indicates, cardiac magnetic resonance imaging; E, peak early mitral inflow velocity; e’, peak early mitral annular relaxation velocity; LA, left atrial; LGE, late gadolinium enhancement; LV, left ventricular; LVEDV, left ventricular end-diastolic volume; LVEF, left ventricular ejection fraction; LVESV, left ventricular end-systolic volume; NYHA, New York Heart Association Class; VO2, peak oxygen uptake. Difference between groups assessed by Chi-Square or Fisher’s exact test or two-sample t test depending on number and type of variable.
The maximum LV wall thickness from CMR is the maximum value across the 16 American Heart Association segments.
At baseline, 178 (84%) participants had phenotypic HCM with LVH (as determined by the core echocardiographic laboratory) and were randomized into the primary analysis cohort, while 34 (16%) had normal LV wall thickness and were randomized into the exploratory cohort as preclinical HCM participants.
Primary Analysis Cohort
Primary cohort participants were at mean age of 23.3 ± 10.1 years (range 8–45) at baseline and 61% were male. Overall mean maximal LV wall thickness by echocardiography was 16 ± 4 mm. Mean maximal LV wall thickness for adult participants was 17 ± 4 mm and mean maximal LV wall thickness z-score in children was 7.0 ± 4.5. Nineteen percent of participants in the primary cohort were on beta-blockers and 3% were on calcium channel blockers. Figure 2 shows the distribution of variants, with most participants carrying variants in myosin binding protein C (MYBPC3) or myosin heavy chain (MYH7) genes. Cardiovascular symptoms were reported by 21 (12%) participants, most commonly palpitations (10%) and exertional dyspnea (6%) (Figure 3). Most participants (n=164, 92%) were classified NYHA functional class I while 14 (8%) were NYHA class II. As shown in Table 2, NYHA class II participants were over a decade older (34.7 ± 10.0 vs 21.3 ± 9.1 years, p<0.001) and more likely to carry a pathogenic variant in MYH7 (71% vs 32%) but less likely to carry a variant in MYBPC3 (14% vs 54%, p=0.02). Additionally, NYHA class II participants had higher CMR LV mass (77 ± 19 vs 62 ± 17 grams/m2, p=0.005) and LV maximal wall thickness (16 ± 4 vs 11 ± 3 mm, p<0.001), more severely impaired diastolic parameters (average e’ 7 ± 3 vs 12 ± 8 cm/sec, p<0.001; E/e’ 11 ± 4 versus 7 ± 2, p=0.004), and larger CMR left atrial volume (44 ± 19 vs 28 ± 19 ml, p=0.01). Moreover, LGE was both more prevalent (present in 55% vs 13%, p<0.001) and more extensive (involving 26 ± 21 vs 10 ± 8 % of LV mass, p=0.009) in NYHA class II and class I participants respectively.
Figure 2. Distribution of pathogenic and likely pathogenic variants in the primary (Panel A, n=178) and exploratory (Panel B, n=34) cohorts.
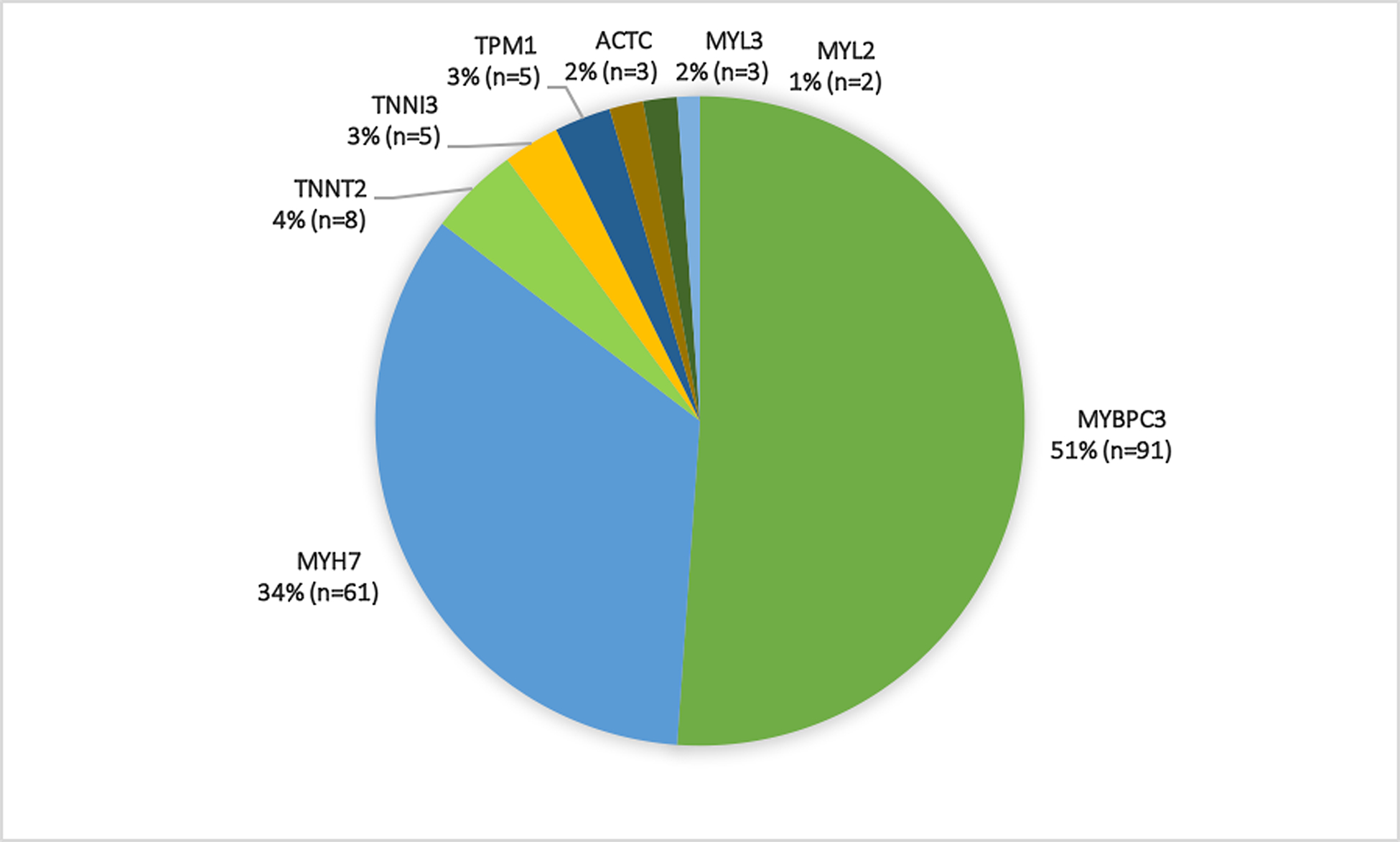
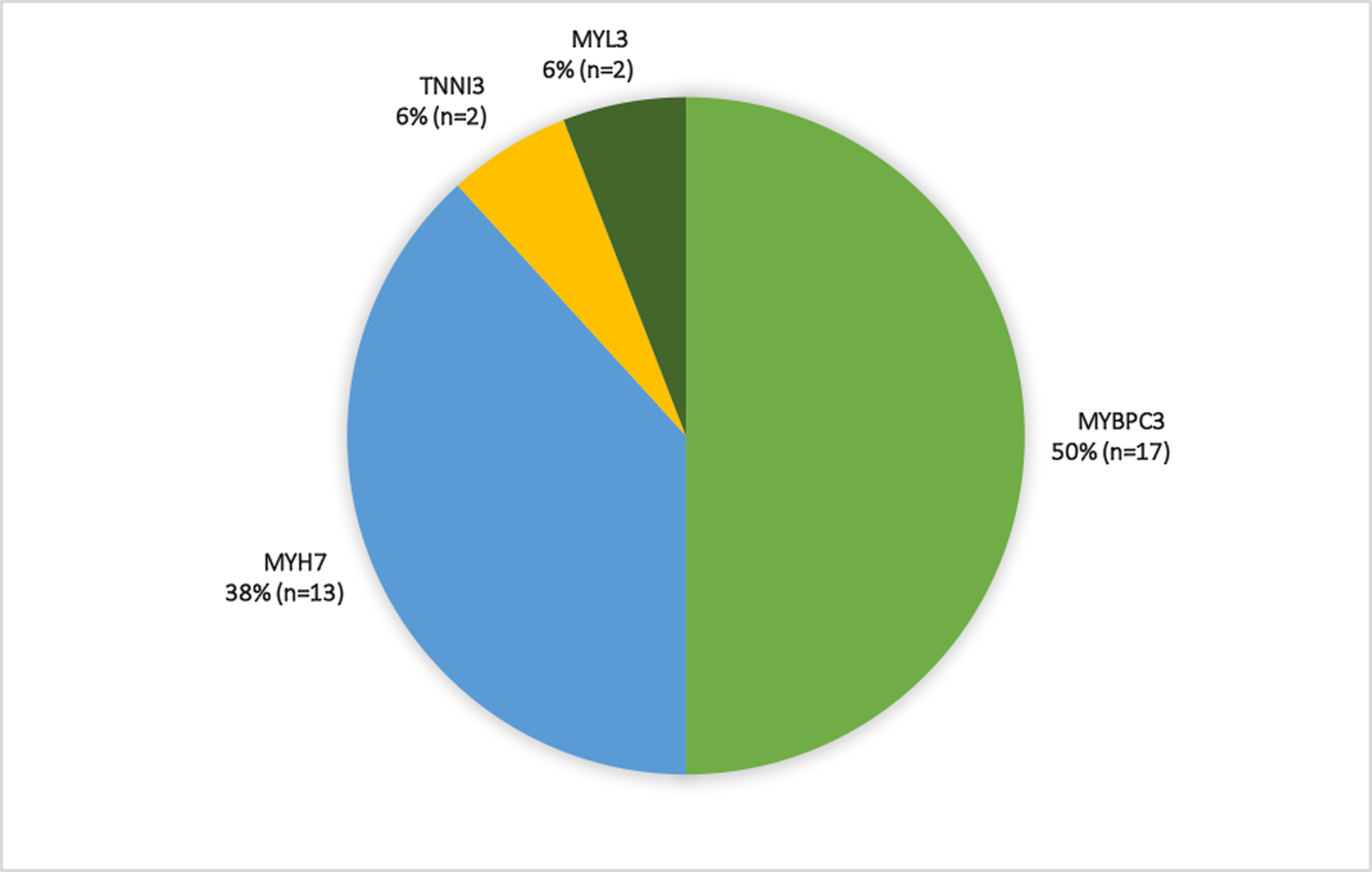
Compound heterozygosity was seen in five primary cohort participants who carried variants in MYBPC3 in addition to MYH7 (n=4) or TNNT2 (n=1) variants.
Figure 3.
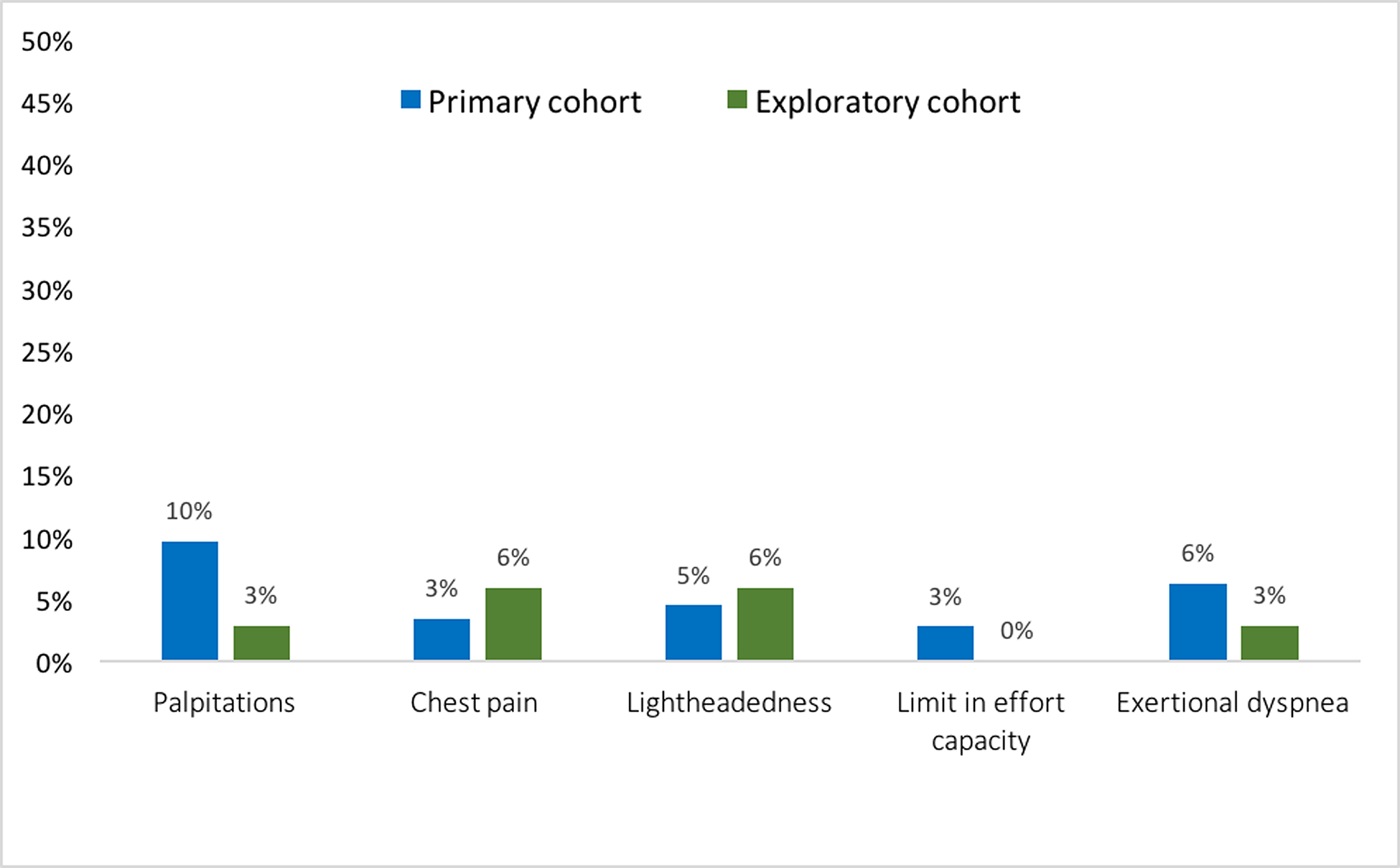
Symptoms at the time of enrollment in the primary (blue, n=178) and exploratory (green, n= 34) cohorts.
Table 2.
Comparison of participants in NYHA class I and II at baseline
| NYHA class I (N=198) | NYHA class II (N=14) | P Value | |
|---|---|---|---|
| Age, years | 21.3 (9.1) | 34.7 (10.0) | <0.001 |
| Female gender (%) | 83 (42) | 3 (21) | 0.13 |
| Race (%) | 1.000 | ||
| White | 189 (95) | 14 (100) | |
| Black | 3 (1) | 0 (0) | |
| Other | 6 (3) | 0 (0) | |
| Sarcomeric Gene (%) | 0.02 | ||
| ACTC | 3 (1.5) | 0 (0) | |
| MYBPC3* | 106 (53.5) | 2 (14.3) | |
| MYH7 | 64 (32.3) | 10 (71.4) | |
| MYL2 | 1 (0.5) | 1 (7.1) | |
| MYL3 | 5 (2.5) | 0 (0) | |
| TNNI3 | 7 (3.5) | 0 (0) | |
| TNNT2 | 7 (3.5) | 1 (7.1) | |
| TPM1 | 5 (2.5) | 0 (0) | |
| Echocardiographic findings | |||
| Average e’, cm/s | 12 (3) | 7 (3) | <0.001 |
| E/e’ average | 7 (2) | 11 (4) | 0.004 |
| Peak left ventricular outflow tract gradient at rest, mmHg | 7 (3) | 9 (3) | 0.472 |
| Peak left ventricular outflow tract gradient during Valsalva maneuver, mmHg | 7 (4) | 10 (6) | 0.126 |
| Cardiopulmonary exercise test | |||
| Peak VO2, ml/kg/min | 33 (9) | 26 (6) | 0.022 |
| % predicted maximal VO2 | 82 (20) | 73 (19) | 0.308 |
| CMR findings | |||
| LGE Present (%) | 23 (13) | 6 (55) | <0.001 |
| LGE mass, g | 15 (12) | 40 (30) | 0.004 |
| LGE mass, % of LV | 10 (8) | 26 (21) | 0.009 |
| LV mass, g | 114 (41) | 159 (46) | <0.001 |
| LV mass indexed to BSA, g/m2 | 62 (17) | 77 (19) | 0.005 |
| Max LV wall thickness, mm** | 11 (3) | 16 (4) | <0.001 |
| LA volume diastolic, ml | 28 (19) | 44 (19) | 0.010 |
Results are presented as % or as mean (SD). CMR indicates, cardiac magnetic resonance imaging; E, peak early mitral inflow velocity; e’, peak early mitral annular relaxation velocity; LA, left atrial; LGE, late gadolinium enhancement; LV, left ventricular; NYHA, New York Heart Association Class; VO2, peak oxygen uptake; ACTC, Alpha cardiac actin gene; MYBPC3, Cardiac myosin binding protein C gene; MYH7, Cardiac β-myosin heavy chain gene; MYL2, Myosin light chain 2 gene; MYL3, Myosin light chain 3 gene; TNNI3, Cardiac troponin I gene; TNNT2, Cardiac troponin T gene; TPM1, Tropomyosin 1 gene. Difference between groups assessed by Chi-Square or Fisher’s exact test or two-sample t test depending on number and type of variable.
5 Class I patients with MYBPC3 mutation also have MYH7 (n=4) or TNNT2 (n=1).
The maximum LV wall thickness from CMR is the maximum value across the 16 American Heart Association segments. Between group differences were assessed using ANOVA, Chi-square test or Fisher’s exact test depending on type of variable and expected frequency.
The distribution and prevalence of ECG and echocardiographic features are presented in Figure 4. All subjects were in sinus rhythm except 1 who had an ectopic atrial rhythm. In addition, Q waves, repolarization changes, and voltage criteria for LVH were seen in 108 (63%) of the primary cohort participants. Regional differences in baseline age were identified with patients from North America (US [20.2 ± 8.8 years] and Canada [14.0 ± 0.8 years]) being younger than patients included in Brazil (33.6 ± 8.2 years) and Denmark (27.6 ± 7.8 years) (Supplemental Table 3).
Figure 4. Distribution of ECG and echocardiographic features in the primary (blue, n=178) and exploratory (green, n=34) cohorts.
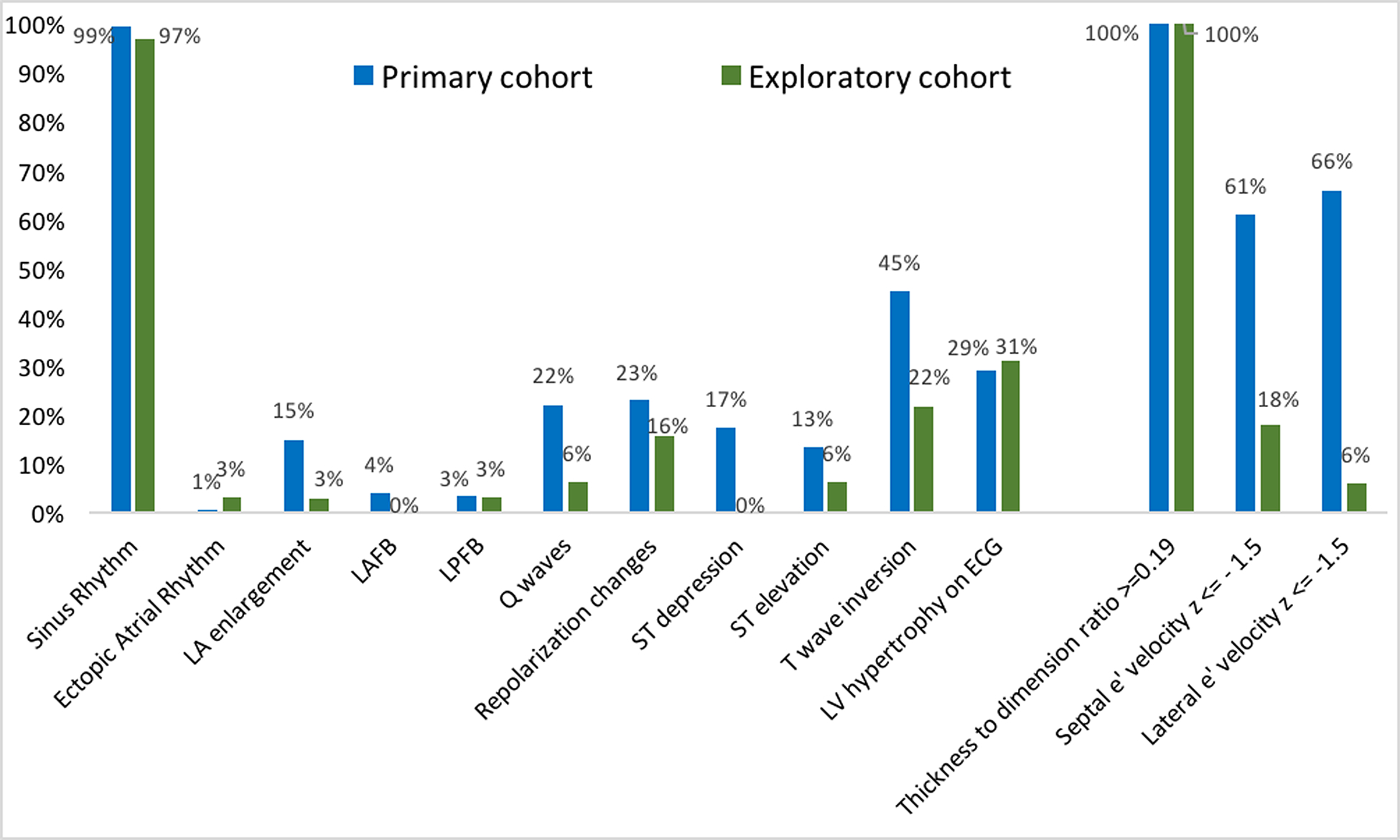
e’ indicates peak early mitral annular relaxation velocity; LA, left atrial; LAFB, left anterior fascicular block; LPFB, left posterior fascicular block; LV, left ventricular.
Exploratory Cohort
The mean age of participants in the exploratory cohort was 16.5 ± 4.9 years (range 10–25) and 50% were male. Participants in the exploratory cohort qualified for inclusion based on 1) LV wall thickness z-score between 1.5 and 2.9 combined with LV thickness to dimension ratio >=0.19 (68%), 2) qualifying ECG abnormality (38%) or 3) e’ z-score<1.5 (21%). Nine (27%) participants fulfilled two of these criteria, whereas the remaining participants fulfilled one. Maximal wall thickness for the exploratory cohort was 10 ± 1 mm. Adult participants had a mean maximal wall thickness of 10 ± 1 mm; children had a mean maximal LV wall thickness z-score of 2.3 ± 0.4. Most participants carried variants in MYBPC3 or MYH7 genes (Figure 2). Only two of 34 participants in the exploratory cohort were taking cardioactive medications, both beta-blockers. As shown in Figure 3, cardiovascular symptoms were reported by 4 (12%) participants, most commonly lightheadedness and chest pain (6% each). All exploratory cohort participants were NYHA class I. The distribution and prevalence of ECG and echocardiographic findings are presented in Figure 4. All subjects were in sinus rhythm except one with ectopic atrial rhythm. Q waves, repolarization changes, and voltage criteria for LVH were seen in 13 (38%) exploratory cohort participants. None of the subjects in the exploratory cohort had LGE on CMR.
Comparison to prior ARB Trials in HCM
Table 3 lists the key entry criteria of VANISH compared to the six previously published trials testing ARBs in HCM. In contrast to previous trials, a pathogenic or likely pathogenic sarcomeric gene mutation was necessary for inclusion in VANISH. VANISH was the only trial to include children and adolescents, as well as the only trial to include participants with preclinical HCM (no LVH) for exploratory study. The duration of study is at least twice as long. Furthermore, VANISH is the only multinational trial and is almost ten times the size of all but one of the previous trials.
Table 3.
Design of VANISH and other clinical trials assessing the effect of Angiotensin II Receptor Blockers in Hypertrophic Cardiomyopathy
| VANISH (N=212)* | INHERIT21 (N=133) | SHIMADA ET AL20 (N=20) | CHANCE19 (N=24) | YAMAZAKI ET AL17 (N=19) | KAWANO ET AL16 (N=23) | ARAUJO ET AL18 (N=30) | |
|---|---|---|---|---|---|---|---|
| YEAR PUBLISHED | -- | 2014 | 2013 | 2009 | 2007 | 2005 | 2005 |
| STUDY DESIGN | |||||||
| Randomized | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Placebo-controlled | Yes | Yes | Yes | Yes | No | No | No |
| Double-blind | Yes | Yes | Yes | Yes | No | No | No |
| INCLUSION CRITERIA | |||||||
| Sarcomeric mutation | Pathogenic or likely pathogenic | -- | -- | -- | -- | -- | -- |
| LV wall thickness | Primary Cohort: 12–25mm or z score 3–18 Exploratory Cohort: <12 mm and Z <3† |
≥15mm or 13–14 mm in first-degree relatives | -- | ≥15 mm | ≥15mm | -- | ≥15 mm |
| Age (years) | Primary 8–45; Preclinical 10–25 | ≥18 | ≥18 | ≥18 | -- | -- | 18–50 |
| NYHA | I-II | -- | -- | -- | -- | -- | I-III |
| Obstruction | Peak gradient ≤30 mmHg at rest or with provocation | No limit for peak gradient | Peak gradient ≤30 mmHg at rest or with provocation | Peak gradient ≤30 mmHg at rest | No obstruction | No obstruction | Peak gradient ≤30 mmHg |
| LVEF | ≥55% | ≥50% | ≥55% | ≥60% | -- | -- | Normal |
| INTERVENTION | Valsartan (adults 320mg/d, children ≥ 35 kg: 160 mg/d; children <35 kg: 80 mg/d) | Losartan (100 mg per day) | Losartan (100 mg per day) | Candesartan (target 32 mg per day) | Losartan (50 mg per day) | Valsartan (80 mg per day) | Losartan (100 mg per day) |
| RECRUITMENT PERIOD | April 2014 - Feb 2017 | Dec 2011 - May 2013 | April 2007 -March 2010 | -- | -- | -- | -- |
| FOLLOW-UP TIME | 24 months | 12 months | 12 months | 12 months | 12 months | 12 months | 6 months |
| PRIMARY ENDPOINT | Composite | LV mass by CMR/CT | LGE by CMR | Not specified | LV mass by CMR | Not specified | Not specified |
| SECONDARY END-POINTS | Safety | LGE by CMR, LV wall thickness, LV mass, diastolic indices, exercise capacity, LA volume, NT-pro-BNP | LV mass by CMR, symptoms, diastolic indices, LA volume, collagen markers | LV wall thickness, LV mass, diastolic indices, exercise time. | -- | LV metrics, collagen markers | LV metrics, diastolic indices, LA diameter, symptoms, NT-pro-BNP |
| COUNTRY | US, Canada, Brazil, Denmark | Denmark | US | Czech republic | Japan | Japan | Brazil |
| SINGLE OR MULTICENTER | Multi | Single | SIngle | Multi | SIngle | Single | SIngle |
178 participants with left ventricular hypertrophy in the primary cohort and 34 participants with preclinical disease in the exploratory cohort.
Not reported.
E’ z score ≤ −1.5 OR electrocardiographic abnormalities other than non-specific ST-T wave changes (Q waves, T wave inversion, repolarization changes) OR LV wall thickness z-score 1.5–2.9 combined with LV thickness to dimension ratio ≥0.19 for preclinical subjects. BP indicates blood pressure; CMR, cardiovascular magnetic resonance imaging; CT, cardiac computed tomography; HCM, hypertrophic cardiomyopathy; LA, left atrial; LGE, late gadolinium enhancement; LV, left ventricular; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association Class.
Participant characteristics are compared to those in previous trials in Table 4. By design, VANISH Primary Analysis participants were younger and had features consistent with earlier-stage disease. The mean age of randomized participants in the primary cohort of VANISH was 23 years; 30 years younger than the mean age in all but 1 of the previous trials. Furthermore, participants in VANISH had less LVH than previous trials of ARB in HCM. Mean maximal wall thickness, measured by echocardiography in the primary analysis cohort in VANISH was 16 mm, whereas in the six previous trials, the combined mean maximal wall thickness was 20.7 mm. Similar to the most recent trials, the vast majority of participants in VANISH (96%) were NYHA functional class I at baseline compared to one third of patients in INHERIT who were NYHA functional class II20, 21. Older trials included an equal distribution of patients in NYHA functional classes I, II and III18, 19. None of the participants in VANISH had atrial fibrillation. Although atrial fibrillation at the time of enrollment was an exclusion criterion in INHERIT, the proportion of patients with a history of atrial fibrillation was 12%21. In two other previous trials atrial fibrillation was an exclusion criterion and in the three remaining trials the prevalence was not reported16–20. The use of calcium channel blockers in VANISH was much lower (2%) than in prior studies (14–44%) whereas the use of beta-blockers in VANISH was comparable (17%) to that in prior studies (11–57%)
Table 4.
Baseline characteristics of primary analysis participants in VANISH as compared to other clinical trials assessing Angiotensin Receptor Blockers in Hypertrophic Cardiomyopathy.
| VANISH (n=212) | INHERIT21 (n=133) | Shimada et al20(n=20) | CHANCE19 (n=24) | Yamazaki et al17 (n=19) | Kawano et al16 (n=23) | Araujo et al18 (n=30) | |
|---|---|---|---|---|---|---|---|
| Age (yr) | 22.2 (9.7) | 52 (13) | 51 (13) | 53 (13) | 55 (6)/58 (9) | 64 (range 31–79) | 34(12)/32(13) |
| Female gender (%) | 40.6 | 35.3 | 15.0 | 54.0 | 0 | 21.7 | 50.0 |
| Race (%) | |||||||
| White | 95.8 | 94.0 | -- | -- | -- | -- | -- |
| Black | 1.4 | 0 | -- | -- | -- | -- | -- |
| Other | 2.8 | 6.0 | -- | -- | -- | -- | -- |
| NYHA class (%) | |||||||
| I | 93.4 | 63.9 | 90.0 | 35.0 | -- | -- | 33.3 |
| II | 6.6 | 30.0 | 10.0 | 35.0 | -- | -- | 46.7 |
| III and IV | 0 | 6.0 | 0 | 30.0 | -- | -- | 20.0 |
| Pathogenic or likely pathogenic variant (%)* | 100 | 42.9# | -- | 81.8† | -- | -- | -- |
| MYBPC3 | 51.4 | 50.9 | -- | 33.3 | -- | -- | -- |
| MYH7 | 34.4 | 28.1 | -- | 50.0 | -- | -- | -- |
| TNNI3 | 3.3 | 10.5 | -- | 16.0 | -- | -- | -- |
| TNNT2 | 3.8 | 5.3 | -- | - | -- | -- | -- |
| Cardiac medications (%) | |||||||
| Beta-blocker | 17.0 | 57.1 | 35.0 | 33.0 | 10.5 | 17.4 | -- |
| Calcium channel blocker | 2.4 | 14.2 | 20.0 | 33.0 | 15.7 | 43.5 | -- |
| Heart rate (bpm) | 70 (14) | 65 (11) | 63 (9)/65 (10) | 65/66 | -- | 64 (7)/65 (7) | -- |
| Blood pressure (mmHg) | -- | ||||||
| Systolic | 117 (12) | 128 (12) | 118 (13)/123 (8) | 113/119 | 126 (9)/128 (11) | 133 (16)/123 (13) | |
| Diastolic | 68 (10) | 79 (9) | -- | -- | 79 (8)/79 (6) | 76 (14)/76 (6) | |
| Imaging findings | |||||||
| Max. LV wall thickness (mm) | 15 (4) | 23 (6) | 16 (4)/15 (3)‡ | 20 (4)/20 (3) | 22 (6)/20(3) | 18 (4)/17 (4) ‡ | 26 (6)/24 (8) ‡ |
| LVEF (%) | 66 (8) | 69 (8) | 73 (7)/71 (8) | 69 (5)/70 (6) | -- | 72 (10)/70 (17) | -- |
| LA diameter (mm) | 34 (6) | 41 (7) | -- | -- | -- | 41 (5)/44 (7) | 44 (6)/44 (5) |
| LV mass (g) | 116 (42) | -- | 155 (135–222)/139 (112–205) | 407 (139)/451 (228) | 203 (47)/177 (48) | -- | -- |
| LV mass (g/m2) | 63 (17) | 103 (36) | -- | -- | -- | -- |
Results are presented as % or as mean (SD) or median (IQR). Where parameters appear twice they were only reported separately for the two treatment arms.
indicates Not reported. CMR indicates cardiac magnetic resonance imaging; LA, left atrial; LV, left ventricular; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association Class.
5 participants with MYBPC3 also have MYH7 (n=4) or TNNT2 (n=1).
108 (81%) patients had genetic testing performed prior to inclusion in the INHERIT trial, four patients were compound heterozygous.
22 (92%) patients had genetic testing performed in the CHANCE trial.
Diameter of interventricular septum (maximum wall thickness not reported).
Discussion
The VANISH trial was designed to test the hypothesis that ARB administration can attenuate disease progression in early sarcomeric HCM. Inclusion criteria were designed to capture younger mutation carriers anticipated to have less advanced disease. This population is thought to potentially be more responsive to disease-modifying therapy than older patients with established myocardial hypertrophy and fibrosis. The rationale for VANISH draws upon findings in studies on animal models of sarcomeric HCM suggesting a modifiable period early in disease evolution, but once advanced phenotypic manifestations are present, they may be irreversible and unresponsive to pharmacologic manipulation. Thus, our goal was to randomize a cohort of participants at an earlier stage of disease than previously attempted12.
The baseline characteristics of participants randomized into VANISH indicate that we succeeded in enrolling a young cohort of participants with less advanced disease than previously assessed. Per protocol, participants in the primary cohort had LVH supporting a diagnosis of HCM, but had a mean maximal wall thickness of 16 mm; 35% less than in prior trials. Also, in keeping with the young age and less advanced disease of the primary cohort, mean peak oxygen consumption was nearly normal (mean peak VO2 81% predicted) and LGE on CMR was present in only 19% of participants, rather than ~60% typically seen in adult HCM cohorts29. Although most participants reported NYHA I functional class, those with NYHA class II symptoms (8%) were on average over 13 years older, more likely to carry an MYH7 variant, and had more advanced myocardial changes, with more hypertrophy, LGE, left atrial enlargement, and diastolic abnormalities, as well as lower peak oxygen consumption. Collectively, these findings demonstrate that phenotype, genotype and age contribute to disease experience. Further study is needed to better characterize interactions between genetic variation, cardiac structure and function, and symptom burden throughout an individual’s lifetime.
Participants in the exploratory cohort were required to have subtle echocardiographic or ECG changes to indicate some level of phenotypic expression of their sarcomeric gene variant, despite essentially normal LV wall thickness. All had increased (≥0.19) LV thickness:dimension ratio (normal range [mean +/− 2SD] 0.16 +/− 0.02). Borderline LVH (LV wall thickness z score 1.5–2.9) was the most common early phenotypic feature of variant carriers in the exploratory cohort, present in 68%. ECG abnormalities were present in 18 (53%) participants, 72% of which were T-wave inversion/repolarization changes and criteria for LVH being the most prevalent - present in 13 (38%). Notably, the proportion of participants with ECG criteria for LVH was similar between primary (50 [29%]) and the exploratory (10 [29%]) cohorts, in contrast with their echocardiographic findings. Low tissue Doppler relaxation velocities (e’ z-score<1.5) were present in 21% of the exploratory cohort.
Compared to prior studies using ARBs in HCM, the baseline characteristics of participants randomized in the VANISH trial were distinctly different, reflecting our specific aims and entry criteria. First, participants in VANISH were nearly 30 years younger. Secondly, participants randomized into the primary analysis cohort of VANISH had substantially less hypertrophy at baseline. Thirdly, we included only individuals carrying a pathogenic or likely-pathogenic sarcomeric variants to more fully mirror the preclinical studies (performed on animal models of HCM due to sarcomeric gene variants) and ensure that the study population had a more uniform disease etiology. In previous trials, the presence of a sarcomeric gene variant was not an entry criterion. The INHERIT21 (n=133) and CHANCE19 studies (n=24) reported that 43% and 82% of the cohorts, respectively carried pathogenic or likely pathogenic variants. The distribution of sarcomeric gene variants in VANISH was similar to those two studies and reflects the typical distribution in clinical practice, with variants in MYH7 and MYBPC3 being the most abundant6, 7. Finally, all prior trials but the INHERIT study included fewer than 30 patients treated for 6 to 12 months, contrasting with the 178 participants randomized for 24 months of treatment in the VANISH primary analysis cohort16–21.
In summary, the VANISH trial is the largest and longest trial to date in HCM and tests a novel hypothesis that ARBs can attenuate disease progression in HCM if administered early, during a more modifiable period in disease evolution. Randomized participants all carried clinically significant sarcomeric gene variants, were much younger, and at an earlier stage in disease development compared to previous studies. In participants with clinically overt HCM, symptom burden was associated with age, phenotype and genotype. The results of the VANISH trial will provide patients and their physicians with further insights on disease progression in HCM and the potential impact of ARBs, thus advancing the care of patients and families with inherited cardiomyopathies.
Supplementary Material
What is new?
The Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) Trial is the first trial to test the hypothesis that valsartan attenuates disease progression in sarcomeric mutation carriers with early stage disease, based on young age and absence of pronounced left ventricular hypertrophy or substantial symptoms.
Compared to patients enrolled in previous trials evaluating angiotensin receptor blockers in hypertrophic cardiomyopathy, the subjects enrolled in VANISH were much younger, all carried a pathogenic or likely pathogenic sarcomere variant, 92% were NYHA class I and 88% had no cardiovascular symptoms at all.
Symptom burden (NYHA II functional class) was associated with age, more prominent imaging abnormalities and genotype.
What are the clinical implications?
The results of the VANISH trial will address whether it is possible to modify the progression of disease with angiotensin receptor blockers in early stage hypertrophic cardiomyopathy, thus impacting clinical practice for treatment of this group of patients.
Acknowledgments
Executive Committee: Harold Dietz, MD; Steven Lipshultz, MD; William McKenna, MD.
Clinical Events Committee: Chair: Akshay Desai, MD; Members: Neal Lakdawala, MD; Elizabeth Blume, MD.
Sources of funding
VANISH is funded by the National Institutes of Health (NIH 1P50HL112349 to CYH). Study drug was donated by Novartis Pharmaceuticals Corporation who had no part in development of the protocol and are not involved in data analysis or publication.
Nonstandard abbreviations
- ARB
Angiotensin II receptor blockers
- CHANCE
Candesartan use in Hypertrophic And Nonobstructive Cardiomyopathy Estate
- CMR
Cardiac magnetic resonance imaging
- CPET
Cardiopulmonary exercise test
- CT
Cardiac computed tomography
- E
Peak early mitral inflow velocity
- e’
Peak early mitral annular relaxation velocity
- ECG
Electrocardiogram
- HCM
Hypertrophic cardiomyopathy
- LA
Left atrial
- LGE
Late gadolinium enhancement
- LV
Left ventricle
- LVH
left ventricular hypertrophy
- LVEF
Left ventricular ejection fraction
- NT-pro-BNP
N-terminal of the pro brain natriuretic peptide
- NYHA
New York Heart Association, heart failure class
- TGF- β
Tissue growth factor β
- VO2 max
Peak oxygen uptake
Footnotes
Independent data access and analysis
LS had full access to all the data in the study and takes responsibility for its integrity and the data analysis.
Disclosures
There are no conflicts of interest directly relevant to this work.
Drs. Ho, Colan, Jefferies and Day have received research support from Myokardia, Inc. Dr. Ho has consulted for MyoKardia. Dr. Solomon has received research grants from Alnylam, Amgen, AstraZeneca, Bellerophon, Bayer, BMS, Celladon, Cytokinetics, Eidos, Gilead, GSK, Ionis, Lone Star Heart, Mesoblast, MyoKardia, NIH/NHLBI, Novartis, Sanofi Pasteur, Theracos, and has consulted for Akros, Alnylam, Amgen, Arena, AstraZeneca, Bayer, BMS, Cardior, Corvia, Cytokinetics, Daiichi-Sankyo, Gilead, GSK, Ironwood, Merck, Myokardia, Novartis, Roche, Takeda, Theracos, Quantum Genetics, Cardurion, AoBiome, Janssen, Cardiac Dimensions, and Tenaya. Dr Bundggard has received lecture fees from Amgen, Astra-Zeneca, Pfizer and MSD. Dr McMurray declares that his employer, Glasgow University, has been paid by Novartis for his time spent as Executive Committee member and co-principal investigator of clinical trials as well as for his participation in a company advisory board. Dr Braunwald reports research grants through the Brigham and Women’s Hospital from Astra Zeneca, Daiichi Sankyo, Merck, and Novartis; consultancies with Cardurion, MyoKardia, Sanofi, and Verve; fees for lectures from Medscape, and uncompensated consultancies and lectures from Merck, Novartis, and The Medicines Company. Dr McRae reports research funding from Novartis, Array BioPharma, Sanofi, Merck, Microsoft, American Heart Association, Verily, Astra Zeneca, Quest Diagnostics, NHGRI, NINDS, and NIGMS as well as in kind support from Illumina and has consulted fir AtlasVentures, Vertex, Biosymetrics and Foresite Labs.
References
- 1.Maron BJ, Spirito P, Roman MJ, Paranicas M, Okin PM, Best LG, Lee ET and Devereux RB. Prevalence of hypertrophic cardiomyopathy in a population-based sample of American Indians aged 51 to 77 years (the Strong Heart Study). The American journal of cardiology. 2004;93:1510–4. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT and Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–9. [DOI] [PubMed] [Google Scholar]
- 3.Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H, Lu S, Wu P, Zhang Y, Shen L, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. The American journal of medicine. 2004;116:14–8. [DOI] [PubMed] [Google Scholar]
- 4.Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). European heart journal. 2014;35:2733–79. [DOI] [PubMed] [Google Scholar]
- 5.American College of Cardiology Foundation/American Heart Association Task Force on P, American Association for Thoracic S, American Society of E, American Society of Nuclear C, Heart Failure Society of A, Heart Rhythm S, Society for Cardiovascular A, Interventions, Society of Thoracic S, Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE and Yancy CW. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. The Journal of thoracic and cardiovascular surgery. 2011;142:e153–203. [DOI] [PubMed] [Google Scholar]
- 6.Alcalai R, Seidman JG and Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. Journal of cardiovascular electrophysiology. 2008;19:104–10. [DOI] [PubMed] [Google Scholar]
- 7.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG and Seidman CE. Shared genetic causes of cardiac hypertrophy in children and adults. The New England journal of medicine. 2008;358:1899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP, et al. Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation. 2010;122:1130–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cowan BR and Young AA. Left ventricular hypertrophy and renin-angiotensin system blockade. Current hypertension reports. 2009;11:167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH and Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. The Journal of clinical investigation. 2010;120:3520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsybouleva N, Zhang L, Chen S, Patel R, Lutucuta S, Nemoto S, DeFreitas G, Entman M, Carabello BA, Roberts R, et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation. 2004;109:1284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim DS, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, Roberts R and Marian AJ. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vignier N, Le Corvoisier P, Blard C, Sambin L, Azibani F, Schlossarek S, Delcayre C, Carrier L, Hittinger L and Su JB. AT1 blockade abolishes left ventricular hypertrophy in heterozygous cMyBP-C null mice: role of FHL1. Fundamental & clinical pharmacology. 2014;28:249–56. [DOI] [PubMed] [Google Scholar]
- 16.Kawano H, Toda G, Nakamizo R, Koide Y, Seto S and Yano K. Valsartan decreases type I collagen synthesis in patients with hypertrophic cardiomyopathy. Circulation journal : official journal of the Japanese Circulation Society. 2005;69:1244–8. [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki T, Suzuki J, Shimamoto R, Tsuji T, Ohmoto-Sekine Y, Ohtomo K and Nagai R. A new therapeutic strategy for hypertrophic nonobstructive cardiomyopathy in humans. A randomized and prospective study with an Angiotensin II receptor blocker. International heart journal. 2007;48:715–24. [DOI] [PubMed] [Google Scholar]
- 18.Araujo AQ, Arteaga E, Ianni BM, Buck PC, Rabello R and Mady C. Effect of Losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. The American journal of cardiology. 2005;96:1563–7. [DOI] [PubMed] [Google Scholar]
- 19.Penicka M, Gregor P, Kerekes R, Marek D, Curila K, Krupicka J, Candesartan use in H and Non-obstructive Cardiomyopathy Estate Study I. The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy: a pilot, randomized study. The Journal of molecular diagnostics : JMD. 2009;11:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimada YJ, Passeri JJ, Baggish AL, O’Callaghan C, Lowry PA, Yannekis G, Abbara S, Ghoshhajra BB, Rothman RD, Ho CY, et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart failure. 2013;1:480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Axelsson A, Iversen K, Vejlstrup N, Ho C, Norsk J, Langhoff L, Ahtarovski K, Corell P, Havndrup O, Jensen M, et al. Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. The lancet Diabetes & endocrinology. 2015;3:123–31. [DOI] [PubMed] [Google Scholar]
- 22.Ho CY, McMurray JJV, Cirino AL, Colan SD, Day SM, Desai AS, Lipshultz SE, MacRae CA, Shi L, Solomon SD, et al. , VANISH trial investigators and executive committee. The Design of the Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) Trial. American heart journal. 2017;187:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. , ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research. 2016;44:D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang JW, Guo J, Hung CY, Lu S, Shin SJ, Quek R, Ying A, Ho GF, Nguyen HS, Dhabhar B, et al. Sunrise in melanoma management: Time to focus on melanoma burden in Asia. Asia Pac J Clin Oncol. 2017;13:423–427. [DOI] [PubMed] [Google Scholar]
- 26.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2015;28:1–39 e14. [DOI] [PubMed] [Google Scholar]
- 27.Nagueh SF, Smiseth OA, Appleton CP, Byrd BF 3rd, Dokainish H, Edvardsen T, Flachskampf FA, Gillebert TC, Klein AL, Lancellotti P, et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2016;29:277–314. [DOI] [PubMed] [Google Scholar]
- 28.Sluysmans T and Colan SD. Theoretical and empirical derivation of cardiovascular allometric relationships in children. Journal of applied physiology. 2005;99:445–57. [DOI] [PubMed] [Google Scholar]
- 29.Green JJ, Berger JS, Kramer CM and Salerno M. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovascular imaging. 2012;5:370–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.