

Abstract
Histone methylation is critical for the formation and maintenance of long-term memories. Long noncoding RNAs (lncRNAs) are regulators of histone methyltransferases and other chromatin-modifying enzymes (CMEs), thereby epigenetically modifying gene expression. Here, we investigated how the lncRNA NEAT1 may epigenetically contribute to hippocampus-dependent, long-term memory formation using a combination of transcriptomics, RNA-binding protein immunoprecipitation, CRISPR-mediated gene activation (CRISPRa), and behavioral approaches. Knockdown of the lncRNA Neat1 revealed widespread changes in gene transcription, as well as perturbations of histone 3 lysine 9 dimethylation (H3K9me2), a repressive histone modification mark that was increased in the hippocampus of aging rodents. We identified a NEAT1-dependent mechanism of transcriptional repression by H3K9me2 at the c-Fos promoter, corresponding with observed changes in c-Fos mRNA expression. Overexpression of hippocampal NEAT1 using CRISPRa was sufficient to impair memory formation in young adult mice, recapitulating observed memory deficits in old adult mice, whereas knocking down NEAT1 in both young and old adult mice improved behavior test–associated memory. These results suggest that the lncRNA NEAT1 is an epigenetic suppressor of hippocampus-dependent, long-term memory formation.
INTRODUCTION
While thousands of long noncoding RNAs (lncRNAs) in the human and murine genomes have been characterized, few lncRNAs are as well-studied as the human nuclear-enriched abundant transcript 1 (NEAT1). NEAT1 is evolutionarily conserved between rodents and humans, particularly within the 5’ region of the transcript (1). Multiple isoforms of NEAT1 exist in rodents and in humans, with the longer of the major isoforms proving essential for phase separation and induction of nuclear paraspeckle assembly (2, 3), whereas the shorter NEAT1 isoforms do not appear to be a major regulator of paraspeckle formation (4). Several studies have characterized a number of molecular pathways by which NEAT1 regulates the epigenome, including both paraspeckle-dependent sequestration of transcription factors as well as paraspeckle-independent scaffolding of chromatin modifying enzymes (CMEs) (5–7). Additionally, NEAT1 itself has been observed to bind numerous genomic loci and to effect the regulation of transcription (8, 9).
Research on the human NEAT1 has been largely focused on its role as an oncogene in various cancers (10)), which is mediated largely through its regulation of epigenetic mechanisms. However, the abundance of NEAT1 and its rodent homolog (Neat1) are increased in the brains of aging humans and mice (11–13) and are linked to multiple cognitive and neurodegenerative disorders, including schizophrenia (14), Huntington’s Disease (15), Parkinson’s Disease (16, 17), Alzheimer’s Disease (18), and epilepsy (19, 20). Furthermore, evidence from rodent and human samples suggests that NEAT1 may play a role in neuroplasticity (19); however, despite such extensive relevance to physiology and health, the role of NEAT1 in the epigenetic regulation of genes within hippocampal neurons, particularly during long-term memory formation, is not clear. We used RNA-sequencing in mouse and human tissue, CRISPR-mediated gene activation (CRISPRa) in vivo, and behavioral memory tests in mice to investigate the functional role of NEAT1 in gene expression dynamics and the role that age-related changes in its expression might play in memory deficits seen in older adults.
RESULTS
Expression of the long noncoding RNA NEAT1 is relatively low in human CNS tissue
Expression of NEAT1 is abundant in many cultured cell lines including those characterized in the ENCODE project (21) (Fig. 1A). However, we observed that in contrast to the abundant expression of NEAT1 observed in most tissues, the human central nervous system (CNS) as a whole, as well as specifically the hippocampus therein (outlined in red, Fig. 1, B and C), expresses minimal quantities of NEAT1 (22, 23). Unsupervised hierarchical clustering based on tissue expression of NEAT1 supports this observation, as CNS tissues segregate cleanly when sorted based on NEAT1 transcript expression (Fig. 1D; fig. S1, A and B; data file S1).
Figure 1. Restricted expression of lncRNA NEAT1 in human CNS tissues.
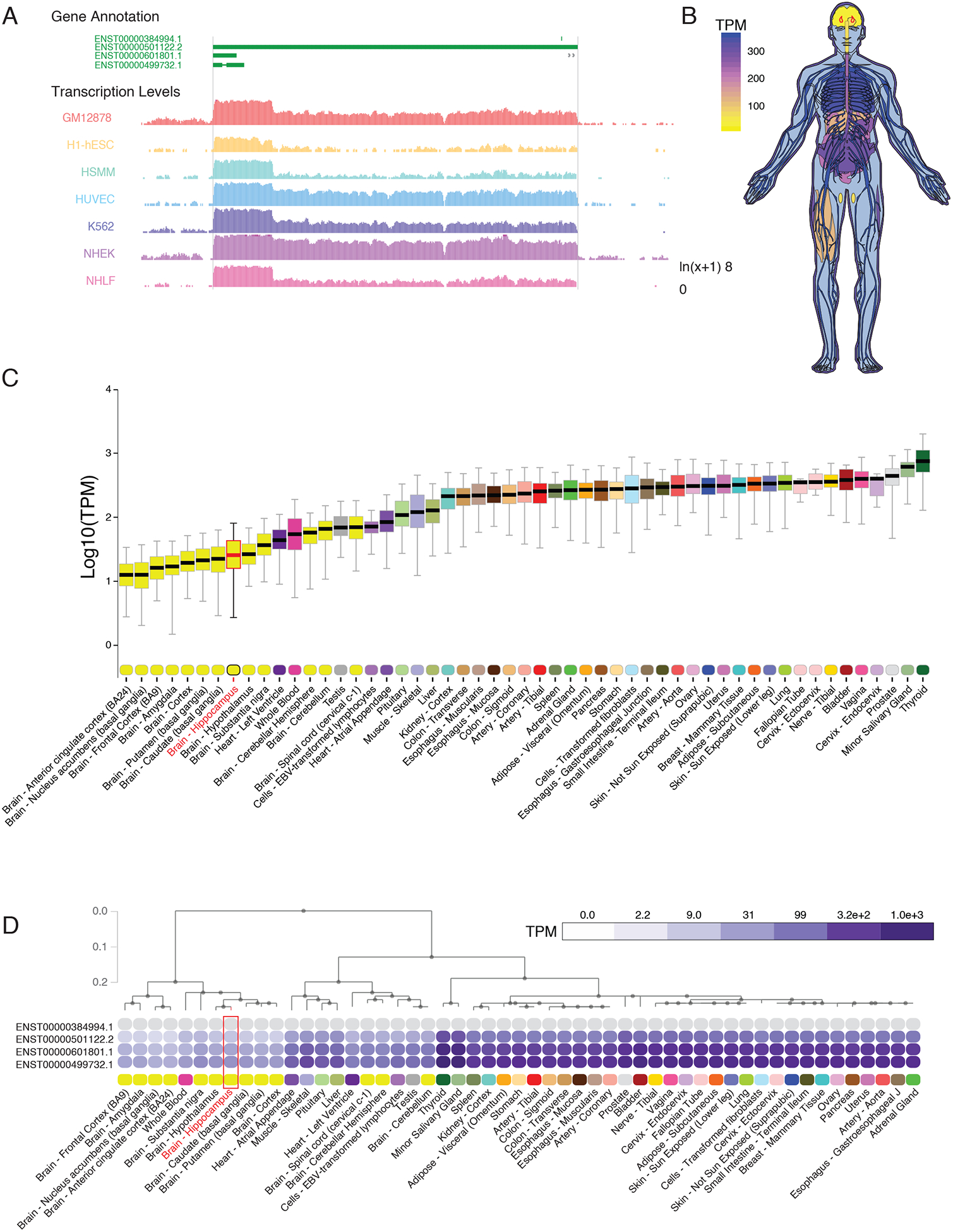
(A) University of California, Santa Cruz (UCSC) Genome browser track export showing expression of NEAT1 in seven cell types from ENCODE. (B) Human body plot illustrating the expression of NEAT1 in 53 human tissues from the GTEx project, values shown are the median transcripts per million (TPM) values by tissue, hippocampus outlined in red. (C) Bar plots showing median, upper quartile, and lower quartile expression of the NEAT1 gene (ENSG00000245532.4) in 53 human tissues from the GTEx project; hippocampal expression outlined in red. (D) Hierarchical clustering of NEAT1 based on transcript isoform level expression in 53 human tissues from the GTEx project. Dendrogram scale shows cluster distance. Expression values displayed in the heatmap are the median expression values in TPM for each isoform in each tissue. See data file S1 for complete n values for all tissue types.
Examination of single-cell RNA-sequencing data from resected human CNS tissue and glioblastoma (24) further suggested that expression of NEAT1 within CNS cells is restricted (low) in neurons, whereas other cell types including astrocytes, oligodendrocytes, and vascular cells, express NEAT1 at higher amounts. (fig. S1, C and D). This is in contrast to the lncRNA transcript MALAT1, which is transcribed from a region adjacent to the NEAT1 gene locus, and which appears to be ubiquitously expressed at high abundance in all CNS cell types (fig. S1E). Given the growing body of literature that has noted increased expression of NEAT1 in the aging brain (12, 13) as well as the established role of NEAT1 as a regulator of epigenetic mechanisms and the recently-described role of NEAT1 in cognitive disorders, such as schizophrenia (14), we sought to further investigate the role of NEAT1 on the neuroepigenetic mechanisms of cognition.
NEAT1 suppresses the immediate-early and synaptic plasticity-related gene c-Fos
To investigate the role of NEAT1 at the transcriptomic level, we analyzed a publicly available RNA-seq dataset from induced pluripotent stem cell (iPSC)-derived human neurons. Antisense oligo (ASO) knockdown of NEAT1 in KCl-treated human neurons revealed an extensive cohort of differentially expressed messenger RNAs (mRNAs). Knockdown alone was not sufficient to perturb the transcriptome in resting iPSC-derived human neurons, as evidenced by an imperfect separation after unsupervised hierarchical clustering prior to KCl stimulation (Fig. 2A). In contrast, NEAT1 knockdown appeared to potentiate KCl-induced differential expression of many genes (Fig. 2 A and B).
Figure 2. NEAT1 regulates expression of c-FOS mRNA and the AP-1 complex in iPSC-derived human neurons.
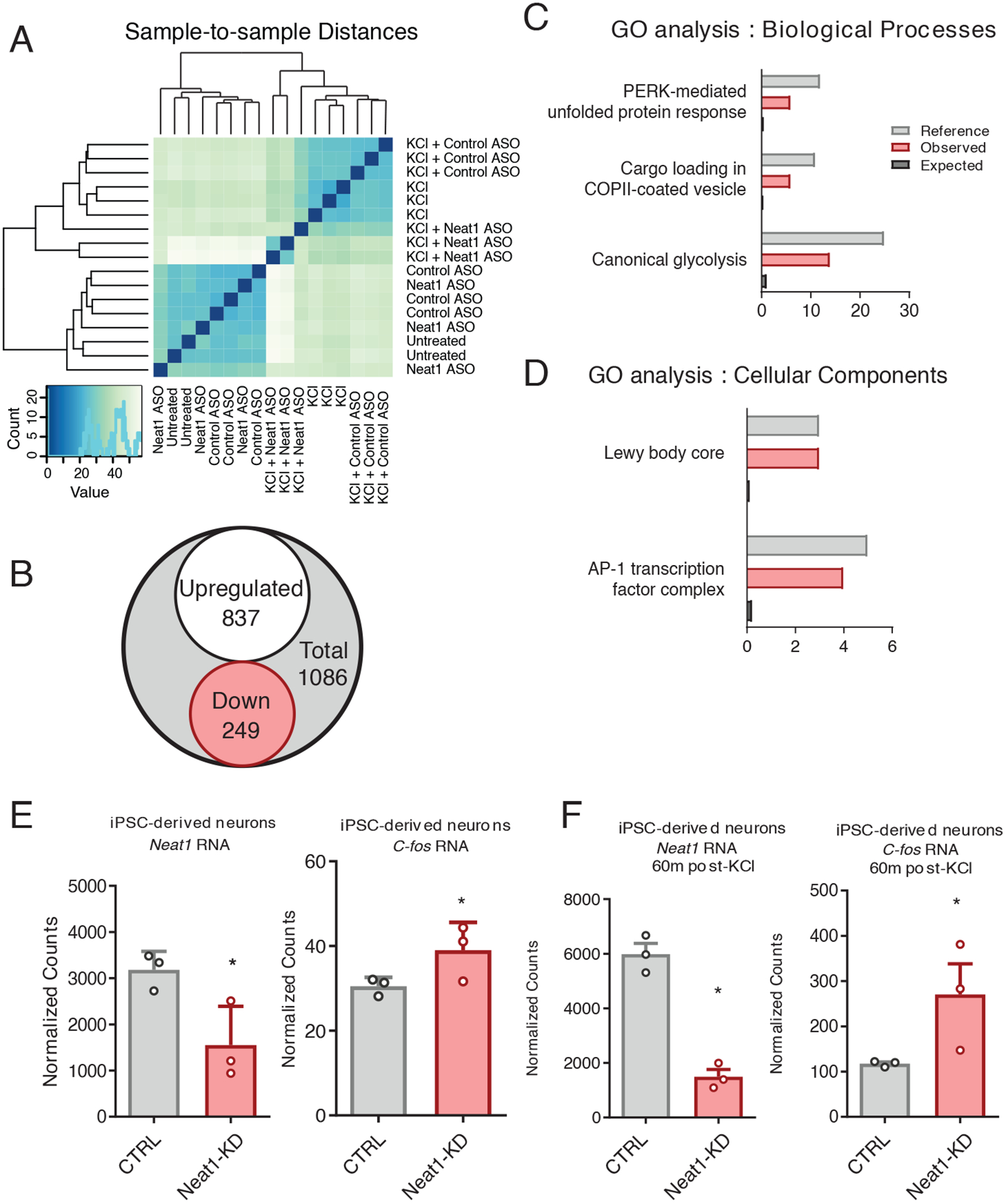
(A) Unsupervised hierarchical clustering transcriptomes from Neat1/Control ASO and KCl-treated iPSC-derived human neurons, based on DESeq2-normalized counts. (B) Venn diagram depicting the total number of differentially expressed genes detected between KCl+Control antisense oligonucleotide (ASO) and KCl+Neat1_ASO groups detected by DESeq2. (C and D) Gene Ontology (GO) term enrichment for DE genes depicted in (B). All GO terms shown showed statistically significant enrichment (BH corrected, P < 0.05). (E and F) Normalized count values for lncRNA NEAT1 and C-FOS mRNA either prior to (E) or after (F) KCl treatment of iPSC-derived neurons. Count values significantly different between groups (BH corrected, P < 0.05).
To gain some insight into the physiological relevance for the observed NEAT1-mediated changes in gene expression in human neurons, we queried the annotated disease classes from the Genetic Association Database using the Database for Annotation, Visualization and Integrated Discovery tool (DAVID) (25), and observed significant enrichment for three disease classes: cancer, renal, and aging. (Data File S2). Gene ontology (GO) enrichment was then assessed using a PANTHER (Protein ANalysis THrough Evolutionary Relationships) Overrepresentation Test, and differentially expressed genes appeared to be non-randomly distributed among annotated biological processes (Fig. 2C), molecular functions (Data File S2) and cellular components (Fig. 2D). Significant GO term enrichment was partially consistent with previous observations of the NEAT1 regulatory axis (26), as we observed significant regulation of GO terms associated with viral gene expression; however, we also observed significant enrichment of GO terms important for hippocampal function, including the transcription factor AP-1 complex (GO:0035976, Fig. 2, C and D).
The human Fos proto-oncogene (FOS, also known as c-FOS), a critical component of the AP-1 transcription factor subunit, appeared to be overexpressed in human neurons after knocking down NEAT1 both in quiescent and KCl-stimulated neurons (Fig. 2, E and F). Because c-Fos has known relevance to hippocampus-dependent memory formation (27), we selected the murine homolog c-Fos as a candidate gene to further study Neat1’s regulatory potential.
NEAT1 is downregulated by neuronal excitation.
Because modeling NEAT1 expression changes in response to in vivo neuronal activity and behavioral experience required the use of mammalian model organisms, we next sought to examine the regulatory capacity of NEAT1 in rodent neurons. For this purpose, we knocked down murine NEAT1 in the mouse Neuro-2a (N2a) cell line using small interfering RNAs (siRNAs). We observed that 24 hours after treatment with Neat1-targeting siRNAs (Fig. 3A), the abundance of the c-Fos mRNA was significantly increased (Fig. 3B).
Figure 3. NEAT1 represses expression of c-Fos mRNA in murine neuronal cells.
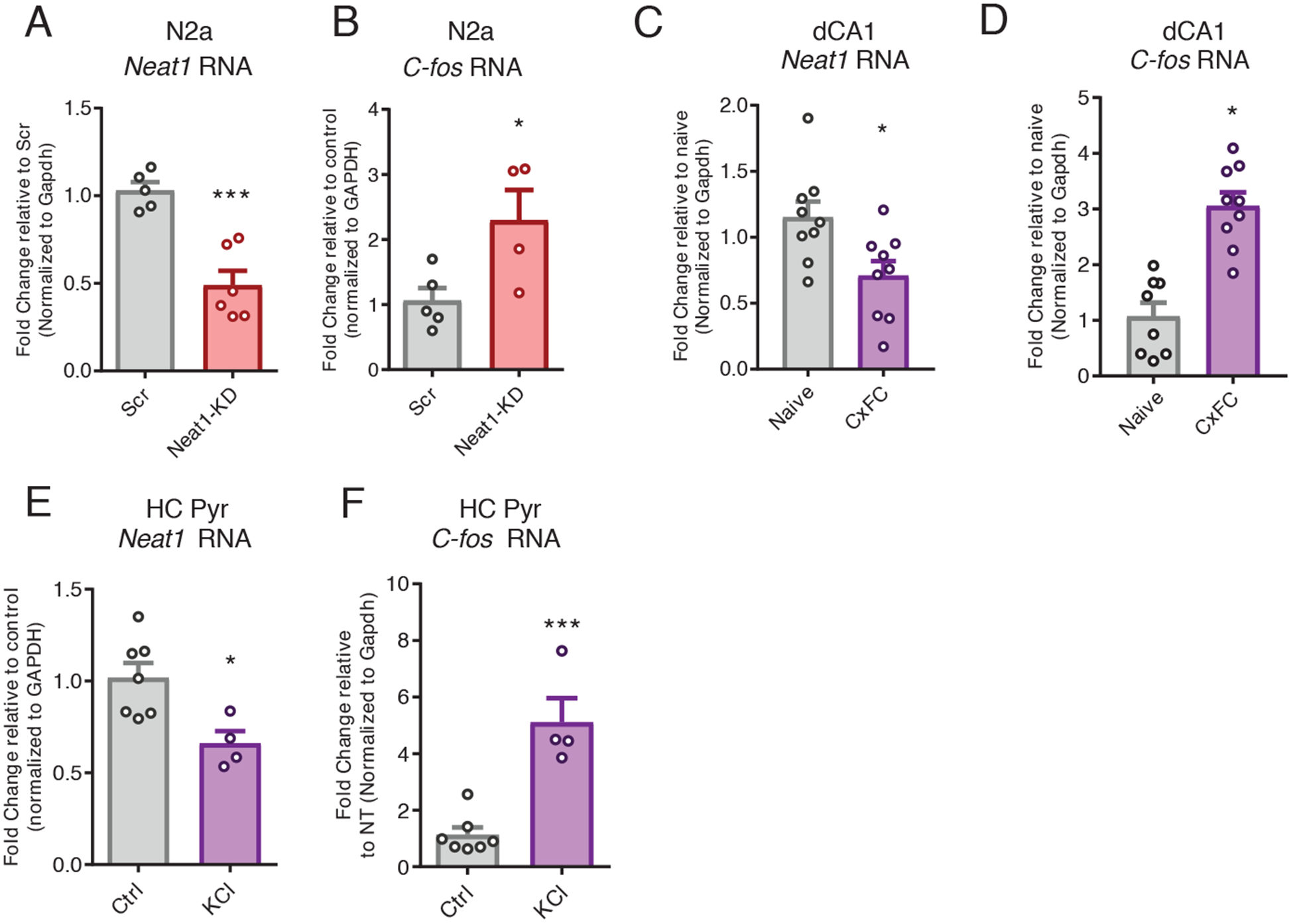
(A) Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis of the abundance of Neat1 transcript in control or Neat1- targeted siRNA-treated murine N2a cells. Data are n = 5 to 6 independent cultures of N2a cells. ***P < 0.0005 by Student’s t test. (B) RT-qPCR analysis of the abundance of c-Fos mRNA in control or Neat1-targeted siRNA-treated murine N2a cells. Data are cultured cells from n = 3 to 4 experiments; *P < 0.05 by Student’s t-test. (C and D) RT-qPCR analysis of Neat1 transcript (C) and c-Fos mRNA (D) abundance in dorsal CA1 (dCA1) from naïve or contextual fear conditioning (CxFC) trained mice. Data are from n = 8 to 9 mice; *P < 0.05 by Student’s t-test. (E and F) RT-qPCR analysis of Neat1 transcript (E) and c-Fos mRNA (F) abundance in (CxFC) trained mice. Data are cultured primary neurons from n = 4 to 7 experiments; *P < 0.05 by Student’s t-test, ***P < 0.0005 by Student’s t-test.
Although our observations of the c-Fos transcript expression in murine neurons recapitulated observations from human neurons, we observed that mRNA expression of the associated immediate-early genes Egr1 and Btg2 and others were not increased in mouse (fig. S2, A and B) as they were in human neurons (data file S2), suggesting that there are species-specific regulatory differences in the NEAT1 regulatory axis.
Given that mice lacking c-Fos expression in the CNS show a specific loss of hippocampus-dependent spatial and associative learning tasks (27), we next sought to investigate the relevance of Neat1 expression during memory consolidation after a hippocampus-dependent learning task. One hour after training in contextual fear conditioning, we observed a significant reduction in the expression of Neat1 in the dorsal hippocampus coinciding with previously reported increases in expression of the c-Fos mRNA (Fig. 3, C and D). Because baseline expression of Neat1 in neurons is expected to be quite restricted compared to other cell types, we next stimulated neurons with KCl to ascertain the effect of activity on the expression of Neat1. Consistent with recent reports (19), we observed that KCl stimulation induced a rapid reduction in Neat1 expression in both N2a cells (fig. S2E) and primary hippocampal pyramidal neurons (Fig. 3, E and F), consistent with the effects of context fear conditioning in vivo.
NEAT1 regulates H3K9me2 globally and controls both H3K9me2 of the c-Fos promoter and c-Fos expression.
We next sought to investigate the c-Fos-relevant mechanisms of NEAT1-orchestrated transcriptional control. To accomplish this, we used publicly available data assaying NEAT1 binding to chromatin with capture hybridization analysis of RNA targets and high-throughput sequencing (CHART-seq) in human MCF7 cells (8). After mapping NEAT1-bound peaks to the nearest transcription start sites, we observed that only a small subset of genes directly bound by NEAT1 are differentially expressed either after NEAT1 knockdown or in the context of neuronal activation. However, we observed significant enrichment of NEAT1 binding near genes associated with histone methyltransferase activity, including the histone H3 lysine 9 (H3K9) dimethyltransferase EHMT1 (also known as GLP) (Fig. 4, A and B; fig S5,Data File S3) (28).
Figure 4. NEAT1 modulates neuronal H3K9me2.
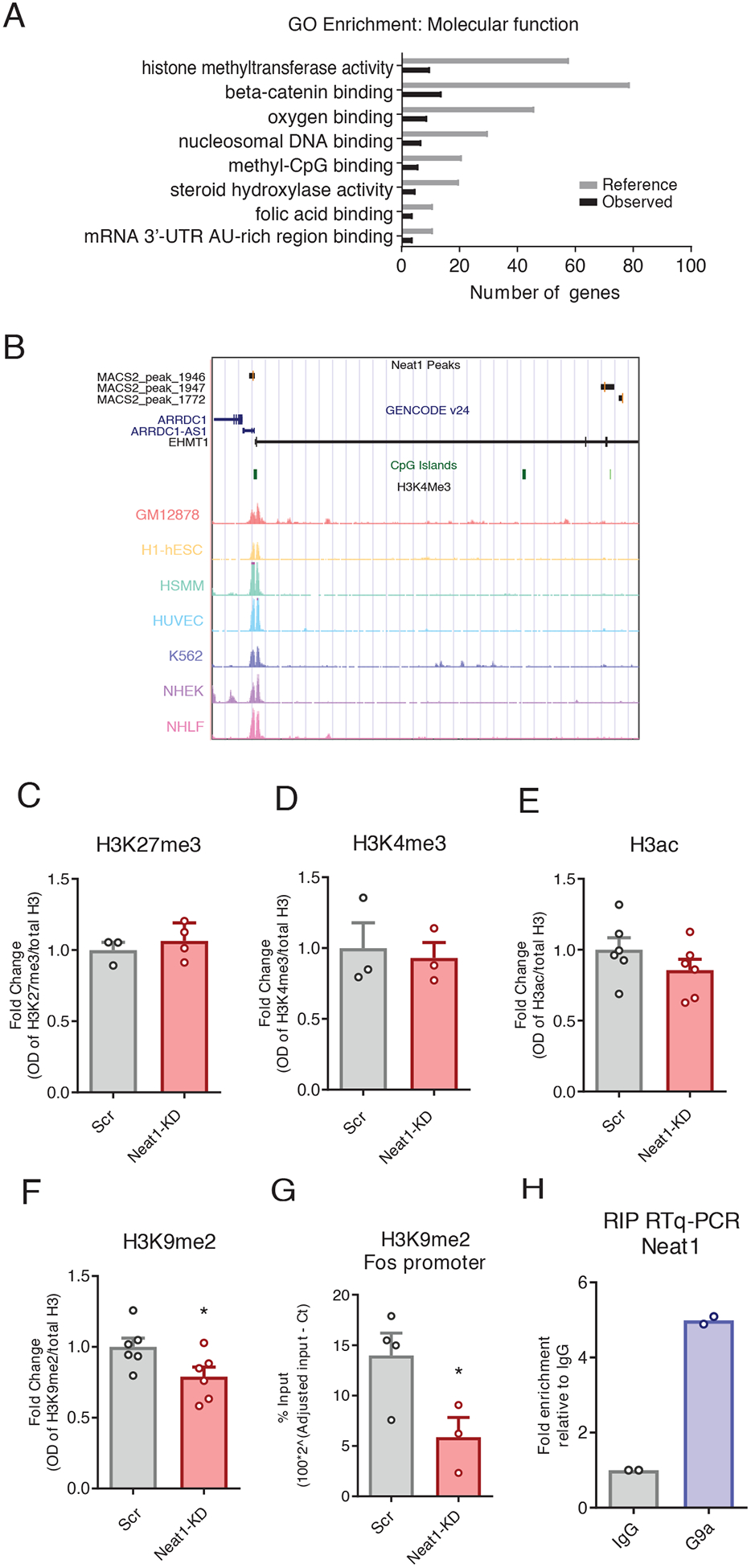
(A) NEAT1 CHART-seq peaks were mapped to the nearest gene transcription start site, and functional enrichment was assessed using ChIP-ENRICH, with histone methyltransferase activity being noted as a significantly enriched GO term (BH corrected, *P < 0.05 for all terms shown). (B) UCSC genome browser plot showing NEAT1-binding peaks overlapping the human EHMT1 gene. (C to G) Analysis of Western blots assessing histone modifications H3K27me3 (C), H3K4me3 (D), H3ac (E), and H3K9me2 (F) globally, and ChIP-qPCR assays at the H3K9me2 at the c-Fos gene promoter (G) in N2a cells after siRNA-mediated knockdown of NEAT1. Data are cultured cells from at least 3 independent experiments, *P< 0.05 by Student’s t-test (F, P=0.0456; G, P=0.0472. (H) RNA-binding protein immunoprecipitation followed by RT-qPCR to assess for Ehmt2/Neat1 interaction in murine N2a cells. Data are means of 2 independent experiments, each represented by a data point.
The c-Fos locus has previously been observed to be methylated by the EHMT1/2 complex in the context of hippocampus-dependent memory formation (29). Therefore, we next sought to investigate the role of NEAT1 in the regulation of histone methylation and H3K9me2 specifically. After knockdown of NEAT1 in neuronal cells, we observed that H3K9me2 is reduced at a global scale, whereas we did not observe such changes in the expression of several histone marks previously associated with NEAT1 in other cell types (Fig. 4, C to F).
To assess the functional relevance of the NEAT1-H3K9me2 regulatory axis on the expression of c-Fos mRNA, we performed chromatin immunoprecipitation assays followed by qPCR (ChIP-qPCR) at the c-Fos promoter. We observed that after NEAT1 knockdown with siRNAs, H3K9me2 at the c-Fos promoter was significantly depleted (Fig. 4G), consistent with observed changes in gene expression (Fig. 3B), whereas H3K9me2 within the c-Fos gene body was not significantly changed (figs. S2F).
To ascertain whether NEAT1 physically associates with the H3K9me2 methyltransferase complex in neurons, we performed RNA-binding protein immunoprecipitations (RIP assays) against the EHMT2 subunit of the obligatory EHMT1/2 heterodimer (Fig. 4H) (29–31). Consistent with published results (6), we observed interaction between NEAT1 and EHMT2, which suggests that NEAT1 may bind both protein components of EHMT1/2 complex, as well as the Ehmt1 locus (Fig. 4, A and B).
NEAT1 knockdown improves hippocampal memory formation and de-represses the epigenetic landscape at the c-Fos promoter in vivo
Having demonstrated that NEAT1 alters the epigenetic landscape and represses neuronal expression of the memory-critical c-Fos gene in cultured cells, we next sought to investigate the functional role of NEAT1 expression on c-Fos promoter methylation and memory formation in vivo. To explore whether Neat1 expression impacts hippocampus-dependent memory formation, we knocked down the abundance of NEAT1 in the hippocampal area Cornu Ammonis 1 (CA1) by directly infusing Neat1-targeting siRNAs or, as a control, non-targeted siRNAs and assayed longterm memory function by means of contextual fear conditioning, a hippocampus-dependent memory task (Fig. 5A). We observed that five days after intra-CA1 injection of Neat1-targeting siRNAs, a time when we observed significantly reduced expression of Neat1 (fig. S3A), mice had no substantial difference in freezing behavior during the training phase of contextual fear conditioning, either before or after delivery of the foot shock (Fig. 5B). However, when returned to the training context 24 hours later, mice that had been injected with Neat1-targeting siRNAs displayed significantly increased freezing behavior relative to mice that had been injected with non-targeting siRNAs (Fig. 5C).
Figure 5. Neat1 knockdown regulates c-Fos promoter methylation in vivo and improves long-term memory.
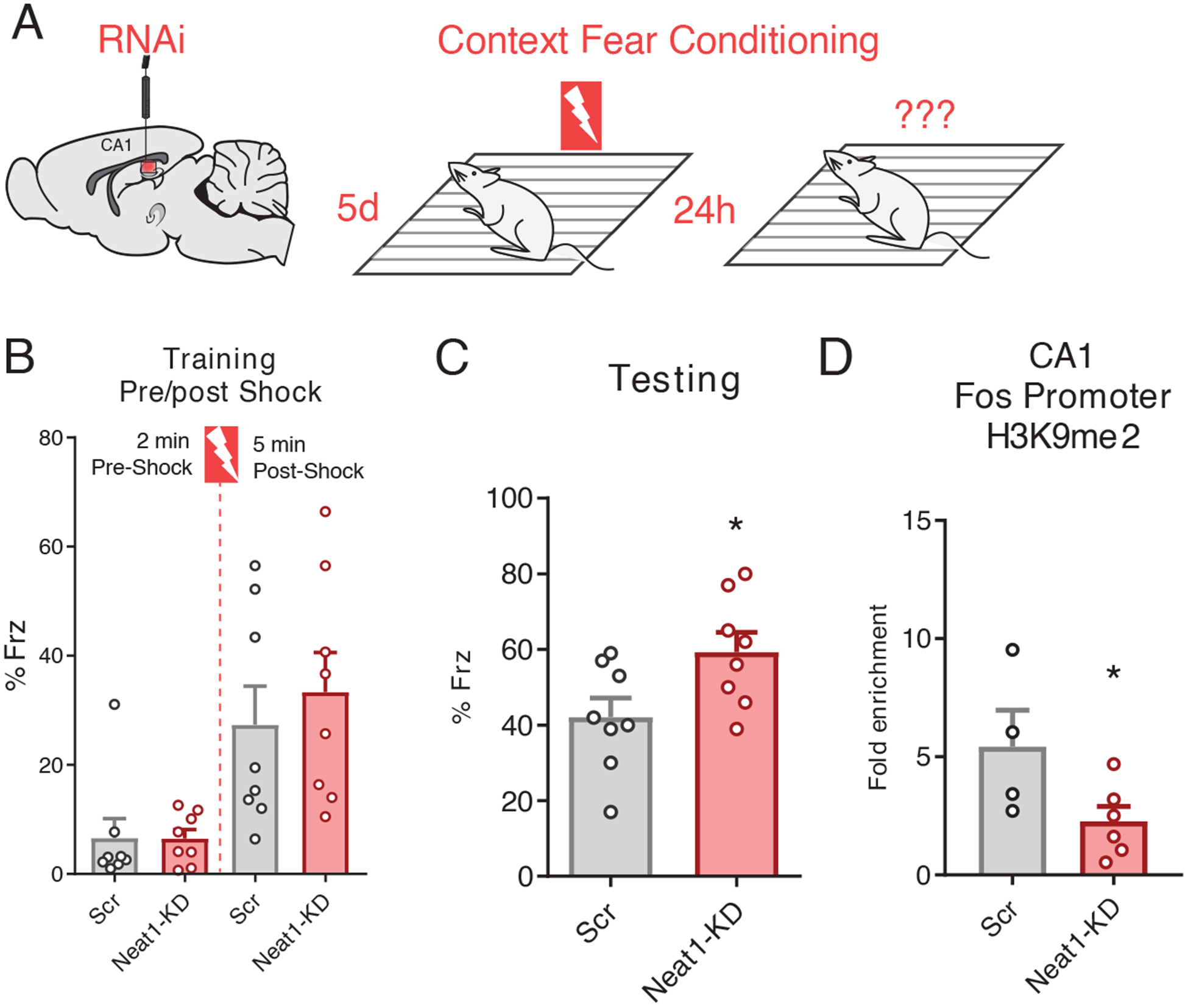
(A) Graphic depiction of siRNA infusion into hippocampal area CA1 and single-pairing contextual fear conditioning paradigm. Briefly, 3- to 7-month-old male C57BL/6 mice were trained 5 days after bilateral infusion of siRNAs and tested 24 hours after training. (B) Freezing behavior of mice described in (A) as a percent of epoch during training phases of the contextual fear conditioning paradigm. No significant difference detected for either the “pre-shock” or “post-shock” epochs. Data are means ± SEM from n = 8 mice, assessed by Student’s t-test. (C) Freezing behavior of mice described in (A) as a percent of total time during the 5-min test trial. Data are means ± SEM from n = 8 mice, *p = 0.0307 by Student’s t-test. (D) ChIP-qPCR analysis of H3K9me2 at the c-Fos promoter in dCA1 tissue from animals sacrificed 5 days after the conclusion of the behavior experiments described in (A). Data are from dCA1 tissue of 4 to 6 mice, *p = 0.0450 by Student’s t-test.
To determine whether NEAT1 abundance impacts c-Fos promoter methylation in vivo, we sacrificed an additional cohort of behaviorally naïve animals five days after injection of siRNAs and performed ChIP-qPCR on one hemisphere of dorsal CA1 tissue collected from around the injection site. Consistent with our results in cultured neurons described above (Fig. 4G), we observed that concurrent with NEAT1 knockdown five days after infusion with siRNAs, the amount of H3K9me2 at the c-Fos promoter in dorsal area CA1 was significantly reduced (Fig. 5D). Thus, we hypothesized a model in which NEAT1 abundance might be regulating memory formation through epigenetic repression of c-Fos.
Mimicking age-related upregulation of Neat1 expression impairs, and knocking down Neat1 restores, hippocampus-dependent memory formation
Numerous studies have reported overexpression of NEAT1 in senescing cells, as well as in aging CNS tissues in both humans and mice (11, 32). Upon comparing publicly available hippocampus RNA-seq datasets from 3-month-old (young) mice versus 24-month-old (aged) mice, we observed increased expression of Neat1 in the hippocampus of aged relative to that of young animals (Fig. 6A) as well as decreased expression of c-Fos (Fig. 6B), both consistent with previously reported results and age-associated hippocampus-dependent memory impairments, respectively (11).
Figure 6. Neat1-knockdown improves long-term memory in aged animals.
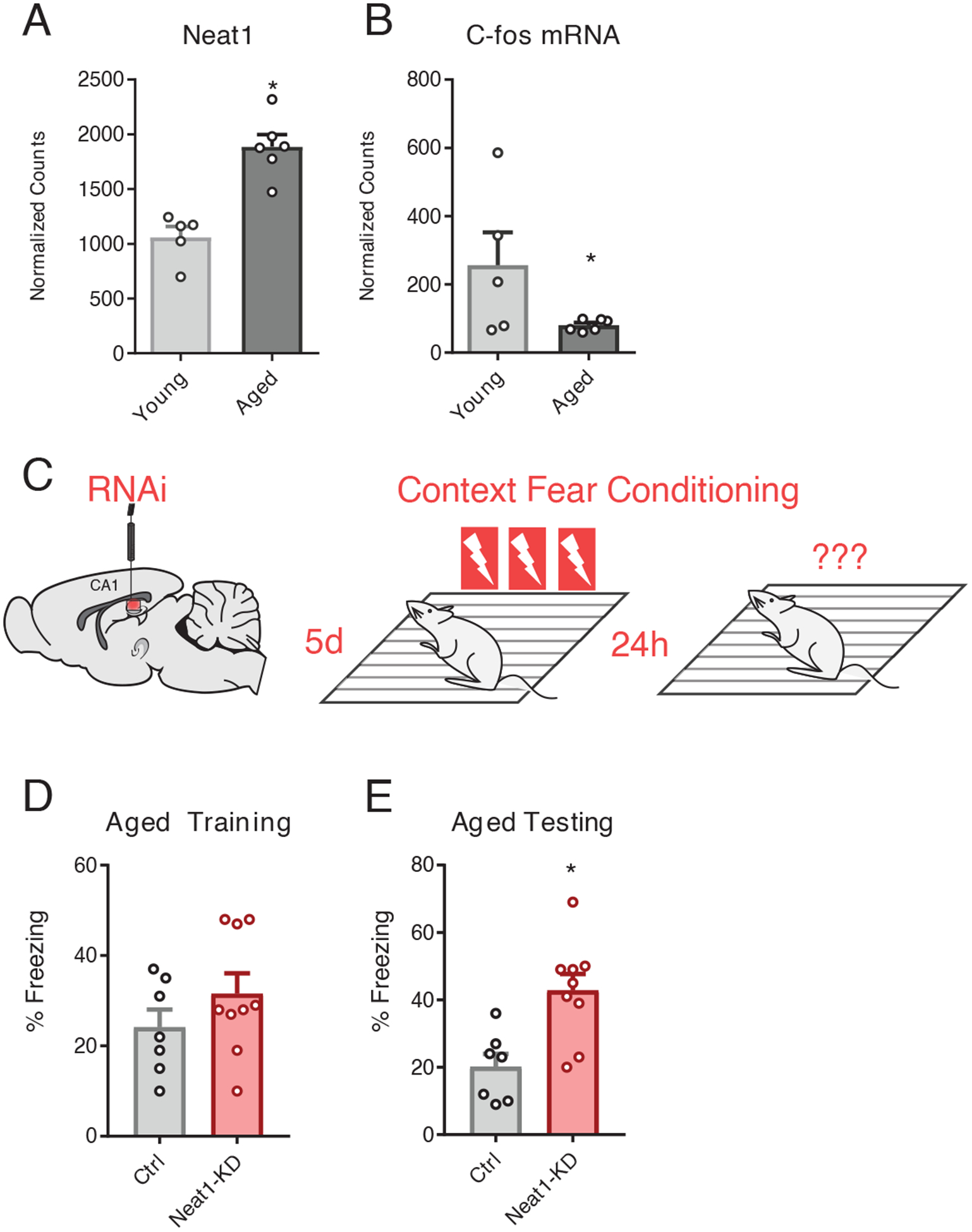
(A and B) DESeq2-generated normalized counts for Neat1 (A) and c-Fos (B) from RNAseq assay of hippocampi from 3mo and 24mo C57/B6 mice. Data are means ± SEM from n = 5 to 6 mice, *Benjamini-Hochberg (BH) adjusted p < 0.05. Neat1 abundance (A) and c-Fos mRNA abundance were (*BH corrected p < 0.05) (B) was significantly repressed in aged hippocampi relative to the hippocampi of young mice. (*BH corrected p < 0.05) (C) Graphic depiction of siRNA infusion into hippocampal area CA1 and three shock-pairing contextual fear conditioning paradigm. Briefly, 18mo old male C57/B6 mice were trained 5 days after bilateral infusion of siRNAs and tested 24 hours after training. (D) Freezing behavior of mice described in (C) as a percent of time during the training phase of the contextual fear conditioning paradigm. No significant difference detected; data are means ± SEM from n = 7 to 9 mice, assessed by Student’s t-test. (E) Freezing behavior of mice described in (C) as a percent of time during the testing phase of the contextual fear conditioning paradigm. Data are means ± SEM from n = 7 to 9 mice, *p = 0.0039 by Student’s t-test.
We next tested whether hippocampus-dependent memory formation might be improved in aged mice by knocking down NEAT1. To this end, we knocked down NEAT1 in the hippocampal area CA1 of 18- to 19-month-old mice (fig. S4), an age at which we have previously observed significant upregulation of H3K9me2 in the aging rat hippocampus (33), by directly infusing Neat1-targeting siRNAs or non-targeted siRNAs. We then assayed longterm memory function using contextual fear conditioning, with three pairings of the shock to the novel context (Fig. 6C). We observed that knockdown of NEAT1 in the dorsal hippocampus of aged mice resulted in significant improvements in freezing after 24 hours but not during training (Fig. 6, D and E), similar to the results we had observed in young mice (described above, Fig. 5C).
We next sought to test the sufficiency of NEAT1 overexpression to impair performance in memory tasks. To this end, we designed a single guide RNA (sgRNA) targeting Neat1 for overexpression from the endogenous locus (Fig. 7, A and B; fig. S3, B and C), and delivered a Neat1-targeting CRISPRa system in vivo into dorsal CA1 through in vivo transfection (Fig. 7A). Mice were then trained in contextual fear conditioning with three pairings of the shock to the novel context (Fig. 7C). Animals overexpressing NEAT1 from the endogenous locus (NEAT1-OE) had no significant differences in freezing during the training period, either before or after exposure to the unconditioned stimulus (Fig. 7C); however, when returned to the training context 24 hours after training, NEAT1-OE animals froze significantly less than control animals which received only the sgRNA plasmid (Fig. 7D), suggesting that increased NEAT1 abundance in area CA1 is sufficient to impair hippocampus-dependent memory formation.
Figure 7. Mimicking age-related Neat1 overexpression using CRISPRa impairs hippocampal memory formation.
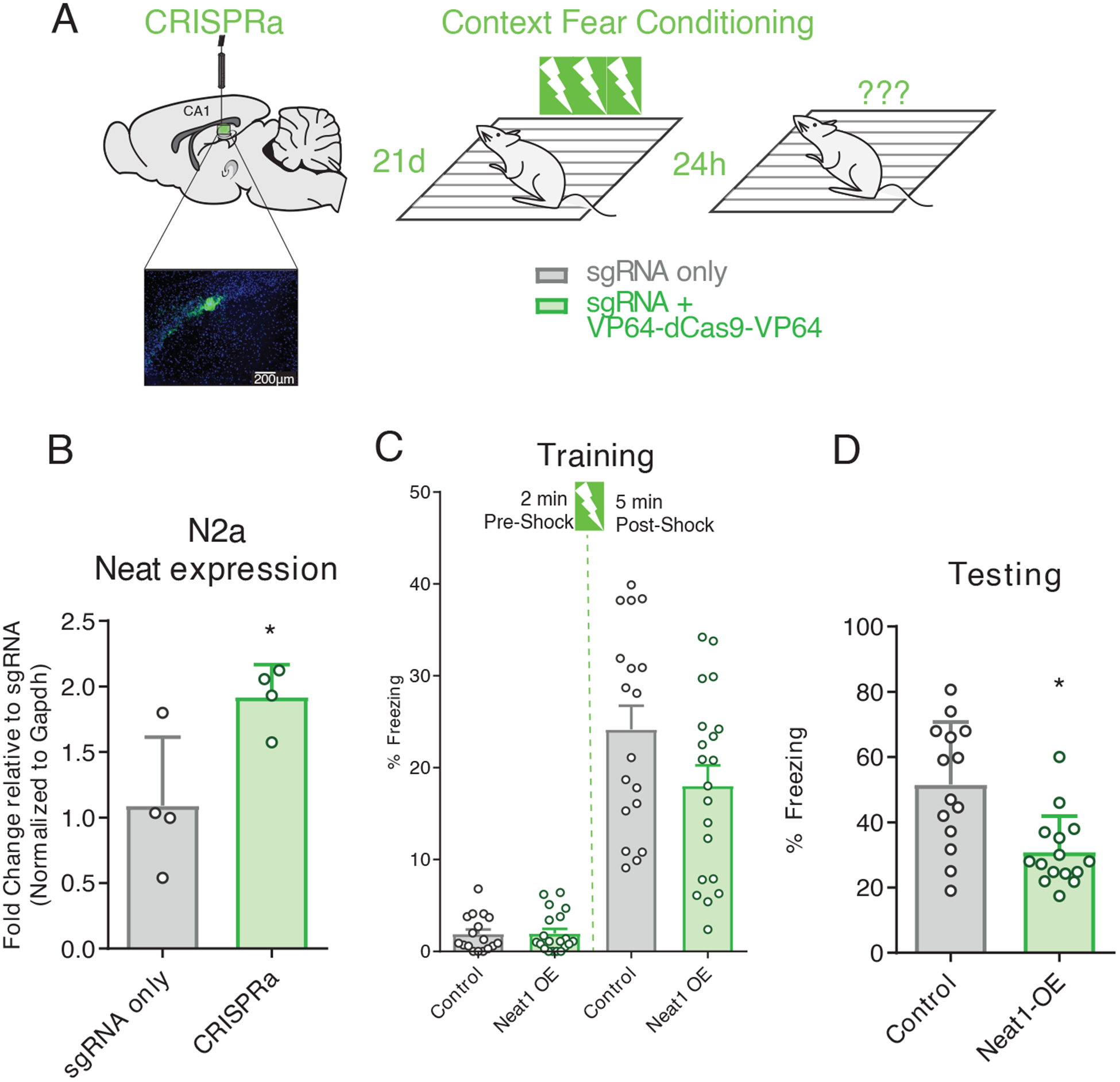
(A) Graphic depiction CRISPRa system infusion into hippocampal area CA1, with visualization of hippocampal expression of EGFP fluorescent marker, and three shock-pairing contextual fear conditioning paradigm. Briefly, male C57BL/6 mice (3- to 7-month-old) were trained 21 days after bilateral infusion of either sgRNA plasmid alone or co-delivered with a transcription-activating dCas9-effector protein and tested 24 hours after training. (B) Confirmation of efficacy of CRISPRa system to upregulate Neat1 expression in murine N2a cells (Student’s t-test: n = 4, p = 0.0283). (C) Freezing behavior of mice described in (A) as a percent of time during the training phase of the contextual fear conditioning paradigm. No significant difference detected for either the pre- or post-shock epochs; data are means ± SEM from n = 18 mice, assessed by Student’s t-test. (D)) Freezing behavior of mice described in (A) as a percent of time during the testing phase of the contextual fear conditioning paradigm. Data are means ± SEM from n = 18 mice, *p = 0.450 assessed by Student’s t-test.
DISCUSSION
Previous studies have observed regulatory roles for the lncRNA NEAT1, including that NEAT1 localizes to chromatin and governs chromatin modification (8, 9). Here, we aimed to resolve this regulatory role of NEAT1 in the context of long-term memory formation.Our data revealed that murine NEAT1 acts as a potent regulator of H3K9me2 both in cultured cells and in vivo. Due to a previously reported observation that NEAT1 interacts directly with EHMT2 (6), which we reproduced here, we cannot yet ascertain whether transcriptional control of EHMT1 or direct interaction with the repressor complex is the rate-limiting factor for H3K9me2 abundance. This intricate multipoint interaction is perhaps illustrative of the intricate systems of regulatory feedback which are thought to control epigenetic mechanisms. Nonetheless, knockdown of NEAT1 was sufficient to perturb this system and to result in both bulk and site-specific changes in H3K9me2, an epigenetic mark that has been observed to play a crucial role in the in neurons.
Previous studies investigating NEAT1 in the context of epilepsy have reported that, in the excitotoxic conditions of this neurodegenerative disorder, activity-dependent downregulation of NEAT1 expression is impaired (19). Thus, we hypothesized that similar mechanisms might play a role in brain regions which become hyperexcitable during aging. Although our results indicate that NEAT1 upregulation impairs memory performance, we wonder whether increased abundance of NEAT1 might be involved in neuroprotective feedback in the context of neurodgeneration or excitotoxicity. However, observations from studies on other neurodegenerative diseases would suggest that NEAT1 upregulation is deleterious to neuronal survival (17). Our observations here also suggest that NEAT1 upregulation might be sufficient to explain some memory impairments observed in aging, as well as rodent models of temporal lobe epilepsy and other neurodegenerative diseases.
Investigations as to the epigenetic regulatory role of NEAT1 have resulted in paradoxical observations to the effect that NEAT1 binds to genomic loci and mediates transcription activation (9), but that suppression of Neat1 expression results primarily in increased neuronal gene expression (our results, above). We showed here that knockdown of NEAT1 nduced widespread downregulation of neuronal H3K9me2, potentially explaining observed increases in gene expression, and further explaining age-related increases in H3K9me2 previously observed in the hippocampus. Moreover, we observed that in mice Neat1 expression was correlated with H3K9me2 globally as well as at the promoter of the aging-repressed memory-related gene c-Fos. While NEAT1 has been observed to act on numerous epigenetic mechanisms (6, 8, 9, 34, 35), this finding suggests that NEAT1-mediated epigenetic mechanisms may be sufficient to govern cognitive function.
Studies of the neuronal impact of NEAT1 expression have thus far been limited to the context of neurological disorders, and in many cases to cultured neuronal cells. Our observations suggest that NEAT1 plays a regulatory role in neuronal H3K9me2 both in cultured neurons and in vivo, and that increased NEAT1 abundance might play a notable role in the age-related decline of hippocampus-dependent memory formation. In humans, expression of NEAT1 is generally limited to a low amount in the CNS, and overexpression is a common hallmark of several neurological disorders. While experimental reduction of NEAT1 has been shown to alleviate MPTP-induced neurogeneration in human neuronal cell lines and may have therapeutic potential in mouse models of Parkinson’s disease, the impact of age-related changes in expression has remained unexplored until now. Although our experiments were designed to investigate the age-related impact of Neat1, we note that other studies have described neuroinflammation-mediated increases in Neat1 expression (36); hence, the findings described in this manuscript implicate NEAT1 as a potential mechanism by which neuroinflammation might impact memory.
Although the experiments described here may explain prior observations of increased H3K9me2 in the aging hippocampus (33), our experiments indicate that increased expression of NEAT1 is not sufficient to explain all of the aging-related neuroepigenetic changes observed in this region. It is likely that many hippocampal lncRNAs have distinct or overlapping roles in the regulation of the neuroepigenetic aging process. Indeed, human NEAT1 itself has been observed to associate with multiple chromatin modifying enzymes (6, 26, 37, 38). Although we did not detect significant regulation of histone modifications other than H3K9me2 at the global level after knockdown of NEAT1, the absence of such observations does not preclude the existence of biologically or behaviorally meaningful epigenetic regulation of marks not explored in this manuscript, or site-specific epigenetic changes which might be uncovered in future studies with a large-scale sequencing approach.
In summary, our findings demonstrate that the lncRNA NEAT1 regulates a critical transcriptional pathway for hippocampus-dependent memory in rodent neurons in vitro, in vivo, and likewise in iPSC-derived human neurons and suggest that NEAT1 may serve as an endogenous molecular brake on the formation of hippocampus-dependent spatial memories.
MATERIALS AND METHODS
Animal housing
Naïve 3–7 month-old or 18 month-old C57BL/6 mice were group housed (2–7 animals/cage) in plastic cages with ad libitum access to food and water and were maintained on a 12-h light/dark cycle. All behavioral tests were conducted during the light cycle, and all procedures were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee and done in accordance with the National Institute of Health ethical guidelines.
Cell culture
N2A cells were maintained in DMEM supplemented with 10% FBS. After thawing, the cells were passaged a minimum of two times prior to use in experiments. The cells were kept at 37℃ in a 5% CO2 incubator. Dissociated cultures of hippocampal pyramidal cells were obtained from embryonic day 18 rat embryos as described previously (39). Briefly, timed-pregnancy female Sprague-Dawley rats were terminally anesthetized and embryos were removed from the uterus, then transferred to Hank’s balanced salt solution (HBSS, Gibco) for dissection. Primary rat hippocampal neurons were dissociated using incubation with papain for 20 min at 37°C, rinsed in HBSS, then resuspended in Neurobasal medium (Gibco) and further mechanically dissociated by passing through a series of progressively smaller fire-polished glass Pasteur pipettes. The resulting suspension was passed through a 70-μm cell strainer and plated on poly-L-lysine coated 24-well plates (~7.5×104 cells per well). Cells were maintained for 2 weeks in Neurobasal supplemented with B-27 and Glutamax (Thermo Fisher Scientific) at 37°C and 5% CO2. For KCl stimulation, 6.25μL 1M KCl (Sigma) was added to two-week in vitro cultures, for a final concentration of 12.5mM KCl.
siRNA delivery
Young, 3–7 month-old mice were anesthetized with an intraperitoneal injection of ketamine-dexmedetomidine and received bilateral intra-CA1 injections of Lincode SMARTpool siRNAs (Dharmacon) targeting the murine Neat1 (#R-160022-00-0005) or a negative control (#D-001320-10-05), conjugated with in vivo JetPEI (PolyPlus Transfection), an in vivo transfection reagent, at the stereotaxic coordinates (AP −2.0mm, ML ±1.5 mm, DV−1.7 mm) with respect to bregma. Aliquots of siRNA stocks (100μM) were diluted to a concentration of ~2.5μM and conjugated with in vivo JetPEI on the day of surgery. Infusions were given over a 10 min period (0.1μL per min) for a total volume of 1μL per hemisphere. After a 48-hour recovery period, mice were handled daily for >3min and trained in contextual fear conditioning at five days post-surgery. Aged (18–19 month old) mice were treated similarly but were anesthetized with vaporized isoflurane (3% induction, 2% maintenance). Mice were sacrificed at ten days post-surgery and dorsal area CA1 was harvested from each hemisphere.
CRISPRa delivery
Mice were anesthetized with an intraperitoneal injection of ketamine-dexmedetomidine and received bilateral intra-CA1 injections of a guide RNA expression vector driven by the murine U6 promoter and targeting the murine Neat1 promoter region (Addgene #44248) either alone or in conjunction with an expression vector coding for the S. pyogenes dCas9 fused to two copies of the VP64 transactivator domain (Addgene #59791,). Endotoxin-free plasmids were purified using an endotoxin-free plasmid DNA purification kit (Machery-Nagel) and aliquoted to minimize freeze-thaw cycles. Endotoxin-free plasmid stocks were diluted to a final concentration of ~500ng/uL in sterile 10% glucose and incubated with in vivo JetPEI for 15 min at room temperature on the day of surgery. The resulting transfection complex was delivered by direct infusion at the stereotaxic coordinates (AP −2.0mm, ML ±1.5 mm, DV−1.4 mm) with respect to bregma. Infusions were given over a 10 min period (0.1μL per min) for a total volume of 1μL (~500ng plasmid DNA) per hemisphere.
Contextual fear conditioning
Mice were trained to either a weak or strong contextual fear conditioning (CFC) paradigm in a novel context, and long term memory was assessed upon returning the animals to the training context 24h after training. The weak CFC paradigm consisted of a 118s baseline followed by a single shock (0.5mA, 2 sec) pairing in the novel context, while the strong CFC paradigm consisted of a 119-sec baseline followed by three shock pairings (0.5mA, 1s) with interleaved rest periods of 59 sec each. Twenty-four hours after training, animals were placed back into the training context for five min to test retention. Freezing behavior was scored by Med Associates software.
Collection of whole area CA1
One hour after training, the whole brain was removed by gross dissection and placed in oxygenated (95%/5% O2/CO2) ice-cold cutting solution (110mM sucrose, 60mM NaCl, 3mM KCl, 1.25mM NaH2PO4, 28mM NaHCO3, 0.5mM CaCl2, 7mM MgCl2, 5mM glucose, and 0.6mM ascorbate). The CA1 region of the hippocampus was then microdissected from each hemisphere and flash frozen on dry ice.
Collection of dorsal area CA1
Animals were sacrificed by cervical dislocation after overdosing with isoflurane at experiment-specific time points, and the whole brain was rapidly removed and immediately frozen on dry ice. The CA1 region of the dorsal hippocampus was then dissected out with the aid of a mouse brain matrix (Harvard Apparatus) to collect the area of CA1 targeted by siRNA or CRISPRa infusions. All tissue was stored at −80°C prior to processing.
Western blotting
Normalized proteins (2–10μg) were separated by electrophoresis on either 10% or 20% polyacrylamide gels, transferred onto an Immobilon-FL membrane using a turbo transfer system (Biorad). Membranes were blocked in Licor blocking buffer and probed with the following primary antibodies for histone H3 (1:1000; Abcam #ab1791), H3K9me2 (1:1000; Millipore #07–441), H3K27me3 (1:1000; Millipore #07–449), H3K4me3 (1:1000; Millipore #04–745). Secondary goat anti-rabbit 700CW antibody (1:20,000; Licor Biosciences) was used for detection of proteins using the Licor Odyssey system. All Western blot quantification was done using ImageStudio Lite software (Licor).
Reverse transcription qPCR (RT-qPCR)
RNA was extracted from isolated CA1 or cultured cells using Trizol reagent according to the manufacturer’s recommended protocol (Fisher). RNA yield was quantified spectrophotometrically (Nanodrop 2000c), and ~200ng of RNA was DNAse treated (Amplification grade DNAse I, Sigma), converted to cDNA (iScript cDNA synthesis kit; Biorad), and PCR amplified on the CFX1000 real-time PCR system (BioRad), with primer annealing temperatures of 60°C. Full descriptions of primers used are in the supplementary materials (Data File S4). All data were analyzed using the delta delta Ct method (40).
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously (33, 41). Briefly, samples were fixed in PBS with 1% formaldehyde for ten minutes at room temperature, chromatin was sheared using a Bioruptor XL on high power, lysates cleared by centrifugation and diluted in TE and RIPA buffer. Extracts were mixed with MagnaChIP protein A/G beads and immunoprecipitations were carried out at 4°C overnight with 5μg primary antibody (anti-H3, Abcam #ab40542; anti-Ezh2, #ab3748) or no antibody (control). Immune complexes were sequentially washed with low salt buffer (20 mM Tris, pH 8.0, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl), high salt buffer (20 mM Tris, pH 8.1, 0.1% SDS, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA), LiCl immune complex buffer (0.25 M LiCl, 10 mM Tris, pH 8.1, 1% deoxycholic acid, 1% IGEPAL-CA630, 500 mM NaCl, 2 mM EDTA), and TE buffer, and eluted into 1xTE containing 1% SDS. Protein-DNA crosslinks were reversed by heating at 65°C overnight. After proteinase K digestion (100μg; 2h at 37°C), DNA was purified by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation. Immunoprecipitated DNA was quantified by spectrophotometry (Nanodrop 2000c) and ~15ng of DNA from each sample was assayed using quantitative real-time PCR using primers specific to mouse genes of interest. Full descriptions of primers used are in the supplementary material (Data File S4).
RNA binding protein immunoprecipitation (RIP)
RIP was performed as described previously(42). Briefly, ~5ug of primary antibody against Ehmt2 (Abcam #ab40542), Ezh2 (Abcam #ab3748) or normal rabbit IgG (Cell signaling) were conjugated with 25uL MagnaChIP protein A/G beads (EMD Millipore). Freshly harvested nuclear pellets from at least 10^6 N2a cells were sheared by Dounce homogenization (15–20 strokes) in RIP buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP40, 1x Protease Inhibitor Cocktail (Sigma), 100 U/ml SUPERASin (Ambion), cleared using centrifugation at 13,000 RPM to remove nuclear membrane and debris, and split into fractions for IP. Sheared nuclear extracts were mixed with antibody conjugated MagnaChIP protein A/G beads and immunoprecipitations were carried out at 4°C for four hours. Beads were then immobilized on a magnetic tube rack, and immune complexes were sequentially washed three times with RIP buffer. Beads were then resuspended in 1mL of Trizol (Thermo Fisher), and coprecipitated RNAs were isolated according to the manufacturer’s recommended protocol. RT-qPCR for Neat1 was then performed as described above.
Statistical analyses
Data from all experiments were analyzed using Analysis of Variance (ANOVA) with Fisher LSD post hoc test or with Student’s t-test unless otherwise noted in the figure legend. Values reported in the text and error bars are the mean ± SEM unless otherwise noted. All datasets were screened for outliers prior to analysis using Grubb’s test (α=0.05) and outliers were subsequently excluded. Statistical tests were performed in R or Prism 7 (GraphPad). Nonparametric tests were used where appropriate and tests were 2-tailed unless otherwise noted. For all experiments, n indicates the number of biological replicates. For cell culture experiments, this indicates the number of independently growing flasks or wells. For experiments involving animal behavior, this indicates the number of animals used. For experiments involving tissue collection from animals, this indicates the number of animals we collected the tissue from.
Human tissue expression data and analysis
Data from the GTEx Analysis Release V7 (dbGaP Accession phs000424.v7.p2) were obtained using the GTEx portal web tool. Expression values plotted are in transcripts per million (TPM), using the GENCODE-annotated transcript for isoforms or a gene-level model based on the GENCODE model with isoforms collapsed to single genes. Isoform expression values were hierarchically clustered using Euclidean distance and average linkage; dendrogram scale shows cluster distance. Body plot was generated in R from median TPM using the gganatogram package (43).
Analysis of bulk RNA-seq and ChIP-seq data
Single or paired-end RNA-seq data was imported into the public Galaxy server at usegalaxy.org directly from the European Nucleotide Archive (study accession numbers PRJEB9006 and PRJNA262674) in FASTQ format and run through a standardized workflow consisting of quality trimming using Trim Galore! (44) (Galaxy Version 0.4.2), read alignment using HISAT (45) (Galaxy Version 2.0.3), and feature counting using featureCounts (Galaxy Version 1.4.6.p5). Individual count files were grouped by treatment and differential expression testing was performed using DESeq2 (46) (Galaxy Version 2.11.39). All reference genomes and annotations were obtained from Gencode releases current at the time of analysis, including the Genome Reference Consortium Mouse Build 38 patch release 5 (GRCm38.p5) and evidence-based annotation of the mouse genome (GRCm38), version M16 (Ensembl 91), human build GRCh38 and the human annotation Release 25 (GRCh38.p7). Gene ontology (GO) enrichment was assessed using a PANTHER Overrepresentation Test web tool provided by the Gene Ontology Consortium (47, 48) (release date 2017-11-28). DAVID functional annotation was used to assess gene set enrichment for GAD_DISEASE_CLASS using default settings (DAVID 6.8).
CHART-seq data was accessed using the NIH SRA Toolkit from accession PRJNA252626 and analyzed using similar read quality control and alignment tools as described above. CHART-seq peaks were called using the MACS2 algorithm (49, 50). Overlapping peaks were combined into a single peak, as recommended for input into ChIP-Enrich package. Using the ChIP-Enrich R package (28) (version 2.4.0), CHART-seq peaks from MACS2 were assigned to the nearest transcription start site and GO Enrichment was assessed for Biological Processes and Molecular Functions.
scRNA-seq analysis
Data were obtained from the European Bioinformatics Institute’s Single-cell Expression Atlas. T-distributed Stochastic Neighbor Embedding (t-SNE) plots were constructed using transcript per million (TPM) values from the transcriptomes of 3,589 single cells biopsied from four glioblastoma patients (24). Unbiased clusters were generated using a t-SNE perplexity of 10; plots were colored according to biased inferred cell type, as reported by the authors of the dataset. Biopsied tissue included cells from the tumor core as well as peripheral tissue; however, all cells inferred to be neurons were collected from noncancerous tissue adjacent to the glioblastoma.
Supplementary Material
Fig. S1. NEAT1 expression is uniquely reduced in the human CNS, and baseline expression is low in human neurons relative to other cell types
Fig. S2. Neuronal regulation of immediate early genes after NEAT1 knockdown
Fig. S3. Validation of NEAT1 abundance manipulation using RNAi and CRISPRa
Fig. S4. Age-related increase of H3K9me2 in dCA1.
Fig. S5. Quality control plots from Chip-Enrich.
Data File S1. GTex tissue data.
Data File S2. iPSC differentially expressed genes.
Data File S3. ChIP-Enrich GO results.
Data File S4. Primers and oligos.
Acknowledgments:
We would like to thank Peng Li at the UAB for providing statistical review. We would also like to thank Andrew Arrant, Anh Tran, and Anita Hjelmeland at UAB for logistic and troubleshooting support.
Funding: This work was supported in part by National Institute of Health (NIH) grants MH097909 to F.D.L., the Evelyn F. McKnight Brain Institute at UAB, and the UAB Neuroscience Behavior Assessment Core P30 NS47466, and A.A.B. was supported by an NIH Institutional Training Grant (NS061788).
Footnotes
Data and materials availability: pLV hUbC-VP64 dCas9 VP64-T2A-GFP was a gift from Charles Gersbach (Addgene plasmid # 59791; http://n2t.net/addgene:59791; RRID:Addgene_59791). pgRNA-humanized was a gift from Stanley Qi (Addgene plasmid # 44248; http://n2t.net/addgene:44248; RRID:Addgene 44248). RNA-seq data are available at BioProject study accession numbers PRJEB9006 for iPSC data and PRJNA262674 for aging hippocampus RNA-seq. CHART-seq data are available at study accession number PRJNA252626. All data needed to evaluate the conclusions in the paper are present in the paper or the supplementary materials.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, and Chess A. 2007. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, and Lawrence JB. 2009. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell 33: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, and Hirose T. 2018. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol Cell 70: 1038–1053.e7. [DOI] [PubMed] [Google Scholar]
- 4.Li R, Harvey AR, Hodgetts SI, and Fox AH. 2017. Functional dissection of NEAT1 using genome editing reveals substantial localization of the NEAT1_1 isoform outside paraspeckles. RNA 23: 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, Kato A, Kawaguchi Y, Sato H, Yoneda M, Kai C, Yada T, Suzuki Y, Yamada T, Ozawa T, Kaneki K, Inoue T, Kobayashi M, Kodama T, Wada Y, Sekimizu K, and Akimitsu N. 2014. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell 53: 393–406. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, and Cheng C. 2018. Long noncoding RNA NEAT1 promotes the metastasis of osteosarcoma via interaction with the G9a-DNMT1-Snail complex. Am J Cancer Res 8: 81–90. [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang J, Zhou J, Kang C, Li M, and Jiang C. 2018. Long Noncoding RNA NEAT1, Regulated by the EGFR Pathway, Contributes to Glioblastoma Progression Through the WNT/β-Catenin Pathway by Scaffolding EZH2. Clin Cancer Res 24: 684–695. [DOI] [PubMed] [Google Scholar]
- 8.West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, Tolstorukov MY, and Kingston RE. 2014. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell 55: 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakravarty D, Sboner A, Nair SS, Giannopoulou E, Li R, Hennig S, Mosquera JM, Pauwels J, Park K, Kossai M, MacDonald TY, Fontugne J, Erho N, Vergara IA, Ghadessi M, Davicioni E, Jenkins RB, Palanisamy N, Chen Z, Nakagawa S, Hirose T, Bander NH, Beltran H, Fox AH, Elemento O, and Rubin MA. 2014. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun 5: 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X, Li Z, Zheng H, Chan MTV, and Wu WKK. 2017. NEAT1: A novel cancer-related long non-coding RNA. Cell Prolif 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stilling RM, Benito E, Gertig M, Barth J, Capece V, Burkhardt S, Bonn S, and Fischer A. 2014. De-regulation of gene expression and alternative splicing affects distinct cellular pathways in the aging hippocampus. Front Cell Neurosci 8: 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pereira Fernandes D, Bitar M, Jacobs FMJ, and Barry G. 2018. Long Non-Coding RNAs in Neuronal Aging. Non-coding RNA 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barry G, Guennewig B, Fung S, Kaczorowski D, and Weickert CS. 2015. Long Non-Coding RNA Expression during Aging in the Human Subependymal Zone. Front Neurol 6: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Zhu L, Guan F, Yan Z, Liu D, Han W, and Chen T. 2018. Relationship between schizophrenia and changes in the expression of the long non-coding RNAs Meg3, Miat, Neat1 and Neat2. J Psychiatr Res 106: 22–30. [DOI] [PubMed] [Google Scholar]
- 15.Sunwoo J-S, Lee S-T, Im W, Lee M, Byun J-I, Jung K-H, Park K-I, Jung K-Y, Lee SK, Chu K, and Kim M. 2017. Altered expression of the long noncoding RNA NEAT1 in huntington’s disease. Mol Neurobiol 54: 1577–1586. [DOI] [PubMed] [Google Scholar]
- 16.Yan W, Chen Z-Y, Chen J-Q, and Chen H-M. 2018. LncRNA NEAT1 promotes autophagy in MPTP-induced Parkinson’s disease through stabilizing PINK1 protein. Biochem Biophys Res Commun 496: 1019–1024. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, and Lu Z. 2018. Long non-coding RNA NEAT1 mediates the toxic of Parkinson’s disease induced by MPTP/MPP+ via regulation of gene expression. Clin Exp Pharmacol Physiol 45: 841–848. [DOI] [PubMed] [Google Scholar]
- 18.Puthiyedth N, Riveros C, Berretta R, and Moscato P. 2016. Identification of Differentially Expressed Genes through Integrated Study of Alzheimer’s Disease Affected Brain Regions. PLoS ONE 11: e0152342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barry G, Briggs JA, Hwang DW, Nayler SP, Fortuna PRJ, Jonkhout N, Dachet F, Maag JLV, Mestdagh P, Singh EM, Avesson L, Kaczorowski DC, Ozturk E, Jones NC, Vetter I, Arriola-Martinez L, Hu J, Franco GR, Warn VM, Gong A, Dinger ME, Rigo F, Lipovich L, Morris MJ, O’Brien TJ, Lee DS, Loeb JA, Blackshaw S, Mattick JS, and Wolvetang EJ. 2017. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci Rep 7: 40127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipovich L, Dachet F, Cai J, Bagla S, Balan K, Jia H, and Loeb JA. 2012. Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics 192: 1133–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, Guan P, Korzeniewski GE, Lockhart NC, Rabiner CA, Rao AK, Robinson KL, Roche NV, Sawyer SJ, Segrè AV, Shive CE, Smith AM, Sobin LH, Undale AH, Valentino KM, Vaught J, Young TR, Moore HM, and GTEx Consortium. 2015. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and biobanking 13: 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Consortium GTEx. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, Li G, Chang SD, Connolly ID, Li Y, Barres BA, Gephart MH, and Quake SR. 2017. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep 21: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang DW, Sherman BT, and Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 26.Murthy UMS, and Rangarajan PN. 2010. Identification of protein interaction regions of VINC/NEAT1/Men epsilon RNA. FEBS Lett 584: 1531–1535. [DOI] [PubMed] [Google Scholar]
- 27.Fleischmann A, Hvalby O, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Reschke M, Spanagel R, Sprengel R, Wagner EF, and Gass P. 2003. Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J Neurosci 23: 9116–9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welch RP, Lee C, Imbriano PM, Patil S, Weymouth TE, Smith RA, Scott LJ, and Sartor MA. 2014. ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res 42: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL, and Lubin FD. 2012. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J Neurosci 32: 5440–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma M, Razali NB, and Sajikumar S. 2017. Inhibition of G9a/GLP Complex Promotes Long-Term Potentiation and Synaptic Tagging/Capture in Hippocampal CA1 Pyramidal Neurons. Cereb Cortex 27: 3161–3171. [DOI] [PubMed] [Google Scholar]
- 31.Tachibana M, Matsumura Y, Fukuda M, Kimura H, and Shinkai Y. 2008. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 27: 2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akbarian S 2010. Epigenetics of schizophrenia. Current topics in behavioral neurosciences 4: 611–628. [DOI] [PubMed] [Google Scholar]
- 33.Morse SJ, Butler AA, Davis RL, Soller IJ, and Lubin FD. 2015. Environmental enrichment reverses histone methylation changes in the aged hippocampus and restores age-related memory deficits. Biology 4: 298–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian K, Liu G, Tang Z, Hu Y, Fang Y, Chen Z, and Xu X. 2017. The long non-coding RNA NEAT1 interacted with miR-101 modulates breast cancer growth by targeting EZH2. Arch Biochem Biophys 615: 1–9. [DOI] [PubMed] [Google Scholar]
- 35.Kawaguchi T, Tanigawa A, Naganuma T, Ohkawa Y, Souquere S, Pierron G, and Hirose T. 2015. SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc Natl Acad Sci U S A 112: 4304–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, Li X, Chen X, Li S, Ho IHT, Liu X, Chan MTV, and Wu WKK. 2018. Emerging roles of long non-coding RNAs in neuropathic pain. Cell Prolif e12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirose T, Virnicchi G, Tanigawa A, Naganuma T, Li R, Kimura H, Yokoi T, Nakagawa S, Bénard M, Fox AH, and Pierron G. 2014. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol Biol Cell 25: 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spiniello M, Knoener RA, Steinbrink MI, Yang B, Cesnik AJ, Buxton KE, Scalf M, Jarrard DF, and Smith LM. 2018. HyPR-MS for multiplexed discovery of MALAT1, NEAT1, and NORAD lncRNA protein interactomes. J Proteome Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meadows JP, Guzman-Karlsson MC, Phillips S, Holleman C, Posey JL, Day JJ, Hablitz JJ, and Sweatt JD. 2015. DNA methylation regulates neuronal glutamatergic synaptic scaling. Sci Signal 8: ra61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livak KJ, and Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 41.Jarome TJ, Butler AA, Nichols JN, Pacheco NL, and Lubin FD. 2015. NF-κB mediates Gadd45β expression and DNA demethylation in the hippocampus during fear memory formation. Front Mol Neurosci 8: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, and Chang HY. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, Füllgrabe A, Fuentes AM-P, Jupp S, Koskinen S, Mannion O, Huerta L, Megy K, Snow C, Williams E, Barzine M, Hastings E, Weisser H, Wright J, Jaiswal P, Huber W, Choudhary J, Parkinson HE, and Brazma A. 2016. Expression Atlas update--an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res 44: D746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krueger F 2015. Trim Galore!: A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files.
- 45.Kim D, Langmead B, and Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and’ ‘ dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, and Sherlock G. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.The Gene Ontology Consortium. 2017. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45: D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chalei V, Sansom SN, Kong L, Lee S, Montiel JF, Vance KW, and Ponting CP. 2014. The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. elife 3: e04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng J, Liu T, Qin B, Zhang Y, and Liu XS. 2012. Identifying ChIP-seq enrichment using MACS. Nat Protoc 7: 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. NEAT1 expression is uniquely reduced in the human CNS, and baseline expression is low in human neurons relative to other cell types
Fig. S2. Neuronal regulation of immediate early genes after NEAT1 knockdown
Fig. S3. Validation of NEAT1 abundance manipulation using RNAi and CRISPRa
Fig. S4. Age-related increase of H3K9me2 in dCA1.
Fig. S5. Quality control plots from Chip-Enrich.
Data File S1. GTex tissue data.
Data File S2. iPSC differentially expressed genes.
Data File S3. ChIP-Enrich GO results.
Data File S4. Primers and oligos.